

Abstract
Gram-negative bacteria including Escherichia coli, Salmonella typhimurium, and Pseudomonas aeruginosa can modify the structure of lipid A in their outer membrane with 4-amino-4-deoxy-L-arabinose (Ara4N). Such modification results in resistance to cationic antimicrobial peptides of the innate immune system and antibiotics such as polymyxin. ArnA is a key enzyme in the lipid A modification pathway, and its deletion abolishes both the Ara4N-lipid A modification and polymyxin resistance. ArnA is a bifunctional enzyme. It can catalyze (i) the NAD+-dependent decarboxylation of UDP-glucuronic acid to UDP-4-keto-arabinose and (ii) the N-10-formyltetrahydrofolate-dependent formylation of UDP-4-amino-4-deoxy-L-arabinose. We show that the NAD+-dependent decarboxylating activity is contained in the 360 amino acid C-terminal domain of ArnA. This domain is separable from the N-terminal fragment, and its activity is identical to that of the full-length enzyme. The crystal structure of the ArnA decarboxylase domain from E. coli is presented here. The structure confirms that the enzyme belongs to the short-chain dehydrogenase/reductase (SDR) family. On the basis of sequence and structure comparisons of the ArnA decarboxylase domain with other members of the short-chain dehydrogenase/reductase (SDR) family, we propose a binding model for NAD+ and UDP-glucuronic acid and the involvement of residues T432, Y463, K467,R619, and S433 in the mechanism of NAD+-dependent oxidation of the 4″-OH of the UDP-glucuronic acid and decarboxylation of the UDP-4-keto-glucuronic acid intermediate.
In the process of establishing infections, bacteria must overcome the host defense mechanism including the bactericidal action of cationic antimicrobial peptides (CAMPs).1 These are small, amphipathic, positively charged peptides that destroy bacteria through membrane permeabilization and constitute a phylogenetically conserved branch of the innate immune system (1-4). In the case of Gram-negative bacteria, CAMPs bind to the bacterial cell surface through electrostatic interactions with the negatively charged groups of the lipopolysaccharide (LPS), the immunogenic glycolipid in the outer membrane in Gram-negative bacteria (5, 6). They then traverse to the inner membrane and form a pore, which leads to membrane permeabilization and cell death (7-9). In addition to their function as a key member of the innate immune system, CAMPs represent an important class of clinical antimicrobials. They have both intrinsic bactericidal activity and appear to enhance the activity of other antibiotics, presumably by facilitating their entry into the microbe (3, 10, 11).
Most Gram-negative bacteria, including Salmonella typhimurium, Escherichia coli, and the main cystic fibrosis (CF) pathogen Pseudomonas aeruginosa, have evolved mechanisms to resist the bactericidal action of CAMPs (5, 6, 12). These pathogens can modify the structure of lipid A, the anionic, conserved component of LPS in the bacterial outer membrane. The modifications include lipid A acylation and addition of the positively charged sugar 4-amino-arabinose (Ara4N) to lipid A (5). Addition of Ara4N to lipid A results in a less negatively charged cell surface, which reduces the electrostatic interactions and, therefore, binding of CAMPs to the bacterial cell surface. It has been clearly shown that the addition of Ara4N to lipid A is responsible for the resistance to polymyxin (an acylated cyclic CAMP) and other CAMPs, such as azurocidin and the bactericidal/permeability-increasing protein (13-15). Importantly, lipid A from P. aeruginosa isolated from CF patients showed modifications with Ara4N (16). These modifications confer resistance to the bactericidal action of CAMPs, thus helping the proliferation of the bacteria in the CF lung (17).
Modification of lipid A with Ara4N occurs through transcriptional activation of the pmrE gene and the seven protein operon pmrHFIJKLM. All of these gene products except pmrM are essential for the biosynthesis of Ara4N-lipid A and for resistance to CAMPs (18). In vitro studies by Raetz and co-workers have shown a pathway for the biosynthesis of UDP-L-Ara4N from UDP-glucose (UDP-Glc) (19-21). The pathway begins with the oxidation of UDP-Glc to UDP-glucuronic acid (UDP-GlcA) catalyzed by the well-characterized UDP-Glc dehydrogenase (PmrE/Ugd) (Figure 1). UDP-GlcA is then oxidatively decarboxylated by ArnA (PmrI) to yield UDP-4-keto-arabinose (UDPAra4O), which in turn is transaminated to produce UDP-4-amino-arabinose (UDP-Ara4N) in a reaction catalyzed by ArnB (PmrH). On the basis of sequence similarity to enzymes with known activities, additional gene products of the pmrHFIJKLM operon have been proposed to catalyze the transfer of Ara4N from the UDP intermediate to lipid A (19, 21-24). The enzymes in this pathway are potential targets for antibacterial drug design. Inhibitors of the pathway would abolish microbial resistance to both CAMPs and cationic peptide antibiotics. Such inhibitors may prove particularly useful in treating chronic infections such as those caused by P. aeruginosa in CF patients.
FIGURE 1.

Proposed pathway for the biosynthesis of UDP-Ara4N. The pathway starts with Ugd/PmrE oxidizing UDP-Glc to UDP-GlcA. UDP-GlcA is then oxidized at position 4 by the C-terminal domain of ArnA to yield the UDP-4-keto glucuronic acid intermediate that is then decarboxylated to UDP-4-keto arabinose by the same enzyme. UDP-Ara4O is transaminated by ArnB yielding the novel sugar-nucleotide UDP-4-amino-4-deoxy-arabinose (UDP-Ara4N). The N-terminal domain of ArnA can formylate UDP-Ara4N and has been proposed to help displace the reaction catalyzed by ArnB toward UDP-Ara4N synthesis and generate a transiently formylated product (21).
ArnA (formerly, PmrI) is encoded by the pmrHFIJKLM operon and is a 74-kDa bifunctional protein. The enzyme can catalyze the transfer of a formyl group from N10-formyltetrahydrofolate to UDP-L-Ara4N (20, 21). The N-terminal domain (residues 1-313) is similar in sequence to other enzymes involved in formyl transfer. However, the relevance of this reaction in the biosynthesis of Ara4N-lipid A is unclear. ArnA is also responsible for the C-4″ oxidation of UDP-GlcA to UDP-4-keto-glucuronic acid and its decarboxylation to yield UDP-4-keto-arabinose (boxed in Figure 1) (20). The C terminus of ArnA (amino acids 314-660) has sequence similarity to other enzymes that oxidize the C-4″ position of UDP sugars, such as UDP-galactose epimerase, dTDP-glucose-4,6-dehydratase, and UDP-glucuronic acid decarboxylase, all members of the short-chain dehydrogenase/reductase (SDR) superfamily (25, 26). These enzymes use NAD+ to oxidize the C-4″ hydroxyl of a sugar-nucleotide and recycle the NADH generated to reduce the 4-keto intermediate back to an alcohol (Figure 2). ArnA on the other hand, utilizes NAD+ as a true substrate, releasing NADH and the UDP-4-keto-sugar as products (20) (Figure 2A).
FIGURE 2.

Schematic representation of the reactions catalyzed by some SDR enzymes. (A) ArnA decarboxylase domain. (B) UDP-Galactose epimerase. (C) UDP-Glucuronic acid decarboxylase. (D) dTDP glucose-4,6-dehydratase.
A clear understanding of the ArnA mechanism is crucial for both design and evaluation of selective inhibitors. Here, we show that the C-terminal fragment of ArnA is wholly responsible for the decarboxylation of UDP-GlcA. We thus designate it as the ArnA decarboxylase domain and describe its high-resolution crystal structure. The sequence and structural comparison with other members of the SDR family highlight unique features in ArnA and suggest putative catalytic residues responsible for the decarboxylation step in the conversion of UDP-GlcA to UDP-Ara4O.
MATERIALS AND METHODS
Purification of Full-Length ArnA
The plasmid (pETArnA) for ArnA overexpression was a generous gift from Prof. C. Raetz (20). pETArnA was transformed into E. coli Nova Blue (DE3) cells (Novagen). A 100 mL overnight culture from a single colony containing 30 μg/mL kanamycin was used to inoculate 6 × 1 L LB medium supplemented with 50 μg/mL kanamycin. Cultures were grown at 37 °C to an OD600 of 0.6 and cooled to room temperature before induction with 1 mM IPTG. Cultures were allowed to grow for an additional 3.5 h at room temperature. Cells were harvested by centrifugation at 6000 rpm for 10 min at 4 °C. The cell pellet was resuspended in 100 mL of lysis buffer containing 100 mM HEPES at pH 7.1, 10% glycerol, 500 mM KCl, 5 mM MgCl2, 1 mM PMSF, and 5 mM 2-mercaptoethanol. Lysis was achieved by sonication on ice. Cell debris was removed by centrifugation at 15 000 rpm for 30 min at 4 °C. The supernatant was then applied to a 10 mL Ni-NTA column (Qiagen) previously equilibrated with the above buffer. The column was washed with 5 column volumes of wash buffer containing 50 mM HEPES at pH 7.1, 200 mM KCl, 10% glycerol, 5 mM 2-mercaptoethanol, and 25 mM imidazol at pH 8.0. Elution of the protein from the column was achieved by increasing the concentration of imidazol in the above buffer to 300 mM. Fractions containing the protein were loaded on a size-exclusion (HiLoad 26/60 Superdex 200, Amersham Pharmacia Biotech) column pre-equilibrated with 25 mM Tris-HCl at pH 8.0, 150 mM KCl, 10% glycerol, 1 mM EDTA at pH 8.0, and 5 mM 2-mercaptoethanol and eluted in the same buffer. Elution was monitored by measuring the absorption at 280 nm. The fractions containing protein were combined, and the protein was concentrated to approximately 14 mg/mL (Bio-Rad Protein Assay, Bio-Rad Laboratories). The protein was stored at −80 °C until needed.
Cloning of the ArnA Decarboxylase Domain
The E. coli ArnA C terminus was amplified by polymerase chain reaction (PCR) from genomic E. coli DNA using the following primers: sense primer, 5′-GTT CAC GCC ATA TGA GCC AGC CTG CCT GCA CCG, containing a NdeI restriction site; and antisense primer, 5′-AAG CCT AGA GCT CTC ATG ATG GTT TAT CCG TAA GAT C, containing a SacI restriction site. The PCR amplification was performed with Pfu Turbo polymerase (Invitrogen) according to the instructions of the manufacturer. The PCR product was purified with the QIquick PCR Purification Kit (Qiagen) followed by digestion with NdeI and SacI overnight at 37 °C. The digested gene was purified on a 1% agarose gel using the QIquick Gel Purification Kit (Qiagen) and the purified gene ligated into the pMS122 vector [an engineered variant of the pET28 vector that generates an N-terminal His-tag fusion that can be efficiently and specifically cleaved with the Tobacco etch virus (TEV) protease]. The resulting plasmid, pMS159, was sequenced to confirm that no mutations had been introduced in the ArnA C-terminal sequence.
Protein Expression and Purification
The plasmid pMS159 was transformed into E. coli Rosetta (DE3) cells (Novagen) and plated on LB media supplemented with 50 μg/mL kanamycin. A total of 100 mL of overnight culture from a single colony containing 50 μg/mL kanamycin was used to inoculate 10 × 1 L LB medium supplemented with 50 μg/ mL kanamycin. Cultures were grown at 37 °C to an OD600 of 0.6 and cooled on ice to approximately 4 °C. Expression was induced with 0.4 mM isopropyl-β-D-thiogalactopyranoside (IPTG, Gold Bio Technology Inc.), and cultures were allowed to grow overnight at room temperature. Cells were harvested by centrifugation at 6000 rpm for 10 min at 4 °C, and the cell pellet was resuspended in lysis buffer containing 25 mM Tris-HCl at pH 8.0, 5 mM 2-mercaptoethanol, and complete EDTA-free protease inhibitor cocktail used at 1 tablet per 100 mL of buffer (Roche). Cells were lysed on ice by sonication. After lysis, KCl was added to a final concentration of 300 mM and cell debris was removed by centrifugation at 16 000 rpm for 30 min at 4 °C. The supernatant was applied to a 7 mL Ni-NTA column (Qiagen) pre-equilibrated with the lysis buffer containing 300 mM KCl. The column was washed with 5 column volumes of the above buffer, followed by 5 column volumes of wash buffer (25 mM HEPES-KOH at pH 7.5, 300 mM KCl, 10% glycerol, 5 mM 2-mercaptoethanol, and 25 mM imidazole at pH 8.0). The protein was eluted using a 25-300 mM imidazole gradient at pH 8.0 (70 mL final volume). Fractions containing the protein were loaded on a size-exclusion (HiLoad 26/60 Superdex 200, Amersham Pharmacia Biotech) column pre-equilibrated with 25 mM Tris-HCl at pH 8.0, 150 mM KCl, 10% glycerol, 1 mM EDTA at pH 8.0, and 5 mM 2-mercaptoethanol and eluted in the same buffer. Elution was monitored by measuring the absorption at 280 nm. Fractions containing the protein were dialyzed against 25 mM Tris-HCl at pH 8.0 and 5 mM 2-mercaptoethanol and loaded on a MonoQ HR5 column (Pharmacia Biotech) equilibrated in the same buffer. The protein was eluted in buffer containing 25 mM Tris-HCl at pH 8.0, 5 mM 2-mercaptoethanol, and 0-1 M NaCl gradient. The fractions containing protein were combined, and the 6× His tag was removed by overnight incubation at 4 °C with TEV protease (1:50 TEV protease/ArnA) and 10 mM dithiothreitol (DTT). The protein was separated from the protease and the cleaved tag by size-exclusion chromatography with the column and buffers specified above. The ArnA C terminus was eluted as a monomer from the column. The fractions containing protein were combined, and the protein was concentrated to approximately 10 mg/mL (Bio-Rad Protein Assay, Bio-Rad Laboratories). This protein stock was used for crystallization experiments.
Protein Crystallization and Data Collection
Crystals of the ArnA C terminus were grown by the hanging drop method of vapor diffusion at 16 °C (protein/precipitant, 1.5: 1.5 μL). The precipitant was 2.0 M (NH4)2SO4, 5 mM DTT, and 100 mM MES at pH 6.75. Crystal growth generally required 4-6 days with crystals having approximate dimensions of 0.3 × 0.3 × 0.3 mm. All of the crystals belonged to the P4132 space group with typical unit-cell dimensions of a = b = c = 150.5 Å, α = β = γ = 90°, and 1 molecule per asymmetric unit. Prior to X-ray data collection, the crystals were transferred to cryo-protecting solutions composed of 2.0 M (NH4)2SO4, 5 mM DTT, 100 mM MES at pH 6.75, and 5-25% glycerol and flash-cooled in a nitrogen stream. Data were collected with a rotating anode generator using Cu Kα radiation and a Rigaku RAXIS IV2+ detector. Data were indexed and integrated with DENZO and scaled with SCALEPACK (27). X-ray data collection statistics are shown in Table 1.
Table 1.
Data Collection and Refinement Statisticsa
| Data Collection Statistics | |
| wavelength (Å) | 1.54 |
| space group | P4132 |
| cell parameters (Å) | a = b = c = 150.5 |
| resolution (Å) | 30.0-2.40 (2.49-2.40) |
| measured reflections | 317 340 (27 399) |
| unique reflections | 23 500 (2302) |
| I/σ | 35.0 (5.2) |
| redundancy | 13.5 (11.9) |
| data completeness (%) | 99.3 (100.0) |
| Rmerge (%) | 7.1 (53.1) |
| Refinement Statistics | |
| Rwork | 20.5 (23.5) |
| Rfree | 24.0 (27.8) |
| rms deviation from ideal values | |
| bond lengths (Å) | 0.0094 |
| bond angles (deg) | 1.4822 |
| dihedrals (deg) | 23.1836 |
| improper dihedrals (deg) | 0.9882 |
| mean B value (Å2) | 47.4 |
| B factor deviation bonds (Å2) | 1.36 |
| B factor deviation angles (Å2) | 2.19 |
| Ramachandran | |
| residues in most favored region (%) | 90.0 |
| residues in allowed regions (%) | 10.0 |
Rwork = Σ|Fobs - Fcalc|/ΣFobs, where Fobs = the observed structure factor amplitude and Fcalc = the structure factor calculated from the model. Rfree is computed in the same manner as Rwork, using the test set of reflections.
Structure Determination and Refinement
The structure of the ArnA C terminus was solved by molecular replacement. The phasing model used was the 2.15 Å refined structure of UDP-galactose 4-epimerase from E. coli (PDB ID: 1KVS) (28). All non-glycine side chains of the model were set to Ala. Rotation/translation searches, performed with the program AMoRe (29) and data between 15 and 5 Å, yielded a solution clearly above the noise level in the space group P4132 but not in the enantiomorphic spacegroup P4332. Inspection of the crystal packing revealed no unfavorable molecular contacts. Using CNS (30), 10% of the data was removed for cross validation, and the model was subjected to a round of simulated annealing with torsion-angle dynamics (31, 32). An electron-density map was calculated with data to 2.6 Å resolution. Several side chains were visible in the map and were incorporated into the model using the program O (33). The map also revealed sections of the model for which density was not clearly visible, and thus the sections were removed. This new model was again subjected to a round of simulated annealing with torsion-angle dynamics, and model phases were improved by solvent flipping as implemented in CNS (solvent content of 63%). A new map calculated with the improved phases showed unambiguous density for most side chains and connectivity for most of the molecule. The amino acid sequence was readily assigned in this map. The model was subjected to a round of simulated annealing with Cartesian dynamics followed by positional and B-factor refinement with data to 2.4 Å. Manual rebuilding was effected with the program O, and the refinement was continued until no further improvement of the Rwork and Rfree was observed (Rwork of 24.6% and Rfree of 27.9%). At this point, electron-density maps showed clear density for several solvent molecules and a sulfate ion and were added to the model. Iterative steps of positional and atomic B-factor refinement followed by manual rebuilding were performed until no further improvement of R factors was achieved. The final model (Rwork of 20.5% and Rfree of 24.0%) has good stereochemistry as determined using PROCHEK (34), with all amino acids laying in the most favorable or allowed regions on the Ramachandran plot. No electron density was observed for residues S605-V616, which are assumed to belong to a conformationally flexible loop. Refinement statistics and model stereochemistry are summarized in Table 1.
Enzyme Assays and Kinetic Studies
The standard reaction mixture contained 25 mM Tris at pH 8.0, 5 mM 2-mercaptoethanol, 0.2 mg/mL BSA, 10% glycerol, 100 mM KCl, 4 mM NAD+, and 1 mM UDP-glucuronic acid. The reaction was started with the addition of the 200 nM ArnA full-length or C-terminal domain. Enzyme activity was measured by following the absorbance of the produced NADH at 340 nm. All enzyme assays were carried out at 37 °C in a final volume of 800 μL. The initial velocity studies were performed by varying the concentration of NAD+ from 0.125 to 4.0 mM and keeping UDP-glucuronic acid constant at 1 mM (Figure 3A) or by changing the concentration of UDP-glucuronic acid from 0.031 to 1.0 mM and keeping NAD+ at 4 mM (Figure 3A).
FIGURE 3.

Decarboxylase activity of full-length ArnA and its C-terminal domain. (A) Plots of initial velocity versus substrate concentration for full-length ArnA (○) and ArnA C-terminal domain (●). The enzyme activity was measured by monitoring the formation of NADH. (B) Detection of the reaction product UDP-Ara4O by thin-layer chromatography. Lanes 1 and 2, Full-length ArnA; lanes 3 and 4, ArnA C-terminal domain. No formation of UDP-Ara4O is observed in the absence of NAD+ in the reaction mixture.
NAD+-Dependent Conversion of UDP-GlcA to UDP-Ara4O
Assays were performed as described before with minor modifications (20). Briefly, a solution contained 25 mM Tris at pH 8.0 and 5 mM 2-mercaptoethanol with the 0.5 mg/mL purified ArnA full-length or C-terminal domain, 11 μM UDP-GlcA (glucuronyl-14C(U), Perkin-Elmer), and 3 mM NAD+ (Sigma) at room temperature for 40 min. The above reaction mixture without NAD+ was used as a control. A total of 0.5 μL of each reaction mixture was then spotted on polyethyleneimine (PEI) cellulose plate prewashed in methanol. The plate was developed in a solvent system containing 0.25 M acetic acid and 0.4 M LiCl. Radioactivity in the plate was visualized with a PhosphorImager.
RESULTS AND DISCUSSION
The C-Terminal Domain of ArnA Is a UDP-GlcA Decarboxylase
As mentioned previously, ArnA is a bifunctional enzyme with both formyl-transferase and UDP-GlcA decarboxylase activities (20). Sequence analysis suggested that the C-terminal fragment of ArnA may represent a separable domain with UDP-GlcA decarboxylase activity. A fragment of E. coli ArnA comprising amino acids 306-660 was cloned and overexpressed as a His-tag fusion and purified to homogeneity as described in the Materials and Methods. The His tag was cleaved with TEV protease resulting in an ArnA C-terminal fragment with a three additional amino acids (HGM) at the N terminus.
The UDP-GlcA acid decarboxylation reaction catalyzed by ArnA can be followed spectrophotometrically by measuring the production of NADH (20). A comparison of the decarboxylase activity of the ArnA full-length enzyme and its C-terminal fragment shows identical behavior for the two proteins (parts A and B of Figure 3). The apparent Km values for NAD+ and UDP-GlcA were 0.76 ± 0.09 and 0.086 ± 0.006 mM for the full-length enzyme and 0.57 ± 0.09 and 0.054 ± 0.003 mM for the C-terminal fragment. The decarboxylated product released by the ArnA full-length enzyme is UDP-4-keto-arabinose (UDP-Ara4O). The conversion of UDP-GlcA into UDP-Ara4O can be detected by thin-layer chromatography, where UDP-Ara4O migrates faster than UDP-GlcA. Using NMR analysis, Breazeale et al. confirmed that the fast-migrating species correspond to UDP-Ara4O (20). As shown in Figure 3B, the C-terminal domain of ArnA also produces UDP-Ara4O as detected by thin-layer chromatography. We therefore conclude that the C-terminal fragment of ArnA is responsible for the oxidative decarboxylation of UDP-GlcA. This C-terminal fragment is a separable, functional domain, and from here onward, we shall refer to it as the ArnA decarboxylase domain.
Crystal Structure of the ArnA Decarboxylase Domain
The decarboxylase domain of ArnA described above was crystallized, and the structure was determined to 2.4 Å resolution as described in the Materials and Methods. Data collection and refinement statistics are shown in Table 1.
The overall structure of ArnA decarboxylase domain is distinctly bilobal. It contains a larger N-terminal subdomain formed by amino acids R315-R510 and R541-G566 folding into a 7-stranded parallel β sheet sandwiched by three α helices on either side (Figure 4). This represents a modified version of the classic Rossmann fold observed in many dinucleotide-binding proteins in that an α helix and a β strand are donated to the Rossmann fold by the C-terminal subdomain (Figure 4). The smaller C-terminal subdomain is formed by residues A511-I540 and N567-T656 and consists of four strands of pleated β sheet and three α helices.
FIGURE 4.
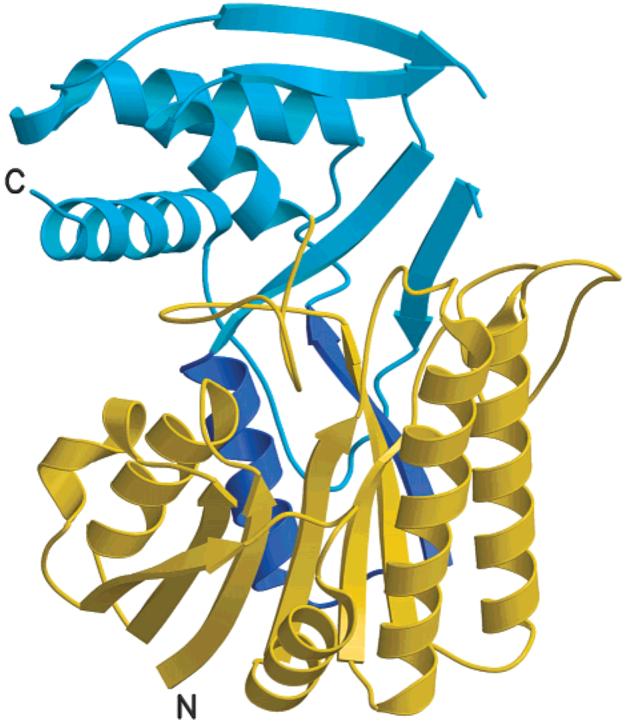
Overall structure of the ArnA decarboxylase domain. The N-terminal subdomain (in gold) is formed by residues R315-R510 and R541-G566. It adopts a modified version of the classic Rossmann fold in that an α helix and a β strand are donated by the C-terminal subdomain (shown in dark blue). The C-terminal subdomain (in blue) is formed by residues A511-I540 and N567-T656. All molecular diagrams were prepared with Molscript (52) and rendered with Raster 3D (53).
The structure of ArnA decarboxylase shown here represents the unliganded form of the enzyme and clearly shows that ArnA decarboxylase belongs to the SDR superfamily (25, 26). This group of proteins is characterized by high structural similarity and the presence of specific sequence motifs despite low overall sequence identity (15-30%). ArnA decarboxylase retains the classical glycine-rich NAD+-binding motif GX(X)GXXG represented by amino acids G322VNG325FIG328. The structure also reveals the presence of a conserved water molecule (HOH 32 in the coordinate file) that normally bridges the dinucleotide with the glycine-rich region and is proposed to be important for dinucleotide binding (35). Also important for NAD+ binding is the presence of a conserved acidic amino acid (D/E) (D347 in ArnA) that interacts with adenine ribose hydroxyls and is present in all NAD+- and FAD-binding members of the SDR family. The characteristic signature sequence YXXXK, which together with a conserved T/S residue forms a catalytic triad that catalyzes the NAD+-dependent oxidation of a sugar hydroxyl is also present in ArnA, represented by residues T432 and Y463SVSK467.
Comparison to UDP-Gal Epimerase and Substrate-Binding Model
The E. coli UDP-galactose 4-epimerase (ec-GALE) has been extensively studied both structurally and kinetically and represents one of the best characterized members of the SDR family (28, 36-42). The structures of ArnA and ecGALE share the same overall fold and topology. Despite relatively low overall sequence identity (27%), the structures of the two enzymes superimpose with a root-mean-square (rms) deviation of 1.53 Å for 216 α-carbon atoms in structurally conserved regions (see the Supporting Information for a movie with a superposition of both enzymes). The residues determined to be crucial for NAD+ binding, as well as those responsible for the 4″-OH oxidation in ecGALE, are conserved in ArnA, both in terms of sequence and structure (in ArnA, G322G325G328 and D347 for NAD+ binding; T432, Y463, and K467 for UDP-GlcA 4″-OH oxidation). ArnA and ecGALE catalyze identical first steps in their reactions, namely, the NAD+-mediated oxidation of the hydroxyl group at position C4″ in the UDP-sugar (Figure 2). On the basis of these similarities, we have modeled the substrates NAD+ and UDP-GlcA in the active site of ArnA decarboxylase, using the positions of NAD+ and UDP-Glc in ecGALE [PDB ID: 1A9Y, (42)] as a guide (Figure 5).
FIGURE 5.
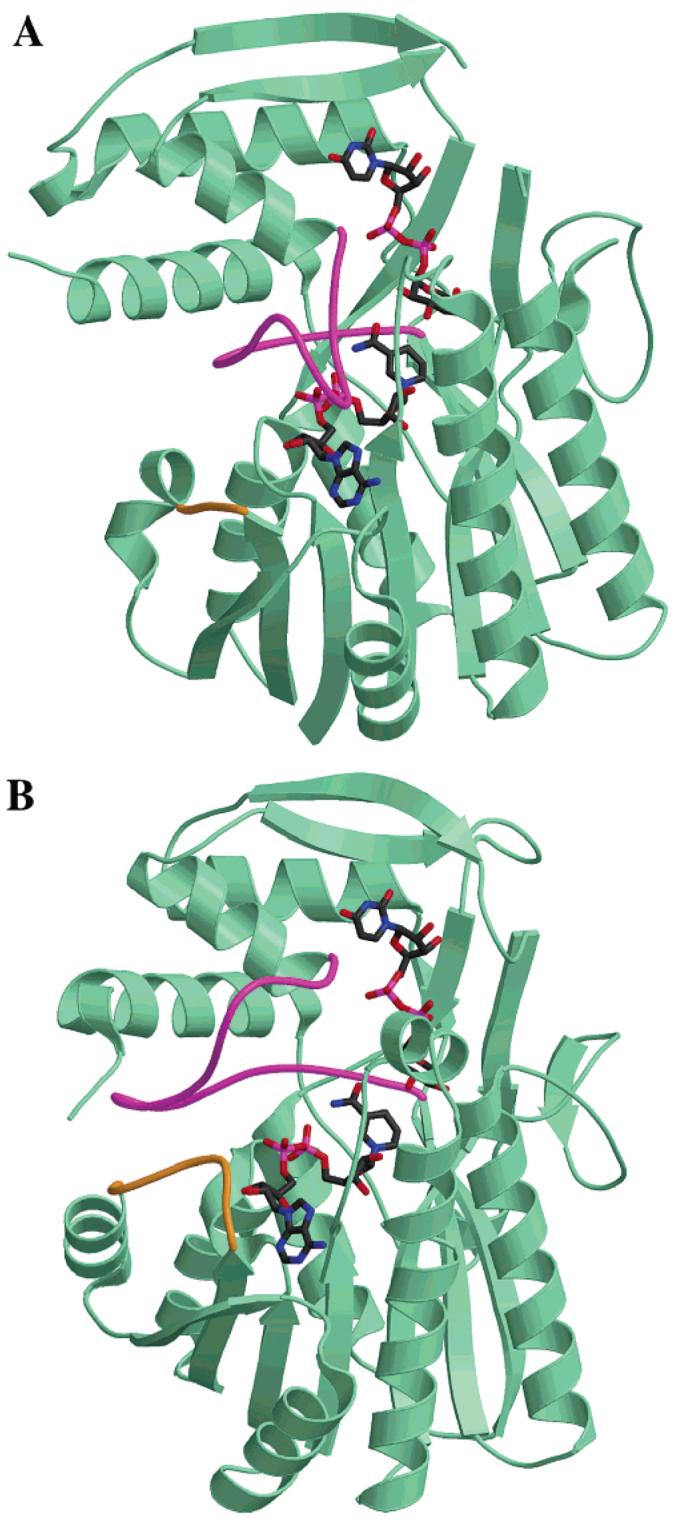
(A) Crystal structure of the E. coli ArnA decarboxylase domain with substrates modeled in the active site. (B) Crystal structure of E. coli UDP-galactose 4-epimerase with its substrates bound in the active site (PDB ID: 1A9Y). The two loops highlighted in magenta and gold in both proteins reveal structural differences likely to be important in substrate binding.
The comparison between the structure of ecGALE in complex with its substrates NAD+·UDP-Glc and ArnA decarboxylase with NAD+ and UDP-GlcA modeled in the active site highlights many similarities but also reveals some striking differences. Residues 178-200 in ecGALE define a long loop that lines the NAD+-binding site but makes no contacts with the ligand. The same region in ArnA (residues 491-510) is in a completely different conformation with the loop occupying the space where NAD+ would bind (highlighted in magenta in parts A and B of Figure 5). Thus, the NAD+-binding site appears closed in ArnA, while the UDPGlcA-binding site appears open. In contrast to ecGALE, where the NAD+ is tightly bound to the enzyme at all times (36), the structure of ArnA decarboxylase suggests a model in which UDP-GlcA binds first and induces a conformational change in the enzyme that opens the binding site for NAD+. Further experimentation is needed to test this hypothesis.
The loop defined by amino acids 31-38 in ecGALE makes close contacts with the adenine base, the ribose, and the α phosphate of NAD+ with residues D31, N32, N35, and S36, contributing several hydrogen bonds for its binding (Figure 6) (36-38). In the corresponding region of ArnA, however, there is a four amino acid deletion in the loop (residues 348-350) that would prevent contact with the NAD+ (colored in gold in Figures 5 and 6). This would result in lower affinity binding of NAD+ to ArnA, which is in agreement with the different use of NAD+ by the two enzymes. In ecGALE, the NAD+ is used as a cofactor that is regenerated during the reaction cycle (Figure 2) and remains tightly bound to the enzyme [there are 35 protein/dinucleotide contacts in ecGALE (38)]. In contrast, ArnA uses NAD+ as a substrate for the oxidation of UDP-GlcA and releases NADH as a product that would require weaker binding of NADH relative to NAD+.
FIGURE 6.
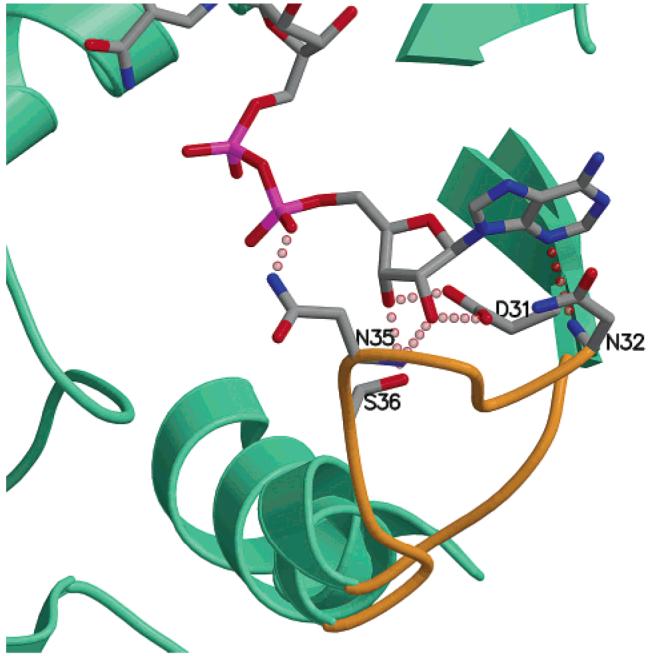
Conformational differences between the ArnA decarboxylase domain and UDP-galactose epimerase in the NAD+-binding loop that contacts the adenine. The loop is highlighted in gold and shows relevant residues in ecGALE hydrogen bonding with the adenine base. There is a four amino acid residue deletion in this loop in ArnA, and its conformation would prevent contact with NAD+.
Other conformational differences between ecGALE and ArnA decarboxylase, which include amino acid insertions and deletions, occur in areas distant from the active site and do not have any obvious functional significance at this time.
Putative Catalytic Residues in ArnA Decarboxylase
The current model for the catalytic mechanism of UDP-sugar 4-epimerases, such as ecGALE, requires ring flipping of the 4-keto-sugar intermediate, which is accomplished by rotation about the bond linking the sugar α anomeric oxygen and the β phosphorus of UDP (Figure 2B). The active site of these enzymes is large enough to accommodate the reorientation of the sugar. The decarboxylation reaction catalyzed by ArnA does not require ring flipping (Figure 2A). Only one orientation of the sugar ring is likely to place the C6″ carboxylate in the active site of the enzyme. In our model of UDP-GlcA bound to ArnA, the side chain of residue E434 is positioned such that it would prevent flipping of the glucuronic acid ring. This glutamate is strictly conserved in all UDP-GlcA decarboxylases (see the Supporting Information). The smaller residues (serine, threonine, or alanine) found at this position in UDP-sugar 4-epimerases provide additional space in the active site, which would allow ring flipping.
When the structures of the GALE enzymes from various sources were compared with the structure of WbpP (a UDPGlcNAc 4-epimerase), Berghuis and co-workers identified residues that determine UDP-sugar specificity in these SDR enzymes (43). They found that the relatively large side chains of residues K84, N199, and Y299 in ecGALE form a binding pocket, which accommodates UDP-Glc/Gal but not the larger substrates UDP-GlcNAc/GalNAc. The corresponding positions in WbpP are occupied by the smaller residues G102, A209, and S306, making for a larger binding site, which can accommodate both UDP-GlcNAc/GalNAc and UDP-Glc/ Gal. In fact, WbpP is much more efficient at catalyzing the epimerization of the N-acetylated sugars than the smaller, non-N-acetylated ones, suggesting that the binding site in WbpP is too big to constrain the conformation of UDP-Glc/ Gal to a catalytically favorable orientation (43). In ArnA decarboxylase, the three positions discussed above are occupied by A393, R510, and R619. Our modeling of UDPGlcA binding to ArnA suggest that the large side chains of the arginines would not permit the binding of 2-N-acetylated substrates and are likely to constrain the conformation of the UDP-GlcA substrate.
The residue Y299 in ecGALE interacts with the C6″ position of UDP-Glc and, as discussed above, is important to define the sugar-nucleotide specificity of the enzyme. Mutational studies revealed that a Y299C substitution (Cys is the residue at that position in human GALE) decreased the activity with regard to UDP-galactose about 5-fold and increased the activity toward UDP-GalNAc about 230-fold (40). The human GALE naturally has a cysteine at that position and can also catalyze the epimerization of UDPGalNAc (the human epimerase substrate) (44). A multiple sequence alignment of various SDR family members with known structures shows that the position occupied by Y299 in ecGALE is typically a noncharged residue. However, that position is an arginine in ArnA decarboxylase (R619) and in UDP-GlcA decarboxylases (highlighted in blue and green in Figure 7). These UDP-GlcA decarboxylases (also known as UDP-xylose synthases) catalyze a reaction slightly different from ArnA in that they use NAD+ for a complete reaction cycle (45-48). After the 4″-OH of the sugar is oxidized and the resulting 4-keto acid is decarboxylated (as in ArnA), the eukaryotic UDP-GlcA decarboxylases use NADH to re-reduce the 4-keto back to an alcohol producing UDP-xylose (Figure 2C).
FIGURE 7.

Sequence alignment of members of the SDR family and secondary-structure assignment of ArnA decarboxylase. α helices are shown as green cylinders and β strands, as red arrows. The proteins are E. coli UDP-galactose 4-epimerase (UDP-Gal epimerase); E. coli dTDP-glucose-4,6-dehydratase (dTDP-Glc dehydratase); E. coli ADP-glycero-mannoheptose 6-epimerase (AGM epimerase); S. thyphi CDP-tyvelose 2-epimerase (CDP-tyvelose epimerase); A. thaliana UDP-glucose sulfotransferase also known as UDP-sulfoquinovose synthase (UDP-Glc sulfotransferase); Homo sapiens UDP-glucuronic acid decarboxylase (UDP-GlcA decarboxylase); and E. coli ArnA C-terminal (decarboxylase) domain (ArnA C terminus). The catalytic residues S/T, Y, and K, and the NAD-binding glycine-rich motif GXXGXXG are shaded in red. Other strictly conserved residues are shaded in dark orange, while less conserved residues are shaded in light orange and yellow. The serine and arginine residues that we propose to be involved in decarboxylation are shaded in green. The corresponding residues in other proteins are shaded in blue.
In our model of ArnA decarboxylase with UDP-GlcA bound, R619 is well-positioned to interact with the C6″-carboxylate of UDP-glucuronic acid (Figure 8). According to our model, the side chain of S433 would also be poised to interact with the carboxylate of UDP-GlcA. The S433 position in ArnA is also a serine in UDP-GlcA decarboxylases (highlighted in blue and green in Figure 7). That position is occupied by an aspartate in dTDP-Glc-4,6-dehydratases and has been shown to play an important role in the reaction mechanism of these enzymes (49-51).
FIGURE 8.
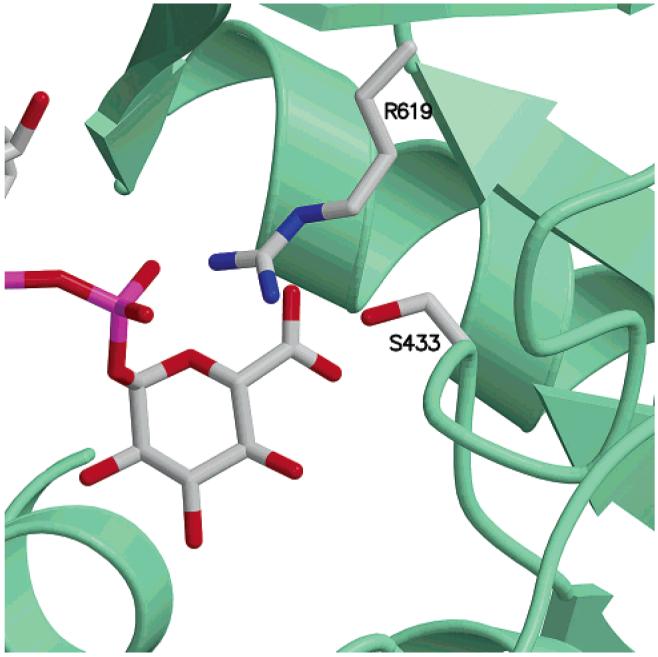
Arrangement of R619 and S433 in the vicinity of the UDPGlcA carboxylate. The strict conservation of these residues in decarboxylases reveals their potential importance for the decarboxylation reaction.
A multiple sequence alignment of ArnA decarboxylase with every enzyme in the GeneBank annotated as UDP-GlcA decarboxylase shows that only 13.6% of the residues are conserved across all proteins (see the Supporting Information). In this context, it is striking that R619 and S433 in ArnA are among the strictly conserved residues. We therefore propose that residues R619 and S433 are important for the decarboxylase activity of ArnA. This hypothesis is supported by (i) the strict conservation of these residues in all enzymes catalyzing UDP-GlcA decarboxylation, (ii) the correct positioning of the side chains for interaction with the carboxylate of UDP-GlcA in our model of ArnA with substrates bound, and (iii) the important roles in substrate binding and catalysis played by residues in the same positions in other SDR enzymes.
Given the requirement of ArnA in the biosynthesis of Ara4N-lipid A and bacterial resistance to CAMP antimicrobials, a detailed understanding of the enzyme structure and mechanism is important. The structure of ArnA decarboxylase presented here and the accompanying hypothesis provide an excellent platform for detailed structure-function studies that may help in the design of selective inhibitors.
Supplementary Material
ACKNOWLEDGMENT
We thank Sandra Metzner for excellent technical assistance. We thank Dr. C. R. Raetz for generously providing the expression plasmid for full-length ArnA. This work is supported in part by a grant from the William M. Keck Foundation to the University of Colorado.
This work was supported by a grant from the Cystic Fibrosis Foundation and NIH Grant (1 R01 AI060841-01) to M.C.S. Support for P.Z.G.-T. was provided by NIH Training Grant (GM65103).
Footnotes
Abbreviations: SDR, short-chain dehydrogenase/reductase; CAMPs, cationic antimicrobial peptides; LPS, lipopolysaccharide.
Coordinates and structure factors for ArnA decarboxylase have been deposited in the Protein Data Bank as entry 1U9J.
REFERENCES
- 1.Hoffmann JA, Kafatos FC, Janeway CA, Ezekowitz RA. Phylogenetic perspectives in innate immunity. Science. 1999;284:1313–1318. doi: 10.1126/science.284.5418.1313. [DOI] [PubMed] [Google Scholar]
- 2.Scott MG, Hancock RE. Cationic antimicrobial peptides and their multifunctional role in the immune system. Crit. Rev. Immunol. 2000;20:407–431. [PubMed] [Google Scholar]
- 3.Zasloff M. Antimicrobial peptides of multicellular organisms. Nature. 2002;415:389–395. doi: 10.1038/415389a. [DOI] [PubMed] [Google Scholar]
- 4.Zasloff M. Antibiotic peptides as mediators of innate immunity. Curr. Opin. Immunol. 1992;4:3–7. doi: 10.1016/0952-7915(92)90115-u. [DOI] [PubMed] [Google Scholar]
- 5.Gunn JS, Ryan SS, Van Velkinburgh JC, Ernst RK, Miller SI. Genetic and functional analysis of a PmrA-PmrB-regulated locus necessary for lipopolysaccharide modification, antimicrobial peptide resistance, and oral virulence of Salmonella enterica serovar typhimurium. Infect Immun. 2000;68:6139–6146. doi: 10.1128/iai.68.11.6139-6146.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gunn JS. Bacterial modification of LPS and resistance to antimicrobial peptides. J. Endotoxin Res. 2001;7:57–62. [PubMed] [Google Scholar]
- 7.Matsuzaki K. Why and how are peptide-lipid interactions utilized for self-defense? Magainins and tachyplesins as archetypes. Biochim. Biophys. Acta. 1999;1462:1–10. doi: 10.1016/s0005-2736(99)00197-2. [DOI] [PubMed] [Google Scholar]
- 8.Yang L, Weiss TM, Lehrer RI, Huang HW. Crystallization of antimicrobial pores in membranes: Magainin and protegrin. Biophys. J. 2000;79:2002–2009. doi: 10.1016/S0006-3495(00)76448-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shai Y. Mechanism of the binding, insertion, and destabilization of phospholipid bilayer membranes by α-helical antimicrobial and cell non-selective membrane-lytic peptides. Biochim. Biophys. Acta. 1999;1462:55–70. doi: 10.1016/s0005-2736(99)00200-x. [DOI] [PubMed] [Google Scholar]
- 10.Giacometti A, Cirioni O, Barchiesi F, Scalise G. In-vitro activity and killing effect of polycationic peptides on methicillin-resistant Staphylococcus aureus and interactions with clinically used antibiotics. Diagn. Microbiol. Infect. Dis. 2000;38:115–118. doi: 10.1016/s0732-8893(00)00175-9. [DOI] [PubMed] [Google Scholar]
- 11.Darveau RP, Cunningham MD, Seachord CL, Cassiano-Clough L, Cosand WL, Blake J, Watkins CS. β-lactam antibiotics potentiate magainin 2 antimicrobial activity in vitro and in vivo. Antimicrob. Agents Chemother. 1991;35:1153–1159. doi: 10.1128/aac.35.6.1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guo L, Lim KB, Gunn JS, Bainbridge B, Darveau RP, Hackett M, Miller SI. Regulation of lipid A modifications by Salmonella typhimurium virulence genes phoP-phoQ. Science. 1997;276:250–253. doi: 10.1126/science.276.5310.250. [DOI] [PubMed] [Google Scholar]
- 13.Roland KL, Martin LE, Esther CR, Spitznagel JK. Spontaneous pmrA mutants of Salmonella typhimurium LT2 define a new two-component regulatory system with a possible role in virulence. J. Bacteriol. 1993;175:4154–4164. doi: 10.1128/jb.175.13.4154-4164.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roland KL, Spitznagel JK. Molecular genetics of polymyxin resistance in Salmonella typhimurium. Prog. Clin. Biol. Res. 1995;392:3–14. [PubMed] [Google Scholar]
- 15.Shafer WM, Casey SG, Spitznagel JK. Lipid A and resistance of Salmonella typhimurium to antimicrobial granule proteins of human neutrophil granulocytes. Infect. Immun. 1984;43:834–838. doi: 10.1128/iai.43.3.834-838.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ernst RK, Yi EC, Guo L, Lim KB, Burns JL, Hackett M, Miller SI. Specific lipopolysaccharide found in cystic fibrosis airway Pseudomonas aeruginosa. Science. 1999;286:1561–1565. doi: 10.1126/science.286.5444.1561. [DOI] [PubMed] [Google Scholar]
- 17.Peschel A. How do bacteria resist human antimicrobial peptides? Trends Microbiol. 2002;10:179–186. doi: 10.1016/s0966-842x(02)02333-8. [DOI] [PubMed] [Google Scholar]
- 18.Gunn JS, Ernst RK, McCoy AJ, Miller SI. Constitutive mutations of the Salmonella enterica serovar Typhimurium transcriptional virulence regulator phoP. Infect. Immun. 2000;68:3758–3762. doi: 10.1128/iai.68.6.3758-3762.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou Z, Lin S, Cotter RJ, Raetz CR. Lipid A modifications characteristic of Salmonella typhimurium are induced by NH4VO3 in Escherichia coli K12. Detection of 4-amino-4-deoxy-L-arabinose, phosphoethanolamine, and palmitate. J. Biol. Chem. 1999;274:18503–18514. doi: 10.1074/jbc.274.26.18503. [DOI] [PubMed] [Google Scholar]
- 20.Breazeale SD, Ribeiro AA, Raetz CR. Oxidative decarboxylation of UDP-glucuronic acid in extracts of polymyxin-resistant Escherichia coli. Origin of lipid a species modified with 4-amino-4-deoxy-L-arabinose. J. Biol. Chem. 2002;277:2886–2896. doi: 10.1074/jbc.M109377200. [DOI] [PubMed] [Google Scholar]
- 21.Breazeale SD, Ribeiro AA, Raetz CR. Origin of lipid A species modified with 4-amino-4-deoxy-L-arabinose in polymyxin-resistant mutants of Escherichia coli. An aminotransferase (ArnB) that generates UDP-4-deoxyl-L-arabinose. J. Biol. Chem. 2003;278:24731–24739. doi: 10.1074/jbc.M304043200. [DOI] [PubMed] [Google Scholar]
- 22.Trent MS, Ribeiro AA, Doerrler WT, Lin S, Cotter RJ, Raetz CR. Accumulation of a polyisoprene-linked amino sugar in polymyxin-resistant Salmonella typhimurium and Escherichia coli: Structural characterization and transfer to lipid A in the periplasm. J. Biol. Chem. 2001;276:43132–43144. doi: 10.1074/jbc.M106962200. [DOI] [PubMed] [Google Scholar]
- 23.Trent MS, Ribeiro AA, Lin S, Cotter RJ, Raetz CR. An inner membrane enzyme in Salmonella and Escherichia coli that transfers 4-amino-4-deoxy-L-arabinose to lipid A: Induction on polymyxin-resistant mutants and role of a novel lipid-linked donor. J. Biol. Chem. 2001;276:43122–43131. doi: 10.1074/jbc.M106961200. [DOI] [PubMed] [Google Scholar]
- 24.Baker SJ, Gunn JS, Morona R. The Salmonella typhi melittin resistance gene pqaB affects intracellular growth in PMA-differentiated U937 cells, polymyxin B resistance, and lipopolysaccharide. Microbiology. 1999;145(Part 2):367–378. doi: 10.1099/13500872-145-2-367. [DOI] [PubMed] [Google Scholar]
- 25.Jornvall H. Multiplicity and complexity of SDR and MDR enzymes. Adv. Exp. Med. Biol. 1999;463:359–364. doi: 10.1007/978-1-4615-4735-8_44. [DOI] [PubMed] [Google Scholar]
- 26.Jornvall H, Persson B, Krook M, Atrian S, Gonzalez-Duarte R, Jeffery J, Ghosh D. Short-chain dehydrogenases/reductases (SDR) Biochemistry. 1995;34:6003–6013. doi: 10.1021/bi00018a001. [DOI] [PubMed] [Google Scholar]
- 27.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 28.Thoden JB, Gulick AM, Holden HM. Molecular structures of the S124A, S124T, and S124V site-directed mutants of UDP-galactose 4-epimerase from Escherichia coli. Biochemistry. 1997;36:10685–10695. doi: 10.1021/bi9704313. [DOI] [PubMed] [Google Scholar]
- 29.Navaza J. Implementation of molecular replacement in AMoRe. Acta Crystallogr., Sect. D. 2001;57:1367–1372. doi: 10.1107/s0907444901012422. [DOI] [PubMed] [Google Scholar]
- 30.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography and NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr., Sect. D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 31.Brunger AT, Adams PD, Rice LM. New applications of simulated annealing in X-ray crystallography and solution NMR. Structure. 1997;5:325–336. doi: 10.1016/s0969-2126(97)00190-1. [DOI] [PubMed] [Google Scholar]
- 32.Brünger AT, Krukowski A, Erickson J. Slow-cooling protocols for crystallographic refinement by simulated annealing. Acta Crystallogr., Sect. A. 1990;46:585–593. doi: 10.1107/s0108767390002355. [DOI] [PubMed] [Google Scholar]
- 33.Jones A. A graphics model building and refinement system for macromolecules. J. Appl. Crystallogr. 1978;11:268–272. [Google Scholar]
- 34.Laskowski RA, Macarthur MW, Moss DS, Thornton JM. ProchecksA program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993;26:283–291. [Google Scholar]
- 35.Bottoms CA, Smith PE, Tanner JJ. A structurally conserved water molecule in Rossmann dinucleotide-binding domains. Protein Sci. 2002;11:2125–2137. doi: 10.1110/ps.0213502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thoden JB, Frey PA, Holden HM. High-resolution X-ray structure of UDP-galactose 4-epimerase complexed with UDP-phenol. Protein Sci. 1996;5:2149–2161. doi: 10.1002/pro.5560051102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thoden JB, Frey PA, Holden HM. Molecular structure of the NADH/UDP-glucose abortive complex of UDP-galactose 4-epimerase from Escherichia coli: Implications for the catalytic mechanism. Biochemistry. 1996;35:5137–5144. doi: 10.1021/bi9601114. [DOI] [PubMed] [Google Scholar]
- 38.Thoden JB, Frey PA, Holden HM. Crystal structures of the oxidized and reduced forms of UDP-galactose 4-epimerase isolated from Escherichia coli. Biochemistry. 1996;35:2557–2566. doi: 10.1021/bi952715y. [DOI] [PubMed] [Google Scholar]
- 39.Thoden JB, Hegeman AD, Wesenberg G, Chapeau MC, Frey PA, Holden HM. Structural analysis of UDP-sugar binding to UDP-galactose 4-epimerase from Escherichia coli. Biochemistry. 1997;36:6294–6304. doi: 10.1021/bi970025j. [DOI] [PubMed] [Google Scholar]
- 40.Thoden JB, Henderson JM, Fridovich-Keil JL, Holden HM. Structural analysis of the Y299C mutant of Escherichia coli UDP-galactose 4-epimerase. Teaching an old dog new tricks. J. Biol. Chem. 2002;277:27528–27534. doi: 10.1074/jbc.M204413200. [DOI] [PubMed] [Google Scholar]
- 41.Thoden JB, Wohlers TM, Fridovich-Keil JL, Holden HM. Crystallographic evidence for Tyr 157 functioning as the active site base in human UDP-galactose 4-epimerase. Biochemistry. 2000;39:5691–5701. doi: 10.1021/bi000215l. [DOI] [PubMed] [Google Scholar]
- 42.Thoden JB, Holden HM. Dramatic differences in the binding of UDP-galactose and UDP-glucose to UDP-galactose 4-epimerase from Escherichia coli. Biochemistry. 1998;37:11469–11477. doi: 10.1021/bi9808969. [DOI] [PubMed] [Google Scholar]
- 43.Ishiyama N, Creuzenet C, Lam JS, Berghuis AM. Crystal structure of WbpP, a genuine UDP-N-acetylglucosamine 4-epimerase from Pseudomonas aeruginosa: Substrate specificity in UDP-hexose 4-epimerases. J. Biol. Chem. 2004;279:22635–22642. doi: 10.1074/jbc.M401642200. [DOI] [PubMed] [Google Scholar]
- 44.Thoden JB, Wohlers TM, Fridovich-Keil JL, Holden HM. Human UDP-galactose 4-epimerase. Accommodation of UDP-N-acetylglucosamine within the active site. J. Biol. Chem. 2001;276:15131–15136. doi: 10.1074/jbc.M100220200. [DOI] [PubMed] [Google Scholar]
- 45.Moriarity JL, Hurt KJ, Resnick AC, Storm PB, Laroy W, Schnaar RL, Snyder SH. UDP-glucuronate decarboxylase, a key enzyme in proteoglycan synthesis: Cloning, characterization, and localization. J. Biol. Chem. 2002;277:16968–16975. doi: 10.1074/jbc.M109316200. [DOI] [PubMed] [Google Scholar]
- 46.Kearns AE, Vertel BM, Schwartz NB. Topography of glycosylation and UDP-xylose production. J. Biol. Chem. 1993;268:11097–11104. [PubMed] [Google Scholar]
- 47.John KV, Schutzbach JS, Ankel H. Separation and allosteric properties of two forms of UDP-glucuronate carboxy-lyase. J. Biol. Chem. 1977;252:8013–8017. [PubMed] [Google Scholar]
- 48.John KV, Schwartz NB, Ankel H. UDP-glucuronate carboxy-lyase in cultured chondrocytes. J. Biol. Chem. 1977;252:6707–6710. [PubMed] [Google Scholar]
- 49.Vogan EM, Bellamacina C, He X, Liu HW, Ringe D, Petsko GA. Crystal structure at 1.8 Å resolution of CDP-D-glucose 4,6-dehydratase from Yersinia pseudotuberculosis. Biochemistry. 2004;43:3057–3067. doi: 10.1021/bi035547f. [DOI] [PubMed] [Google Scholar]
- 50.Babbitt PC, Mrachko GT, Hasson MS, Huisman GW, Kolter R, Ringe D, Petsko GA, Kenyon GL, Gerlt JA. A functionally diverse enzyme superfamily that abstracts the α protons of carboxylic acids. Science. 1995;267:1159–1161. doi: 10.1126/science.7855594. [DOI] [PubMed] [Google Scholar]
- 51.Allard ST, Giraud MF, Whitfield C, Graninger M, Messner P, Naismith JH. The crystal structure of dTDP-D-glucose 4,6-dehydratase (RmlB) from Salmonella enterica serovar Typhimurium, the second enzyme in the dTDP-L-rhamnose pathway. J. Mol. Biol. 2001;307:283–295. doi: 10.1006/jmbi.2000.4470. [DOI] [PubMed] [Google Scholar]
- 52.Kraulis P. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 1991;24:946–950. [Google Scholar]
- 53.Merritt EA, Bacon DJ. Raster3D: Photorealistic Molecular Graphics. Methods Enzymol. 1997;277:505–524. doi: 10.1016/s0076-6879(97)77028-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.