

Summary
Autophagy has recently been shown to be an important component of the innate immune response by degrading foreign microbial invaders. The signaling pathways leading to activation of autophagy in innate immunity are not known. Here, we show that Toll-like receptor 4 (TLR4) serves as a previously unrecognized environmental sensor for autophagy. We discovered that autophagy is induced by LPS in primary human macrophages and in murine macrophage RAW264.7 cell line. We define a new molecular pathway in which LPS-induced autophagy is regulated through a TRIF-dependent MyD88-independent TLR4 signaling. Receptor-interacting protein (RIP1) and p38 mitogen-activated protein-kinase are downstream components of this pathway. This signaling pathway does not affect cell viability and is not mediated by c-Jun amino terminal kinase (JNK), indicating that it is distinct from autophagic death signaling pathway. We further show that LPS-induced autophagy can overcome mycobacterial phagosome arrest and enhance its co-localization with the autophagosomes. This study links two ancient processes, autophagy and innate immunity, together through a shared signaling pathway.
Introduction
Autophagy is an evolutionary highly conserved process in virtually all eukaryotic cells. It involves the sequestration of regions of the cytosol within double-membrane bound compartments and delivery of the contents to the lysosome for degradation. Recently, autophagy has been shown to be an important player in many critical biological processes such as cellular response to starvation, cell survival or death, cancer, clearance of inclusion bodies in neurodegenerative diseases and more recently host defense (Levine and Klionsky 2004; Levine 2005). Rapidly accumulating functional evidence has shown that autophagy is a component of innate immunity (Kirkegaard et al., 2004) and is involved in the host defense elimination of bacterial pathogens including Group A Streptococcus (Nakagawa et al., 2004), Shigella flexneri (Ogawa et al., 2005) and Mycobacterium tuberculosis (Gutierrez et al., 2004; Singh et al., 2006). The signaling pathways leading to activation of pathogen induced-autophagy remain to be elucidated.
Although, the process of autophagy has been described decades ago, its genetic components have been only recently identified through extensive studies using yeast genetics (Kirkegaard et al., 2004). Significant progress has been made in understanding genetic factors contributing to the formation of the autophagic vesicles but the molecular mechanisms and the signaling pathways leading to induction of autophagy are still enigmatic. Although it has been shown that the signals that contribute to induction of autophagy are mediated by the target of rapamycin (TOR), the phosphatidylinositol 3-kinases (PI3Ks), protein phosphatases and trimeric G proteins, the upstream signaling events, are incompletely understood. Furthermore, whereas it is clear that class III PI3Ks are essential for starvation induced autophagy, it is not known what signaling pathway mediate pathogen-induced autophagy. In immune cells, Toll-like receptors (TLR) are charged with microbe detection. These receptors represent a conserved family of innate immune recognition receptors that play key roles in detecting microbes, initiating innate immune responses, and linking innate and adaptive immunity (Takeda and Akira, 2005). Among these stimuli, the cell wall of Gram-negative bacteria constituent lipopolysaccharide (LPS), a potent pro-inflammatory pathogen-associated molecular pattern, is the ligand for TLR4 receptor. In this study we show that TLR4 serves as a previously unrecognized environmental sensor for autophagy. Autophagy is regulated through a TRIF-dependent MyD88-independent TLR4 signaling pathway. Receptor-interacting protein (RIP1) and p38 mitogen-activated protein-kinase (MAPK) are downstream components of this pathway. This signaling pathway does not affect cell viability and is not mediated by c-Jun amino terminal kinase (JNK), indicating that it is distinct from autophagic death signaling pathway. We further show that LPS-induced autophagy can overcome mycobacterial phagosome arrest and force its co-localization with the autophagosomes. The LPS-induced autophagic signaling pathway further facilitates autophagy vesicle formation by promoting the incorporation of the PI3-Kinase VPS34 association with the membrane. Furthermore, LPS upregulated LRG47, a GTPase that has been recently shown to be involved in anti-mycobacterial effects through contribution to autophagy induction (MacMicking et al., 2003; Gutierrez et al., 2004). This study links two ancient processes, autophagy and innate immunity together through a shared signaling pathway, the toll-like receptor.
Results and Discussion
We hypothesized the existence of a potential link between autophagy and LPS-induced innate immune response. To test this hypothesis, we studied autophagy in the murine macrophage cell line RAW264.7 and in primary human alveolar macrophages. Incubation of RAW264.7 cells with LPS (100 ng/ml) led to the redistribution of microtubule-associated protein 1 light chain (LC3/Atg8) from diffuse to punctate staining, typical of autophagosomes (Figure 1A) (Gutierrez et al., 2004; Ogawa et al., 2005). The LPS treatment did not result in any significant effect on cell viability (Figure 1B). We then conducted detailed quantitative analysis of LPS-induced autophagy. LPS induced a significant increase in the percentage of cells expressing autophagosomes (Figure 2A). The increase in type II LC3 was biochemically confirmed by Western analysis with anti-LC3 antibody. LC3 type II, the indicator of autophagosome formation (Nakagawa et al., 2004), was increased in RAW264.7 cells with LPS treatment (Figure 2A, lower panel). Moreover, incubation of RAW264.7 cells, stably expressing GFP-LC3, with LPS also led to the redistribution of GFP-LC3 from diffuse to punctate staining (Figure 2B) (Gutierrez et al., 2004; Ogawa et al., 2005). GFP-LC3 positive autophagosomes were lower compared to autophagosomes detected by immunostaining for endogenous LC3 (compare Figures 2A and 2B). This may be due to inability of GFP-LC3 to get incorporated into all cellular autophagosomes. LPS-induced autophagy was prevented by the co-addition of Polymyxin B (PMB), an antibiotic that blocks biological effects of LPS by binding to lipid A, the component of LPS responsible for receptor binding and cellular signaling (Duff and Atkins 1982; Palsson-McDermott and O’Neill 2004). Evidence of cellular signaling of LPS was demonstrated through monitoring inducible nitric oxide synthase (iNOS) expression by Western analysis and nitric oxide (NO) production, important downstream products of LPS signaling in RAW264.7 cells (Musial and Eissa 2001) (Figure 2B, lower panel). Time course experiments revealed that LPS-induced autophagy could be detected at 8 h and was maximal at 12–16 h, following LPS stimulation (supplementary Figure 1). Effect of LPS on autophagy was also examined in primary human alveolar macrophages by immunofluorescence. With LPS treatment, there were 61.8% of cells with autophagosomes compared to 10.3% for control cells (Figure 2C). Furthermore, transmission electron microscopy (EM) was used to examine LPS effect on autophagy. The number of double membrane vacuoles typical of autophagosomes was markedly increased in RAW264.7 cells treated with LPS compared to control cells (Figure 2D) (Levine and Klionsky 2004; Ogawa et al., 2005). The above set of data establishes the induction of autophagic response in human and murine macrophages following LPS stimulation.
Figure 1. LPS induces formation of autophagosomes.

(A) RAW264.7 cells were incubated in the absence or presence of LPS for 16 h, fixed, stained with DAPI to visualize the nuclei (blue), and immunolabled with anti- LC3 antibody followed by Alexa Fluor 488-conjugated goat anti-rabbit IgG (green). Representative images are shown. (B) Cell viability analysis of RAW264.7 cells, stably expressing LC3-GFP, following incubation in the presence or absence of LPS (100 ng/ml) for 16 h. Cell viability was determined using Cell Viability Analyzer (Vi-Cell, BECKMAN COULTER) based on trypan blue reagent (Mean±SEM, n=4).
Figure 2. Induction of autophagy in primary human alveolar macrophages and murine macrophages RAW264.7 cells by LPS Stimulation.

Cells were incubated the in the absence (control cells) or presence of LPS (100 ng/ml) or in the presence of LPS plus PMB (25 μg/ml) for 16 h. (A) Upper panel- Representative immunofluorescence images with LC3 antibody staining in RAW264.7 cells. Middle panel- Quantitation of the percentage of cells with autophagosomes. Lower panel- Western analysis using antibodies against LC3 or β-actin. (B) GFP-LC3 fluorescence images and quantitation analyses are shown in upper and middle panels, respectively. Lower panel- Western analysis with anti-iNOS antibody. Activity of iNOS was evaluated by measuring nitrite accumulation in culture media. (C) Upper panel- Representative immunofluorescence images of LC3 antibody staining in primary human alveolar macrophages. Lower panel-Quantitation of the percentage of cells with autophagosomes. (D) Ultrastructural analysis of LPS-induced autophagy by transmission electron microscopy in RAW264.7 cells. av, autophagic vacuole; n, nucleus; m, mitochondria. Graph represents quantitation of the number of autophagosomes per cross-sectioned cell. Data are mean±SEM from three (A-B) or two (C-D) independent experiments. Scale bar, 10 μm. * and ** denote p < 0.05 and p < 0.001, respectively, when compared to control condition.
As maturation of the autophagosomes proceeds, autophagosomes fuse with endosomal vesicles and acquire lysosome-associated membrane proteins (LAMP1 and LAMP2), thus becoming late autophagosomes that subsequently fuse with lysosomes. Consequently, the colocalization of GFP-LC3 with LAMP1 indicates the presence of late autophagosomes (Kirkegaard et al., 2004). We investigated whether or not LPS-induced autophagy ultimately leads to the formation of mature autophagosomes. In RAW264.7 cells, LPS promoted LAMP1 expression and the maturation of autophagosomes as evidenced by an increase in the co-localization of GFP-LC3 with LAMP1 (Data not shown). These results indicate that LPS-induced autophagosomes proceed to mature forms.
LPS exerts multiple cellular effects by inducing signaling through TLR4 receptor (Beutler and Rietschel 2003; Palsson-McDermott and O’Neill 2004). To determine whether or not induction of autophagy by LPS is also mediated through TLR4, a dominant negative TLR4 mutant and siRNA against TLR4 were used. RAW264.7 cells stably expressing GFP-LC3 were transfected with either vector only or with a plasmid expressing TLR4 dominant negative mutant. Cells were then subjected to LPS treatment and evaluated for autophagy. LPS-induced autophagy was markedly inhibited in cells transfected with TLR4 dominant-negative compared to cells transfected with vector only (Figure 3A and Supplementary Figure 2A). In addition, knockdown of TLR4 with specific siRNA completely abrogated the induction of autophagy by LPS (Figure 3B and Supplementary Figure 2B). These results indicate that LPS-induced autophagy is mediated through signaling by TLR4. Additional experiments were done using cell line 23 ScCr (ATCC) which is a bone marrow derived macrophage cell line developed from a mouse strain C57BL/10ScN with a deletion of the TLR4 locus. These mice are naturally deficient in TLR4 gene (Lorenz et al., 2002). These cells could not form autophagosomes in response to LPS (data not shown).
Figure 3. TRIF-dependent TLR-4 signaling is required for LPS-induced autophagy.

Quantitation analysis of the percentage of cells with GFP-LC3-positive autophagosomes in RAW264.7 cells, stably expressing GFP-LC3, after incubation in the presence of LPS (100 ng/ml) for 16 h. In (A), cells were transfected with vector only or with a plasmid expressing TLR4-dominant negative mutant. In (B, D and E), cells were transfected with control siRNA or siRNA specific for TLR4, MyD88 or TRIF, respectively. All transfections were done for 32 h, prior to LPS treatment. In (C), cells stably expressed both GFP-LC3 and MyD88 dominant negative mutant. Right panel in (B)- results of RT-PCR confirming deletion of TLR4 by siRNA. Lower panel in (C)- evaluation of iNOS protein expression by Western analysis and iNOS activity by measuring nitrite in culture media. Lower panel in (D)- Western analysis of MyD88 in cell lysates. Lower panel in (E)- Western analysis of TRIF in cell lysates. Data represent means±SEM of three independent experiments. * and ** denote p < 0.05 and p < 0.001, respectively, when compared to control condition.
LPS-induced TLR4 signaling pathway uses several adaptors, including myeloid differentiation factor 88 (MyD88), TIR domain-containing adaptor protein (TIRAP), and TIR-domain-containing adaptor-inducing interferon-β (TRIF). Recent emerging evidence divides the TLR4 signaling pathway into two categories: MyD88-dpendent and MyD88-independent arms (Kawai and Akira, 2005). To determine the role of MyD88-dependent TLR4 signaling in LPS-induced autophagy, we established a RAW264.7 stable cell line that expresses both GFP-LC3 and a MyD88 dominant negative mutant (MyD88-DN). The percentage of cells with GFP-LC3 positive autophagosomes, following LPS treatment, in cells expressing both GFP-LC3 and MyD88-DN was similar to that of cells expressing GFP-LC3 only (Figure 3C and Supplementary Figure 2C). In cells expressing MyD88-DN, inhibition of MyD88-dependent signaling was confirmed by observing the reduction in iNOS expression and NO production by Western analysis and nitrite measurements, respectively (Figure 3C, lower panel). These results suggest that LPS-induced autophagy is not mediated via MyD88-dependent pathway. We confirmed this finding using siRNA against MyD88. Knockdown of MyD88 by siRNA had no significant effect on LPS-induced autophagy in RAW264.7 cells stably expressing GFP-LC3 (Figure 3D and Supplementary Figures 2D and 3A). In additional experiments, a peptide inhibitor of TIRAP, another adaptor in the TLR4-MyD88 signaling pathway, failed to block LPS-induced autophagy (data not shown). These data indicate that MyD88 and TIRAP adaptors are not required for LPS-induced autophagy. In contrast to the experiments above, deletion of TRIF by siRNA completely prevented LPS-induced autophagy in RAW264.7 cells stably expressing GFP-LC3 (Figure 3E and Supplementary Figures 2E and 3B). These results indicate that LPS induces autophagy through a TRIF-dependent TLR4 signaling pathway.
Receptor-interacting protein (RIP1) has been characterized as a signaling component of the tumor necrosis factor (TNF)-induced activation of mitogen-activated protein kinases (Devin et al., 2003). Recent evidence suggests that RIP1 also participates in TLR3/TRIF mediated induction of NF-κb activation (Meylan et al., 2004), and in LPS-induced caspase 8-independent cell death in the presence of caspase inhibitors (Holler et al., 2000; Yu et al., 2004; Xu et al., 2006). To examine whether or not RIP1 mediates LPS-induced autophagy through TLR4/TRIF pathway, siRNA against RIP1 was used to knockdown RIP1 in RAW264.7 cells stably expressing GFP-LC3. Knockdown of RIP1 prevented LPS-induced autophagy (Figure 4A and supplementary Figures 2F and 3C), suggesting that RIP1 is required for this cellular process in RAW264.7 cells. Additional siRNA experiments for MyD88, TRIF and RIP1 were performed using gene specific scrambled siRNA as a control siRNA (supplementary Figure 3). The results were very consistent with above data using siRNA against luciferase, as a control siRNA.
Figure 4. IP1 and p38 MAPK are required for LPS-induced autophagy.

(A) RAW264.7 cells, stably expressing GFP-LC3, were transfected with control siRNA or siRNA specific for RIP1 for 32 h, followed by LPS treatment (100 ng/ml) for 16 h. Upper panel- Western analysis of cell lysates with antibodies against RIP1 or β-actin. Lower panel- Quantitation analysis of the percentage of cells with GFP-LC3-positive autophagosomes. (B) RAW264.7 cells, stably expressing GFP-LC3, were incubated for 16 h in the absence (Control) or in the presence of LPS+vehicle (DMSO), LPS+JNK inhibitor (20 μM), or LPS+p38 inhibitor (20 μM). Quantitation of percentage of cells with GFP-LC3 positive autophagosomes is shown. Data in A-B represent means±SEM of three independent experiments. * and ** denote p < 0.05 and p < 0.001, respectively, compared to control.
Previous work suggested a critical role for RIP1 in mediating TNF-induced activation of MAP kinases including JNK and p38 MAPK (Devin et al., 2003). We reasoned that these same pathways might be involved in downstream signaling of TLR4/TRIF/RIP1 pathway. We examined the effects of inhibition of JNK or p38 on LPS-induced autophagy in RAW264.7 stably expressing GFP-LC3 (Figure 4B and supplementary Figure 2G). Inhibition of JNK did not have any significant effect on LPS-induced autophagy. In contrast, p38 MAPK inhibition blocked LPS-induced autophagy (Figure 4B). The effect of p38 MAPK inhibitor on LPS-induced autophagy was dose dependent (supplementary Figure 4). These data suggest that p38 MAPK is involved in LPS-induced autophagy. Functional assays of JNK and p38 MAPK inhibitors were done by assaying the phosphorylation of the downstream targets of JNK and p38 MAPK in RAW264.7 cells. JNK activity was assayed by analyzing phosphorylation of c-jun. P38 MAPK activity was assayed by evaluating phosphorylation of MAP kinase-activated protein kinase 2 (supplementary Figure 5).
There has been an intense controversy regarding the role of autophagy as a pro-survival or pro-death mechanism. Whereas inhibition of apoptosis enhances autophagic cell death (Yu et al., 2004), autophagy is essential for restricting cell death to infected sites in plants (Liu et al., 2005). The majority of evidence (Levine and Yuan 2005) suggests that in cells with intact apoptotic machinery and particularly in the context of innate immunity, autophagy is primarily a pro-survival pathway. Our study provides the molecular mechanisms for such a role. JNK pathway has been recently shown to mediate RIP induced-autophagic cell death following caspase 8 inhibition (Yu et al., 2004). In our experiments, done without caspase inhibition, LPS-induced autophagy had no effect on cell viability (Figure 1B). The above results suggest that the signaling pathway for autophagy through RIP1 may undergo bifurcation downstream of RIP1. In the absence of caspase inhibition, RIP1 signaling is mediated through p38 MAPK and maintains cell survival. Under conditions favoring autophagic cell death, e.g., following caspace inhibition, RIP1 signaling is mediated through JNK, as previously shown (Yu et al., 2004). Thus our data suggest that RIP1 is emerging as a critical mediator for autophagy in innate immunity and in controlling cell survival and death.
Inhibition of the class III phosphoinositide 3-kinase (PI3 K) by 3-methyladenine (3MA) or Wortmannin (Wm) has been shown to inhibit starvation-induced autophagy (Seglen and Gordon 1982; Lum et al., 2005). Our results showed that 3MA (5 mM) or Wm (100 nM) was able to block LPS-induced autophagy as detected by Monodansylcadaverine (MDC) staining and GFP-LC3 fluorescence (Figure 5A–B).
Figure 5. Inhibition of PI3-kinase class III activity blocks LPS-induced autophagy.

(A) RAW264.7 cells were incubated for 16 h in the absence (control), or presence of LPS, LPS plus 3MA (5 mM) or LPS plus Wortmannin (Wm, 100 nM). (B) RAW264.7 cells stably expressing GFP-LC3 were subjected to the same conditions as in (A). Cells were fixed, stained with DAPI to visualize the nuclei (blue). (C) RAW264.7 cells stably expressing a dominant negative (DN) mutant of VPS34 were incubated with LPS for 16 h in parallel with RAW264.7 cells incubated with or without LPS. Autophagic vacuoles were stained in fixed cells using MDC (blue). Deconvolution microscopy images are shown. Scale bar, 10 μm. Graphs represent quantitation analysis of the number of MDC-positive autophagosomes per cell (A and C) or the percentage of cells with GFP-LC3-positive autophagosomes (B). Data represent mean ±SEM of three independent experiments. * and ** denote p< 0.05 and p< 0.001, respectively, when compared to control condition.
VPS34 is a class III PI3-kinase that functions in the regulation of autophagy as a catalytic subunit in a complex with Beclin 1 and regulatory subunit p150. This complex participates in autophagosome formation through mediating the recruitment of other autophagy proteins to the pre-autophagosomal membrane (Kihara et al., 2001). To determine whether or not VPS34 is required for promotion of LPS-induced autophagy, we established a RAW264.7 cell line stably expressing a human VPS34 dominant-negative mutant (Stein et al., 2003). In this cell line, there was no increase in autophagy in response to LPS (Figure 5C), indicating that VPS34 activity is required for the execution of LPS-induction of autophagy. The VPS34 complex described above functions in the promotion of autophagy, as a membrane-associated complex (Kihara et al., 2001; Stein et al., 2003). We evaluated the amount of VPS34 associated with membrane by Western analysis following fractionation of cell lysates into soluble and particulate fractions. In RAW264.7 cells incubated with LPS, there was an increase in the percentage of VPS34 in the particulate fraction (supplementary Figure 6), suggesting that LPS promoted the incorporation of VPS34 complex into the membrane.
In host phagocytic cells, Mycobacterium tuberculosis is known to reside intracellularly in the phagosome and block its maturation along the phagosome-lysosome pathway. Induction of autophagy by starvation, rapamycin or LRG47 expression resulted in increased co-localization of the mycobacterial phagosomes with the autophagosomes, and reduced mycobacterial survival (Gutierrez et al., 2004; Singh et al., 2006). We hypothesized that LPS-induced autophagy can overcome the mycobacterial phagosome block. In RAW264.7 cells, infected with M. tuberculosis, LPS treatment enhanced co-localization of M. tuberculosis with the autophagic vacuoles, compared to infected cells not treated with LPS (Figure 6A). The effects of LPS-induced autophagy on mycobacterial phagosomes were also examined at the ultrastructural level. Electron microscopy analysis showed that in the absence of LPS, M. tuberculosis bacilli were seen in single membrane-bound phagosomes (Figure 6B, 1–2). In cells treated with LPS, M. tuberculosis bacilli were seen inside double membrane-bound vacuoles typical of autophagosomes (Figure 6B, 3–4) (Gutierrez et al., 2004; Ogawa et al., 2005; Lum et al., 2005). Quantitation results showed that 79% of mycobacterial bacilli were found in autophagosomes in LPS-treated cells, compared to only 11% of mycobacterial bacilli residing in autophagosomes in control cells (Figure 6B, lower panel). Potential fusion events of autophagosomes with endosomal structures were seen (Figure 6B, 5) (Gutierrez et al., 2004). We could also detect autophagosomes with onion-like structures (Figure 6B, 6), previously reported to occur during autophagy of M. tuberculosis and of Shigella (Gutierrez et al., 2004; Ogawa et al., 2005).
Figure 6. LPS-induced autophagy promotes the co-localization of mycobacterial phagosomes with the autophagosomes.

(A) RAW264.7 cells were infected with PKH-26-stained Mycobacterium tuberculosis H37Rv (Mt, red) for 1 hr. Following phagocytosis, cells were incubated in the presence or absence of LPS for 16 hr. Cells were fixed and autophagic vacuoles were stained using Monodansylcadaverine (MDC, blue). The colocalization of Mt and the MDC-positive autophagosomes is represented in the merge panels by the pink color and quantitated in the graph in the lower panel (mean±SEM, n=2). (B) Ultrastructural analysis of Mycobacterium tuberculosis localization by transmission electron microscopy using RAW264.7 cells subjected to experiments as described in (A). In the absence of LPS (control), Mt bacilli were found inside typical single-membrane mycobacterial phagosome compartment, indicated by black arrows in images 1 and 2. By contrast, in the presence of LPS, Mt bacilli were found in typical double-membrane autophagosomes (images 3 and 4). Potential fusion events between mycobacterial phagosome and autophagosome were observed (image 5). Image 6 illustrates the presence of onion-like multilamellar structures containing mycobacteria. Black arrows, outer membranes; white arrows, internal membranes; double black arrows, onion-like multilamellar structures; white asterisks, Mycobacterium tuberculosis. Graph represents the quantitation of 100 internalized mycobacteria per experimental condition.
We tested if p38 MAPK inhibition could block LPS-induced co-localization of Mycobacterium tuberculosis with autophagosomes. RAW264.7 cells stably expressing GFP-LC3 were infected with Mycobacterium tuberculosis expressing red fluorescent protein (RFP) 1 h prior to incubation for 16 h in the presence or absence of LPS plus vehicle (DMSO) or LPS plus p38 MAPK inhibitor (Figure 7). LPS treatment resulted in significant increased co-localization of M. tuberculosis with GFP-LC3 positive autophagosomes. Recent reports suggested that LRG47 exerts its anti-mycobacterial action by enhancing autophagy (Gutierrez et al., 2004; Singh et al., 2006). We observed that LPS induced upregulation of LRG47 expression in RAW264.7 and that LPS-induced LRG47 was almost exclusively membrane-bound (Supplementary Figure 7).
Figure 7. p38 MAPK inhibition blocks LPS-induced co-localization of Mycobacterium tuberculosis with autophagosomes.
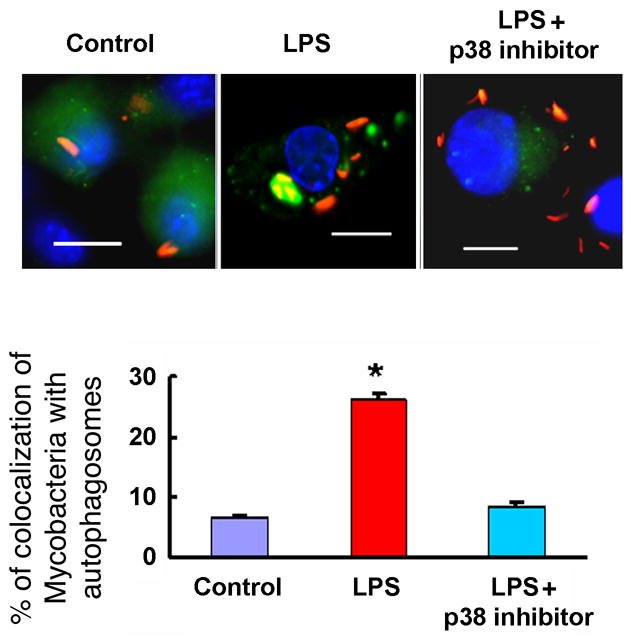
RAW264.7 cells stably expressing GFP-LC3 were infected with Mycobacterium tuberculosis expressing red fluorescent protein (RFP) 1 h prior to a 30 min incubation with p38 MAPK inhibitor or vehicle (DMSO) followed by further incubation for 16 h in the presence or absence of LPS. Upper panel: representative fluorescence images. Lower panel: Quantitation of percentage of colocalization of Mycobacterium tuberculosis with GFP-LC3-positive autophagosomes GFP-LC3-positive autophagosomes. Graph represents the quantitation of 100 internalized mycobacteria per experimental condition. Data denote means ± SEM from two independent experiments. Scale bar, 10 μm. *denotes p< 0.05, when compared to control condition.
Our study reveals that TLR4 serves as an environmental sensor for autophagy. LPS induces autophagy through TRIF/RIP1/p38MAPK dependent TLR4 signaling pathway that maintains cell survival. Our study places RIP1 as a critical signaling adaptor at the intersection of innate immunity pathway and cell survival/death pathway.
It is intriguing to note, that among all TLRs, only TLR4 uses both the MyD88-dependent and MyD88-independent pathways. It had been unclear why both pathways are necessary. Our discovery of TRIF-dependent induction of autophagy may, thus, offer a mechanistic rationale for the need for two TLR4 mediated pathways. A plausible explanation could be that a close cooperation between the two arms for TLR4 is needed. For a mammalian cell to control a pathogen, two processes will need to be performed in sequence; phagocytosis for pathogen internalization and then autophagy which would involve fusion of the phagosome with autophagosome which matures and fuses with lysosomes. A recent report suggested that TLR4 MyD88 dependent pathway is needed for phagocytosis (Blander and Medzhitove 2004). Phagocytosis of gram-negative bacteria E. coli was impaired in MyD88 deficient cells. A cooperation model would suggest that TLR4 MyD88, a fast response pathway, would be in charge of phagocytosis and that TLR4 TRIF, a slower response pathway, would be in charge of autophagy.
In macrophages infected with M. tuberculosis, induction of autophagy by LPS had a marked effect on overcoming M. tuberculosis-phagosome block and promoting its co-localization with autophagosome. M. tuberculosis do not have LPS in their wall and do not directly activate TLR4. Activation of autophagy by stimulating TRIF pathway may offer an unexpected therapeutic approach against M. tuberculosis. Our study unravels an important aspect of autophagy regulation and provides a link between stimulation of autophagy and pattern recognition receptors of innate immunity. Thus, it opens a new window for potential therapeutic interventions for modulation of autophagy through the TLRs. These new therapeutic strategies might be able to outsmart pathogens, which rely on suppressing autophagy for their survival.
Experimental Procedures
Cell and Bacterial Cultures
RAW264.7 cells were maintained in Dulbecco’s modified Eagle’s medium containing 10% FBS. Mycobacterium tuberculosis H37Rv was grown in Middlebrook 7H9 broth medium supplemented with 0.2% glycerol and 0.25% Tween 80 or on 7H11 agar plates and homogenized to generate a single cell suspension. In some experiments, M. tuberculosis was stained with 10% PKH26 Red Fluorescent dye according to Manufacturer’s instructions. Primary human macrophages were from Bronchoalveolar lavage (BAL) fluid of subjects undergoing a medically indicated bronchoscopy for a localized pulmonary nodule. BAL was performed before diagnostic evaluation of pulmonary lesion and BAL fluid was obtained from either right middle or lingual lobe. The research protocol was approved by institutional review board of Baylor College of Medicine.
Antibodies and Reagents
Anti-LC3 antibody used for immunoblot was purchased from ANASpec; anti-LC3 antibody used for immunofluorescence was a gift from Dr. Takashi Ueno; anti-MyD88 antibody was from Stressgen; anti-TRIF antibody was from abcam; anti-LAMP1 antibody was from ABR; anti-VPS34 antibody was from Invitrogen, anti-LRG-47 antibody was from SantaCruz; anti-iNOS antibody was from Research & Diagnostic Antibodies. LC3A cDNA was cloned from human brain RNA and subcloned into C-terminal end of enhanced green fluorescent protein (EGFP, BD Biosciences) to produce GFP-LC3. Mouse MyD88-dominant negative (DN) plasmid was from InvivoGen. TLR4-DN (P712H) plasmid was a Gift from Dr. Sankar Ghosh. Human VPS34-DN (N743A/N748I) plasmid was a gift from Dr. Robert Moore. LPS of Escherichia coli serotype 0111:B4, 3-methyladenine, Wortmannin, and PKH26 red fluorescent dye were from Sigma. p38 MAPK inhibitor (SB 203580) and JNK inhibitor II were from Calbiochem.
Transfection and Establishment of Stable Cell Lines
RAW264.7 cells were transfected by electroporation using program D-32 with of Amaxa Nucleofector (Amaxa). RAW264.7 cells stably expressing GFP-LC3 or VPS34 were selected and maintained in G418. A double stable cell line clone expressing both GFP-LC3 and a dominant negative MyD88 mutant was selected and maintained in the presence of both G418 (600 μg/ml) and Zeocin (50 μg/ml).
Cell Lysis and Fractionation
Cells were lysed on ice for 30 min in 40 mM Bis-Tris propane buffer, pH 7.7, 150 mM NaCl, 10% glycerol, 1% Triton-X100 and protease inhibitors. Fractionation of cellular proteins into cytosolic soluble fraction and membrane precipitate fractions was performed as follows. Cells were homogenized with a sonicator and lysates were ultracentrifuged for 30 min at 100,000 × g at 4°C. The supernatant was removed (soluble fraction). The remaining pellet was subjected to a second round of sonication and ultracentrifugation as above. The final pellet was resuspended in PBS buffer (precipitate fraction).
Autophagy Assays
Autophagy was evaluated in cells by fluorescence microscopy, electron microscopy or by immunoblot. In fluorescence microscopy experiments, autophagy was evaluated by examining the punctate forms (type II) of the autophagy marker LC3, based on GFP-LC3 or endogenous LC3 immunofluorescence using LC3 antibody. Quantitation of autophagy was done based on the percentage of GFP-LC3-positive autophagic vacuoles or cells with LC3 punctate dots. In some experiments autophagy was evaluated by observing the MDC-positive autophagic vacuoles. In EM experiments, autophagy was evaluated by observing the typical double membrane vesicles (Ogawa et al., 2004; Gutierrez et al., 2004; Liu et al., 2005). In experiments using EM, autophagy was quantitated by determining the number of autophagic vacuoles per cell. In all experiments, a minimum of 100 cells per sample were counted, and duplicate or triplicate samples were counted per experimental condition. For EM experiments, data obtained from a minimum of 50 independent sectioned cells were used. Statistical analysis was done using two-tailed student T test (Liu et al., 2005).
Fluorescence Microscopy
Cells were grown on collagen pre-coated glass coverslips in 6-well plates. Cells were fixed with 4% formaldehyde and mounted using SlowFade Antifade Kit and the blue nuclear chromatin stain 4′, 6-diamidino-2-phenylindole, dihydrochloride (DAPI) and viewed using a Zeiss Axiovert 200 M Microscope. Z sections were collected at an optical depth of 0.2–.8 μm and Images were optimized with deconvolution software. In immunofluorescence experiments, following fixation, cells were permeabilized with 0.2% Triton X 100, blocked with 10% normal goat serum and incubated with primary antibodies followed by secondary antibodies before mounting (Kolodziejska et al., 2005).
RNA Interference
Short interfering RNAs (siRNA) against TLR4 were made using SureSilencing Mouse TLR4 kit (SuperArray Bioscience Corporation). siRNA, designed against MyD88, TRIF, and RIP1 were from Dharmacon using the following sequences. TRIF siRNA: sense strand 5′GGAAAGCAGUGGCCUAUUA 3′, anti-sense strand 5′-UAAUAGGCCACUGCUUUCC 3′; MyD88 siRNA: sense strand 5′ GCCUAUC GCUGUUCUUGAAUU 3′, antisense 5′-UUCAAGAACAGCGAUAGGC 3′. RIP1 siRNA: sense strand 5′ CCACUAGUCUGACUGAUGA 3′, antisense 5′ UCAUCAGUCAGACUAGUGG 3′. siRNA were transiently transfected using Lipofectamine 2000 (Invitrogen). As a control we used either siRNA against luciferase (Dharmacon) or gene specific scrambled siRNA.
Transmission Electron Microscopy
RAW264.7 cells were fixed in 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer (pH 7.0) for 1 hr, postfixed in 1% osmium tetroxide in 0.1 M cacodylate buffer for 1 hr, dehydrated with increasing concentrations of ethanol, and gradually infiltrated with Araldite resin. Ultrathin sections (70–80 nm) were obtained using an ultramicrotome (RMC MT6000-XL). Sections were stained with uranyl acetate and lead citrate and examined using a Hitachi H-7500 transmission electron microscope equipped with Gatan digital camera.
Functional assays of JNK and p38 MAPK inhibitors
Functional assays of JNK and p38 MAPK inhibitors were done by assaying the phosphorylation of the downstream targets of JNK and p38 MAPK in RAW264.7 cells incubated in the presence or absence of the corresponding inhibitors. JNK activity was assayed by analyzing phosophorylation (Ser63) of c-jun. P38 MAPK activity was assayed by evaluating phosphorylation (Thr334) of MAP kinase-activated protein kinase 2. MAP kinase-activated protein kinase 2, is a downstream target of p38 MAPK (Zarubin and Han, 2005) and c-jun is a downstream target for JNK (Holzberg et al, 2003).
Supplementary Material
Supplementary Figure 1. Time-course for LPS-induced autophagy in RAW264.7 cells. RAW264.7 cells stably expressing GFP-LC3 were treated with LPS (100 ng/ml) for the indicated time points and the percentage of cells with autophagosomes were calculated. Data are means ± SEM from three independent experiments.
Supplementary Figure 2. LPS induces autophagy through TRIF-dependent TLR4 signaling pathway. Representative fluorescence images are shown of RAW264.7 cells, stably expressing GFP-LC3 (green), following their incubation in the presence of LPS (100 ng/ml) for 16 h in the experiments described in Figures 3 and 4. Scale bar, 10 μm.
Supplementary Figure 3. TRIF-dependent TLR-4 signaling is required for LPS-induced autophagy. Quantitation analysis of the percentage of cells with GFP-LC3-positive autophagosomes in RAW264.7 cells, stably expressing GFP-LC3, after incubation in the presence of LPS (100 ng/ml) for 16 h. In (A, B and C), cells were transfected with siRNA specific for MyD88, TRIF, or RIP1, respectively or with the corresponding gene specific scrambled siRNA. All transfections were done for 32 h, prior to LPS treatment. Lower panels represent Western analysis of targeted genes or β-actin. Data represent means±SEM of three independent experiments. ** denotes p < 0.001, respectively, when compared to control condition.
Supplementary Figure 4. Dose-dependent inhibition of LPS-induced autophagy by p38 MAPK inhibitor. RAW264.7 cells, stably expressing GFP-LC3, were incubated for 16 h in the absence or in the presence of LPS (100 ng/ml) and various concentrations of the p38 MAPK inhibitor (SB203580). Quantitation of percentage of cells with GFP-LC3 positive autophagosomes is shown. Data in A–B represent means±SEM of three independent experiments.
Supplementary Figure 5. Functional assays of JNK and p38 MAPK inhibitors. RAW264.7 cells were treated with 20 μM of JNK or p38 MAPK inhibitors, for 30 min prior to further incubation in the presence of LPS (100 ng/ml) for 2 h. Cell lysates were analyzed by Western blot for the phosphorylation of either c-jun or MAP kinase-activated protein kinase 2 (MAPKAPK-2). Analysis of β-actin was used as a loading control.
Supplementary Figure 6. LPS promotes VPS34 membrane association. Upper panel- RAW264.7 cells were incubated in the presence or absence of LPS for 16 h, lysed by sonication and analyzed by Western analysis using anti VPS34 antibody. Lower panel- Portion of the lysates was further, fractionated into soluble fraction (S) or particulate faction (P) and analyzed as above.
Supplementary Figure 7. LPS promotes LRG47 expression. RAW264.7 cells were incubated for 16 h in the absence or the presence of LPS (100 ng/ml). Upper panel- Cell lysates were subjected to Western analysis using antibody for LRG47. Lower panel-Cells treated with or without LPS as described above were lysed and fractionated into soluble (S) and particulate (P) fractions and analyzed by Western analysis for LRG47.
Acknowledgments
This work was supported by NHLBI, NIAID, AHA, and Alpha One Foundation to NTE and by NIH Kirschstein National Research Service Award (1F32 HL078520-01) to Y.X. We thank providers of plasmids and reagents (as detailed throughout the text), Drs., Margie Moczygemba and Li-Yuan Yu-Lee for critical review of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6:463–477. doi: 10.1016/s1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- 2.Levine B. Eating oneself and uninvited guest: autophagy-related pathways in cellular defense. Cell. 2005;120:159–162. doi: 10.1016/j.cell.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 3.Kirkegaard K, Taylor MP, Jackson WT. Cellular Autophagy: surrender, avoidance and subversion by microorganisms. Nat Rev Microbiol. 2004;2:301–314. doi: 10.1038/nrmicro865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nakagawa I, Amano A, Mizushima N, Yamamoto A, Yamaguchi H, Kamimoto T, Nara A, Funao J, Nakata M, Tsuda K, et al. Autophagy defends cells against invading group A Streptococcus. Science. 2004;306:1037–1040. doi: 10.1126/science.1103966. [DOI] [PubMed] [Google Scholar]
- 5.Ogawa M, Yoshimori T, Suzuki T, Sagara H, Mizushima N, Sasakawa C. Escape of intracellular Shigella from autophagy. Science. 2005;307:727–731. doi: 10.1126/science.1106036. [DOI] [PubMed] [Google Scholar]
- 6.Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo ML, Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. 2004;119:753–766. doi: 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- 7.Singh SB, Davis AS, Taylor GA, Deretic V. Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science. 2006;313:1438–1441. doi: 10.1126/science.1129577. [DOI] [PubMed] [Google Scholar]
- 8.Beutler B, Rietschel ET. Innate immune sensing and its roots: the story of endotoxin. Nat Rev Immunol. 2003;3:169–176. doi: 10.1038/nri1004. [DOI] [PubMed] [Google Scholar]
- 9.Duff GW, Atkins E. The inhibitory effect of polymyxin B on endotoxin-induced endogenous pyrogen production. J Immunol Methods. 1982;52:333–340. doi: 10.1016/0022-1759(82)90005-9. [DOI] [PubMed] [Google Scholar]
- 10.Palsson-McDermott EM, O’Neill LA. Signal transduction by the lipopolysaccharide receptor, Toll-like receptor-4. Immunology. 2004;113:153–162. doi: 10.1111/j.1365-2567.2004.01976.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Musial A, Eissa NT. Inducible nitric oxide synthase is regulated by the proteasome degradation pathway. J Biol Chem. 2001;276:24268–24273. doi: 10.1074/jbc.M100725200. [DOI] [PubMed] [Google Scholar]
- 12.Kawai T, Akira S. Toll-like receptor downstream signaling. Arthritis Res Ther. 2005;7:12–19. doi: 10.1186/ar1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Devin A, Lin Y, Liu ZG. The role of the death-domain kinase RIP in tumour-necrosis-factor-induced activation of mitogen-activated protein kinases. EMBO Rep. 2003;4:623–627. doi: 10.1038/sj.embor.embor854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meylan E, Burns K, Hofmann K, Blancheteau V, Martinon F, Kelliher M, Tschopp J. RIP1 is an essential mediator of Toll-like receptor 3-induced NF-kappa B activation. Nat Immunol. 2004;5:503–507. doi: 10.1038/ni1061. [DOI] [PubMed] [Google Scholar]
- 15.Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, Bodmer JL, Schneider P, Seed B, Tschopp J. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol. 2000;1:489–495. doi: 10.1038/82732. [DOI] [PubMed] [Google Scholar]
- 16.Yu L, Alva A, Su H, Dutt P, Freundt E, Welsh S, Baehrecke EH, Lenardo MJ. Regulation of an ATG7-beclin1 program of autophagic cell death by caspase-8. Science. 2004;304:1500–1502. doi: 10.1126/science.1096645. [DOI] [PubMed] [Google Scholar]
- 17.Xu Y, Kim SO, Li Y, Han J. Autophagy contributes to caspase-independent macrophage cell death. J Biol Chem. 2006;281:8788–8795. doi: 10.1074/jbc.M513377200. [DOI] [PubMed] [Google Scholar]
- 18.Liu Y, Schiff M, Czymmek K, Talloczy Z, Levine B, Dinesh-Kumar SP. Autophagy regulates programmed cell death during the plant innate immune response. Cell. 2005;121:567–577. doi: 10.1016/j.cell.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 19.Levine B, Yuan J. Autophagy in cell death: an innocent convict? J Clin Invest. 2005;115:2679–2688. doi: 10.1172/JCI26390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seglen PO, Gordon PB. 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc Natl Acad Sci U S A. 1982;79:1889–1892. doi: 10.1073/pnas.79.6.1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, Thompson CB. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell. 2005;120:237–248. doi: 10.1016/j.cell.2004.11.046. [DOI] [PubMed] [Google Scholar]
- 22.Kihara A, Noda T, Ishihara N, Ohsumi Y. Two distinct Vps34 phosphatidylinositol 3-kinase complexes function in autophagy and carboxypeptidase Y sorting in Saccharomyces cerevisiae. J Cell Biol. 2001;152:519–530. doi: 10.1083/jcb.152.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stein MP, Feng Y, Cooper KL, Welford AM, Wandinger-Ness A. Human VPS34 and p150 are Rab7 interacting partners. Traffic. 2003;4:754–771. doi: 10.1034/j.1600-0854.2003.00133.x. [DOI] [PubMed] [Google Scholar]
- 24.Blander JM, Medzhitove R. Regulation of phagosome maturation by signals from toll-like receptors. Science. 2004;304:1014–1018. doi: 10.1126/science.1096158. [DOI] [PubMed] [Google Scholar]
- 25.Kolodziejska KE, Burns AR, Moore RH, Stenoien DL, Eissa NT. Regulation of Inducible Nitric Oxide Synthase by Aggresome Formation. Proc Natl Acad Sci USA. 2005;102:4854–4859. doi: 10.1073/pnas.0500485102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Takeda K, Akira S. Toll-like receptors in innate immunity. Int Immunol. 2005;17:1–14. doi: 10.1093/intimm/dxh186. [DOI] [PubMed] [Google Scholar]
- 27.MacMicking JD, Taylor GA, Mickinney JD. Immune control of tuberculosis by IFN-γ-inducible LRG-47. Science. 2003;302:654–659. doi: 10.1126/science.1088063. [DOI] [PubMed] [Google Scholar]
- 28.Holzberg D, Knight CG, Dittrich-Breiholz O, Schneider H, Dorrie A, Hoffmann E, Resch K, Kracht M. Disruption of the c-JUN-JNK complex by a cell-permeable peptide containing the c-JUN delta domain induces apoptosis and affects a distinct set of interleukin-1-induced inflammatory genes. J Biol Chem. 2003;278:40213–23. doi: 10.1074/jbc.M304058200. [DOI] [PubMed] [Google Scholar]
- 29.Zarubin T, Han J. Activation and signaling of the p38 MAP kinase pathway. Cell Res. 2005;15:11–18. doi: 10.1038/sj.cr.7290257. [DOI] [PubMed] [Google Scholar]
- 30.Lorenz E, Patel DD, Hartung T, Schwartz DA. Toll-like receptor 4 (TLR4)-deficient murine macrophage cell line as an in vitro assay system to show TLR4-independent signaling of Bacteroides fragilis lipopolysaccharide. Infect Immun. 2002;70:4892–4896. doi: 10.1128/IAI.70.9.4892-4896.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Time-course for LPS-induced autophagy in RAW264.7 cells. RAW264.7 cells stably expressing GFP-LC3 were treated with LPS (100 ng/ml) for the indicated time points and the percentage of cells with autophagosomes were calculated. Data are means ± SEM from three independent experiments.
Supplementary Figure 2. LPS induces autophagy through TRIF-dependent TLR4 signaling pathway. Representative fluorescence images are shown of RAW264.7 cells, stably expressing GFP-LC3 (green), following their incubation in the presence of LPS (100 ng/ml) for 16 h in the experiments described in Figures 3 and 4. Scale bar, 10 μm.
Supplementary Figure 3. TRIF-dependent TLR-4 signaling is required for LPS-induced autophagy. Quantitation analysis of the percentage of cells with GFP-LC3-positive autophagosomes in RAW264.7 cells, stably expressing GFP-LC3, after incubation in the presence of LPS (100 ng/ml) for 16 h. In (A, B and C), cells were transfected with siRNA specific for MyD88, TRIF, or RIP1, respectively or with the corresponding gene specific scrambled siRNA. All transfections were done for 32 h, prior to LPS treatment. Lower panels represent Western analysis of targeted genes or β-actin. Data represent means±SEM of three independent experiments. ** denotes p < 0.001, respectively, when compared to control condition.
Supplementary Figure 4. Dose-dependent inhibition of LPS-induced autophagy by p38 MAPK inhibitor. RAW264.7 cells, stably expressing GFP-LC3, were incubated for 16 h in the absence or in the presence of LPS (100 ng/ml) and various concentrations of the p38 MAPK inhibitor (SB203580). Quantitation of percentage of cells with GFP-LC3 positive autophagosomes is shown. Data in A–B represent means±SEM of three independent experiments.
Supplementary Figure 5. Functional assays of JNK and p38 MAPK inhibitors. RAW264.7 cells were treated with 20 μM of JNK or p38 MAPK inhibitors, for 30 min prior to further incubation in the presence of LPS (100 ng/ml) for 2 h. Cell lysates were analyzed by Western blot for the phosphorylation of either c-jun or MAP kinase-activated protein kinase 2 (MAPKAPK-2). Analysis of β-actin was used as a loading control.
Supplementary Figure 6. LPS promotes VPS34 membrane association. Upper panel- RAW264.7 cells were incubated in the presence or absence of LPS for 16 h, lysed by sonication and analyzed by Western analysis using anti VPS34 antibody. Lower panel- Portion of the lysates was further, fractionated into soluble fraction (S) or particulate faction (P) and analyzed as above.
Supplementary Figure 7. LPS promotes LRG47 expression. RAW264.7 cells were incubated for 16 h in the absence or the presence of LPS (100 ng/ml). Upper panel- Cell lysates were subjected to Western analysis using antibody for LRG47. Lower panel-Cells treated with or without LPS as described above were lysed and fractionated into soluble (S) and particulate (P) fractions and analyzed by Western analysis for LRG47.