

Abstract
The patterning of the cardiovascular system into systemic and pulmonic circulations is a complex morphogenetic process, the failure of which results in clinically important congenital defects. This process involves extensive vascular remodeling and coordinated division of the cardiac outflow tract (OFT). We demonstrate that the homeodomain transcription factor Pbx1 orchestrates separate transcriptional pathways to control great-artery patterning and cardiac OFT septation in mice. Pbx1-null embryos display anomalous great arteries owing to a failure to establish the initial complement of branchial arch arteries in the caudal pharyngeal region. Pbx1 deficiency also results in the failure of cardiac OFT septation. Pbx1-null embryos lose a transient burst of Pax3 expression in premigratory cardiac neural crest cells (NCCs) that ultimately specifies cardiac NCC function for OFT development, but does not regulate NCC migration to the heart. We show that Pbx1 directly activates Pax3, leading to repression of its target gene Msx2 in NCCs. Compound Msx2/Pbx1-null embryos display significant rescue of cardiac septation, demonstrating that disruption of this Pbx1-Pax3-Msx2 regulatory pathway partially underlies the OFT defects in Pbx1-null mice. Conversely, the great-artery anomalies of compound Msx2/Pbx1-null embryos remain within the same spectrum as those of Pbx1-null embryos. Thus, Pbx1 makes a crucial contribution to distinct regulatory pathways in cardiovascular development.
Keywords: Pbx, Hox, Pax3, Msx2, Heart development, Vascular patterning, Mouse
INTRODUCTION
Anomalies of the cardiac outflow tract (OFT) are among the most common congenital malformations in humans. They account for 20-30% of congenital heart anomalies (Sandler, 2004), which occur in at least 1% of live births and lead to significant morbidity and mortality. The high incidence of anomalies in part reflects the fact that cardiac OFT formation is a complex developmental process that requires several elaborate morphogenetic steps, including the division of a common arterial trunk, alignment of the divided arteries to their respective cardiac chambers, and the formation of valves for each arterial channel (Harvey and Rosenthal, 1999). Development of the great arteries that supply the head, neck and upper limbs is also a challenging task for developing embryos. This process involves extensive vascular remodeling of five pairs of primitive branchial arch arteries to form a distinctive arterial network. Perturbations of branchial arch artery patterning in humans result in a variety of vascular anomalies that often require surgical correction.
Development of the cardiac OFT and branchial arch arteries requires a specific subpopulation of neural crest cells (NCCs), the cardiac NCCs, which originate from rhombomeres 6, 7 and 8 in the hindbrain and migrate to the branchial arches and heart to regulate patterning of the branchial arch arteries and septation of the OFT, respectively (Kirby et al., 1983). Ablation of cardiac NCCs in the chick leads to characteristic cardiac and vascular anomalies, including persistent truncus arteriosus (PTA) and aberrant branchial artery patterning. Loss-of-function genetic experiments in mice have provided several models that recapitulate all or part of the NCC ablation phenotype in chick (reviewed by Kirby, 2007). Among a variety of signaling and transcriptional regulators, these studies have demonstrated crucial roles for several homeodomain transcription factors. Mice deficient for Hoxa3 have defects in branchial arch arteries consistent with a NCC defect (Chisaka and Capecchi, 1991; Chisaka and Kameda, 2005; Kameda et al., 2003). Similarly, disrupted Hox expression in chick embryos is associated with abnormal patterning of the great arteries, but not with cardiac OFT defects (Kirby et al., 1997). With the exception of Hoxa3, however, single Hox gene deficiencies in mice have not been found to affect cardiovascular development, possibly reflecting redundancy in their contributions. Conversely, mutation of the Pax3 gene, which encodes a paired-homeodomain transcription factor, results in abnormal patterning of the branchial arch arteries and cardiac OFT (Conway et al., 1997; Epstein, 1996). Msx2, a homeodomain transcription factor, is an obligate repressed target of Pax3 in heart development (Kwang et al., 2002) as loss-of-function of Msx2 rescues the cardiac defects of Splotch (Pax3 mutant) mice.
Pbx1 is a TALE-class homeodomain transcription factor that forms heterodimeric complexes with a subset of Hox homeodomain proteins that are essential for regulating segmental identities during development (Chang et al., 1996; Chang et al., 1995; Knoepfler and Kamps, 1995; Peltenburg and Murre, 1996; Phelan et al., 1995). Interactions with Pbx1 confer a significant increase in the otherwise modest DNA-binding specificities and affinities of Hox proteins in vitro (Chang et al., 1996), and Pbx1 deficiency compromises Hox (Selleri et al., 2001) and para-Hox (Kim et al., 2002) protein functions in vivo. Pbx1 also partners with Meis/Prep proteins, members of the TALE class of homeodomain transcription factors (Abu-Shaar et al., 1999; Chang et al., 1997), which facilitate the formation of trimeric transcriptional complexes with Hox proteins (Jacobs et al., 1999). Consistent with the roles of Hox genes in specifying rhombomere identities, both Pbx and Meis orthologs regulate hindbrain development in zebrafish (Choe et al., 2002; Waskiewicz et al., 2001; Waskiewicz et al., 2002). However, as the cardiac OFT in zebrafish does not normally divide into separate circulations, these previous studies did not address whether Pbx1 is required for the contribution of rhombomere-derived cardiac NCCs to OFT septation or branchial arch artery patterning.
In the current study, we demonstrate that Pbx1 impacts branchial arch artery patterning by controlling formation of the fourth and sixth branchial arches. Additionally, Pbx1 cooperates with Meis1 and/or Hox proteins to induce a high, but transient, activation of Pax3 in premigratory cardiac NCCs that ultimately governs their function, but not migration, during OFT septation.
MATERIALS AND METHODS
Mice
Pbx1-null, Wnt1Cre and Pax3Cre mice have been described previously (Jiang et al., 2000; Li et al., 2000; Selleri et al., 2001). Pbx1+/- mice were maintained in a C57BL/6 background. Phenotypes of Pbx1-null mice were typically analyzed in embryos (E10.5-15.5) resulting from intercrossed parental mice that had been backcrossed to the C57BL/6 strain background for at least eight generations. Gestational age was determined by the date of observing a vaginal plug [set at embryonic day (E) 0.5], as confirmed by ultrasonography (Chang et al., 2003).
Angiography and vascular casting
Chest walls of mouse embryos were opened under microscopic visualization. A 33-gauge needle (Hamilton) mounted on a 1 ml tuberculin syringe was used to inject an acrylic resin (Batson no. 17) containing blue dye (Methyl Methacrylate Casting Kit, Polyscience) into the right ventricle. The dynamic flow of the blue dye filling the right ventricle, main pulmonary artery, ductus arteriosus, aortic arch and ascending aorta was carefully observed. Following angiography, embryos were held at 4°C for 2-6 hours to allow the resin to polymerize and cast the vasculature. Soft tissues of the embryos were subsequently dissolved in potassium hydroxide (Maceration Solution, Polysciences) at 55°C for 1-3 hours to expose the vascular casts, which were then cleaned and photographed under a dissecting microscope. For India ink-based angiography, embryos were harvested at E10.5 or E11.5 and fixed overnight in 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS). India ink (undiluted, water-insoluble form) was injected into the ventricles using a fine glass micropipette while the embryos rested in PBS. For E11.5 embryos, the branchial arches and surrounding tissues were carefully dissected to expose the branchial arch arteries prior to imaging.
Histology
Paraffin sections of mouse embryos were prepared as described previously (Stankunas et al., 2008a). Consecutive sections of 5-7 μm through the chest cavity were collected and stained with Hematoxylin and Eosin.
RNA in situ hybridization and β-galactosidase staining
These procedures were performed as described previously (Stankunas et al., 2008a). The plexin A2, Pax3 and Msx2 antisense probes were as described previously (Brown et al., 2001; Kwang et al., 2002).
Immunostaining
Fluorescent immunostaining and immunohistochemistry on paraffin tissue sections (7 μm) were performed as previously described (Chang et al., 2004). The anti-Pbx1b monoclonal antibody (clone 41.1) (Chang et al., 1997) and anti-Pax3 monoclonal antibody (concentrate, Developmental Studies Hybridoma Bank) were used at 1:300 and 1:500 dilution, respectively, for immunostaining.
Electrophoretic mobility shift assay (EMSA)
EMSA was performed as described previously (Chang et al., 1995; Wu et al., 2007). Proteins (Pbx1a, Pbx2, Pbx3, Meis1, HoxB2, HoxB4 and HoxB7) were prepared using a TnT Quick Coupled Transcription/Translation System (Promega) following the manufacturer's instructions. Oligonucleotides probes corresponding to sequences from the Pax3 promoter were (5′ to 3′): Site A, CTCTACATCAAAACTGTCAAAGGCTCT; Site B, CTCTCCTTTTGATTGATTAAGCTCT.
Luciferase reporter assays
The 1.6 kb promoter region of the Pax3 gene was amplified from mouse genomic DNA and cloned into the pGL3-basic vector (Promega). Expression plasmids for Pbx1b, Meis1 and HoxB4 were described previously (Chang et al., 1997; Chang et al., 1995). PC12 cells were co-transfected with expression plasmids, a Pax3 luciferase reporter construct and a constitutively expressing Renilla luciferase construct for normalization of transfection efficiency using Fugene 6 Transfection Reagent (Roche). Luciferase activities were analyzed using a Dual Luciferase Reporter Assay System (Promega). Fold activation was calculated relative to reporter baseline activities, and data presented as mean ± one s.d. P-values were determined using Student's t-test.
RESULTS
Pbx1 is required for cardiac OFT septation
Septation of the cardiac OFT in mice occurs between E11.5 and E12.5, and the interventricular septum is sealed by E14.5 (Fig. 1A,D,F). By contrast, angiographic and serial histological analysis of Pbx1-/- embryos at E14.5 showed a single common arterial trunk (truncus arteriosus) that arose directly from the right ventricle with no arterial or OFT septation and no ductus arteriosus (Fig. 1B,E). The absence of the conal septum, which normally contributes to the sealing of the interventricular septum, resulted in a large, non-restrictive ventricular septal defect approaching the size of the normal aortic root (Fig. 1G).
Fig. 1. Pbx1 is required for cardiac OFT septation.

(A-C) Angiographic casting of wild-type (A) and Pbx1-/- (B,C) E14.5 mouse embryos. (D-G) Hematoxylin and Eosin-stained transverse sections through the outflow tract (D,E) and ventricular septal regions (F,G) of wild-type (D,F) and Pbx1-/- (E,G) E14.5 embryos. The arrow indicates the interventricular septum. Ao, aorta; MPA, main pulmonary artery; RPA and LPA, right and left pulmonary arteries; PTA, persistent truncus arteriosus; RCA and LCA, right and left coronary arteries; DAo, descending artery; VSD, ventricular septal defect.
The PTA in Pbx1-/- embryos displayed dual features of the aorta and main pulmonary artery in that it gave rise to coronary, pulmonary and systemic arteries. The right and left coronary arteries arose anteriorly, whereas a short stump of the main pulmonary artery arose posteriorly from the truncus (Fig. 1C). This short main pulmonary artery divided into the right and left pulmonary arteries (Fig. 1C), which were of similar size to wild-type pulmonary arteries (Fig. 1, compare A with C). After the coronary and pulmonary artery branching points, the truncus continued as the ascending aorta and generally arched to the left to form the descending aorta (Fig. 1B). These data demonstrate a requirement for Pbx1 in septation of the cardiac OFT.
Pbx1 regulates patterning of the branchial arch arteries
The aortic arch originates in the cervical region of the embryo during early development and, along with the developing heart, descends to its normal intrathoracic location by E13.5 (Fig. 2A). In Pbx1-/- embryos, the aortic arch failed to descend into the thorax and remained cervical in location (Fig. 2B), leading to a phenotype reminiscent of the human anomaly `cervical aortic arch'. As a consequence, the peripheral pulmonary arteries descended to the thorax to reach the lungs (Fig. 1C, Fig. 2E), in contrast to the normal course of pulmonary arteries within the thorax. In 18% (n=3/17) of Pbx1-/- embryos, the aorta arched to the right side, creating a mirror image of the arteries branching from the aorta (Fig. 2F).
Fig. 2. Pbx1 contributes to patterning of the branchial arch arteries.

(A-F) Angiographic casting of wild-type (A,C) and Pbx1-/- (B,D-F) E14.5 mouse embryos. (A,B) The position of the aortic arch (arrow) is indicated relative to the forelimbs (arrowheads). (G,H) India ink casting of the branchial arch arteries in wild-type (G) and Pbx1-/- (H) E11.5 embryos. The branchial arch arteries are numbered according to branchial arch origin. (I,J) Whole-mount in situ hybridization staining for Msx2 (blue) on wild-type (I) and Pbx1-/- (J) E10.5 embryos. The arrowhead indicates the groove separating the third and fourth branchial arches in the wild-type embryo. The arrow indicates Msx2 expression in the fourth branchial arch. CCA, common carotid artery; ICA, internal carotid artery; ECA, external carotid artery; DA, ductus arteriosus; BCA, brachiocephalic artery; RSA and LSA, right and left subclavian arteries; LPA, left pulmonary artery; PA, right and left pulmonary arteries; IVA, internal vertebral artery; AA, axillary artery; IMA, internal mammary artery; SVC, superior vena cava; DLSCA, developmentally left, but anatomically right subclavian artery; DRSCA, developmentally right, but anatomically left subclavian artery.
The great arteries, which supply the head, neck and upper limbs, were aberrantly patterned in Pbx1-/- embryos (n=17). The left common carotid artery (CCA), which normally arises from the aortic arch, and the right CCA, which branches off the brachiocephalic artery (BCA), were generally absent in Pbx1-/- embryos (Fig. 2C,D). In the absence of the CCA, the external carotid artery (ECA) and internal carotid artery (ICA) arose directly from the aortic arch (Fig. 2D). Occasionally, a residual stump of CCA was present, connecting the ECA and ICA to the aortic arch (Fig. 2D and data not shown). The identity of the ECA was confirmed by its branching into facial and superficial temporal arteries (not shown); the ICA identity by its continued course into the cranium. The right subclavian artery (RSA), which normally arises from the BCA (Fig. 2C), originated instead from the descending aorta distal to the origin of the left subclavian artery (LSA) in Pbx1-/- embryos (Fig. 2E). The LSA, instead of arising from the aortic arch, arose from the descending aorta in Pbx1-/- embryos (Fig. 2C,E). The identities of the RSA and LSA were confirmed by their branching into internal vertebral (IVA), internal mammary (IMA) and axillary arteries (AA) (Fig. 2E), and by their destination in the right and left forelimbs, respectively.
Pbx1 is required for caudal branchial arch development
To examine whether the abnormal great-artery patterning seen at E14.5 in Pbx1-/- embryos reflected aberrant remodeling or a failure to establish the initial complement of branchial arches, India ink injections were used to mark the arterial systems of E10.5 and E11.5 embryos, prior to branchial arch artery regression. Instead of possessing three branchial arch arteries on each side (Fig. 2G), Pbx1-/- embryos (n=5) had only one or two patent branchial arch arteries (on both left and right sides) (Fig. 2H). When two arch arteries were present, the caudal-most artery was always narrow. The anatomic position of the aortic arches relative to the branchial arches was consistent with a failure to establish the sixth, and frequently the fourth, branchial arch arteries.
The arch arteries are derived from mesodermal cells of the branchial arches, and Pbx1-/- embryos have abnormalities in the development of the pharyngeal pouches of the caudal branchial arches (Manley et al., 2004). We therefore examined whether the branchial arches formed normally in the absence of Pbx1 by using Msx2 whole-mount in situ hybridization to mark mesenchymal cells of the arches (MacKenzie et al., 1992). At E10.5, the pharyngeal groove separating arch 3 and 4 was absent in Pbx1-/- embryos (Fig. 2, J versus I), and the fourth branchial arch was smaller than normal and showed reduced Msx2 staining. Therefore, the great-artery patterning defects in Pbx1-/- embryos are at least in part due to a failure to develop a full set of branchial arches, which might also underlie the absence or reduction of organs derived from the caudal pharyngeal region (Manley et al., 2004).
Pbx1 is widely present in cells regulating branchial arch artery and cardiac OFT development
The OFT septation and great-artery patterning defects observed in Pbx1-deficient embryos are similar to those previously associated with defects in cardiac NCC migration and/or function (Kirby et al., 1983), suggesting a possible role in these processes. Alternatively, the OFT defects could represent a failure of secondary heart field (SHF)-derived cells to populate the OFT or aberrations in endocardial cushion development. The presence of Pbx1b, the major isoform of Pbx1 during development (Schnabel et al., 2001), was examined by immunostaining in each of these relevant tissues. At E8.75, Pbx1b was present in all cells of the hindbrain, including the premigratory NCCs at the extreme dorsal tip of the neural tube (the neuroectodermal junction) (Fig. 3A). By E9.5, Pbx1b was found in many, but not all, cells of the neural tube, including the premigratory NCCs. Pbx1b was also detected in many paraxial mesoderm cells flanking the neural tube at both E8.75 and E9.5 (Fig. 3A,B). In the septating OFT at E11.0, Pbx1b was abundant in vascular smooth muscle cells (Fig. 3C). Pbx1b was also present throughout the mesenchyme of the endocardial cushion, most likely in cells derived from both endocardial cells (by an epithelial-to-mesenchymal transformation) and NCCs (Fig. 3C). Whereas Pbx1b was decreased in cardiomyocytes of the OFT, it remained present at low levels in cushion endocardial cells (Fig. 3C). An immunofluorescence based-staining method for Pbx1b demonstrated its broad, but not ubiquitous, presence in E9.5 embryos (Fig. 3D-F). A higher magnification view of the region encompassing the E9.5 aortic sac and OFT revealed abundant Pbx1b in SHF cells entering the OFT and in the ectoderm, with lower Pbx1b levels in endocardial cells (Fig. 3G). Pbx1b was also found in many, but not all, endoderm-derived cells of the pharyngeal pouches and in mesenchymal cells throughout the embryo, including in the branchial arches (Fig. 3H). Pbx1b was present only in the endocardium (at low levels), among cells of the atria and left ventricle (Fig. 3I). Our staining results are consistent with other studies (Manley et al., 2004; Schnabel et al., 2001; Selleri et al., 2001; Stankunas et al., 2008b) and indicate many possible sites of action for Pbx1b in branchial arch artery and OFT development, including cardiac NCCs.
Fig. 3. Pbx1 is present in multiple cell types that influence OFT septation and artery patterning, including premigratory neural crest cells.

(A-C) Immunohistochemical analysis of the Pbx1b isoform of Pbx1 (brown) in sections of mouse neural tube at E8.75 (A) and E9.5 (B) and the outflow tract (OFT) at E11.0 (C). Sections are counterstained with Hematoxylin. (A,B) Arrowheads indicate the neuroectodermal junction where neural crest cells (NCCs) originate. Arrows indicate paraxial mesodermal cells. (C) Arrowheads show vascular smooth muscle cells of the OFT. The asterisk indicates the endocardial cushion, the arrow cushion endocardium. (D-I) Immunofluorescent staining for Pbx1b in sections of a wild-type E9.5 embryo. (D,G,H,I) Pbx1b is in green and the nuclei are purple (stained with Hoechst). (E,F) The Pbx1b and Hoechst channels are shown separately in grayscale. (G,H). Higher-magnification views of specific regions of the embryo shown in D. (G) Arrowheads indicate secondary heart field and/or NCCs entering the OFT. The arrow points to ectodermal cells. The double arrow indicates endothelial cells of the OFT. (H) The arrowheads indicate endodermal cells lining the pharyngeal pouch. The arrow indicates a paraxial mesodermal cell. The double arrow indicates mesenchymal cells of the branchial arches. (I) Pbx1b immunofluorescent staining of a section of an E9.5 embryonic heart. Arrows mark endocardial cells and arrowheads indicate pericardial cells. RV, right ventricle; NT, neural tube; AS, aortic sac.
Pbx1 is not required for migration of cardiac NCCs into the outflow tract
NCC migration was assessed by whole-mount RNA in situ hybridization with a plexin A2 probe that stains post-migratory NCCs (Brown et al., 2001). In Pbx1-/- embryos, plexin A2 staining highlighted two streams of NCCs migrating into the cardiac OFT, as seen in wild-type embryos (Fig. 4A,B). The absence of a generalized NCC migration defect in Pbx1-/- embryos was confirmed by plexin A2 staining of the dorsal root ganglia and sympathetic chains (Fig. 4C,D). Cell fate mapping, using Wnt1 promoter-driven Cre activity and the Rosa26RlacZ line to mark NCCs and their derivatives (Jiang et al., 2000), showed that NCCs migrated into the cardiac OFT of both wild-type and Pbx1-null embryos (Fig. 4E,F). Thus, although Pbx1 is normally present in cardiac NCCs and their derivatives, its absence does not detectably affect their migration and appropriate localization to the cardiac OFT.
Fig. 4. Pbx1 deficiency has no effect on the migration of cardiac NCCs but abolishes Pax3 proximal promoter activity in rhombomere 6.

(A-D) Whole-mount in situ hybridization for plexin A2 transcripts (blue) in Pbx1+/+ (A,C) and Pbx1-/- (B,D) E11.5 mouse embryos. Frontal heart views (A,B) and dorsal views (C,D) of the embryos are shown. Black arrows indicate dorsal root ganglia and white arrows indicate sympathetic chains. (E-L) Whole-mount b-galactosidase (lacZ) staining (blue) of Pbx1+/+ (E,G,I,K) and Pbx1-null (F,H,J,L) embryos of the indicated ages and genotypes. lacZ expression is driven by the Rosa26RlacZ (R26RlacZ) allele in cell lineages that express Cre recombinase driven by either the Wnt1 promoter (Wnt1Cre, E,F,I,J) or the Pax3 1.6 kb proximal promoter (Pax3Cre, G,H,K,L). (M-P) Transverse sections of the embryos shown in I-L at the level of R6. lacZ-expressing cells, derived from progenitors expressing either Wnt1Cre (M,N) or Pax3Cre (O,P), stain blue. Sections are counterstained with Nuclear Fast Red (pink). DRG, dorsal root ganglia; OFT, outflow tract; R, rhombomere; O, otic vesicle; RV, right ventricle; LV, left ventricle; LA, left atrium; RA, right atrium.
Pbx1 is required for Pax3 promoter activity in rhombomeres where cardiac NCCs originate
To further assess the impact of Pbx1 deficiency on NCCs, fate mapping studies were performed using Pax3Cre transgenic mice, which express Cre under the control of the Pax3 1.6 kb proximal promoter (Li et al., 2000). Like Wnt1Cre, Pax3Cre targets Cre activity to cardiac NCCs at their rhombomeric origins, prior to delamination from the neural tube (Li et al., 2000). In marked contrast to wild-type embryos, Pax3 promoter-driven Cre activity was not detected in the cardiac OFT of Pbx1-/- embryos at E12.5 (Fig. 4G,H), even though cardiac NCCs successfully migrated into the OFT (Fig. 4B,F). To determine whether the absence of Pax3Cre-marked cells in the cardiac OFT results from inactivity of the Pax3 promoter in premigratory cardiac NCCs, we examined the expression of β-galactosidase (lacZ) in the rhombomeres of E10.5 Pax3Cre;R26RlacZ;Pbx1-/- mice. In Pbx1+/+ embryos, Pax3Cre drove lacZ expression in rhombomere (R) 2, 4 and 6, and in the streams of NCCs migrating from these regions (Fig. 4K). By contrast, Pax3Cre activity in Pbx1-/- embryos was selectively absent from R6 (Fig. 4L). Conversely, similar studies using the Wnt1Cre;R26RlacZ combination showed that Wnt1 promoter activity was maintained in cardiac NCCs from R6, R7 and R8 in Pbx1-/- embryos (Fig. 4I,J). The absence of Pax3Cre and preservation of Wnt1Cre activity at the dorsal end of R6 was confirmed by histology of consecutive transverse sections in which R6 was marked by the caudal end of the otic vesicle (Fig. 4M-P). The difference between Wnt1Cre and Pax3Cre promoter activity in Pbx1-/- embryos does not reflect earlier activity of the Pax3 promoter because, if any difference is present, Wnt1Cre activity initiated prior to that of Pax3Cre (see Fig. S1 in the supplementary material). Similarly, the Wnt1Cre domain in R6 through R8 appeared to entirely overlap with the missing R6 expression normally driven by the Pax3 promoter. Therefore, the absence of Pax3Cre-marked cells in the OFT of Pbx1-/- embryos is not due to the loss of a subpopulation of Pax3-positive Wnt1-negative cardiac NCCs, but rather suggests a failure to activate the Pax3 promoter in premigratory cardiac NCCs.
Pbx1 regulates transient expression ofPax3 in premigratory NCCs
The loss of R6 expression driven by Pax3Cre but not Wnt1Cre implied that Pbx1 regulates transcription of the Pax3 1.6 kb proximal promoter, a possibility consistent with the requirement for a Pbx1 binding site within the Pax3 proximal promoter for its full activity in embryos (Pruitt et al., 2004). However, using whole-mount in situ hybridization for Pax3 transcripts, we found no clear change in Pax3 in the neural tube of E9.5 Pbx1-/- embryos (Fig. 5A). This surprising result led us to examine the presence of Pax3 in more detail using immunostaining of paraffin sections of wild-type and Pbx1-null embryos. In an 18-somite wild-type embryo (equivalent to E8.75), Pax3 appeared in a broad dorsal pattern that was overlaid with considerably more abundant Pax3 in a subset of cells at the extreme dorsal end of the neural tube of R6 (Fig. 5B). Interestingly, this cluster of cells that strongly expresses Pax3, which corresponds to the premigratory neural crest, was absent in a littermate Pbx1-/- embryo (Fig. 5C). However, Pax3 was still found in migrating NCCs in the absence of Pbx1 (Fig. 5C). We examined the presence of Pax3 in the dorsal neural tube of the hindbrain from E8.0 through E9.5. Pax3 was detected in a broad dorsal band in the neural tube of a zero-somite (E8.0) embryo (Fig. 5D), a pattern that was retained throughout the developmental stages examined (Fig. 5E-G). By contrast, the robust Pbx1-dependent presence of Pax3 in premigratory NCCs was found only transiently, between E8.5 and E9.0 (Fig. 5B,E,F). At E9.5, Pax3 was retained in a broad band of cells of the R6 dorsal neural tube, whereas the premigratory NCCs had relatively low levels of Pax3 protein (Fig. 5G). Taken together, these results show that Pbx1 is required specifically for a transient burst of Pax3 expression in premigratory NCCs of R6 prior to their delamination from the neural tube.
Fig. 5. Pbx1 is required for transient high expression of Pax3 in premigratory NCCs.
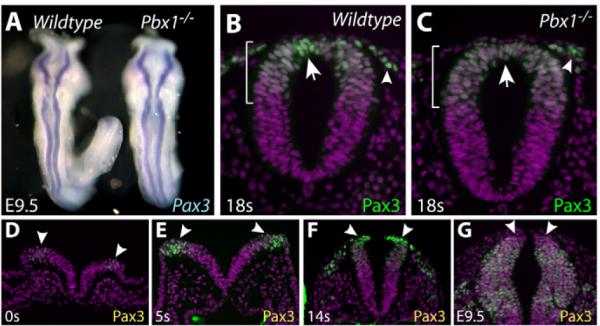
(A) Dorsal view of wild-type (left) and Pbx1-/- (right) E9.5 mouse embryos stained by whole-mount in situ hybridization for Pax3 transcripts (blue). (B-G) Transverse sections of embryos of the indicated ages [by somite (s) number (between E7.5 and E9.0) or embryonic date] immunostained for Pax3 (green) and with Hoechst (purple). (B,C) Sections through R6 of wild-type (B) and Pbx1-/- (C) 18s embryos. The extent of the Pax3+ domain in the dorsal neural tube is marked by a bracket. Arrows indicate a cluster of premigratory NCCs at the extreme dorsal end of the neural tube. Arrowheads mark migrating NCCs. (D-G) A time course of Pax3 expression in the hindbrain between E7.5 and E9.5. Arrowheads mark the dorsal end of the neural tube.
Pbx1 transcriptional complexes activate thePax3 promoter
In vitro studies were conducted to further assess the potential role of Pbx1 in the transcriptional regulation of Pax3, which contains Pbx1 binding sites in its promoter (Pruitt et al., 2004) (Fig. 6A). Electrophoretic mobility shift assays (EMSA) (Chang et al., 2004; Chang et al., 1995) confirmed that Site A, which contains a consensus Pbx1/Meis1 binding sequence (5′-TGACAGTT-3′) (Chang et al., 1997), supported robust cooperative binding by Pbx1 and Meis1, but not binding by either protein alone (Fig. 6B). By contrast, Pbx1 did not form binding complexes with several representative Hox proteins (HoxB2, HoxB4 or HoxB7) on Site A (Fig. 6B and data not shown). Site B, which is located 1.1 kb upstream of the Pax3 transcriptional start site, was bound robustly by HoxB4 or Meis1 in the presence of Pbx1 (Fig. 6C). DNA binding by HoxB2 and HoxB7 on Site B was also dependent on Pbx1 (data not shown). Pbx1-Meis1-Hox trimeric complexes did not form on either isolated Site A or Site B (Fig. 6B,C). The requirement for Pbx1 in regulating Pax3 promoter activity was assessed using a reporter gene containing the 1.6 kb Pax3 promoter fragment (Li et al., 2000) in PC12 pheochromocytoma cells, which are derivatives of NCCs (Greene and Tischler, 1976). Whereas HoxB4 alone produced a modest increase in Pax3 promoter activity, co-transfection of Pbx1 and HoxB4 strongly activated transcription (Fig. 6D). Consistent with the binding studies, adding Meis1 to the transfection mixture did not further enhance the Pax3 transcriptional response (Fig. 6D). Thus, Pbx1 partners with Meis and Hox proteins to directly activate expression of Pax3 through its proximal promoter elements. Taken together, our results show that Pbx1 is essential for Pax3 proximal promoter activity and for transient premigratory cardiac NCC expression of Pax3. We propose that activity of the Pax3 1.6 kb proximal promoter in vivo partially reflects Pbx1-dependent Pax3 expression in premigratory cardiac NCCs. The broader and sustained dorsal neural tube expression of Pax3 is likely to require additional regulatory elements outside the 1.6 kb region that are not under Pbx1 control.
Fig. 6. Pbx1 transcriptional complexes activate the Pax3 promoter and are required for repression of Msx2 expression.

(A) Schematic of the mouse Pax3 promoter showing the location of Pbx, Meis and Hox binding sites within Sites A and B. Numbers denote the distance (in bp) from the transcription start site (arrow) of the Pax3 gene. (B,C) Electrophoretic mobility shift assays using the indicated in vitro translated transcription factors (Pbx1, Meis1 and HoxB4) and radiolabeled DNA fragments from Site A (B) or Site B (C) of the Pax3 promoter. (D) Transient reporter assays using a transfected plasmid containing the Pax3 1.6 kb proximal promoter driving luciferase expression and co-transfected plasmids expressing the indicated transcription factors. Fold activation was calculated relative to reporter baseline activity following normalization and is presented as the mean ± one s.d. P-values were determined using Student's t-test. (E,F) Whole-mount in situ hybridization for Msx2 (blue) in the hindbrain (dorsal view) in Pbx1+/+ (E) and Pbx1-/- (F) E9.5 embryos. The bracketed regions indicate R6-8. (G,H) Msx2 in situ hybridization (brown) on sections through R6 of Pbx1+/+ (G) and Pbx1-/- (H) E9.5 embryos. The sections are counterstained with Hematoxylin (blue). The arrows indicate premigratory neural crest. O, otic vesicle; R, rhombomere.
Pax3 misexpression contributes to outflow tract defects inPbx1-/- embryos
Cardiac OFT defects in Pax3 mutant embryos arise from derepression of its downstream target gene Msx2, as demonstrated by rescue of the PTA in Pax3-/-;Msx2-/- embryos (Kwang et al., 2002). Since Msx2 is repressed by Pax3, the activation of which requires Pbx1, we examined Pbx1-/- embryos for misexpression of Msx2. By whole-mount RNA in situ hybridization, Msx2 transcripts were detected in the neural tube of E9.5 wild-type embryos (Kwang et al., 2002), with high levels in R5 and lower levels in the cardiac NCC-originating R6-R8 (Fig. 6E). By contrast, considerably higher levels of Msx2 transcripts were detected in R6 through R8 of littermate Pbx1-/- embryos (Fig. 6F). RNA in situ hybridization of tissue sections at the R6 level confirmed enhanced expression of Msx2 transcripts in the dorsal neural tube in Pbx1-/- embryos (Fig. 6G,H). These results, which mirror those reported in Pax3-/- embryos, suggest that decreased Pax3 expression in Pbx1-/- embryos results in derepression of Msx2, which then causes the observed OFT septation defects.
To assess whether Msx2 misexpression contributes to the developmental abnormalities in Pbx1-/- embryos, mice with compound deficiencies of Pbx1 and Msx2 were generated by interbreeding. Pbx1-/-;Msx2-/- embryos (Fig. 7D) were grossly similar to Pbx1-/- embryos (Fig. 7B); they showed edema and did not survive past E14.5, in contrast to the normal appearance of Msx2-/- embryos (Fig. 7A,C). Thus, increased rhombomeric Msx2 expression resulting from Pbx1 deficiency does not account for the gross defects observed in Pbx1-/- embryos. Furthermore, angiography revealed that Pbx1-/-;Msx2+/- and Pbx1-/-;Msx2-/-embryos exhibited the same spectrum of great-artery malformations as seen in Pbx1-/- embryos, including cervical aortic arch, aberrant carotid and subclavian arteries (data not shown). This indicates that Msx2 misexpression is not the main cause of the arterial patterning defects in Pbx1-/- mice.
Fig. 7. Msx2 deficiency rescues the truncal septation defect of Pbx1-/- embryos.

(A-D) Gross morphology of E14.5 mouse embryos of the indicated genotypes with Pbx1 and Msx2 loss-of-function alleles. Arrows indicate generalized edema. (E-M) Hematoxylin and Eosin-stained transverse sections through E14.5 embryos of the indicated genotypes showing that loss of one allele of Msx2 rescues truncal but not conal septation in Pbx1-/- embryos. Ao, aorta; DA, ductus arteriosus; MPA, main pulmonary artery; RV, right ventricle; RVOT and LVOT, right and left ventricular OFTs; LV, left ventricle; PTA, persistent truncus arteriosus; VSD, ventricular septal defect.
The potential recovery of NCC contributions to OFT septation in Pbx1-/- embryos lacking Msx2 was assessed by examining consecutive histologic sections through the cardiac OFT. This revealed significant rescue of septation of the distal (truncal) portion of the OFT in Pbx1-/-;Msx2+/- embryos (Fig. 7H,K) (4/6 embryos; mean length of septation, 75.8 μm) and Pbx1-/-;Msx2-/- embryos (data not shown) (3/4; mean, 47.5 μm), as compared with littermate Pbx1-/- embryos (0/3; mean, 6.7 μm) (P<0.015). Since rescue did not extend to the proximal (conal) region of the OFT (Fig. 7I,J,L,M), these Pbx1-/-;Msx2+/- embryos had a milder form of PTA, arising from the right ventricle with an associated ventricular septal defect. By comparison, Msx2-/- embryos had no defects in OFT septation (Fig. 7E-G). Septation of the distal truncal region of the OFT is provided by the NCC-derived aorticopulmonary septal complex (Hutson and Kirby, 2007). Thus, Pbx1-/- embryos deficient for one or both Msx2 alleles had significant recovery of NCC function, with septation of the truncal, but not conal, region. These results demonstrate that dysfunction of the Pax3-Msx2 transcriptional hierarchy contributes to septation defects in Pbx1-/- embryos, although it does not entirely account for the role of Pbx1, suggesting that Pbx1 impacts additional pathways to regulate cardiac NCCs or other tissues contributing to OFT development.
DISCUSSION
Pbx1 is a global developmental regulator implicated in the formation of many organ systems (Brendolan et al., 2005; DiMartino et al., 2001; Manley et al., 2004; Schnabel et al., 2003a; Schnabel et al., 2003b; Zhang et al., 2006). However, the specific subordinate pathways that mediate its developmental contributions have not been extensively characterized. Our current studies extend the roles of Pbx1 to major morphogenetic events underlying the patterning of branchial arch arteries and formation of the cardiac OFT (Fig. 8). Vascular abnormalities in Pbx1-/- embryos include cervical aortic arch, right-sided aortic arch, and retroesophageal vascular ring, each of which is frequently encountered in human patients (De la Cruz and Markwald, 1998; Sandler, 2004). The absence of cardiac OFT septation in Pbx1-/- embryos, resulting in PTA, is partially accounted for by loss of Pax3 expression in premigratory NCCs, culminating in a failure of aorticopulmonary septation. Interestingly, the cardiovascular defects in Pbx1-/- embryos, combined with craniofacial abnormalities (Selleri et al., 2001) and hypoplastic thymus, thyroid and parathyroid glands (Manley et al., 2004), resemble the anomalies observed in patients with DiGeorge syndrome (Epstein, 2001), which results from a deletion of chromosome 22q11 that includes TBX1, CRKL and other genes (Merscher et al., 2001; Moon et al., 2006). These features underscore the contributions of Pbx1 to the development of the caudal branchial arches and their derived organs, in addition to the cardiac OFT.
Fig. 8. A working model of Pbx1 function during branchial arch artery and conotruncal development in the mouse.

In the absence of Pbx1, the right and left internal and external carotid arteries branch directly off the aortic arch, the brachiocephalic artery is missing, and the right and left subclavian arteries have aberrant origins from the descending aorta. These defects originate in a failure to establish the fourth and sixth aortic arches owing to small or absent fourth and sixth branchial arches. In premigratory NCCs, Pbx1-Meis and/or Pbx1-Hox transcriptional complexes activate a transient but robust Pax3 expression that is seen on E8. This induction is required to repress Msx2 and ultimately guide establishment of the aorticopulmonary septum by cardiac NCCs. Pbx1 complexes have additional, uncharacterized functions within NCCs or other cells in which Pbx1 is expressed, including those of the secondary heart field, to regulate conotruncal septation at the base of the great arteries (aorta and main pulmonary artery) and conal region of the heart. APS, aorticopulmonary septum; DA, ductus arteriosus; T, truncus; C, conus; TA, truncus arteriosus; RBCA, right brachiocephalic artery; LCCA, left common carotid artery; LSCA, left subclavian artery; Desc.A. descending aorta; LICA and RICA, left and right internal carotid arteries; LECA and RECA, left and right external carotid arteries.
Failure of Pbx1-/- embryos to establish the caudal branchial arches might result in the absence of the fourth and sixth arch arteries, eventually producing an abnormal great-artery pattern. Loss of the fourth and sixth arch arteries in Pbx1-/- embryos accounts for the anomalous derivation of the aortic arch from the third branchial arch and for the absence of the ductus arteriosus, which is normally derived from the sixth arch artery. The consequently more-rostral location of the aortic arch prevents the heart from descending into the thoracic cavity, resulting in a cervical position of the aortic arch and the heart. The third branchial arch artery might not remodel as normal, owing to increased blood flow in the absence of the fourth arch artery, as hemodynamics have recently been shown to cooperate in branchial arch artery remodeling (Yashiro et al., 2007). The anomalous aortic arch derived from the third arch artery lies at the position where the ICA and ECA normally branch from the CCA, resulting in all four carotid arteries emerging from the aortic arch. The change in position of the RSA to an origin off the descending aorta is likewise explained by the absence of the right fourth arch artery, which normally connects the RSA to the BCA. The LSA appears more caudal in origin because the heart has moved rostrally to a cervical location in Pbx1-/- embryos.
Misregulation of Hox activity, which depends on Pbx function, might contribute to the arch artery defects in Pbx1-/- embryos as Hox genes are known to regulate branchial arch artery patterning. Hoxa3-null mice exhibit regression of the third arch artery (Kameda et al., 2003), and antisense targeted to Hox transcripts causes aberrant arch arteries in chick embryos (Kirby et al., 1997). Despite the evidence for a role of Pbx/Hox genes in branchial artery development, chemical targeting of Hox mRNAs in the chick was not accompanied by cardiac OFT defects (Kirby et al., 1997). Nor have studies of Hox-deficient mice shown cardiac malformations, as seen in Pbx1-deficient embryos. This is likely to reflect redundancy in the contributions of Hox genes, which is circumvented by the broader Hox compromise induced by Pbx1 deficiency. Further studies of Pbx1-deficient mice are likely to yield novel insights into the contributions of Pbx1 and Hox genes to various regulatory pathways in cardiac development that might not be apparent from studies of Hox-deficient mice.
Our studies showing the requirement of a Pbx1-Pax3-Msx2 pathway in cardiac development provide an additional example that Pbx and Pax genes act together to regulate organ development. Pbx proteins are known to regulate the expression of Pax6 during pancreatic development (Zhang et al., 2006). Here, we demonstrate that Pbx1 regulates Pax3 expression to control development of the cardiac OFT and involving the function of NCCs. Besides OFT defects, Splotch mice, which are deficient for Pax3, exhibit defects in thymus, thyroid, parathyroid and branchial arch artery development, resembling the malformations observed in Pbx1-/- embryos and chicks ablated for NCCs (Conway et al., 1997; Epstein, 1996; Franz, 1989; Kirby et al., 1983; Kwang et al., 2002; Li et al., 1999). Similarities in these NCC-derived organ defects between Splotch and Pbx1-/- mice suggest that Pax3 misregulation might underlie the phenotypes observed in Pbx1-/- embryos, including branchial arch artery defects. The arch artery defects, however, do not involve Msx2 because Msx2-/- mutations fail to rescue the great-artery malformations of the Pbx1-/- embryos, despite the rescue of cardiac OFT development.
Cardiac OFT defects seen in Pax3 mutants arise from derepression of its downstream target gene, Msx2, in rhombomeres where cardiac NCCs originate. This was demonstrated by rescue of PTA in Pax3-/-;Msx2-/- embryos (Kwang et al., 2002). In Pbx1-/-embryos, we observed a significant reduction of Pax3 and enhancement of Msx2 gene expression in rhombomeres contributing to cardiac NCCs. These observations, together with DNA-binding, cellular transactivation and transgenic reporter assays, indicate that Pax3 is a direct in vivo transcriptional target of Pbx1, and establish a Pbx1-Pax3-Msx2 transcriptional cascade in heart development. Genetic support for this conclusion is provided by a significant rescue of aorticopulmonary septation in 70% of embryos containing both Pbx1 and Msx2 mutations (n=10), as evidenced by reduction of the PTA to milder conal defects, which we have never observed in Pbx1-/- embryos (n=28). Given that Pax3 and Msx2 function cell-autonomously in NCCs to regulate cardiac OFT development (Kwang et al., 2002; Li et al., 1999), our rescue experiments suggest that misregulation of the Pbx1-Pax3-Msx2 pathway in NCCs contributes to cardiac defects in Pbx1-/- embryos.
Although the partial rescue of PTA in Pbx1-/- embryos by Msx2 deficiency points to Pax3 misexpression within premigratory NCCs as underlying the truncal septation defects, we did not observe a widespread change in Pax3 expression within the neural tube. Rather, Pbx1-null embryos lack a transient `burst' of Pax3 in premigratory NCCs prior to their delamination. By contrast, the newly emigrated NCCs retain normal Pax3 levels in the absence of Pbx1. This is consistent with a lack of NCC emigration defects from the neural tube in both Pbx1- and Pax3-null embryos (Conway et al., 1997; Epstein et al., 2000). We propose that the Pbx1-dependent Pax3 expression in premigratory NCCs confers a cellular identity to R6-derived cardiac NCCs that does not affect their migration, but specifies their ability to participate in OFT septation. BMP and FGF signaling pathways might cooperate with Pbx1 to induce the transient expression of Pax3 in premigratory NCCs, given their roles in regulating Pax3 expression in the dorsal neural tube of Xenopus embryos (Monsoro-Burq et al., 2005; Sato et al., 2005). It will be of interest to test these possibilities by early-targeted deletion of Pbx1 and Pax3 in NCCs prior to their emigration from the neural tube.
The rescue of truncal, but not conal, septation in Pbx1-/-;Msx2+/-embryos indicates that Pbx1 might regulate other, unidentified genes in NCCs independent of the Pax3-Msx2 pathway (Fig. 7). Alternatively, Pbx1 might be required in other cell types that regulate OFT development and in which it is expressed, including SHF cells that provide the smooth muscle, endocardium and myocardium of the cardiac OFT (Kelly et al., 2001; Mjaatvedt et al., 2001; Verzi et al., 2005; Waldo et al., 2005). An additional possibility is the pharyngeal endoderm, where both Fgf8 and Tbx1, the null phenotypes of which resemble that of Pbx1-/- embryos (Abu-Issa et al., 2002; Frank et al., 2002; Jerome and Papaioannou, 2001; Lindsay et al., 2001; Merscher et al., 2001), have been proposed to be required for OFT septation (Arnold et al., 2006; Brown et al., 2004; Park et al., 2006; Zhang et al., 2005). These potential non-NCC functions of Pbx1 require further investigations that will involve its tissue-specific deletion in the mesoderm, endoderm or SHF cells that express Pbx1 and contribute to the development of the cardiac OFT.
Supplementary Material
Acknowledgments
We thank M. Ambrus, C. Nicolas, C. Hang and Dr M. Zeini for assistance; Dr J. Epstein for providing the plexin A2 probe and the Pax3Cre mice; Dr R. Maas for providing the Msx2 probe; Dr A. McMahon for providing Wnt1Cre mice; and Drs G. Crabtree, M. Rabinovitch, D. Bernstein, T. Quertermous and members of the Chang laboratory for their advice. This work was supported by funds from the National Cancer Institute (CA90735, CA42971) to M.L.C., and by funds from the National Heart Lung and Blood Institute (HL085345), American Heart Association (AHA), Children's Heart Foundation and March of Dimes Foundation to C.-P.C. K.S. is supported by an AHA postdoctoral fellowship, C.S. by an NIH NRSA fellowship, and K.Y.T. by the McCormick fellowship.
Footnotes
Supplementary material Supplementary material for this article is available at http://dev.biologists.org/cgi/content/full/135/21/3577/DC1
References
- Abu-Issa R, Smyth G, Smoak I, Yamamura K, Meyers EN. Fgf8 is required for pharyngeal arch and cardiovascular development in the mouse. Development. 2002;129:4613–4625. doi: 10.1242/dev.129.19.4613. [DOI] [PubMed] [Google Scholar]
- Abu-Shaar M, Ryoo HD, Mann RS. Control of the nuclear localization of Extradenticle by competing nuclear import and export signals. Genes Dev. 1999;13:935–945. doi: 10.1101/gad.13.8.935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold JS, Werling U, Braunstein EM, Liao J, Nowotschin S, Edelmann W, Hebert JM, Morrow BE. Inactivation of Tbx1 in the pharyngeal endoderm results in 22q11DS malformations. Development. 2006;133:977–987. doi: 10.1242/dev.02264. [DOI] [PubMed] [Google Scholar]
- Brendolan A, Ferretti E, Salsi V, Moses K, Quaggin S, Blasi F, Cleary ML, Selleri L. A Pbx1-dependent genetic and transcriptional network regulates spleen ontogeny. Development. 2005;132:3113–3126. doi: 10.1242/dev.01884. [DOI] [PubMed] [Google Scholar]
- Brown CB, Feiner L, Lu MM, Li J, Ma X, Webber AL, Jia L, Raper JA, Epstein JA. PlexinA2 and semaphorin signaling during cardiac neural crest development. Development. 2001;128:3071–3080. doi: 10.1242/dev.128.16.3071. [DOI] [PubMed] [Google Scholar]
- Brown CB, Wenning JM, Lu MM, Epstein DJ, Meyers EN, Epstein JA. Cre-mediated excision of Fgf8 in the Tbx1 expression domain reveals a critical role for Fgf8 in cardiovascular development in the mouse. Dev. Biol. 2004;267:190–202. doi: 10.1016/j.ydbio.2003.10.024. [DOI] [PubMed] [Google Scholar]
- Chang CP, Shen WF, Rozenfeld S, Lawrence HJ, Largman C, Cleary ML. Pbx proteins display hexapeptide-dependent cooperative DNA binding with a subset of Hox proteins. Genes Dev. 1995;9:663–674. doi: 10.1101/gad.9.6.663. [DOI] [PubMed] [Google Scholar]
- Chang CP, Brocchieri L, Shen WF, Largman C, Cleary ML. Pbx modulation of Hox homeodomain amino-terminal arms establishes different DNA-binding specificities across the Hox locus. Mol. Cell. Biol. 1996;16:1734–1745. doi: 10.1128/mcb.16.4.1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CP, Jacobs Y, Nakamura T, Jenkins NA, Copeland NG, Cleary ML. Meis proteins are major in vivo DNA binding partners for wild-type but not chimeric Pbx proteins. Mol. Cell. Biol. 1997;17:5679–5687. doi: 10.1128/mcb.17.10.5679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CP, Chen L, Crabtree GR. Sonographic staging of the developmental status of mouse embryos in utero. Genesis. 2003;36:7–11. doi: 10.1002/gene.10186. [DOI] [PubMed] [Google Scholar]
- Chang CP, Neilson JR, Bayle JH, Gestwicki JE, Kuo A, Stankunas K, Graef IA, Crabtree GR. A field of myocardial-endocardial NFAT signaling underlies heart valve morphogenesis. Cell. 2004;118:649–663. doi: 10.1016/j.cell.2004.08.010. [DOI] [PubMed] [Google Scholar]
- Chisaka O, Capecchi MR. Regionally restricted developmental defects resulting from targeted disruption of the mouse homeobox gene hox-1.5. Nature. 1991;350:473–479. doi: 10.1038/350473a0. [DOI] [PubMed] [Google Scholar]
- Chisaka O, Kameda Y. Hoxa3 regulates the proliferation and differentiation of the third pharyngeal arch mesenchyme in mice. Cell Tissue Res. 2005;320:77–89. doi: 10.1007/s00441-004-1042-z. [DOI] [PubMed] [Google Scholar]
- Choe SK, Vlachakis N, Sagerstrom CG. Meis family proteins are required for hindbrain development in the zebrafish. Development. 2002;129:585–595. doi: 10.1242/dev.129.3.585. [DOI] [PubMed] [Google Scholar]
- Conway SJ, Henderson DJ, Kirby ML, Anderson RH, Copp AJ. Development of a lethal congenital heart defect in the splotch (Pax3) mutant mouse. Cardiovasc. Res. 1997;36:163–173. doi: 10.1016/s0008-6363(97)00172-7. [DOI] [PubMed] [Google Scholar]
- De la Cruz M, Markwald RR. Living Morphogenesis of the Heart. Birkhauser; Boston, MA: 1998. [Google Scholar]
- DiMartino JF, Selleri L, Traver D, Firpo MT, Rhee J, Warnke R, O'Gorman S, Weissman IL, Cleary ML. The Hox cofactor and proto-oncogene Pbx1 is required for maintenance of definitive hematopoiesis in the fetal liver. Blood. 2001;98:618–626. doi: 10.1182/blood.v98.3.618. [DOI] [PubMed] [Google Scholar]
- Epstein J. Pax3, neural crest and cardiovascular development. Trends Cardiovasc. Med. 1996;6:255–261. doi: 10.1016/S1050-1738(96)00110-7. [DOI] [PubMed] [Google Scholar]
- Epstein JA. Developing models of DiGeorge syndrome. Trends Genet. 2001;17:S13–S17. doi: 10.1016/s0168-9525(01)02450-7. [DOI] [PubMed] [Google Scholar]
- Epstein JA, Li J, Lang D, Chen F, Brown CB, Jin F, Lu MM, Thomas M, Liu E, Wessels A, et al. Migration of cardiac neural crest cells in Splotch embryos. Development. 2000;127:1869–1878. doi: 10.1242/dev.127.9.1869. [DOI] [PubMed] [Google Scholar]
- Frank DU, Fotheringham LK, Brewer JA, Muglia LJ, Tristani-Firouzi M, Capecchi MR, Moon AM. An Fgf8 mouse mutant phenocopies human 22q11 deletion syndrome. Development. 2002;129:4591–4603. doi: 10.1242/dev.129.19.4591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franz T. Persistent truncus arteriosus in the Splotch mutant mouse. Anat. Embryol. 1989;180:457–464. doi: 10.1007/BF00305120. [DOI] [PubMed] [Google Scholar]
- Greene LA, Tischler AS. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc. Natl. Acad. Sci. USA. 1976;73:2424–2428. doi: 10.1073/pnas.73.7.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey RP, Rosenthal N. Heart Development. Academic Press; New York, NY: 1999. [Google Scholar]
- Hutson MR, Kirby ML. Model systems for the study of heart development and disease. Cardiac neural crest and conotruncal malformations. Semin. Cell Dev. Biol. 2007;18:101–110. doi: 10.1016/j.semcdb.2006.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs Y, Schnabel C, Cleary M. Trimeric association of Hox and TALE homeodomain proteins mediates Hoxb2 hindbrain enhancer activity. Mol. Cell. Biol. 1999;19:5134–5142. doi: 10.1128/mcb.19.7.5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerome LA, Papaioannou VE. DiGeorge syndrome phenotype in mice mutant for the T-box gene, Tbx1. Nat. Genet. 2001;27:286–291. doi: 10.1038/85845. [DOI] [PubMed] [Google Scholar]
- Jiang X, Rowitch DH, Soriano P, McMahon AP, Sucov HM. Fate of the mammalian cardiac neural crest. Development. 2000;127:1607–1616. doi: 10.1242/dev.127.8.1607. [DOI] [PubMed] [Google Scholar]
- Kameda Y, Watari-Goshima N, Nishimaki T, Chisaka O. Disruption of the Hoxa3 homeobox gene results in anomalies of the carotid artery system and the arterial baroreceptors. Cell Tissue Res. 2003;311:343–352. doi: 10.1007/s00441-002-0681-1. [DOI] [PubMed] [Google Scholar]
- Kelly RG, Brown NA, Buckingham ME. The arterial pole of the mouse heart forms from Fgf10-expressing cells in pharyngeal mesoderm. Dev. Cell. 2001;1:435–440. doi: 10.1016/s1534-5807(01)00040-5. [DOI] [PubMed] [Google Scholar]
- Kim SK, Selleri L, Lee JS, Zhang AY, Gu X, Jacobs Y, Cleary ML. Pbx1 inactivation disrupts pancreas development and in Ipf1-deficient mice promotes diabetes mellitus. Nat. Genet. 2002;30:430–435. doi: 10.1038/ng860. [DOI] [PubMed] [Google Scholar]
- Kirby ML. Cardiac Development. Oxford University Press; New York, NY: 2007. [Google Scholar]
- Kirby ML, Gale TF, Stewart DE. Neural crest cells contribute to normal aorticopulmonary septation. Science. 1983;220:1059–1061. doi: 10.1126/science.6844926. [DOI] [PubMed] [Google Scholar]
- Kirby ML, Hunt P, Wallis K, Thorogood P. Abnormal patterning of the aortic arch arteries does not evoke cardiac malformations. Dev. Dyn. 1997;208:34–47. doi: 10.1002/(SICI)1097-0177(199701)208:1<34::AID-AJA4>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Knoepfler PS, Kamps MP. The pentapeptide motif of Hox proteins is required for cooperative DNA binding with Pbx1, physically contacts Pbx1, and enhances DNA binding by Pbx1. Mol. Cell. Biol. 1995;15:5811–5819. doi: 10.1128/mcb.15.10.5811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwang SJ, Brugger SM, Lazik A, Merrill AE, Wu LY, Liu YH, Ishii M, Sangiorgi FO, Rauchman M, Sucov HM, et al. Msx2 is an immediate downstream effector of Pax3 in the development of the murine cardiac neural crest. Development. 2002;129:527–538. doi: 10.1242/dev.129.2.527. [DOI] [PubMed] [Google Scholar]
- Li J, Liu KC, Jin F, Lu MM, Epstein JA. Transgenic rescue of congenital heart disease and spina bifida in Splotch mice. Development. 1999;126:2495–2503. doi: 10.1242/dev.126.11.2495. [DOI] [PubMed] [Google Scholar]
- Li J, Chen F, Epstein JA. Neural crest expression of Cre recombinase directed by the proximal Pax3 promoter in transgenic mice. Genesis. 2000;26:162–164. doi: 10.1002/(sici)1526-968x(200002)26:2<162::aid-gene21>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- Lindsay EA, Vitelli F, Su H, Morishima M, Huynh T, Pramparo T, Jurecic V, Ogunrinu G, Sutherland HF, Scambler PJ, et al. Tbx1 haploinsufficieny in the DiGeorge syndrome region causes aortic arch defects in mice. Nature. 2001;410:97–101. doi: 10.1038/35065105. [DOI] [PubMed] [Google Scholar]
- MacKenzie A, Ferguson MW, Sharpe PT. Expression patterns of the homeobox gene, Hox-8, in the mouse embryo suggest a role in specifying tooth initiation and shape. Development. 1992;115:403–420. doi: 10.1242/dev.115.2.403. [DOI] [PubMed] [Google Scholar]
- Manley NR, Selleri L, Brendolan A, Gordon J, Cleary ML. Abnormalities of caudal pharyngeal pouch development in Pbx1 knockout mice mimic loss of Hox3 paralogs. Dev. Biol. 2004;276:301–312. doi: 10.1016/j.ydbio.2004.08.030. [DOI] [PubMed] [Google Scholar]
- Merscher S, Funke B, Epstein JA, Heyer J, Puech A, Lu MM, Xavier RJ, Demay MB, Russell RG, Factor S, et al. TBX1 is responsible for cardiovascular defects in velo-cardio-facial/DiGeorge syndrome. Cell. 2001;104:619–629. doi: 10.1016/s0092-8674(01)00247-1. [DOI] [PubMed] [Google Scholar]
- Mjaatvedt CH, Nakaoka T, Moreno-Rodriguez R, Norris RA, Kern MJ, Eisenberg CA, Turner D, Markwald RR. The outflow tract of the heart is recruited from a novel heart-forming field. Dev. Biol. 2001;238:97–109. doi: 10.1006/dbio.2001.0409. [DOI] [PubMed] [Google Scholar]
- Monsoro-Burq AH, Wang E, Harland R. Msx1 and Pax3 cooperate to mediate FGF8 and WNT signals during Xenopus neural crest induction. Dev. Cell. 2005;8:167–178. doi: 10.1016/j.devcel.2004.12.017. [DOI] [PubMed] [Google Scholar]
- Moon AM, Guris DL, Seo JH, Li L, Hammond J, Talbot A, Imamoto A. Crkl deficiency disrupts Fgf8 signaling in a mouse model of 22q11 deletion syndromes. Dev. Cell. 2006;10:71–80. doi: 10.1016/j.devcel.2005.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park EJ, Ogden LA, Talbot A, Evans S, Cai CL, Black BL, Frank DU, Moon AM. Required, tissue-specific roles for Fgf8 in outflow tract formation and remodeling. Development. 2006;133:2419–2433. doi: 10.1242/dev.02367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peltenburg LT, Murre C. Engrailed and Hox homeodomain proteins contain a related Pbx interaction motif that recognizes a common structure present in Pbx. EMBO J. 1996;15:3385–3393. [PMC free article] [PubMed] [Google Scholar]
- Phelan ML, Rambaldi I, Featherstone MS. Cooperative interactions between HOX and PBX proteins mediated by a conserved peptide motif. Mol. Cell. Biol. 1995;15:3989–3997. doi: 10.1128/mcb.15.8.3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruitt SC, Bussman A, Maslov AY, Natoli TA, Heinaman R. Hox/Pbx and Brn binding sites mediate Pax3 expression in vitro and in vivo. Gene Expr. Patterns. 2004;4:671–685. doi: 10.1016/j.modgep.2004.04.006. [DOI] [PubMed] [Google Scholar]
- Sandler T. Langman's Medical Embryology. Lippincott Williams & Wilkins; Philadelphia, PA: 2004. [Google Scholar]
- Sato T, Sasai N, Sasai Y. Neural crest determination by co-activation of Pax3 and Zic1 genes in Xenopus ectoderm. Development. 2005;132:2355–2363. doi: 10.1242/dev.01823. [DOI] [PubMed] [Google Scholar]
- Schnabel CA, Selleri L, Jacobs Y, Warnke R, Cleary ML. Expression of Pbx1b during mammalian organogenesis. Mech. Dev. 2001;100:131–135. doi: 10.1016/s0925-4773(00)00516-5. [DOI] [PubMed] [Google Scholar]
- Schnabel CA, Godin RE, Cleary ML. Pbx1 regulates nephrogenesis and ureteric branching in the developing kidney. Dev. Biol. 2003a;254:262–276. doi: 10.1016/s0012-1606(02)00038-6. [DOI] [PubMed] [Google Scholar]
- Schnabel CA, Selleri L, Cleary ML. Pbx1 is essential for adrenal development and urogenital differentiation. Genesis. 2003b;37:123–130. doi: 10.1002/gene.10235. [DOI] [PubMed] [Google Scholar]
- Selleri L, Depew MJ, Jacobs Y, Chanda SK, Tsang KY, Cheah KS, Rubenstein JL, O'Gorman S, Cleary ML. Requirement for Pbx1 in skeletal patterning and programming chondrocyte proliferation and differentiation. Development. 2001;128:3543–3557. doi: 10.1242/dev.128.18.3543. [DOI] [PubMed] [Google Scholar]
- Stankunas K, Hang CT, Tsun ZY, Chen H, Lee NV, Wu JI, Shang C, Bayle JH, Shou W, Iruela-Arispe ML, et al. Endocardial Brg1 represses ADAMTS1 to maintain the microenvironment for myocardial morphogenesis. Dev. Cell. 2008a;14:298–311. doi: 10.1016/j.devcel.2007.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankunas K, Shang C, Twu KY, Kao SC, Jenkins NA, Copeland NG, Sanyal M, Selleri L, Cleary ML, Chang CP. Pbx/Meis deficiencies demonstrate multigenetic origins of congenital heart disease. Circ. Res. 2008b;103:702–709. doi: 10.1161/CIRCRESAHA.108.175489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verzi MP, McCulley DJ, De Val S, Dodou E, Black BL. The right ventricle, outflow tract, and ventricular septum comprise a restricted expression domain within the secondary/anterior heart field. Dev. Biol. 2005;287:134–145. doi: 10.1016/j.ydbio.2005.08.041. [DOI] [PubMed] [Google Scholar]
- Waldo KL, Hutson MR, Ward CC, Zdanowicz M, Stadt HA, Kumiski D, Abu-Issa R, Kirby ML. Secondary heart field contributes myocardium and smooth muscle to the arterial pole of the developing heart. Dev. Biol. 2005;281:78–90. doi: 10.1016/j.ydbio.2005.02.012. [DOI] [PubMed] [Google Scholar]
- Waskiewicz AJ, Rikhof HA, Hernandez RE, Moens CB. Zebrafish Meis functions to stabilize Pbx proteins and regulate hindbrain patterning. Development. 2001;128:4139–4151. doi: 10.1242/dev.128.21.4139. [DOI] [PubMed] [Google Scholar]
- Waskiewicz AJ, Rikhof HA, Moens CB. Eliminating zebrafish pbx proteins reveals a hindbrain ground state. Dev. Cell. 2002;3:723–733. doi: 10.1016/s1534-5807(02)00319-2. [DOI] [PubMed] [Google Scholar]
- Wu H, Kao SC, Barrientos T, Baldwin SH, Olson EN, Crabtree GR, Zhou B, Chang CP. Down syndrome critical region-1 is a transcriptional target of nuclear factor of activated T cells-c1 within the endocardium during heart development. J. Biol. Chem. 2007;282:30673–30679. doi: 10.1074/jbc.M703622200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yashiro K, Shiratori H, Hamada H. Haemodynamics determined by a genetic programme govern asymmetric development of the aortic arch. Nature. 2007;450:285–288. doi: 10.1038/nature06254. [DOI] [PubMed] [Google Scholar]
- Zhang X, Rowan S, Yue Y, Heaney S, Pan Y, Brendolan A, Selleri L, Maas RL. Pax6 is regulated by Meis and Pbx homeoproteins during pancreatic development. Dev. Biol. 2006;300:748–757. doi: 10.1016/j.ydbio.2006.06.030. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Cerrato F, Xu H, Vitelli F, Morishima M, Vincentz J, Furuta Y, Ma L, Martin JF, Baldini A, et al. Tbx1 expression in pharyngeal epithelia is necessary for pharyngeal arch artery development. Development. 2005;132:5307–5315. doi: 10.1242/dev.02086. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.