

Abstract
C1 inhibitor therapy improves outcome in several animal models of inflammatory disease. These include sepsis and gram negative endotoxin shock, vascular leak syndromes, hyperacute transplant rejection, and ischemia-reperfusion injury. Furthermore, some data suggest a beneficial effect in human inflammatory disease. In many inflammatory conditions, complement system activation plays a role in pathogenesis. The contact system also very likely is involved in mediation of damage in inflammatory disease. Therefore, the beneficial effect of C1 inhibitor has been assumed to result from inhibition of one or both of these systems. Over the past several years, several other potential anti-inflammatory effects of C1 inhibitor have been described. These effects do not appear to require protease inhibition and depend on non-covalent interactions with other proteins, cell surfaces or lipids. In the first, C1 inhibitor binds to a variety of extracellular matrix components including type IV collagen, laminin, entactin and fibrinogen. The biologic role of these reactions is unclear, but they may serve to concentrate C1 inhibitor at extravascular inflammatory sites. The second is a noncovalent interaction with C3b that results in inhibition of formation of the alternative pathway C3 convertase, a function analogous to that of factor H. The third is an interaction with E and P selectins on endothelial cells that is mediated by the Lewisx tetrasaccharides that are expressed on C1 inhibitor. These interactions result in suppression of leukocyte rolling and transmigration. The fourth interaction is the binding of C1 inhibitor to gram negative bacterial endotoxin that results in suppression of endotoxin shock by interference with the interaction of endotoxin with its receptor complex on macrophages. Lastly, C1 inhibitor binds directly to gram negative bacteria, which leads to suppression of the development of sepsis, as demonstrated in the cecal ligation and puncture model. These observations suggest that C1 inhibitor is a multi-faceted anti-inflammatory protein that exerts its effects through a variety of mechanisms including both protease inhibition and several different non-covalent interactions that are unrelated to protease inhibition.
Keywords: C1 inhibitor, cecal ligation and puncture model, endotoxin shock, gram negative bacterial sepsis, selectins, serine proteinase inhibitors
Introduction
Treatment with C1 inhibitor improves outcome in a number of animal models of inflammatory disease including gram negative bacterial sepsis and endotoxin shock, vascular leak syndromes, hyperacute transplant rejection, and ischemia-reperfusion injury (Akita et al., 2003; Buerke et al., 1995; Buerke et al., 1998; Dalmasso and Platt, 1993; De Simoni et al., 2003; De Simoni et al., 2004a; Fiane et al., 1999; Fukuta et al., 2003; Giebler et al., 1999; Guerrero et al., 1993; Hecker et al., 2002; Henze et al., 1997; Horstick et al., 1997; Horstick et al., 2001a; Jansen et al., 1998; Khorram-Sefat et al., 1998; Matsunami et al., 2000; Nielsen et al., 2002; Przemeck et al., 2002; Radke et al., 2000; Schelzig et al., 2001; Scherer et al., 1996; Schmidt et al., 1999a; Schmidt et al., 1999b)(Table 1). For example, in myocardial ischemia-reperfusion injury, treatment with C1 inhibitor resulted in decreased infarct size, decreased myocardial neutrophil accumulation, decreased plasma levels of creatine kinase, C3a and C5a, and decreased expression of P-selectin and ICAM-1 within the cardiac endothelium (Buerke et al., 1995; Buerke et al., 1998; Horstick et al., 1997; Horstick et al., 2002). Similar effects have been observed in skeletal muscle and middle cerebral artery ischemia-reperfusion injury (Akita et al., 2003; De Simoni et al., 2003; Nielsen et al., 2002). One study suggested that the C1 inhibitor-mediated effect was independent of inhibition of classical pathway activation; a role for inhibition of lectin pathway activation was not examined (De Simoni et al., 2004b). In addition, limited experience in human disease suggests that C1 inhibitor may prove to be a useful addition to therapy of diseases including vascular leak syndromes, sepsis, and shock syndromes (Bauernschmitt et al., 1998; Bauernschmitt et al., 2000; de Zwaan et al., 2002; Hack et al., 1993; Hack et al., 1994; Niederau et al., 1995; Ogilvie et al., 1994; Tassani et al., 2001) (Table 1). Two studies have suggested a beneficial effect in humans with ischemia-reperfusion injury (Bauernschmitt et al., 1998; de Zwaan et al., 2002). Because complement system activation plays a role in the mediation of many of these conditions, these beneficial effects have been thought to be a product of inhibition of activation of the complement system. Protease inhibition clearly is the major biologic role of C1 inhibitor. It is the only protease inhibitor that inactivates C1r and C1s (Sim et al., 1979; Ziccardi, 1981), the initial proteases activated via the classical complement pathway. It also regulates lectin pathway activation via inhibition of MASP2 (Ambrus et al., 2003; Matsushita et al., 2000a; Matsushita et al., 2000b; Matsushita et al., 2001), the initial protease activated in this pathway. However, C1 inhibitor may share this latter role with α2 macroglobulin (Ambrus et al., 2003). In the contact or kinin system, C1 inhibitor is the primary inhibitor of both plasma kallikrein and factor XIIa (de Agostini et al., 1984; Kaplan et al., 1985; Pixley et al., 1985). In this instance, α2 macroglobulin clearly plays a lesser role than does C1 inhibitor. It is certain that activation of these pathways have important inflammatory effects, and that their inhibition, whether by C1 inhibitor or other inhibitors, is anti-inflammatory. However, over the past several years, several other interactions of C1 inhibitor with other proteins or cells have come to light. These interactions do not require protease inhibition. Initial data suggest that, in some instances, these reactions may be significant contributors to the anti-inflammatory activity of this important protease inhibitor. These include at least four different binding reactions (Table 2): (1) An interaction with a variety of extracellular matrix components (Patston and Schapira, 1997); (2) inhibition of alternative complement pathway activation via binding of C1 inhibitor to C3b which prevents its interaction with factor B, in a manner analogous to that of factor H (Jiang et al., 2001); (3) an interaction with both E and P selectins on endothelial cells that is mediated by one or more Lewisx tetrasaccharides expressed on C1 inhibitor, and which results in suppression of leukocyte rolling and transmigration (Cai and Davis III, 2003; Cai et al., 2005); (4) binding to endotoxin from several different gram negative bacteria that results in suppression of endotoxin shock by interference with the interaction of endotoxin with its receptor complex on macrophages (Liu et al., 2003; Liu et al., 2004; Liu et al., 2005a; Liu et al., 2005b); (5) direct binding to gram negative bacteria that results in suppression of the development of sepsis (unpublished data). This review will briefly summarize the limited information available on the first two reactions and will describe in more detail the data relating to the interactions with selectins and with endotoxin lipopolysaccharide (LPS).
Table 1.
C1 inhibitor-mediated modulation of inflammatory disease.
| Animal Models | Human Disease |
|---|---|
| Ischemia-reperfusion injury (myocardial, brain, skeletal muscle) | Myocardial infarction |
| Vascular leak associated with thermal injury | Capillary leak associated with IL-2 therapy |
| Gram negative bacterial sepsis and endotoxin shock | Sepsis |
| Hyperacute transplant rejection | Cardiopulmonary bypass |
| Traumatic and hemorrhagic shock | |
Table 2.
C1 inhibitor: interactions unrelated to protease inhibition.
| Component | Potential Biologic Function |
|---|---|
| Extracellular matrix proteins (type IV collagen, laminin, entactin, fibrin) | Retain C1 inhibitor at inflammatory sites |
| C3b | Alternative pathway regulation |
| E and P selectins | Inhibition of leukocyte rolling and transmigration |
| Gram negative bacteria and endotoxin | Protection from sepsis and endotoxin shock |
The interaction of C1 inhibitor with extracellular matrix components
C1 inhibitor binds to a number of extracellular matrix components including type IV collagen, laminin, and entactin, as was illustrated by ligand blotting assays and by ELISA (Patston and Schapira, 1997). Half-maximal binding to collagen and laminin was at approximately 8 μg/ml. Digestion of C1 inhibitor with Crotalus atrox α-proteinase, which cleaves at Pro36, had no effect on binding. This implies that this binding has quite different requirements than the binding to gram negative endotoxin (Liu et al., 2005a)(see below). Binding had no effect on the rate constant of inhibition of C1s by C1 inhibitor. The interaction with type IV collagen resulted in a slight increase in the stoichiometry of inhibition in the reaction between C1 inhibitor and C1s, but the other proteins had no effect. Neither the biochemical mechanism nor the biologic relevance of these reactions have been defined. Tissue transglutaminase, but not factor XIII, has been shown to cross-link C1 inhibitor to immobilized fibrin via Gln453 (Hauert et al., 2000). Such cross-linked C1 inhibitor retains the ability to inactivate target proteases. The biology of this reaction also remains to be investigated. It seems likely, however, that these reactions with extracellular matrix proteins serve at least to concentrate C1 inhibitor at extra-vascular sites of inflammation in order to maximize regulation of local complement and contact system activation.
Suppression of alternative complement pathway activation
C1 inhibitor also has been shown to inhibit alternative pathway activation via an incompletely defined mechanism which, however, does not require protease inhibition (Jiang et al., 2001). The data suggested that the findings might be of biologic relevance because removal of C1 inhibitor from plasma resulted in enhanced alternative pathway activation. The mechanism appears to result from inhibition of binding of factor B to C3b via an interaction of C1 inhibitor with C3b. Binding to C3b does not appear to require protease inhibitor activity. This mechanism, if correct, would be quite similar to the mechanism of inhibition of C3 convertase formation by factor H and CR1. The region of the molecule required for this activity remains uninvestigated and it is not known whether the binding to C3b has any effect on protease inhibition by C1 inhibitor. The potential role of this activity in vivo has not been determined.
The interaction of C1 inhibitor with selectins on endothelial cells
The beneficial effect of C1 inhibitor in inflammatory disease is accompanied by a decrease in leukocyte transmigration to the site of inflammation and by decreased selectin expression (Buerke et al., 1998; Horstick et al., 2001b; Lehmann et al., 2000; Lehmann et al., 2004). These effects are probably largely due to a suppression of complement activation; C3a, C5a and C5b-9 all may enhance selectin expression and leukocyte adhesion (Hattori et al., 1989; Jagels et al., 2000; Monsinjon et al., 2003). However, additional potential mechanisms have not been evaluated previously. Selectins are adhesion molecules that are responsible for leukocyte rolling on the endothelium (Butcher and Picker, 1996). E-selectin is expressed on endothelial cells; expression is induced by cytokines (TNF-α, IL-1) and gram negative lipopolysaccharide. P-selectin is expressed on platelets and endothelial cells; it is mobilized to the cell surface from intracellular granules in response to a variety of agents including histamine and thrombin. Selectins bind to fucosylated mucin-like glycoproteins. A number of selectin ligands bind via sialyl Lewisx, which is a fucose-containing tetrasaccharide expressed on N-linked carbohydrate of a number of cell surface and plasma proteins. All selectins recognize the sialyl Lewisx moiety (Frenette and Wagner, 1996a; Frenette and Wagner, 1996b; Vestweber and Blanks, 1999). C1 inhibitor previously has been reported to bind to endothelial cells that have been incubated in the cold (Bergamaschini et al., 2001a; Bergamaschini et al., 2001b). Because of these observations, and because it is heavily glycosylated and is fucosylated (Strecker et al., 1985), we tested the hypothesis that C1 inhibitor might express the sialyl Lewisx tetrasaccharide and thereby bind to selectins. On immunoblots following separation by SDS-polyacrylamide gel electrophoresis, C1 inhibitor reacted with two different monoclonal antibodies that react with sLewisx (Cai and Davis III, 2003). Reactivity was lost following removal of N-linked, but not O-linked, carbohydrate.
C1 inhibitor was shown to bind to both E- and P-selectins, as demonstrated by FACS analysis of CHO/K1 cells transfected with either E- or P- selectin and by co-precipitation with soluble E- or P-selectin IgG chimeras (Cai and Davis III, 2003). Furthermore, C1 inhibitor, following incubation with human umbilical vein endothelial cells, co-precipitated with both E- and P-selectin after lysis of the cells and immunoprecipitation with antiserum to C1 inhibitor (unpublished data)(Fig. 1). The interaction with selectins had no discernible effect on the ability of C1 inhibitor to form stable complexes with C1s, although reaction kinetics have not been evaluated. Using the macrophage-like cell line, U937, C1 inhibitor suppressed both adhesion to, and transmigration across, TNF-α-treated endothelial cell monolayers (Cai and Davis III, 2003). Carcinoembryonic antigen (CEA), which expresses the sialyl Lewisx tetrasaccharide, is an E-selectin ligand. C1 inhibitor suppressed the binding of CEA to E-selectin expressed on the surface of CHO cells in a dose dependent manner (Cai et al., 2005). In addition, reactive center cleaved C1 inhibitor, which retains no protease inhibitor activity, also suppressed the interaction to as great a degree as did active native C1 inhibitor. However, C1 inhibitor with its N-linked carbohydrate removed, lost the ability to interfere with binding.
Fig. 1.
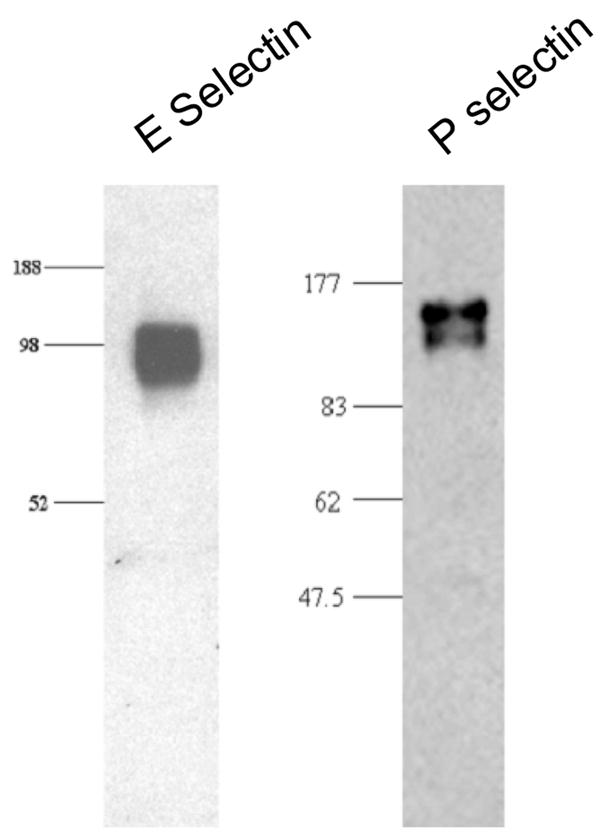
Endothelial cell E- and P-selectin co-immunoprecipitate with C1 inhibitor. Human umbilical vein endothelial cells were treated with TNF-α and H2O2 and were then incubated with C1 inhibitor. The cells then were lysed and immunoprecipitated with rabbit anti-C1 inhibitor antiserum, following which SDS-polyacrylamide gel electrophoresis was performed and blots were probed with antiserum to E-selectin or P-selectin.
The effect of C1 inhibitor on leukocyte rolling was analyzed both in vitro and in vivo. Using a flow chamber in which purified E-selectin was immobilized, C1 inhibitor at concentrations similar to those that occur during acute inflammation (300 μg/ml) increased rolling velocity two fold (Cai et al., 2005)(Fig. 2). Using P-selectin transfected cells cultured on the chamber, rolling velocity was increase by 2.5 fold. With both E- and P-selectin, reactive center cleaved C1 inhibitor, but not N-deglycosylated C1 inhibitor, increased velocity to the same degree. As visualized by intravital microscopy, C1 inhibitor also was shown to suppress TNF-α induced leukocyte rolling in vivo in mice. As with the in vitro experiments, reactive center cleaved C1 inhibitor retained full activity. Lastly, in the thioglycollate peritonitis model, C1 inhibitor (300 μg) suppressed leukocyte infiltration to background levels (Cai et al., 2005). Previous data suggest that leukocyte influx in this model is approximately 50% dependent on the presence of C5 and 50% dependent on leukotriene B(4) (Segal et al., 2002). Therefore, complete suppression of infiltration by C1 inhibitor, together with the effectiveness of inactive C1 inhibitor, suggest that its mechanism is not solely via complement inhibition.
Fig. 2.
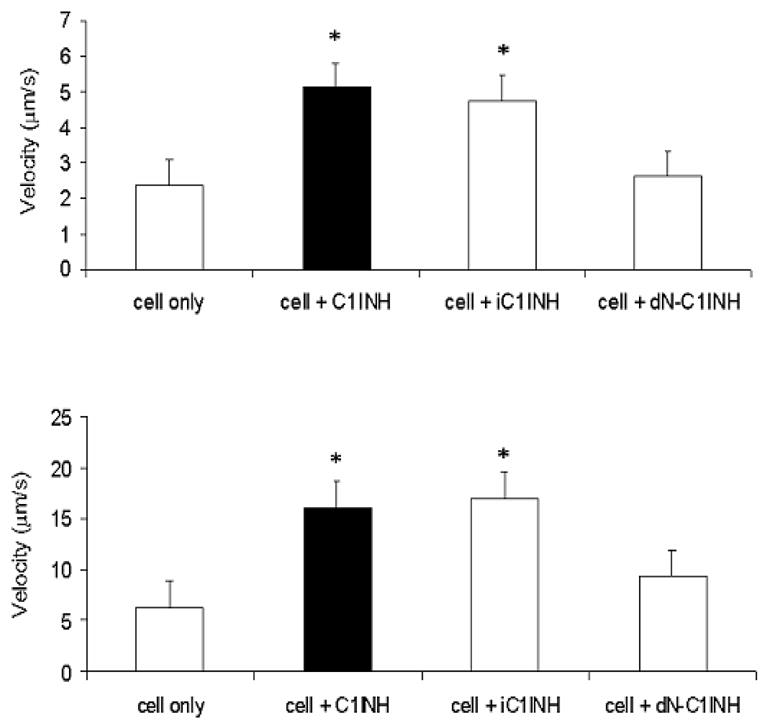
C1 inhibitor blocks the rolling of HL-60 cells on E- and P-selectin under flow conditions in vitro. Human E-selectin (top) was immobilized and CHO cells that express recombinant P-selectin (bottom) were grown to a monolayer on a plate. HL-60 cells (1 × 106/ml) were perfused for a 15 min period in the absence or presence of various forms of C1 inhibitor (300 μg/ml). The velocity of 30 rolling cells was determined.
Several plasma proteins, in addition to C1 inhibitor, express sialyl Lewisx. These include α1-acid glycoprotein, α1-antichymotrypsin and haptoglobin (Brinkman-van der Linden et al., 1998; De Graaf et al., 1993). All these proteins are acute phase reactants. The normal plasma concentration of C1 inhibitor (80–195 μg/ml), for example, may increase up to 2.5-fold during an acute inflammatory response (Kirschfink and Nurnberger, 1999; Tietz, 1995). The data discussed here suggest that, at these concentrations, C1 inhibitor, together with these other acute phase reactants, might interfere with the leukocyte-selectin interaction, resulting in the inhibition of migration of cells to inflammatory sites. This may represent a normal physiologic mechanism that participates in down-regulation of the acute inflammatory response.
The interaction of C1 inhibitor with Gram negative bacterial endotoxin
In a variety of different animal models of Gram negative endotoxin shock and sepsis, survival is significantly improved, together with several physiologic measurements, by treatment with C1 inhibitor (Fischer et al., 1997; Giebler et al., 1999; Guerrero et al., 1993; Jansen et al., 1998; Liu et al., 2003; Scherer et al., 1996; Schmidt et al., 1999a; Schmidt et al., 1999b). C5a very likely plays a role in the pathogenesis of septic shock (Barton and Warren, 1993; Czermak et al., 1999; Strachan et al., 2000). On the other hand, C3 and C4 deficient mice are quite susceptible to endotoxin shock, which supports the hypothesis that C3 may be required for normal endotoxin clearance (Fischer et al., 1997). Interestingly, survival of C3 deficient mice is improved by treatment with C1 inhibitor, suggesting that the effect of C1 inhibitor is not mediated via inhibition of complement activation (Fischer et al., 1997). The contact system may be involved in the mediation of some aspects of endotoxin shock (Colman, 1999; Jansen et al., 1996; Pixley et al., 1993). Other data suggest that contact system activation does not play an important role (Feletou et al., 1996). Our data indicated that not only did intact, active C1 inhibitor protect from endotoxin shock, but that treatment with inactive, reactive center cleaved C1 inhibitor also was effective (Liu et al., 2003). Both active and inactive C1 inhibitor also prevented both local and systemic vascular permeability changes in response to endotoxin (Liu et al., 2005b). C1 inhibitor prevented the binding of Salmonella typhimurium LPS to the murine macrophage cell line, RAW 264.7, and to human blood cells. Direct binding assays showed that C1 inhibitor interacted with LPS, thereby blocking LPS-induced TNF-α expression (Liu et al., 2003). LPS, a component of the outer membrane of Gram negative bacteria, consists of an inner lipid A component, which is responsible for the pathophysiology of endotoxin shock, and an outer polysaccharide component. In blood, LPS binds to LPS-binding protein (LBP), an acute phase plasma protein, that transfers the LPS to CD14 on the surface of monocytes and macrophages (Fenton and Golenbock, 1998; Kitchens, 2000; Poltorak et al., 1998; Schumann and Latz, 2000). The LPS-CD14 complex then initiates intracellular signalling by binding to Toll-like receptor 4 (TLR4) (Aderem and Ulevitch, 2000). At least one other protein, MD-2, is associated with CD14 and TLR4 on the cell surface; together, these make up the LPS receptor (da Silva Correia and Ulevitch, 2002; Juan et al., 1995; Stelter et al., 1997; Stelter et al., 1999; Viriyakosol and Kirkland, 1995; Viriyakosol et al., 2000). The binding of C1 inhibitor to LPS (or lipid A) was inhibited by fetal bovine serum, which contains LBP, and by an LBP peptide that includes the binding site for LBP (Liu et al., 2003).
A recombinant C1 inhibitor from which the amino terminal non-serpin domain was deleted (amino acids 1–97) failed to bind to LPS (Liu et al., 2003). N-deglycosylated C1 inhibitor did not protect mice from endotoxin shock, did not bind to LPS and did not suppress the binding of LPS to RAW 264.7 or to human blood cells (Liu et al., 2004). Therefore, N-linked carbohydrate within the amino terminal domain appears to be required for the interaction with LPS. The lipid A-binding motifs of other LPS-interacting proteins contain positively charged amino acids that interact with the negatively charged phosphate groups within lipid A (Ferguson et al., 2000; Lamping et al., 1996; Mancek et al., 2002; Schumann et al., 1997; Visintin et al., 2003). We, therefore, in addition to analyzing the requirement for N-linked carbohydrate, tested whether the four basic amino acids within the amino terminal domain of C1 inhibitor (Arg18, Lys22, Lys30, Lys55) might be involved in the interaction with lipid A. Using site-directed mutagenesis, single amino acid substitutions and combinations of substitutions were performed in which Ala was introduced at each of the above sites and and the three N-linked sites within the amino terminal domain (Asn3, Asn30 and Asn55). Substitution of Asn3 resulted in virtually complete loss of the ability to interact with LPS, while little or no effect was observed following replacement of the other Asn residues (Liu et al., 2005a)(Fig. 3). However, the effect of the substitution of the individual positively charged residues was additive; replacement of any single residue reduced binding to 75–80% of that of the wild type control protein, replacement of 2, 3 and 4 residues reduced binding progressively to 10–12% of the control with replacement of all four (Fig. 4). These results were confirmed by the observations that the mutants that lost the ability to bind also lost the abilities to inhibit binding of LPS to RAW264.7 cells and to inhibit LPS-induced TNF-α mRNA expression. A likely explanation of these findings is that the binding site for LPS consists of Arg18, Lys22, Lys30, and Lys55, while the carbohydrate at Asn3 is required to maintain a conformation of the amino terminal domain in which the binding site is exposed.
Fig. 3.

Binding of C1 inhibitor Asn substitution mutants to immobilized LPS. LPS was immobilized on the surfaces of polyvinyl chloride plates as described in Materials and Methods. Binding was detected using an anti-human C1 inhibitor antibody followed by goat anti-rabbit IgG conjugated with horseradish peroxidase, with absorbance measured at 490 nm. Results (level of binding as a percentage of the binding of recombinant wild type C1 inhibitor [wt C1INH] were expressed as the means ± SD from at least three independent experiments.
Fig. 4.

Binding of C1 inhibitor Arg and Lys substitution mutants to immobilized LPS. Assays were performed and results expressed as described in Fig. 3.
More recently, experiments have been initiated to analyze the effect of C1 inhibitor in the mouse cecal ligation and puncture (CLP) model of peritonitis and sepsis. These experiments demonstrate that C1 inhibitor also improves survival in this model. A single dose (600 μg) increased survival from approximately 15% to 45%, while three doses at 0, 12 and 24 hours increased survival to 65% (unpublished data). In agreement with the findings in LPS-induced shock, treatment of CLP mice with reactive center cleaved C1 inhibitor was at least as beneficial as was intact, active C1 inhibitor. Fewer bacteria could be recovered from the peritoneal fluid and blood of treated than from untreated mice. C1 inhibitor did not have a direct effect on bacterial growth in vitro, but it did enhance bacterial phagocytosis and/or killing by both neutrophils and macrophages. Furthermore, C1 inhibitor treatment enhanced H2O2 release by neutrophils and macrophages from CLP mice. Although C1 inhibitor does interact with the bacteria from CLP mice, it has not yet been determined whether the binding site is the LPS or if it is some other site on the bacterial surface. Furthermore, the mechanism of the enhanced phagocytosis/killing remains to be defined.
Conclusions
The data discussed here suggest that C1 inhibitor is a multi-faceted anti-inflammatory protein. Protection from inflammatory diseases, sepsis and septic shock very likely is a result of multiple mechanisms. These obviously include inhibition of both complement and contact system activation. In the complement system, the most important effect of C1 inhibitor in inflammatory disease very likely reflects decreased generation of C5a, while in the contact system, decreased generation of bradykinin is probably the most important effect. In addition to protease inhibition, direct effects of C1 inhibitor via interactions with endotoxin, with selectins and possibly with extracellular matrix components very likely also are important.
Abbreviations
- CEA
carcinoembryonic antigen
- CLP
cecal ligation and puncture
- LBP
LPS-binding protein
- LPS
lipopolysaccharide
- TLR4
Toll-like receptor 4
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aderem A, Ulevitch RJ. Toll-like receptors in the induction of the innate immune response. Nature. 2000;406:782–787. doi: 10.1038/35021228. [DOI] [PubMed] [Google Scholar]
- Akita N, Nakase H, Kaido T, Kanemoto Y, Sakaki T. Protective effect of C1 esterase inhibitor on reperfusion injury in the rat middle cerebral artery occlusion model. Neurosurgery. 2003;52:395–400. doi: 10.1227/01.neu.0000043710.61233.b4. [DOI] [PubMed] [Google Scholar]
- Ambrus G, Gal P, Kojima M, Szilagyi K, Balczer J, Antal J, Graf L, Laich A, Moffatt BE, Schwaeble W, Sim RB, Zavodszky P. Natural substrates and inhibitors of mannan-binding lectin-associated serine protease-1 and -2: a study on recombinant catalytic fragments. J Immunol. 2003;170:1374–1382. doi: 10.4049/jimmunol.170.3.1374. [DOI] [PubMed] [Google Scholar]
- Barton PA, Warren JS. Complement component C5 modulates the systemic tumor necrosis factor response in murine endotoxic shock. Infect Immun. 1993;61:1474–1481. doi: 10.1128/iai.61.4.1474-1481.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauernschmitt R, Bohrer H, Hagl S. Rescue therapy with C1-esterase inhibitor concentrate after emergency coronary surgery for failed PTCA. Intensive Care Med. 1998;24:635–638. doi: 10.1007/s001340050629. [DOI] [PubMed] [Google Scholar]
- Bauernschmitt R, Schreiber C, Lange R. C1 esterase inhibitor concentrate during surgery with cardiopulmonary bypass: is there an indication beyond substitution therapy in patients with hereditary angioneurotic edema? J Thorac Cardiovasc Surg. 2000;120:427–428. doi: 10.1067/mtc.2000.107408. [DOI] [PubMed] [Google Scholar]
- Bergamaschini L, Gatti S, Caccamo L, Prato P, Latham L, Trezza P, Maggioni M, Gobbo G, Fassati LR. C1 inhibitor potentiates the protective effect of organ preservation solution on endothelial cells during cold storage. Transplant Proc. 2001a;33:939–941. doi: 10.1016/s0041-1345(00)02277-6. [DOI] [PubMed] [Google Scholar]
- Bergamaschini L, Gobbo G, Gatti S, Caccamo L, Prato P, Maggioni M, Braidotti P, Di Stefano R, Fassati LR. Endothelial targeting with C1--inhibitor reduces complement activation in vitro and during ex vivo reperfusion of pig liver. Clin Exp Immunol. 2001b;126:412–420. doi: 10.1046/j.1365-2249.2001.01695.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkman-van der Linden EC, de Haan PF, Havenaar EC, van Dijk W. Inflammation-induced expression of sialyl Lewisx is not restricted to alpha1-antichymotrypsin and haptoglobin. Glycoconj J. 1998;15:177–182. doi: 10.1023/a:1006972307166. [DOI] [PubMed] [Google Scholar]
- Buerke M, Murohara T, Lefer AM. Cardioprotective effects of a C1 esterase inhibitor in myocardial ischemia and reperfusion. Circulation. 1995;91:393–402. doi: 10.1161/01.cir.91.2.393. [DOI] [PubMed] [Google Scholar]
- Buerke M, Prufer D, Dahm M, Oelert H, Meyer J, Darius H. Blocking of classical complement pathway inhibits endothelial adhesion molecule expression and preserves ischemic myocardium from reperfusion injury. J Pharmacol Exp Ther. 1998;286:429–438. [PubMed] [Google Scholar]
- Butcher EC, Picker LJ. Lymphocyte homing and homeostasis. Science. 1996;272:60–66. doi: 10.1126/science.272.5258.60. [DOI] [PubMed] [Google Scholar]
- Cai S, Davis AE., III Complement regulatory protein C1 inhibitor binds to selectins and interferes with endothelial-leukocyte adhesion. J Immunol. 2003;171:4786–4791. doi: 10.4049/jimmunol.171.9.4786. [DOI] [PubMed] [Google Scholar]
- Cai S, Dole V, Bergmeier W, Scafidi J, Feng H, Wagner DD, Davis AE., III A direct role for C1 inhibitor in leukocyte adhesion. J Immunol. 2005;174:6462–6466. doi: 10.4049/jimmunol.174.10.6462. [DOI] [PubMed] [Google Scholar]
- Colman RW. Biologic activities of the contact factors in vivo. Potentiation of hypotension, inflammation, and fibrinolysis, and inhibition of cell adhesion, angiogenesis and thrombosis. Thromb Haemost. 1999;82:1568–1577. [PubMed] [Google Scholar]
- Czermak BJ, Sarma V, Pierson CL, Warner RL, Huber-Lang M, Bless NM, Schmal H, Priedl HP, Ward PA. Protective effects of C5a blockage in sepsis. Nature Med. 1999;5:788–792. doi: 10.1038/10512. [DOI] [PubMed] [Google Scholar]
- Dalmasso AP, Platt JL. Prevention of complement-mediated activation of xenogeneic endothelial cells in an in vitro model of xenograft hyperacute rejection by C1 inhibitor. Transplantation. 1993;56:1171–1176. doi: 10.1097/00007890-199311000-00024. [DOI] [PubMed] [Google Scholar]
- da Silva Correia J, Ulevitch RJ. MD-2 and TLR4 N-linked glycoproteins are important for a functional lipopolysaccharide receptor. J Biol Chem. 2002;277:1845–1854. doi: 10.1074/jbc.M109910200. [DOI] [PubMed] [Google Scholar]
- de Agostini A, Lijnen HR, Pixley RA, Colman RW, Schapira M. Inactivation of factor-XII active fragment in normal plasma: predominant role of C1-inhibitor. J Clin Invest. 1984;93:1542–1549. doi: 10.1172/JCI111360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Graaf TW, Van der Stelt ME, Anbergen MG, van Dijk W. Inflammation-induced expression of sialyl Lewis X-containing glycan structures on alpha 1-acid glycoprotein orosomucoid in human sera. J Exp Med. 1993;177:657–666. doi: 10.1084/jem.177.3.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Simoni MG, Storini C, Barba M, Catapano L, Arabia AM, Rossi E, Bergamaschini L. Neuroprotection by complement (C1) inhibitor in mouse transient brain ischemia. J Cereb Blood Flow Metab. 2003;23:232–239. doi: 10.1097/01.WCB.0000046146.31247.A1. [DOI] [PubMed] [Google Scholar]
- De Simoni MG, Rossi E, Storini C, Pizzimenti S, Echart C, Bergamaschini L. The powerful neuroprotective action of C1-inhibitor on brain ischemia-reperfusion injury does not require C1q. Am J Pathol. 2004a;164:1857–1863. doi: 10.1016/S0002-9440(10)63744-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Simoni MG, Rossi E, Storini C, Pizzimenti S, Echart C, Bergamaschini L. The powerful neuroprotective action of C1-inhibitor on brain ischemia-reperfusion injury does not require C1q. Am J Pathol. 2004b;164:1857–1863. doi: 10.1016/S0002-9440(10)63744-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Zwaan C, Kleine AH, Diris JH, Glatz JF, Wellens HJ, Strengers PF, Tissings M, Hack CE, van Dieijen-Visser MP, Hermens WT. Continuous 48-h C1-inhibitor treatment, following reperfusion therapy, in patients with acute myocardial infarction. Eur Heart J. 2002;23:1670–1677. doi: 10.1053/euhj.2002.3191. [DOI] [PubMed] [Google Scholar]
- Feletou M, Jamonneau I, Germain M, Thurieau C, Fauchere JL, Villa P, Ghezzi P, Canet E. Bradykinin B2 receptor involvement in rabbit and murine models of septic shock. J Cardiovasc Pharmacol. 1996;27:500–507. doi: 10.1097/00005344-199604000-00008. [DOI] [PubMed] [Google Scholar]
- Fenton MJ, Golenbock DT. LPS-binding proteins and receptors. J Leukocyte Biol. 1998;64:25–32. doi: 10.1002/jlb.64.1.25. [DOI] [PubMed] [Google Scholar]
- Ferguson AD, Welte W, Hofmann E, Lindner B, Holst O, Coulton JW, Diederichs K. A conserved structural motif for lipopolysaccharide recognition by procaryotic and eucaryotic proteins. Structure. 2000;8:585–592. doi: 10.1016/s0969-2126(00)00143-x. [DOI] [PubMed] [Google Scholar]
- Fiane AE, Videm V, Johansen HT, Mellbye OJ, Nielsen EW, Mollnes TE. C1-inhibitor attenuates hyperacute rejection and inhibits complement, leukocyte and platelet activation in an ex vivo pig-to-human perfusion model. Immunopharmacology. 1999;42:231–243. doi: 10.1016/s0162-3109(99)00008-9. [DOI] [PubMed] [Google Scholar]
- Fischer MB, Prodeus AP, Nicholson-Weller A, Ma M, Murrow J, Reid RR, Warren HB, Lage AL, Moore JFD, Rosen FS, Carroll MC. Increased susceptibility to endotoxin shock in complement C3- and C4-deficient mice is corrected by C1 inhibitor replacement. J Immunol. 1997;159:976–982. [PubMed] [Google Scholar]
- Frenette PS, Wagner DD. Adhesion molecules-First of two parts. N Engl J Med. 1996a;334:1526–1529. doi: 10.1056/NEJM199606063342308. [DOI] [PubMed] [Google Scholar]
- Frenette PS, Wagner DD. Adhesion molecules--Part II: Blood vessels and blood cells. N Engl J Med. 1996b;335:43–45. doi: 10.1056/NEJM199607043350108. [DOI] [PubMed] [Google Scholar]
- Fukuta D, Miyagawa S, Yamada M, Matsunami K, Kurihara T, Shirasu A, Hattori H, Shirakura R. Effect of various forms of the C1 esterase inhibitor (C1-INH) and DAF on complement mediated xenogeneic cell lysis. Xenotransplantation. 2003;10:132–141. doi: 10.1034/j.1399-3089.2003.01120.x. [DOI] [PubMed] [Google Scholar]
- Giebler R, Schmidt U, Koch S, Peters J, Scherer R. Combined antithrombin III and C1-esterase inhibitor treatment decreases intravascular fibrin deposition and attenuates cardiorespiratory impairment in rabbits exposed to Escherichia coli endotoxin. Crit Care Med. 1999;27:597–604. doi: 10.1097/00003246-199903000-00042. [DOI] [PubMed] [Google Scholar]
- Guerrero R, Velasco F, Rodriguez M, Lopez A, Rojas R, Alvarez MA, Villalba R, Rubio V, Torres A, Castillo Dd. Endotoxin-induced pulmonary dysfunction is prevented by C1-esterase inhibitor. J Clin Invest. 1993;91:2754–2760. doi: 10.1172/JCI116516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hack CE, Ogilvie AC, Eisele B, Eerenberg AJ, Wagstaff J, Thijs LG. C1-inhibitor substitution therapy in septic shock and in the vascular leak syndrome induced by high doses of interleukin-2. Intensive Care Med. 1993;19(Suppl 1):S19–S28. doi: 10.1007/BF01738946. [DOI] [PubMed] [Google Scholar]
- Hack CE, Ogilvie AC, Eisele B, Jansen PM, Wagstaff J, Thijs LG. Initial studies on the administration of C1-esterase inhibitor to patients with septic shock or with a vascular leak syndrome induced by interleukin-2 therapy. Prog Clin Biol Res. 1994;388:335–357. [PubMed] [Google Scholar]
- Hattori R, Hamilton KK, McEver RP, Sims PJ. Complement proteins C5b-9 induce secretion of high molecular weight multimers of endothelial von Willebrand factor and translocation of granule membrane protein GMP-140 to the cell surface. J Biol Chem. 1989;264:9053–9060. [PubMed] [Google Scholar]
- Hauert J, Patston PA, Schapira M. C1 inhibitor cross-linking by tissue transglutaminase. J Biol Chem. 2000;275:14558–14562. doi: 10.1074/jbc.275.19.14558. [DOI] [PubMed] [Google Scholar]
- Hecker JM, Lorenz R, Appiah R, Vangerow B, Loss M, Kunz R, Schmidtko J, Mengel M, Klempnauer J, Piepenbrock S, Dickneite G, Neidhardt H, Ruckoldt H, Winkler M. C1-inhibitor for prophylaxis of xenograft rejection after pig to cynomolgus monkey kidney transplantation. Transplantation. 2002;73:675–677. doi: 10.1097/00007890-200203150-00006. [DOI] [PubMed] [Google Scholar]
- Henze U, Lennartz A, Hefemann B, Goldmann C, Kirkpatrick CJ, Klosterhalfen B. The influence of the C1-inhibitor BERINERT and the protein-free haemodialsate ACTIHAEMYL20% on the evolution of the depth of scald burns in a porcine model. Burns. 1997;23:473–477. doi: 10.1016/s0305-4179(97)00019-3. [DOI] [PubMed] [Google Scholar]
- Horstick G, Heimann A, Gotze O, Hafner G, Berg O, Bohmer P, Becker P, Darius H, Rupprecht HJ, Loos M, Bhakdi S, Meyer J, Kempski O. Intracoronary application of C1 esterase inhibitor improves cardiac function and reduces necrosis in an experimental model of ischemia reperfusion. Circulation. 1997;95:701–708. doi: 10.1161/01.cir.95.3.701. [DOI] [PubMed] [Google Scholar]
- Horstick G, Berg O, Heimann A, Gotze O, Loos M, Hafner G, Bierbach B, Petersen S, Bhakdi S, Darius H, Horstick M, Meyer J, Kempski O. Application of C1-esterase inhibitor during reperfusion of ischemic myocardium: dose-related beneficial versus detrimental effects. Circulation. 2001a;104:3125–3131. doi: 10.1161/hc5001.100835. [DOI] [PubMed] [Google Scholar]
- Horstick G, Kempf T, Lauterbach M, Bhakdi S, Kopacz L, Heimann A, Malzahn M, Horstick M, Meyer J, Kempski O. C1-esterase-inhibitor treatment at early reperfusion of hemorrhagic shock reduces mesentery leukocyte adhesion and rolling. Microcirculation. 2001b;8:427–433. doi: 10.1038/sj/mn/7800115. [DOI] [PubMed] [Google Scholar]
- Horstick B, Berg O, Heimann A, Gotze O, Loos M, Hafner G, Bierbach B, Petersen S, Bhakdi S, Darius H, Horstick M, Meyer J, Kempski O. Application of C1-esterase inhibitor during reperfusion of ischemic myocardium: dose-related beneficial versus detrimental effects. Circulation. 2002;104:3125–3131. doi: 10.1161/hc5001.100835. [DOI] [PubMed] [Google Scholar]
- Jagels MA, Daffern PJ, Hugli TE. C3a and C5a enhance granulocyte adhesion to endothelial and epithelial cell monolayers: epithelial and endothelial priming is required for C3a-induced eosinophil adhesion. Immunopharmacology. 2000;46:209–222. doi: 10.1016/s0162-3109(99)00178-2. [DOI] [PubMed] [Google Scholar]
- Jansen PM, Pixley RA, Brouwer M, de Jong IW, Chang AC, Hack CE, Taylor FB, Jr, Colman RW. Inhibition of factor XII in septic baboons attenuates the activation of complement and fibrinolytic systems and reduces the release of interleukin-6 and neutrophil elastase. Blood. 1996;87:2337–2344. [PubMed] [Google Scholar]
- Jansen PM, Eisele B, de Jong IW, Chang A, Delvos U, Taylor JFB, Hack CE. Effect of C1 inhibitor on inflammatory and physiologic response patterns in primates suffering from lethal septic shock. J Immunol. 1998;160:475–484. [PubMed] [Google Scholar]
- Jiang H, Wagner E, Zhang H, Frank MM. Complement 1 inhibitor is a regulator of the alternative complement pathway. J Exp Med. 2001;194:1609–1616. doi: 10.1084/jem.194.11.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juan TS, Hailman E, Kelley MJ, Busse LA, Davy E, Empig CJ, Narhi LO, Wright SD, Lichenstein HS. Identification of a lipopolysaccharide binding domain in CD14 between amino acids 57 and 64. J Biol Chem. 1995;270:5219–5224. doi: 10.1074/jbc.270.10.5219. [DOI] [PubMed] [Google Scholar]
- Kaplan AP, Gruber A, Harpel PC. Assessment of Hageman factor activation in human plasma: quantification of activated Hageman factor-C1 inactivator complexes by an enzyme-linked differential antibody immunosorbent assay. Blood. 1985;66:636–641. [PubMed] [Google Scholar]
- Khorram-Sefat R, Goldmann C, Radke A, Lennartz A, Mottaghy K, Afify M, Kupper W, Klosterhalfen B. The therapeutic effect of C1-inhibitor on gut-derived bacterial translocation after thermal injury. Shock. 1998;9:101–108. doi: 10.1097/00024382-199802000-00005. [DOI] [PubMed] [Google Scholar]
- Kirschfink M, Nurnberger W. C1 inhibitor in anti-inflammatory therapy: from animal experiment to clinical application. Mol Immunol. 1999;36:225–232. doi: 10.1016/s0161-5890(99)00048-6. [DOI] [PubMed] [Google Scholar]
- Kitchens RL. Role of CD14 in cellular recognition of bacterial lipopolysaccharides. Chem Immunol. 2000;74:61–82. doi: 10.1159/000058750. [DOI] [PubMed] [Google Scholar]
- Lamping N, Hoess A, Yu B, Park TC, Kirschning CJ, Pfeil C, Reuter D, Wright SD, Herrman F, Schumann RR. Effects of site directed mutagenesis of basic residues (Arg 94, Lys 95, Lys 99) of LPS-binding protein on binding and transfer of LPS and subsequent immune cell activation. J Immunol. 1996;157:4648–4656. [PubMed] [Google Scholar]
- Lehmann TG, Heger M, Munch S, Kirschfink M, Klar E. In vivo microscopy reveals that complement inhibition by C1-esterase inhibitor reduces ischemia/reperfusion injury in the liver. Transpl Int. 2000;13(Suppl 1):S547–550. doi: 10.1007/s001470050399. [DOI] [PubMed] [Google Scholar]
- Lehmann C, Birnbaum J, Luhrs C, Ruckbeil O, Spies C, Ziemer S, Grundling M, Pavlovic D, Usichenko T, Wendt M, Kox WJ. Effects of C1 esterase inhibitor administration on intestinal functional capillary density, leukocyte adherence and mesenteric plasma extravasation during experimental endotoxemia. Intensive Care Med. 2004;30:309–314. doi: 10.1007/s00134-003-2042-2. [DOI] [PubMed] [Google Scholar]
- Liu D, Cai S, Gu X, Scafidi J, Wu X, Davis AE., III C1 inhibitor prevents endotoxin shock via a direct interaction with lipopolysaccharide. J Immunol. 2003;171:2594–2601. doi: 10.4049/jimmunol.171.5.2594. [DOI] [PubMed] [Google Scholar]
- Liu D, Gu X, Scafidi J, Davis AE., III N-linked glycosylation is required for C1 inhibitor-mediated protection from endotoxin shock in mice. Infect Immun. 2004;72:1946–1955. doi: 10.1128/IAI.72.4.1946-1955.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Cramer CC, Scafidi J, Davis AE., III N-linked glycosylation at Asn3 and the positively charged residues within the amino terminal domain of C1 inhibitor are required for its interaction with Salmonella typhimurium lipopolysaccharide and lipid A. Infect Immun. 2005a;73:4478–4487. doi: 10.1128/IAI.73.8.4478-4487.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Zhang D, Scafidi J, Wu X, Cramer CC, Davis AE., III C1 inhibitor prevents gram negative bacterial lipopolysaccharide-induced vascular permeability. Blood. 2005b;72:1946–1955. doi: 10.1182/blood-2004-05-1963. [DOI] [PubMed] [Google Scholar]
- Mancek M, Pristovsek P, Jerala R. Identification of LPS-binding peptide fragment of MD-2, a Toll-receptor accessory protein. Biochem Biophys Res Commun. 2002;292:880–885. doi: 10.1006/bbrc.2002.6748. [DOI] [PubMed] [Google Scholar]
- Matsunami K, Miyagawa S, Yamada M, Yoshitatsu M, Shirakura R. A surface-bound form of human C1 esterase inhibitor improves xenograft rejection. Transplantation. 2000;69:749–755. doi: 10.1097/00007890-200003150-00013. [DOI] [PubMed] [Google Scholar]
- Matsushita M, Endo Y, Fujita T. Cutting edge: complement-activating complex of ficolin and mannose-binding lectin-associated serine protease. J Immunol. 2000a;164:2281–2284. doi: 10.4049/jimmunol.164.5.2281. [DOI] [PubMed] [Google Scholar]
- Matsushita M, Thiel S, Jensenius JC, Terai I, Fujita T. Proteolytic activities of two types of mannose-binding lectin associated serine protease. J Immunol. 2000b;165:2637–2642. doi: 10.4049/jimmunol.165.5.2637. [DOI] [PubMed] [Google Scholar]
- Matsushita M, Endo Y, Hamasaki N, Fujita T. Activation of the lectin complement pathway by ficolins. Int Immunopharmacol. 2001;1:359–363. doi: 10.1016/s1567-5769(00)00045-x. [DOI] [PubMed] [Google Scholar]
- Monsinjon T, Gasque P, Chan P, Ischenko A, Brady JJ, Fontaine MC. Regulation by complement C3a and C5a anaphylatoxins of cytokine production in human umbilical vein endothelial cells. FASEB J. 2003;17:1003–1014. doi: 10.1096/fj.02-0737com. [DOI] [PubMed] [Google Scholar]
- Niederau C, Brinsa R, Niederau M, Luthen R, Strohmeyer G, Ferrell LD. Effects of C1-esterase inhibitor in three models of acute pancreatitis. Int J Pancreatol. 1995;17:189–196. doi: 10.1007/BF02788538. [DOI] [PubMed] [Google Scholar]
- Nielsen EW, Mollnes TE, Harlan JM, Winn RK. C1-inhibitor reduces the ischaemia-reperfusion injury of skeletal muscles in mice after aortic cross-clamping. Scand J Immunol. 2002;56:588–592. doi: 10.1046/j.1365-3083.2002.01173.x. [DOI] [PubMed] [Google Scholar]
- Ogilvie AC, Baars JW, Eerenberg AJM, Hack CE, Pinedo HM, Thijs LG, Wagstaff J. A pilot study to evaluate the effects of C1 esterase inhibitor on the toxicity of high-dose interleukin 2. Br J Cancer. 1994;69:596–598. doi: 10.1038/bjc.1994.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patston PA, Schapira M. Regulation of C1-inhibitor function by binding to type IV collagen and heparin. Biochem Biophys Res Commun. 1997;230:597–601. doi: 10.1006/bbrc.1996.6010. [DOI] [PubMed] [Google Scholar]
- Pixley RA, Schapira M, Colman RW. The regulation of human factor XIIa by plasma proteinase inhibitors. J Biol Chem. 1985;260:1723–1729. [PubMed] [Google Scholar]
- Pixley RA, De La Cadena R, Page JD, Kaufman N, Wyshock EG, Chang A, Taylor FB, Jr, Colman RW. The contact system contributes to hypotension but not disseminated intravascular coagulation in lethal bacteremia. In vivo use of a monoclonal anti-factor XII antibody to block contact activation in baboons. J Clin Invest. 1993;91:61–68. doi: 10.1172/JCI116201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poltorak A, He X, Smirnova I, Liu MY, van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B. Defective LPS signaling in C3H/HeJ and C57Bl/10ScCr mice: mutations in the Tlr4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- Przemeck M, Lorenz R, Vangerow B, Klempnauer J, Winkler M, Piepenbrock S. Pretreatment with C1-esterase inhibitor improves cardiovascular stability in small primates undergoing porcine kidney xenotransplantation. Transplant Proc. 2002;34:2383. doi: 10.1016/s0041-1345(02)03287-6. [DOI] [PubMed] [Google Scholar]
- Radke A, Mottaghy K, Goldmann C, Khorram-Sefat R, Kovacs B, Janssen A, Klosterhalfen B, Hafemann B, Pallua N, Kirschfink M. C1 inhibitor prevents capillary leakage after thermal trauma. Crit Care Med. 2000;28:3224–3232. doi: 10.1097/00003246-200009000-00018. [DOI] [PubMed] [Google Scholar]
- Schelzig H, Simon F, Krischer C, Vogel A, Abendroth D. Ex-vivo hemoperfusion (eHPS) of pig-lungs with whole human blood: effects of complement inhibition with a soluble C1-esterase-inhibitor. Ann Transplant. 2001;6:34–39. [PubMed] [Google Scholar]
- Scherer RU, Giebler RM, Schmidt U, Paar D, Kox WJ. The influence of C1-esterase inhibitor substitution on coagulation and cardiorespiratory parameters in an endotoxin-induced rabbit model of hypercoagulability. Semin Thrombosis Hemostasis. 1996;22:357–366. doi: 10.1055/s-2007-999032. [DOI] [PubMed] [Google Scholar]
- Schmidt W, Stenzel K, Walther A, Gebhard MM, Martin E, Schmidt H. Influence of C1-esterase inhibitor on tissue oxygenation of jejunal mucosa during endotoxemia. Int J Surg Investig. 1999a;1:277–283. [PubMed] [Google Scholar]
- Schmidt W, Stenzel Z, Gebhard MM, Martin E, Schmidt H. C1-esterase inhibitor and its effects on endotoxin-induced leukocyte adherence and plasma extravasation in postcapillary venules. Surgery. 1999b;125:280–287. [PubMed] [Google Scholar]
- Schumann RR, Lamping N, Hoess A. Interchangeable endotoxin-binding domains in proteins with opposite lipopolysaccharide-dependent activities. J Immunol. 1997;159:5599–5605. [PubMed] [Google Scholar]
- Schumann RR, Latz E. Lipopolysaccharide-binding protein. Chem Immunol. 2000;74:42–60. doi: 10.1159/000058760. [DOI] [PubMed] [Google Scholar]
- Segal BH, Kuhns DB, Ding L, Gallin JI, Holland SM. Thioglycollate peritonitis in mice lacking C5, 5-lipoxygenase, or p47(phox): complement, leukotrienes, and reactive oxidants in acute inflammation. J Leukoc Biol. 2002;71:410–416. [PubMed] [Google Scholar]
- Sim RB, Reboul A, Arlaud GJ, Villiers CL, Colomb MG. Interaction of 125I-labelled complement components C1r and C1s with protease inhibitors in plasma. FEBS Lett. 1979;97:111–115. doi: 10.1016/0014-5793(79)80063-0. [DOI] [PubMed] [Google Scholar]
- Stelter F, Bernheiden M, Menzel R, Jack RS, Witt S, Fan X, Pfister M, Schutt C. Mutation of amino acids 39–44 of human CD14 abrogates binding of lipopolysaccharide and Escherichia coli. Eur J Biochem. 1997;243:100–109. doi: 10.1111/j.1432-1033.1997.00100.x. [DOI] [PubMed] [Google Scholar]
- Stelter F, Loppnow H, Menzel R, Grunwald U, Bernheiden M, Jack RS, Ulmer AJ, Schutt C. Differential impact of substitution of amino acids 9–13 and 91–101 of human CD14 on soluble CD14-dependent activation of cells by lipopolysaccharide. J Immunol. 1999;163:6035–6044. [PubMed] [Google Scholar]
- Strachan AJ, Woodruff TM, Haaima G, Fairlie DP, Taylor SM. A new small molecule C5a receptor antagonist inhibits the reverse-passive Arthus reaction and endotoxic shock in rats. J Immunol. 2000;164:6560–6565. doi: 10.4049/jimmunol.164.12.6560. [DOI] [PubMed] [Google Scholar]
- Strecker G, Ollier-Hartmann MP, van Halbeek H, Vliegenthart JF, Montreuil J, Hartmann L. Primary structure of the glycan chains of normal C1 esterase inhibitor (C1-INH) after NMR analysis at 400 MHz. C R Acad Sci III. 1985;301:571–576. [PubMed] [Google Scholar]
- Tassani P, Kunkel R, Richter JA, Oechsler H, Lorenz HP, Braun SL, Eising GP, Haas F, Paek SU, Bauernschmitt R, Jochum M, Lange R. Effect of C1-esterase-inhibitor on capillary leak and inflammatory response syndrome during arterial switch operations in neonates. J Cardiothorac Vasc Anesth. 2001;15:469–473. doi: 10.1053/jcan.2001.24989. [DOI] [PubMed] [Google Scholar]
- Tietz NW. Tietz Clinical Guide to Laboratory Tests. W. B. Saunders; Philadelphia: 1995. [Google Scholar]
- Vestweber D, Blanks JE. Mechanisms that regulate the function of the selectins and their ligands. Physiol Rev. 1999;79:181–213. doi: 10.1152/physrev.1999.79.1.181. [DOI] [PubMed] [Google Scholar]
- Viriyakosol S, Kirkland TN. A region of human CD14 required for lipopolysaccharide binding. J Biol Chem. 1995;270:361–368. doi: 10.1074/jbc.270.1.361. [DOI] [PubMed] [Google Scholar]
- Viriyakosol S, Mathison JC, Tobias PS, Kirkland TN. Structure-function analysis of CD14 as a soluble receptor for lipopolysaccharide. J Biol Chem. 2000;275:3144–3149. doi: 10.1074/jbc.275.5.3144. [DOI] [PubMed] [Google Scholar]
- Visintin A, Latz E, Monks BG, Espevik T, Golenbock DT. Lysines 128 and 132 enable lipopolysaccharide binding to MD-2, leading to toll-like receptor-4 aggregation and signal transduction. J Biol Chem. 2003;278:48313–48320. doi: 10.1074/jbc.M306802200. [DOI] [PubMed] [Google Scholar]
- Ziccardi RJ. Activation of the early components of the classical complement pathway under physiological conditions. J Immunol. 1981;126:1768–1773. [PubMed] [Google Scholar]