

Abstract
Induction of cyclooxygenase-2 (COX-2) with production of prostaglandins occurs in a wide spectrum of acute and chronic neurodegenerative diseases and is associated with neuronal death. Inhibition of the COX-2 pathway and downstream production of prostaglandins protects neurons in rodent models of cerebral ischemia and neurodegeneration. Recent studies investigating the functions of selected prostaglandin receptor pathways in mediating COX-2 neurotoxicity have demonstrated both toxic and paradoxically neuroprotective effects of several receptors in models of excitotoxicity. In this study, we investigate the functions of additional prostaglandin receptors not previously characterized in organotypic models of glutamate excitotoxicity. We find that PGD2, PGI2, and PGF2α receptors protect motor neurons in an organotypic spinal cord model of amyotrophic lateral sclerosis (ALS). In addition, PGI2 and TXA2 receptors rescue CA1 neurons in an organotypic hippocampal model of N-methyl D-aspartate excitotoxicity. However, in a model of inflammation induced by lipopolysaccharide, prostaglandin receptors previously found to be protective in excitotoxicity now cause CA1 neuronal death. Taken together, these studies identify novel eicosanoid receptor signaling pathways that mediate neuronal protection in excitotoxic paradigms; these data also support the emerging hypothesis that the toxic/protective effects of eicosanoid signaling on neuronal viability diverge significantly depending on whether excitotoxicity or inflammation predominates as the underlying toxic stimulus.
INTRODUCTION
The inducible isoform of cyclooxygenase, COX-2, is rapidly upregulated in vivo in hippocampal and cerebral cortical neurons following N-methyl-D-aspartate (NMDA) receptor-dependent synaptic activity [23], consistent with a physiologic role in modulating synaptic plasticity [6]. However, in pathologic conditions caused by either excitotoxicity or inflammation, COX-2 expression and activity are increased in neurons and glial cells, and can promote either primary or secondary neuronal injury, respectively. Thus increased COX-2 activity and prostaglandin production are hallmarks of a wide range neurological disease models, including acute excitotoxic events such as cerebral ischemia, traumatic brain or spinal cord injury, as well as paradigms of chronic neurodegeneration that model human amyotrophic lateral sclerosis (ALS), Parkinson’s disease (PD), and Alzheimer’s disease (AD; reviewed in [9, 18]) and aging [2]. In humans, increased COX-2 and prostaglandin production have been observed in AD, ALS, multiple sclerosis and PD [1, 10, 22, 24]. Thus COX-2 appears to function physiologically in promoting synaptic activity and pathologically in diseases characterized by excitotoxicity or inflammation.
Because inhibition of COX-2 either genetically or pharmacologically decreases neuronal injury in rodent models of NMDA-dependent excitotoxicity, there is considerable interest in defining the downstream mechanisms by which COX-2 exerts its neurotoxicity. A primary focus has been the examination of prostaglandin signaling cascades downstream of COX-2, and the identification of neurotoxic prostaglandin receptors [4]. Prostaglandins are lipid signaling molecules derived from the metabolism of arachidonic acid by COX-1 and the inducible COX-2. The five prostaglandins PGE2, PGF2α, PGD2, PGI2 (prostacyclin), and TXA2 (thromboxane) bind to classes of G protein-coupled receptors designated EP (for E-prostanoid receptor), FP, DP, IP, and TP receptors, respectively, that differ in their effects on cAMP and phosphoinositol turnover and intracellular Ca2+ mobilization [5]. Additional complexity arises because several receptor subtypes exist for the PGE2 EP receptor family, where there are four subtypes (EP1, EP2, EP3, and EP4) and for the PGD2 DP family, where there are two receptors, DP1 and DP2 (aka CRTH2). COX-2 neurotoxicity is presumed to be mediated by one or more of these prostaglandin receptor signaling cascades.
Recent studies using genetic models have begun to shed light on the function of prostaglandin receptor signaling in models of neuronal damage. A toxic effect of the PGE2 EP1 receptor has been demonstrated in a model of focal ischemia where EP1 impairs the Na+-Ca2+ exchange critical in maintaining Ca2+ homeostasis [11]. However, other studies demonstrate a paradoxical neuroprotective effect of certain prostaglandin receptors. We have previously reported that the PGE2 EP2 receptor rescues CA1 pyramidal neurons in organotypic hippocampal cultures treated with NMDA or oxygen glucose deprivation [16], and both the EP2 and EP3 receptors rescue motor neurons in organotypic spinal cord slices subjected to chronic glutamate toxicity [3]. In vivo, the EP2 receptor mediates a significant protective function in models of focal ischemia [15, 16]. PGD2, signaling via its DP1 receptor similarly rescues CA1 and CA3 neurons in organotypic hippocampal slices treated with NMDA [14] and in vivo in a model of postnatal hypoxia-ischemia [21]. Mechanistically, EP2 and DP1 neuroprotection are dependent on intact cAMP signaling, whereas EP3 neuroprotection is associated with increased AKT phosphorylation [3, 16].
These divergent effects of prostaglandin receptor signaling on neuronal survival occur in models of excitotoxicity, where neuronal injury occurs in large part from overactivation of glutamate ionotropic receptors and downstream disruptions in intracellular Ca2+ homeostasis. In models of neurodegeneration where inflammation and glial activation lead to secondary neurotoxicity, the effects of prostaglandin signaling on neuronal viability diverge. The EP2 receptor, neuroprotective in models of glutamate toxicity, enhances the inflammatory response and secondary synaptotoxicity in the lipopolysaccharide (LPS) model of innate immunity [17, 19]. In a murine model of Familial Alzheimer’s disease, deletion of the EP2 receptor results decreased neuronal lipid peroxidation and Aβ peptide load [13].
In this study, we have investigated the functions of additional prostaglandin receptors whose functions have not been examined in models of excitotoxicity. Interestingly, we find that additional prostaglandin receptors mediate neuroprotection. We also demonstrate that prostaglandin receptors can diverge in their effect on neuronal survival, depending on whether the primary stimulus is excitotoxic or inflammatory.
METHODS
Animals
This study was conducted in accordance with the National Institutes of Health guidelines for the use of experimental animals. Protocols were approved by the Institutional Animal Care and Use Committee at Johns Hopkins University. Sprague Dawley rats or C57B/6 mice were obtained from Charles River for organotypic cultures.
Prostaglandin receptor agonists and other reagents
The following reagents were obtained commercially: LPS (Calbiochem, San Diego, CA); N-methyl D-aspartate (NMDA; Sigma, St Louis, MO); D,L-threo-β-hydroxyaspartate (THA; Tocris, Elisville, MO); propidium idodide (PI, Sigma); PGD2, BW245C, fluprostenol, 13,14-dehydro-15-cyclohexyl carbaprostacyclin, U-46619, and 13,14-dihydro-15-keto-PGD2 (DK-PGD2; Cayman Chemicals, Ann Arbor, MI). Prostaglandin receptor agonists were prepared as a 10 mM stock in 100% ethanol and frozen at −70°C until use.
Organotypic spinal cord slice model
Organotypic spinal cord cultures were prepared from lumbar spinal cord of postnatal day 8 rat pups as previously described [3]. Stimulation with D,L-threohydroxyaspartate (THA), a glutamate transport inhibitor, models ALS with motor neuron loss resulting from chronic glutamate toxicity. Prostaglandin agonists or vehicle (0.1% ethanol) were added to culture medium at DIV 7, and medium was changed and replenished with fresh THA and prostaglandin agonists twice a week for three weeks. PGD2 and DP1, DP2, IP, TP, and FP receptor agonists were tested at 1nM to 1μM, consistent with the established Ki and EC50 values for mouse prostaglandin receptors [5, 12]. Slices were fixed in 4% paraformaldehyde in 0.1M phosphate buffer pH 7.4 and assessed for motor neuron survival using SMI-32 monoclonal antibody immunocytochemistry (Sternberger Monoclonals, Lutherville, MD). Large SMI-32 positive neurons (>25μm in diameter in ventral horn) were counted at 400X by a single investigator blinded to the experimental conditions (n=10–15 slices per experimental group; 2–3 experiments/prostaglandin agonist).
Organotypic hippocampal slice model and measurement of neuronal death
Hippocampal organotypic cultures were prepared from rat or mouse 7-day old pups as described previously [16]. At 13 days in culture, fresh medium containing propidium iodide (PI; 5μg/ml) was added for 24 hours, and basal PI fluorescence was measured the following day (time point tb) as an indicator of basal cell death. Slices were then stimulated with 10 μM NMDA for 1 hour in the presence of prostaglandin receptor agonists or vehicle; after stimulation, media was replaced with fresh media containing either receptor agonist or vehicle (ethanol ≤0.1%) and PI (5 μg/ml) for 23 hours. For LPS experiments, LPS (0.1–5 μg/ml) was administered for 24 hours +/− prostaglandin agonists and PI. Slices were imaged at the 24 hour time point (t24h). Medium was then replaced with fresh medium containing a lethal dose of 10 μM NMDA and PI and incubated overnight to induce maximum CA1 neuronal loss and imaged 24 hours later (tmax). Neuronal death was assayed by quantification of mean PI fluorescence in the CA1 subregion of each hippocampal slice for tb, t24h, and tmax as previously described [16]. The percent neuronal death was normalized for each individual slice and was calculated as follows: (t24h− tb)/(tmax− tb).
Statistical Analysis
All data are reported as mean ± standard error of the mean (SEM). The data were analyzed using Student’s t test or one-way analysis of variance (ANOVA), followed by Tukey post hoc analysis. We used p<0.05 as a significance level. T-test was used to assess differences between vehicle and THA in organotypic spinal cord experiments, between vehicle and NMDA and vehicle and LPS in organotypic hippocampal experiments. One way ANOVA was used to assess effect of dose in multiple dose-response experiments.
RESULTS
Activation of PGD2 DP, PGF2α FP, and PGI2 IP receptors rescues motor neurons
In previous studies, we determined that PGE2 signaling via its EP2 and EP3 receptors paradoxically protected motor neurons in a spinal cord model of chronic glutamate toxicity [3]. In this model, lumbar spinal cord slices are treated with THA which blocks glutamate reuptake at astrocytic and neuronal glutamate transporters and induces a chronic state of glutamate toxicity with motor neuron loss over a period of three weeks (Figure 1B). Co-administration of THA and PGD2 protected motor neurons at nM doses; selective agonists of the DP1 and DP2 receptors, BW245C and DK-PGD2, respectively, also significantly rescued motor neurons (Figure 1C). In addition, co-administration of the PGF2α FP receptor agonist fluprostenol (Figure 1D) and the prostacyclin (PGI2) IP receptor agonist carbaprostacyclin (Figure 1E) significantly rescued motor neurons an low nM doses. There was no effect of the thromboxane receptor agonist U-46619 (Figure 1F). Administration of PGD2, DP1, DP2, FP, IP and TP agonists alone had no effect on motor neuron survival (data not shown).
Figure 1. Neuroprotection by multiple receptor agonists in organotypic spinal cord slices treated chronically with the glutamate reuptake inhibitor THA.
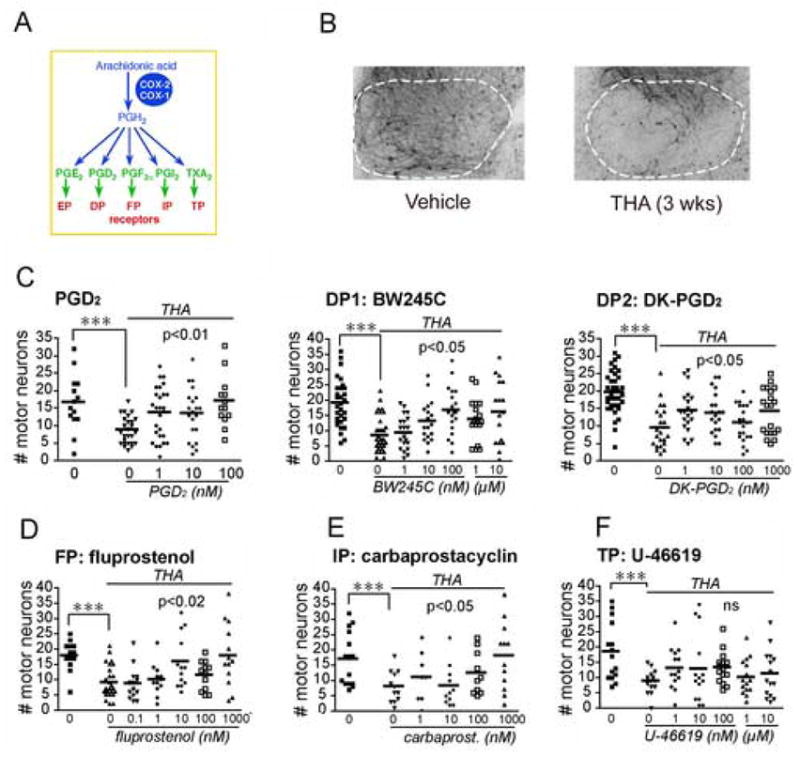
(A) Schema of prostaglandin pathway. (B) Ventral horn (outlined in white) of lumbar spinal cord slices treated with either vehicle or THA (200μM) for 3 weeks. (C) Left panel: co-stimulation of THA treated slices with PGD2 (1nM to 1μM) rescues motor neurons (vehicle vs THA : ***p=0.0009; THA vs THA+PGD2: ANOVA p<0.01, post hoc p<0.05 for 1nM, p<0.01 for 100 nM). Neuroprotection was observed with DP1 agonist BW245C (middle panel; vehicle vs THA : ***p=0.0002; THA vs THA+BW245C: ANOVA p<0.05, post hoc p<0.01 for 100 nM) and DP2 agonist DK-PGD2 (right panel; vehicle vs THA : p=0.0001; THA vs THA+ DK-PGD2: ANOVA p<0.05). (D) The PGF2α FP agonist fluprostenol is protective (vehicle vs THA : p=0.0001; THA vs THA+fluprostenol: ANOVA p<0.02, post hoc p<0.05 for 10 nM and p<0.01 for 1.0 μM) and (E) the prostacyclin PGI2 IP agonist carbaprostacyclin (vehicle vs THA : ***p=0.0068; THA vs THA+carbaprostacyclin: ANOVA p=0.026, post hoc p<0.05 for 1 μM). (F) Stimulation of the thromboxane TP receptor with its agonist U-46619 has no effect.
Activation of PGE2 EP3, PGI2 IP, and thromboxane TP receptors rescues hippocampal CA1 neurons
We have previously determined that activation of the PGE2 EP2 receptor and the PGD2 DP1 receptor protect CA1 neurons in organotypic hippocampal slices treated with NMDA [15, 16]. Here, we examined effects of additional prostaglandin receptors on CA1 neuronal survival in hippocampal slices treated acutely with NMDA. The EP3 agonist sulprostone exerted significant protection at pM-nM concentrations (Figure 2B), reminiscent of its protective role previously described in spinal cord slices [3]. In addition, activation of IP and TP receptors, but not the FP receptor promoted significant neuroprotection (Figure 2C–E). Taken together, these data indicate that additional prostaglandin receptor signaling cascades exert significant neuroprotective effects in excitoxicity. Administration of EP3, FP, IP and TP agonists alone had no effect on CA1 neuronal survival (data not shown).
Figure 2. Neuroprotection by multiple receptor agonists in organotypic hippocampal slices treated acutely with NMDA.
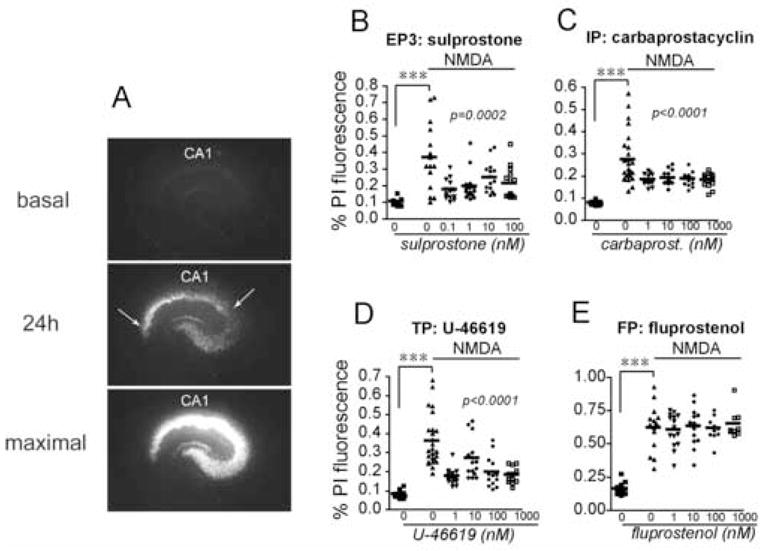
(A) Representative series of images of hippocampal slice treated with NMDA. (Top panel) Basal PI staining (time point tb; see Methods). (Middle panel) Slices were stimulated for one hour with NMDA (10 μM) and allowed to recover for 24 hours (t24h): selective PI uptake occurs in CA1 pyramidal neurons (between arrows), as well as CA2–4. (Bottom panel) Maximal pyramidal neuronal death was induced by incubating slices for an additional 24 hours with NMDA (tmax). PI fluorescence was normalized for each individual slice as follows: (t24h− tb)/(tmax− tb). (B) The PGE2 EP3 agonist sulprostone is protective at pM-nM concentrations (vehicle vs NMDA : ***p=0.0004; NMDA vs NMDA+sulprostone: ANOVA p=0.0002, post hoc p<0.001 for 0.1nM, p<0.01 for 1 nM, p<0.05 for 10 nM, and p<0.01 for 100 nM). (C) The PGI2 IP receptor agonist carbaprostacyclin protected CA1 neurons (vehicle vs NMDA : ***p=0.0001; NMDA vs NMDA+carbaprostacyclin: ANOVA p<0.0001, post hoc p<0.01 for 1nM, p<0.01 for 10 nM, p<0.01 for 100 nM, p<0.01 for 1 μM ). (D) The TXA2 TP receptor agonist U-46619 is protective (vehicle vs NMDA : ***p<0.0001; NMDA vs NMDA+ U-46619: ANOVA p<0.0001, post hoc p<0.001 for 1nM, p<0.05 for 10 nM, p<0.001 for 100 nM, and p<0.001 for 1 μM), but (E) the PGF2α FP receptor agonist fluprostenol has no effect on hippocampal CA1 neuronal survival ( ***p<0.001 control vs NMDA).
Opposing effect on neuronal viability by EP receptor activation in inflammation
Recent studies have demonstrated that the EP2 receptor enhances inflammatory oxidative stress and synaptotoxicity in innate immunity [17, 19] and in a transgenic model of Familial AD [13]. These effects are distinct from the neuroprotective function of the EP2 receptor seen both in vitro in organotypic slices and in vivo in models of cerebral ischemia [15, 16]. We investigated the effect of PGE2 and the EP2 and EP3 receptors in vitro in a model of LPS mediated inflammation. In this model, stimulation of hippocampal slices with LPS (0.1–5 μg/ml) leads to CA1 pyramidal death as measured by PI fluorescence. Co-administration of PGE2 at nM concentrations significantly enhanced the neurotoxicity of LPS (Figure 3A). Selective activation of the EP2 and EP3 receptors also increased LPS-mediated CA1 neurotoxicity (Figure 3B and C). These experiments demonstrate that activation of individual prostaglandin receptors can have divergent effects on neuronal survival depending on whether the pathological stimulus is excitotoxic or inflammatory.
Figure 3. PGE2 agonists are neurotoxic in the setting of LPS-mediated inflammation.
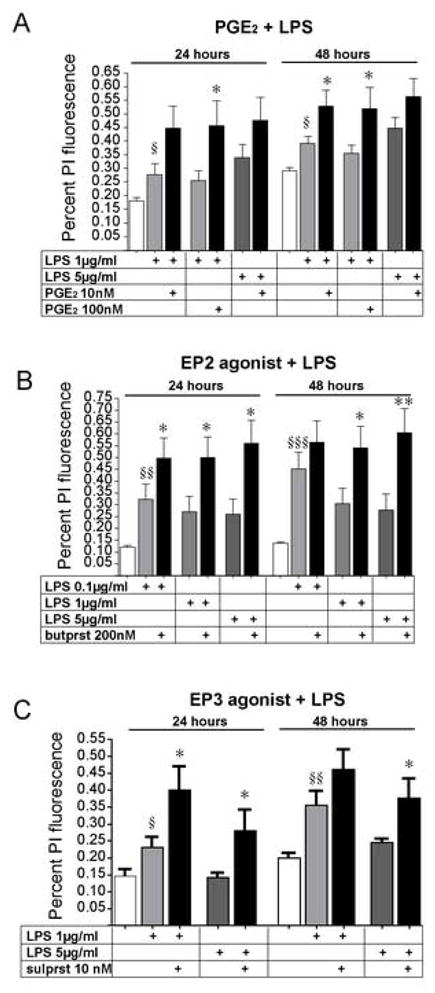
(A) Co-stimulation of hippocampal slices with LPS (1μg/ml–5μg/ml) and PGE2 (10–100nM) results in increased CA1 neuronal death (§p<0.05 control vs LPS; *p<0.05 for LPS vs LPS+ PGE2). (B) The EP2 agonist butaprost and (C) the EP3 agonist sulprostone increase LPS-mediated neurotoxicity (§§p<0.01 for control vs LPS; *p<0.05 and **p<0.01 for LPS vs LPS + EP agonist). Both the EP2 and EP3 agonists have been found in previous studies to be significantly neuroprotective in organotypic models of glutamate toxicity [3, 15, 16].
DISCUSSION
Induction of COX-2 activity and production of downstream prostaglandins is associated in a wide range of neurological disease models with neuronal injury [9, 18]. Previous studies in organotypic excitotocity models have identified paradoxical neuroprotective effects of PGE2 and PGD2 receptors that have been confirmed in vivo in models of ischemia. In this study, we identify additional prostaglandin receptor signaling pathways that rescue neurons in two organotypic models of excitotoxic neuronal injury. In spinal cord slices that are chronically exposed to glutamate reuptake inhibitors, both PGD2 DP receptors, as well as the PGF2α FP and prostacyclin IP receptors rescued motor neurons. COX-2 inhibition has been shown to be protective in this in vitro model, as well as in the transgenic model of Familial ALS [3, 7, 8]. This would suggest that the DP, FP, and IP receptors do not mediate COX-2 toxicity in this in vitro model and from our previous studies [3], the EP2 and EP3 receptors can also be excluded, since they too rescue motor neurons.
In a second model using hippocampal slices, we have identified three additional prostaglandin receptor signaling pathways that confer neuroprotection in the setting of NMDA toxicity. Stimulation of the PGE2 EP3 receptor with picomolar concentrations of agonist resulted in significant rescue of CA1 pyramidal neurons. This finding is consistent with previous data obtained in spinal cord slices, where EP3 activation rescued motor neurons subjected to chronic glutamate toxicity [3]; here EP3 activation was associated with increased levels of the pro-survival phosphorylated form of AKT. Additionally, the prostacyclin IP receptor, which is positively coupled to cAMP production, also conferred significant neuroprotection in hippocampal slices, consistent with its protective effect in spinal cord slices. Although we did not test whether IP neuroprotection is dependent on cAMP signaling, as is the case for the EP2 and DP1 receptors, we hypothesize that this is the case and may represent a common mechanism of neuroprotection for the Gαs coupled prostaglandin receptors. The thromboxane TP receptor mediated a protective role in this model, in contrast to its neutral effect in the spinal cord organotypic model. A similar divergence of effect was found for the PGF2α FP receptor, which had no effect on CA1 neuronal survival, unlike its significant protective effect in the spinal cord model. Taken together, these data demonstrate novel and paradoxical protective effects of additional prostaglandin receptors in organotypic models of glutamate toxicity. The effects of prostaglandin receptors in organotypic models of excitotoxicity so far predicts their roles in vivo in models of cerebral ischemia where glutamate toxicity is an early and acute event [15, 16, 21].
In hippocampal slices, activation of the EP2 [15, 16] and EP3 (Figure 2) receptor rescues CA1 neurons stimulated with NMDA. However, in slices stimulated with LPS, which injures neurons secondarily via microglial production of neurotoxic cytokines and reactive oxygen species, stimulation of the EP2 or EP3 receptor now increases LPS-mediated CA1 toxicity. While we did not find a consistent dose response of LPS and increasing CA1 toxicity in this paradigm, the addition of either PGE2 (10–100 nM), the EP2 agonist butaprost (200nM), or the EP3 agonist sulprostone (10nM) consistently enhanced LPS-induced CA1 PI fluorescence. The increased toxicity of LPS with addition of EP2/EP3 receptor agonists is relevant to recent in vivo studies demonstrating that in the setting of inflammation, prostaglandin receptor signaling can exhibit an opposite and toxic effect on neurons. For example, in the LPS model of innate immunity, the EP2 receptor mediates a major component of inflammatory oxidative stress and secondary synaptotoxicity [17, 19]. One potential mechanism is via induction of iNOS, production of NO, and production of reactive oxygen species; alternatively, microglial-derived NO may lead to presynaptic release of glutamate and excitotoxic injury. In addition, in a model of amyloid deposition, the EP2 receptor similarly promotes an increase in neuronal lipid peroxidation [13]. Taken together, these data suggest a dichotomy of function of the EP2 receptor, in which the EP2 receptor promotes primary neuroprotection in models of glutamate toxicity and ischemia, but secondary neurotoxicity in models where neuroinflammation and glial activation predominate.
One hypothesis to explain this divergent effect on neuronal survival is that prostaglandin signaling will lead to distinct downstream effects depending on the primary cell type involved. In the case of NMDA toxicity, dispersed hippocampal neurons are protected from toxicity with application of EP2 receptor agonist, suggesting that activation of the neuronal EP2 receptor promotes a neuroprotective response; supporting this is the dependence on intact neuronal cAMP signaling for this protection [16]. Conversely, stimulation of primary microglia with LPS results in production of soluble factors that induce paracrine synaptic and neuronal injury, and genetic ablation of the EP2 receptor in this paradigm is protective [20]. Thus the effect of prostaglandin signaling on neuronal viability appears to depend on the dominant cell type affected by the specific injury paradigm, ie neurons in the case of acute excitotoxicity versus microglia in models of inflammation. The protective function of EP2 signaling in vivo in models of excitotoxicity and cerebral ischemia [15, 16] and the neurotoxic effect of EP2 receptor signaling in the LPS [17, 19, 20] and amyloid models [13] lends support to this hypothesis.
A larger question is whether COX inhibition by non-steroidal anti-inflammatory drugs (NSAIDs), which should reduce signaling through the EP2 receptor by decreasing levels of PGE2, would have similar effects on neuronal viability as EP2 inhibition alone. In models of in vitro and in vivo excitotoxicity, COX-2 inhibition is clearly neuroprotective. However, downstream individual prostaglandin receptors mediate paradoxically protective effects in excitotoxic injury. These findings underscore the complexity of the eicosanoid response, and the final neuronal phenotype is likely to be the result of integrated signaling from cell-type specific prostaglandin signaling pathways that are differentially active depending on the type of injury.
Acknowledgments
This work was supported by NINDS (KA), DOD (KA), the MDA and Packard Foundation (KA), and the American Federation for Aging Research (KA).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Almer G, Guegan C, Teismann P, Naini A, Rosoklija G, Hays AP, Chen C, Przedborski S. Increased expression of the pro-inflammatory enzyme cyclooxygenase-2 in amyotrophic lateral sclerosis. Ann Neurol. 2001;49:176–185. [PubMed] [Google Scholar]
- 2.Andreasson KI, Savonenko A, Vidensky S, Goellner JJ, Zhang Y, Shaffer A, Kaufmann WE, Worley PF, Isakson P, Markowska AL. Age-dependent cognitive deficits and neuronal apoptosis in cyclooxygenase-2 transgenic mice. J Neurosci. 2001;21:8198–8209. doi: 10.1523/JNEUROSCI.21-20-08198.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bilak M, Wu L, Wang Q, Haughey N, Conant K, St Hillaire C, Andreasson K. PGE2 receptors rescue motor neurons in a model of amyotrophic lateral sclerosis. Ann Neurol. 2004;56:240–248. doi: 10.1002/ana.20179. [DOI] [PubMed] [Google Scholar]
- 4.Bosetti F. Arachidonic acid metabolism in brain physiology and pathology: lessons from genetically altered mouse models. J Neurochem. 2007 doi: 10.1111/j.1471-4159.2007.04558.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Breyer RM, Bagdassarian CK, Myers SA, Breyer MD. Prostanoid receptors: subtypes and signaling. Annu Rev Pharmacol Toxicol. 2001;41:661–690. doi: 10.1146/annurev.pharmtox.41.1.661. [DOI] [PubMed] [Google Scholar]
- 6.Chen C, Magee JC, Bazan NG. Cyclooxygenase-2 regulates prostaglandin E2 signaling in hippocampal long-term synaptic plasticity. J Neurophysiol. 2002;87:2851–2857. doi: 10.1152/jn.2002.87.6.2851. [DOI] [PubMed] [Google Scholar]
- 7.Drachman DB, Frank K, Dykes-Hoberg M, Teismann P, Almer G, Przedborski S, Rothstein JD. Cyclooxygenase 2 inhibition protects motor neurons and prolongs survival in a transgenic mouse model of ALS. Ann Neurol. 2002;52:771–778. doi: 10.1002/ana.10374. [DOI] [PubMed] [Google Scholar]
- 8.Drachman DB, Rothstein JD. Inhibition of cyclooxygenase-2 protects motor neurons in an organotypic model of amyotrophic lateral sclerosis. Ann Neurol. 2000;48:792–795. [PubMed] [Google Scholar]
- 9.Hewett SJ, Bell SC, Hewett JA. Contributions of cyclooxygenase-2 to neuroplasticity and neuropathology of the central nervous system. Pharmacology & therapeutics. 2006;112:335–357. doi: 10.1016/j.pharmthera.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 10.Hoozemans JJ, O’Banion MK. The role of COX-1 and COX-2 in Alzheimer’s disease pathology and the therapeutic potentials of non-steroidal anti-inflammatory drugs. Current drug targets. 2005;4:307–315. doi: 10.2174/1568007054038201. [DOI] [PubMed] [Google Scholar]
- 11.Kawano T, Anrather J, Zhou P, Park L, Wang G, Frys KA, Kunz A, Cho S, Orio M, Iadecola C. Prostaglandin E2 EP1 receptors: downstream effectors of COX-2 neurotoxicity. Nature medicine. 2006;12:225–229. doi: 10.1038/nm1362. [DOI] [PubMed] [Google Scholar]
- 12.Kiriyama M, Ushikubi F, Kobayashi T, Hirata M, Sugimoto Y, Narumiya S. Ligand binding specificities of the eight types and subtypes of the mouse prostanoid receptors expressed in Chinese hamster ovary cells. Br J Pharmacol. 1997;122:217–224. doi: 10.1038/sj.bjp.0701367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liang X, Wang Q, Hand T, Wu L, Breyer RM, Montine TJ, Andreasson K. Deletion of the prostaglandin E2 EP2 receptor reduces oxidative damage and amyloid burden in a model of Alzheimer’s disease. J Neurosci. 2005;25:10180–10187. doi: 10.1523/JNEUROSCI.3591-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liang X, Wu L, Hand T, Andreasson K. Prostaglandin D2 mediates neuronal protection via the DP1 receptor. J Neurochem. 2005;92:477–486. doi: 10.1111/j.1471-4159.2004.02870.x. [DOI] [PubMed] [Google Scholar]
- 15.Liu D, Wu L, Breyer R, Mattson MP, Andreasson K. Neuroprotection by the PGE2 EP2 receptor in permanent focal cerebral ischemia. Ann Neurol. 2005;57:758–761. doi: 10.1002/ana.20461. [DOI] [PubMed] [Google Scholar]
- 16.McCullough L, Wu L, Haughey N, Liang X, Hand T, Wang Q, Breyer RM, Andreasson K. Neuroprotective function of the PGE2 EP2 receptor in cerebral ischemia. J Neurosci. 2004;24:257–268. doi: 10.1523/JNEUROSCI.4485-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Milatovic D, Zaja-Milatovic S, Montine KS, Shie FS, Montine TJ. Neuronal oxidative damage and dendritic degeneration following activation of CD14-dependent innate immune response in vivo. J Neuroinflammation. 2004;1:20. doi: 10.1186/1742-2094-1-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Minghetti L. Role of inflammation in neurodegenerative diseases. Current opinion in neurology. 2005;18:315–321. doi: 10.1097/01.wco.0000169752.54191.97. [DOI] [PubMed] [Google Scholar]
- 19.Montine TJ, Milatovic D, Gupta RC, Valyi-Nagy T, Morrow JD, Breyer RM. Neuronal oxidative damage from activated innate immunity is EP2 receptor-dependent. J Neurochem. 2002;83:463–470. doi: 10.1046/j.1471-4159.2002.01157.x. [DOI] [PubMed] [Google Scholar]
- 20.Shie FS, Montine KS, Breyer RM, Montine TJ. Microglial EP2 is critical to neurotoxicity from activated cerebral innate immunity. Glia. 2005;52:70–77. doi: 10.1002/glia.20220. [DOI] [PubMed] [Google Scholar]
- 21.Taniguchi H, Mohri I, Okabe-Arahori H, Aritake K, Wada K, Kanekiyo T, Narumiya S, Nakayama M, Ozono K, Urade Y, Taniike M. Prostaglandin D2 protects neonatal mouse brain from hypoxic ischemic injury. J Neurosci. 2007 doi: 10.1523/JNEUROSCI.0321-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Teismann P, Tieu K, Choi DK, Wu DC, Naini A, Hunot S, Vila M, Jackson-Lewis V, Przedborski S. Cyclooxygenase-2 is instrumental in Parkinson’s disease neurodegeneration. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:5473–5478. doi: 10.1073/pnas.0837397100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yamagata K, Andreasson K, Kaufmann WE, Barnes CA, Worley PF. Expression of a mitogen-inducible cyclooxygenase in brain neurons: regulation by synaptic activity and glucocorticoids. Neuron. 1993;11:371–386. doi: 10.1016/0896-6273(93)90192-t. [DOI] [PubMed] [Google Scholar]
- 24.Yasojima K, Tourtellotte WW, McGeer EG, McGeer PL. Marked increase in cyclooxygenase-2 in ALS spinal cord: implications for therapy. Neurology. 2001;57:952–956. doi: 10.1212/wnl.57.6.952. [DOI] [PubMed] [Google Scholar]