

Summary
Symptoms of T-cell hyperactivation shape the course and outcome of HIV-1 infection, but the mechanism(s) underlying this chronic immune activation are not well understood. We find that the viral transactivator Tat promotes hyperactivation of T cells by blocking the nicotinamide adenine dinucleotide (NAD+)-dependent deacetylase SIRT1. Tat directly interacts with the deacetylase domain of SIRT1 and blocks the ability of SIRT1 to deacetylate lysine 310 in the p65 subunit of NF-κB. Because acetylated p65 is more active as a transcription factor, Tat hyperactivates the expression of NF-κB-responsive genes, a function lost in SIRT1−/− cells. These results support a model where the normal function of SIRT1 as a negative regulator of T-cell activation is suppressed by Tat during HIV infection. These events likely contribute to the state of immune cell hyperactivation found in HIV-infected individuals.
Introduction
Immune activation is a hallmark of HIV-1 infection and a significant factor that promotes continuous viral replication and CD4+ T-cell depletion (Douek et al., 2003; Fauci et al., 1996; Lawn et al., 2001). In HIV-infected individuals, levels of circulating activation markers correlate with accelerated disease progression and shortened survival. HIV infection is critically dependent on the activated state of CD4+ T cells since the virus cannot replicate efficiently in resting T cells. Quiescent T cells in blood are refractory to infection because of blocks at the level of reverse transcription and proviral integration (Chiu et al., 2005; Stevenson et al., 1990). In addition, T-cell activation enhances viral transcription through the activation of various transcription factors, notably nuclear factor κB (NF-κB) (Nabel and Baltimore, 1987).
HIV-1 infection itself manipulates the activation status of infected T cells through the expression of viral proteins, including Env, Nef and Tat (Chirmule and Pahwa, 1996; Fackler and Baur, 2002; Peruzzi, 2006; Schindler et al., 2006). The viral transactivator Tat potently activates HIV transcription and binds to an RNA stem-loop structure termed TAR that spontaneously forms at the 5′ extremities of all viral transcripts (Barboric and Peterlin, 2005). Tat also influences the expression of cellular genes in infected T cells. For example, Tat synergizes with signals mediated via the T-cell receptor (TCR) and the CD28 coreceptor to superactivate interleukin (IL)-2 gene expression (Fortin et al., 2004; Ott et al., 1997; Westendorp et al., 1994).
IL-2 is a T-cell growth factor with critical functions in lymphocyte proliferation, cell survival and tolerance (Waldmann, 2006). IL-2 expression is dependent on coordinated signals delivered via the TCR and coreceptors and is tightly regulated at the transcriptional level. IL-2 is important during HIV infection: it primes nonactivated bystander cells for infection in the absence of antigenic stimulation (Kinter et al., 1995; Unutmaz et al., 1994). IL-2, IL-15 and other soluble factors present at lower concentration in the milieu of lymphoid tissue combine to increase permissiveness of naïve CD4+ T cells to HIV infection (Kreisberg et al., 2006).
While the function of Tat in viral transcription is well studied, the molecular mechanism underlying its immunomodulatory effects is less clear. Many prior reports have linked Tat with altered NF-κB action (Buonaguro et al., 1994; Cota-Gomez et al., 2002; Demarchi et al., 1996; Ott et al., 1997; Ott et al., 1998; Scala et al., 1994; Westendorp et al., 1994; Westendorp et al., 1995). In addition to its central role in the regulation of the innate and adaptive immune responses, NF-κB is a critical regulator of HIV transcription. The HIV-1 long terminal repeat (LTR) contains two tandem NF-κB binding sites. The activity of the prototypical NF-κB complex (a heterodimer of p50 and p65 subunits) is regulated through its association with inhibitory IκB molecules and through various posttranslational modifications, including phosphorylation and acetylation. IκB molecules complexed to the NF-κB dimer in the cytoplasm are rapidly phosphorylated, polyubiquitylated and degraded after T-cell activation via the TCR, allowing nuclear translocation of NF-κB. After translocation, the p65 subunit undergoes posttranslational modifications, including reversible acetylation by the histone acetyltransferase p300 (Chen et al., 2002; Kiernan et al., 2003). The acetylation of lysines 218 and 221 enhances DNA binding and prevents assembly with IκBα and nuclear export of p65, while acetylation of lysine 310 (K310) potentiates the transcriptional activity of NF-κB. The transcriptional activity of NF-κB is suppressed by the deacetylase activities of histone deacetylase (HDAC)-3 and SIRT1, two proteins that deacetylate p65 (Chen et al., 2001; Yeung et al., 2004). SIRT1 specifically deacetylates K310 in p65 and inhibits the anti-apoptotic function of NF-κB (Yeung et al., 2004).
SIRT1 is a mammalian homologue of the yeast transcriptional repressor silent information regulator 2 (Sir2), an important factor governing longevity in yeast (Blander and Guarente, 2004). Like Sir2, SIRT1 requires nicotinamide adenine dinucleotide (NAD+) as a cofactor, which links its activity to the metabolic state of the cell. In addition to its enzymatic activity on histone substrates in vitro, recent experimental evidence suggests that SIRT1 predominantly targets nonhistone proteins for deacetylation. We recently reported that Tat is a substrate for the deacetylase activity of SIRT1 (Pagans et al., 2005). Acetylation of lysine 50 (K50) in Tat is mediated by the histone acetyltransferase activities of p300 and human GCN5 and generates binding sites for the bromodomains of PCAF and Brg1 (Agbottah et al., 2006; Col et al., 2001; Dorr et al., 2002; Kiernan et al., 1999; Mahmoudi et al., 2006; Mujtaba et al., 2002; Ott et al., 1999). SIRT1 binds and deacetylates Tat at K50, a process necessary to recycle nonacetylated Tat protein for binding to TAR RNA and the cellular positive transcription elongation factor b (P-TEFb).
Because SIRT1 is an important regulator of both NF-κB and HIV Tat protein, we examined the effects of Tat on the deacetylase activity of SIRT1. Our finding that Tat inhibits SIRT1 activity and potentiates NF-κB transcriptional activity unveils a new molecular mechanism by which hyperactivation of immune cells is promoted during HIV infection.
Results
Tat and nicotinamide hyperactivate T cells via the same cellular pathway
To recapitulate the effect of Tat on immune activation in a manner as close to natural HIV infection as possible, we employed an HIV-based lentiviral vector in which both FLAG-tagged Tat and green fluorescent protein (GFP) are expressed under the control of the HIV promoter in the 5′-LTR. Jurkat T cells were infected with viral particles containing this vector or a control vector expressing only GFP. Infected cultures were stimulated with antibodies specific for the CD3 and CD28 receptors to mimic physiological T-cell activation.
Tat expression was visualized by western blot analysis (Figure 1A). Treatment with α-CD3/28 slightly increased the levels of Tat due to the stimulatory effect of these antibodies on the viral LTR. In cells expressing Tat, a ~1200-fold induction of IL-2 mRNA was detected by real-time PCR in response to α-CD3/28 treatment, whereas IL-2 expression in control cells was only induced ~250 fold (Figure 1B). A similar difference was observed when IL-2 protein was measured in the culture supernatant (Figure 1C). No effect of Tat was observed in nonactivated cultures. These findings recapitulated results previously obtained in peripheral blood lymphocytes and Jurkat T cells infected with infectious HIV and support a model where Tat synergistically stimulates IL-2 production following ligation of the CD3 and CD28 receptors (Fortin et al., 2004; Ott et al., 1997).
Figure 1.

Tat and nicotinamide cause T-cell hyperactivation
(A) Western blot analysis of Tat protein in Jurkat T cells infected with lentiviral vectors expressing LTR-GFP and LTR-Tat-GFP. Infected cultures were activated for 2 h and 8 h with α-CD3/28 antibodies.
(B) Real-time RT-PCR analysis of IL-2 mRNA levels in infected cultures 2 h after activation with α-CD3/28. Results are expressed as fold induction by α-CD3/28 treatment.
(C) IL-2 protein levels measured by ELISA in the supernatant of infected cultures 8 h after α-CD3/28 treatment.
(D) Real-time RT-PCR analysis of IL-2 mRNA levels in noninfected Jurkat T cells after treatment with nicotinamide and activation with α-CD3/28.
(E) Real-time RT-PCR analysis of IL-2 mRNA levels in Jurkat T cells infected with lentiviral vectors after treatment with nicotinamide and activation with α-CD3/28 antibodies.
(F) Western blot analysis of Tat protein in Jurkat T cells infected with lentiviral vectors after treatment with nicotinamide and activation with α-CD3/28. In B–E averages of three independent experiments (±SEM) are shown.
To test the potential regulatory role of SIRT1 in the IL-2 response, we treated Jurkat T cells with nicotinamide, a natural byproduct of the sirtuin deacetylase reaction which functions as a feedback inhibitor of these enzymes (Landry et al., 2000). Treatment with nicotinamide superinduced IL-2 mRNA to similar levels as Tat in α-CD3/28-stimulated cells (Figure 1D). Superinduction of IL-2 mRNA levels was also observed when SIRT1 expression was knocked down in Jurkat T cells via siRNAs (Supplemental Figure S1). To test whether Tat and nicotinamide synergize in the hyperactivation of IL-2 expression, we treated Tat- or control-infected Jurkat T cells with nicotinamide before activation. These studies revealed no additive effect of combinations of Tat and nicotinamide on IL-2 expression in α-CD3/28-stimulated T cells (Figure 1E). Levels of Tat protein were equivalent in infected cultures treated with nicotinamide and control-treated cultures excluding the possibility that nicotinamide suppressed the expression of Tat (Figure 1F). These results suggest that Tat and nicotinamide may target closely related cellular pathways and raised the interesting possibility that Tat hyperactivation of T cells may involve its known assembly with SIRT1.
SIRT1 is required for the hyperactivation of NF-κB target genes by Tat
T-cell activation is coupled to the stimulation of several transcription factors that regulate the transcriptional status of the IL-2 gene. We focused our efforts on NF-κB since its activity is regulated by SIRT1 (Yeung et al., 2004). We and others previously reported that Tat enhanced IL-2 promoter activity via NF-κB binding sites present within the IL-2 promoter (Fortin et al., 2004; Ott et al., 1997; Ott et al., 1998; Westendorp et al., 1994). We speculated that Tat, like nicotinamide, inhibits the SIRT1 deacetylase activity leading to hyperactivation of NF-κB-responsive genes including IL-2.
We tested this hypothesis in mouse embryonic fibroblasts (MEFs) derived from SIRT1−/−mice. MEFs from SIRT1−/− and SIRT1+/+ mice were treated with TNFα to induce NF-κB. Since MEFs do not produce IL-2, we focused instead on the expression of IκBα, a ubiquitous NF-κB-responsive gene product (Sun et al., 1993). Copy numbers of IκBα mRNA were elevated in SIRT1−/− MEFs under basal conditions and in response to TNFα, consistent with the model that SIRT1 suppresses the expression of NF-κB-dependent genes (Supplemental Figure S2A). Introduction of Tat via retroviral infection increased endogenous levels of IκBα mRNA 5 to 11-fold in SIRT1+/+ MEFs, but only 2 to 3-fold in SIRT1−/− MEFs (Supplemental Figure S2B).
To confirm that Tat-mediated superinduction of NF-κB-responsive genes was dependent on SIRT1, we reintroduced SIRT1 in SIRT1−/− MEFs via retroviral infection. Polyclonal populations of reconstituted SIRT1-expressing or SIRT1-null cells were each infected with retroviral vectors expressing Tat or with the empty vector alone (Figure 2A). When we examined the induction of IκBα mRNA in response to TNFα, we found that the presence of Tat increased the induction of IκBα mRNA 2 to 4-fold over control-infected cells (Figure 2B). This effect was only observed in cultures where SIRT1 expression had been reconstituted, demonstrating that Tat requires SIRT1 expression for the superinduction of the IκBα gene.
Figure 2.

Tat-mediated superinduction of NF-κB-responsive genes requires SIRT1
(A) Western blot analysis of SIRT1, Tat and β-actin in SIRT1−/− MEF cells infected with MSCV-based retroviral particles expressing SIRT1 or Tat.
(B–D) Real-time RT-PCR analysis of IκBα (B), E-selectin (C), and GAPDH (D) mRNA levels in the four polyclonal MEF populations described in (A) after incubation with TNFα (20 ng/ml) for 2 h and 8 h. Data are presented relative to values obtained in cells lacking Tat (100%). The mean of three independent experiments (±SEM) is shown.
A similar result was observed with E-selectin, another NF-κB-responsive gene that is also superinduced by Tat (Cota-Gomez et al., 2002; Lee et al., 2004). After TNFα treatment, endogenous E-selectin gene expression was enhanced 6-fold by Tat in SIRT1-expressing, but not in SIRT1-negative cells (Figure 2C). Here, the Tat effect was only observed at 2 h after TNFα treatment. At 8 h, no induction of E-selectin gene expression was evident, consistent with the prior finding that E-selectin mRNA accumulation peaks 2 h after TNFα treatment and then rapidly declines (Edelstein et al., 2005). In contrast to the results with IκBα and E-selectin, Tat had no effect on the constitutive expression of the endogenous glyceraldehyde-3-phosphate dehydrogenase gene (GAPDH) (Figure 2D).
Tat neutralizes the negative effect of SIRT1 on NF-κB function
Next, we assessed the effect of Tat on the IκBα and E-selectin gene promoters. We transfected an IκBα promoter luciferase reporter construct together with combinations of expression vectors encoding p65, SIRT1, and Tat into HeLa cells (Figure 3A). SIRT1 suppressed the activation of the IκBα promoter by p65 in accordance with the previous finding that deacetylation by SIRT1 inactivates p65 activity (Yeung et al., 2004). Coexpression of Tat restored p65 activity, suggesting that Tat interfered with the deacetylation of p65 by SIRT1 (Figure 3A). No change in expression levels of p65 was observed in the presence of SIRT1 or Tat (Supplemental Figure S3). The suppressive activity of SIRT1 was dependent on its intrinsic deacetylase activity, since a catalytically inactive SIRT1 mutant (P447E; shown in Figure 4D) did not suppress the activity of p65 and was unresponsive to the action of Tat. Expression of Tat did not affect basal or p65-mediated activity of the IκBα promoter in the absence of SIRT1 expression (Figure 3A).
Figure 3.
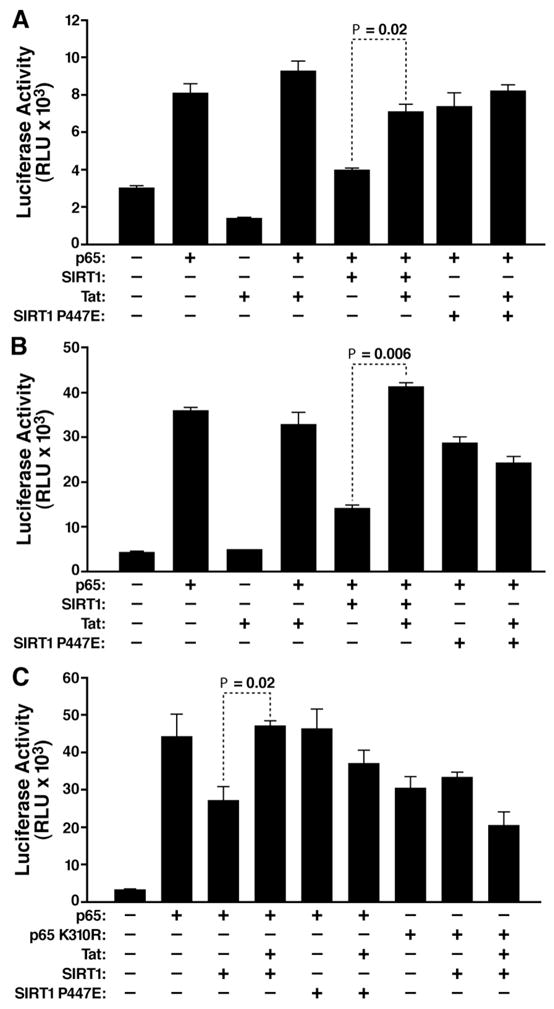
Tat neutralizes the inhibitory effect of SIRT1 on NF-κB-responsive promoters
Promoter reporter assays using (A) the IκBα, (B) the E-selectin, and (C) the 3X-κB luciferase reporter construct. Luciferase constructs (0.2 μg) were transiently cotransfected with expression vectors for p65 or p65 K310R (1 ng), SIRT1 or SIRT1 P447E (0.2 μg), and Tat (50 ng) into HeLa cells. The mean of three independent experiments (±SEM) is shown.
Figure 4.

Tat binds to the acetyl lysine-binding site in SIRT1
(A) Coimmunoprecipitation assay of Tat and SIRT1 deletion mutants. Expression vectors for Myc-tagged murine Sir2α deletion mutants or full length human SIRT1 (each 1 μg) were transiently cotransfected with constructs expressing FLAG-tagged Tat (1 μg) in 293 cells. Immunoprecipitations were performed with α-FLAG agarose and western blotting with α-Myc and α-FLAG antibodies.
(B) Schematic summary of Tat binding to murine Sir2α deletion mutants. The relative localization of the human SIRT1 HDAC domain is shown at the bottom.
(C) Schematic representation of the human SIRT1 HDAC domain with point mutations.
(D) In vitro HDAC assays of immunoprecipitated human SIRT1 mutants with and without NAD+.
(E) Coimmunoprecipitation of SIRT1 mutants and Tat in 293 cells. Cell extracts were immunoprecipitated with α-T7 agarose followed by western blotting with α-FLAG and α-T7 antibodies.
The same results were observed using an E-selectin promoter luciferase construct. Again, SIRT1 reduced p65-activated E-selectin promoter activity, and Tat reversed the negative effect of SIRT1 (Figure 3B). No reduction was observed with the mutant SIRT1, which was also unresponsive to Tat (Figure 3B).
To verify that Tat and SIRT1 modulated the activities of the IκBα and E-selectin promoters through NF-κB, we transfected p65-, Tat-, and SIRT1-expressing constructs together with a 3X-κB luciferase reporter containing triple NF-κB-binding sites corresponding to the high affinity IL-2 receptor promoter. Expression of SIRT1 reduced p65-mediated activation of the 3X-κB reporter, while coexpression of Tat reversed this negative effect (Figure 3C). Again, expression of the catalytically inactive SIRT1 mutant did not interfere with the activity of p65, and additional Tat expression had no effect in this setting (Figure 3C). Likewise, overexpression of a p65 mutant (K310R), which lacked the deacetylation site for SIRT1 and showed reduced activation of the 3X-κB reporter, was unaffected by SIRT1 and Tat (Figure 3C). These results demonstrate that Tat targets the SIRT1 deacetylase activity to hyperactivate NF-κB function.
Tat binds the deacetylase domain of SIRT1
We showed previously that Tat and SIRT1 interact in cells (Pagans et al., 2005). To map the Tat-interacting domain in SIRT1, we used deletion mutants of the murine SIRT1 protein (Sir2α) (Fulco et al., 2003). Progressive N-terminal deletions at amino acids 120, 236, or 341 did not affect coimmunoprecipitation of SIRT1 with Tat (Figure 4A, lanes 1–3). However, deletion of the first 512 amino acids abrogated binding to Tat, indicating that the Tat-interaction domain lies between amino acids 341 and 512 of SIRT1 (Figure 4A, lane 4). In agreement with this conclusion, expression of amino acids 236 to 510 of SIRT1 proved sufficient to support interaction with Tat (Figure 4A, lane 5). The results of the coimmunoprecipitation studies are summarized in Figure 4B.
The region spanning amino acids 341–512 contains the catalytic domain of SIRT1, which is shared among all sirtuins. It is 98% conserved in the murine and human SIRT1 proteins and consists of two distinct domains: a large Rossmann fold domain characteristic of NAD+-binding proteins and a smaller domain containing a structural zinc ion. The NAD+ and acetyl-lysine substrate bind in the active site cleft between the two domains (Avalos et al., 2002). To determine whether Tat interacts with the catalytic domain of SIRT1, point mutations were introduced into human SIRT1 expression construct to disrupt NAD+-binding (R274A, N346A) or acetyl-lysine binding (F414D, E416A, V445E, P447E) (Figure 4C). In addition, we mutated two conserved cysteines (C371/374G) located in a zinc-bound domain predicted to serve as a protein-protein interaction domain (Min et al., 2001).
Prior to analyzing their interaction with Tat, the catalytic activities of these mutants were measured using an in vitro HDAC assay and radioactive acetylated histone peptides as substrates. We transfected wild type and mutant SIRT1 proteins into 293 cells and immunoprecipitated the FLAG-tagged proteins (Figure 4D). The immunoprecipitated material was tested for enzymatic activity. Incubation with wild type SIRT1 or the R274A mutant resulted in deacetylation of the histone peptide in the presence of NAD+ (Figure 4D). The deacetylase activity of the F414D, E416A, and V445E mutants was severely impaired, while no activity was measured for N346A, C371/374G, and P447E mutants.
Next, we cotransfected wild type and SIRT1 mutants together with T7-tagged Tat. Wild type and mutant SIRT1 proteins were expressed at similar levels (Figure 4E, upper panel). After pulldown with T7-agarose, immunoprecipitated complexes were analyzed for the presence of SIRT1 (Figure 4E, lower panels). While binding to Tat was preserved in SIRT1 R274A, N346A, C371/374G, and E416A, no binding to Tat was observed with any of the SIRT1 mutants carrying a mutation within the acetyl-lysine-binding domain (F414D, V445E, P447E). The same results were obtained when SIRT1 proteins were immunoprecipitated, and coimmunoprecipitation of Tat was determined (data not shown). These data demonstrate that Tat interacts with the substrate-binding domain of SIRT1.
Tat inhibits the SIRT1 deacetylase activity and induces hyperacetylation of NF-κB
Since an intact acetyl-lysine binding function is critical for the deacetylase activity of SIRT1, we tested whether Tat binding affected the enzymatic activity of SIRT1. We performed fluorescent deacetylation assays using purified full-length SIRT1 and acetylated p53 peptides (aa 379–382). Addition of synthetic Tat (amino acid 1–72) inhibited deacetylation of p53 by SIRT1 in a dose-dependent manner (Figure 5A). The Tat concentration required for half maximal inhibition (IC50) of SIRT1 was 3.5 ± 0.5 μM. In contrast, the IC50 for nicotinamide in the same experiments was 208 ± 21 μM (data not shown). This result indicates that Tat is a very potent inhibitor of the SIRT1 deacetylase activity.
Figure 5.

Tat inhibits the SIRT1 deacetylase activity
(A) In vitro deacetylation assay of recombinant SIRT1 in the presence of synthetic Tat (aa 1–72). The rate of 1 μM SIRT1 was calculated for 200 μM fluorogenic deacetylase substrate (aa 379–382 of p53) in the presence of twelve concentrations of synthetic Tat. The data were plotted in Prism with X equal to Log10[Tat(μM)] and Y equal to rate. A nonlinear regression was done using the one-site competitive binding equation to determine the IC50.
(B) In vitro deacetylase assay of recombinant SIRT1 and recombinant acetylated p65 in the presence of synthetic Tat or nicotinamide. Western blotting was performed with α-AcK310 p65 and α-p65 antibodies.
(C) In vivo acetylation assay of overexpressed p65. Expression vectors encoding T7-tagged p65 (0.5 μg), p300 (2 μg), SIRT1 (1 μg), and Tat (0.1, 0.25, and 0.5 μg) were transiently cotransfected in 293 cells as indicated. Total cell lysates were analyzed by western blotting with α-AcK310 p65, α-T7, and α-FLAG antibodies. p300 was immunoprecipitated and subjected to western blotting with α-HA antibody.
(D) In vivo acetylation assay of endogenous p65. 293 cells were cotransfected with expression vectors encoding Tat (0.5 μg) and p300 (10 μg) as indicated and treated with TNFα (20 ng/ml) and trichostatin (TSA; 400 nM) over night. TSA is an inhibitor of class I and II HDACs, but not sirtuins and was added to prolong nuclear retention of p65 through hyperacetylation of K218 and K220. Endogenous p65 was immunoprecipitated using α-p65 antibody and subjected to western blotting with α-AcK310 p65 or α-p65 antibodies. Western blotting for p300 was performed with α-HA antibody.
Next, we examined the effect of Tat on deacetylation of p65. In vitro acetylated recombinant p65 protein was incubated with recombinant SIRT1 and synthetic Tat proteins. The extent of deacetylation was determined by western blotting with an antibody specific for acetylated K310 in p65 (α-AcK310 p65). SIRT1 deacetylated p65 in the absence of Tat (Figure 5B, lane 2). In the presence of Tat, deacetylation of p65 by SIRT1 was suppressed, confirming that direct interaction between Tat and SIRT1 inhibits the deacetylase activity of SIRT1 (Figure 5B, lane 3). The same inhibition of p65 deacetylation by SIRT1 was observed with nicotinamide (Figure 5B, lane 4).
To test the effect of Tat on p65 acetylation in cells, we transfected expression vectors for p65, p300, SIRT1 and Tat into 293 cells. Overexpressed p65 translocates spontaneously into the cell nucleus because of limiting amounts of cellular IκBα. To induce efficient p65 acetylation, we coexpressed the acetyltransferase p300 (Figure 5C, lane 2). Coexpression of SIRT1 reduced acetylation of p65 at K310 caused by p300 as expected (Figure 5C, lane 3). Introduction of Tat restored p65 acetylation in a dose-response manner (Figure 5C, lane 4–6). These results demonstrate that Tat reverts deacetylation of p65 by SIRT1 in cells and induces relative hyperacetylation of p65 in the presence of SIRT1.
We also examined the acetylation status of endogenous p65 in Tat-expressing cells. 293 cells were transfected with the Tat expression vector and treated with TNFα to induce nuclear translocation of endogenous p65. Acetylation of endogenous p65 was slightly (1.5-fold) increased in the presence of Tat (Figure 5D, lane 2). A similar increase was observed when p300 was overexpressed (Figure 5D, lane 3). Coexpression of p300 and Tat strongly induced hyperacetylation of endogenous p65 (2.5-fold), consistent with the model that Tat inhibits physiological deacetylation of NF-κB by SIRT1 (Figure 5D, lane 4).
Tat induces T-cell hyperactivation through hyperacetylation of NF-κB
To examine whether the functions of Tat in SIRT1 inhibition and T-cell hyperactivation are linked, we searched for Tat mutants deficient in the induction of p65 hyperacetylation. We screened a series of Tat proteins carrying point mutations in different protein domains of Tat. These point mutants were expressed in 293 cells together with p65 and the p300 acetyltransferase. Expression of p300 induced acetylation of K310 in p65, and wild type Tat (amino acid 1–101) further enhanced this acetylation (Figure 6A, lanes 1–3). Mutations within the so-called cysteine-rich and core domains of Tat (C22G, F38A, K41A) markedly decreased hyperacetylation of p65 induced by Tat, while a point mutation introduced into the N-terminal acidic domain (H13L) still hyperacetylated p65 albeit to a lesser extent (Figure 6A, lanes 4–7). A Tat mutant containing two point mutations within the RNA-binding domain (K50/51A) induced p65 hyperacetylation as efficiently as the wild type protein (Figure 6A, lane 8). Western blotting with FLAG antibody showed that wild type and H13L Tat were equally expressed while the other Tat mutants were slightly less expressed (Figure 6A, lower panel). Of note, expression levels of K41A and K50/51A Tat were equivalent, and both mutants produced similar effects on acetylation of endogenous p65 as observed with overexpressed protein (Supplemental Figure S4). These mutants were chosen for further analysis.
Figure 6.

Hyperacetylation of p65 by Tat contributes to T-cell hyperactivation
(A) Induction of p65 hyperacetylation by wild type and mutant Tat. Expression vectors encoding T7-tagged p65 (0.5 μg), p300 (2 μg), wild type and mutant Tat (1 μg each) were transiently cotransfected in 293 cells as indicated. Immunoprecipitations were performed with α-T7 agarose or α-FLAG agarose to isolate p65 or Tat followed by western blotting with α-AcK310 p65, α-T7, and α-FLAG antibodies. p300 was immunoprecipitated and subjected to western blotting with α-HA antibody.
(B) Flow cytometry of Jurkat T cells infected with lentiviral vectors expressing wild type or mutant Tat and GFP. Data are representative of three independent experiments in which infection efficiencies ranged from 75–85% GFP+ cells.
(C) Real-time RT-PCR analysis of IL-2 mRNA levels induced in infected Jurkat T cells after activation with α-CD3/28 antibodies. Data are presented as fold induction by α-CD3/28 treatment relative to vector (EF-1α-GFP)-infected cells (100%). The average (mean±SEM) of three independent experiments is shown.
To study the effect of Tat mutations on T-cell activation, we generated lentiviral vectors expressing wild type or mutant Tat proteins (K41A and K50/51A Tat). Because both Tat mutants have reduced transcriptional activities on the HIV LTR, we chose a lentiviral construct, which contained the heterologous elongation factor 1α (EF-1α) promoter driving Tat and GFP expression instead of the HIV LTR. High titer viral stocks were produced and used to infect Jurkat T cells. Flow cytometry of GFP showed equivalent infection with wild type or mutant Tat-expressing viruses (Figure 6B). Following treatment with α-CD3/28 antibodies, IL-2 mRNA levels were superinduced 2–3 fold by wild type Tat (Figure 6C). No superinduction was observed with K41A Tat while K50/51A Tat superinduced IL-2 mRNA levels as efficiently as wild type Tat (Figure 6C). These results collectively demonstrate that Tat expressed in the context of lentiviral infection hyperactivates T cells through hyperacetylation of p65.
Discussion
Our finding that Tat inactivates cellular SIRT1 provides a novel molecular mechanism responsible for Tat-mediated T-cell hyperactivation. These studies also reveal a unique role for SIRT1 in T-cell activation.
We find that Tat binds to the SIRT1 catalytic site and impairs the ability of SIRT1 to deacetylate K310 in p65. Consistent with previous data that deacetylation by SIRT1 inactivates p65 function we find NF-κB activity “superactivated” in the presence of Tat. Based on these results, we propose a model where Tat hyperactivates T cells through inhibition of SIRT1 and abnormally sustained action of NF-κB. In the absence of Tat, a balance of p300 and SIRT1 activities regulates p65 acetylation. This balance tightly controls the activation of cellular genes involved in the immune response of T cells (Figure 7A). During HIV infection, Tat expression disrupts the balance between p300 and SIRT1 and increases levels of acetylated p65 through inhibition of SIRT1 (Figure 7B). This increase hyperactivates the function of NF-κB and the expression of NF-κB-responsive genes including IL-2.
Figure 7.
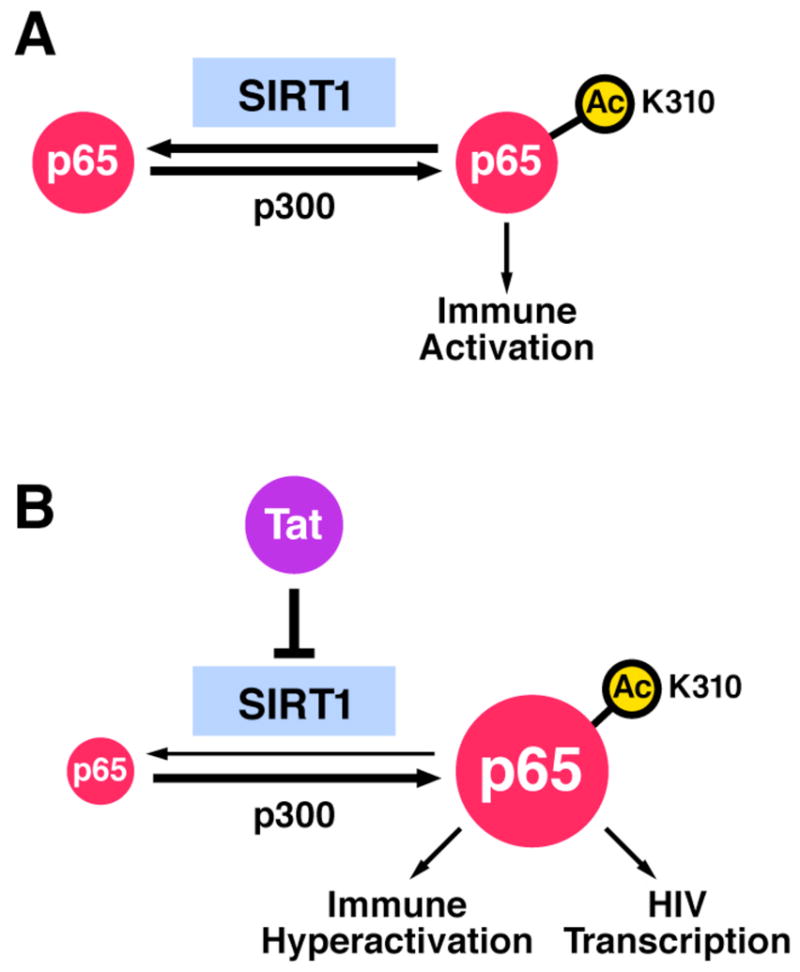
Tat blocks the SIRT1 deacetylase activity and superinduces T-cell activation and HIV transcription via NF-κB. See text for details.
One of the most studied NF-κB-responsive genes is HIV itself. The two NF-κB-binding sites in the HIV enhancer link HIV transcription to the activation status of the infected T cells. This is particularly important at the beginning of the infectious process and during reactivation from latency when Tat levels are limiting. During these times, the action of cellular transcription factors, most importantly NF-κB, is essential for the production of full-length viral transcripts necessary for the initial synthesis of Tat. Once Tat accumulates to sufficient levels, it binds to TAR RNA and dramatically increases the elongation competence of the RNA polymerase II complex through recruitment of the P-TEFb complex. At that time, Tat may also enhance NF-κB function through inhibition of SIRT1 (Figure 7B).
Tat itself is deacetylated by SIRT1. Although SIRT1 is a negative regulator of NF-κB, it functions as a coactivator of Tat transactivation through deacetylation of K50 (Pagans et al., 2005). We favor the model that deacetylation of Tat occurs early in the infectious process and recycles acetylated Tat to the unacetylated form, which is the only form of Tat able to engage in the formation of the Tat/TAR/P-TEFb complex. Interestingly, in vitro binding of Tat to SIRT1 does not require acetylation of K50 (Pagans et al., 2005). Accordingly, we show that hyperacetylation of p65 and T-cell hyperactivation are efficiently induced by a Tat mutant that lacks an intact acetylation site at position 50 (K50/51A Tat). This raises the possibility that Tat acts as a “supersubstrate” of the SIRT1 enzymatic activity (Borgstrom, 1975). Acetylated Tat is efficiently deacetylated by SIRT1, but remains bound to SIRT1 and blocks access of other substrates to the catalytically active site.
We identified Tat mutants (C22G, F38A, K41A) that no longer induce p65 hyperacetylation. The finding that one of these mutants (K41A) also lost its ability to hyperactivate T cells couples the inhibitory function of Tat on SIRT1 with its role in T-cell hyperactivation. Interestingly, C22G, F38A, and K41A Tat mutants are deficient in the transactivation of the HIV LTR. This raises the possibility that Tat-mediated hyperacetylation of p65 may be part of the activatory function of Tat on the HIV promoter. However, other Tat mutations (H13L, K50/51A) also reduce Tat function on the LTR but maintain the ability of Tat to induce p65 hyperacetylation (Emiliani et al., 1998; Ott et al., 1999). C22G, F38A, and K41A mutations lie in the transactivation domain of Tat, which mediates interaction of Tat with the cofactor cyclinT1 (Rice and Carlotti, 1990; Wei et al., 1998). The finding that Tat hyperactivates NF-κB activity in murine cells makes a direct involvement of cyclin T1 in Tat-mediated T-cell hyperactivation unlikely since murine cyclin T1 cannot properly bind Tat (Bieniasz et al., 1998; Garber et al., 1998). However, the fact that both Tat and p65 bind cyclinT1 as well as SIRT1 is striking and could indicate that P-TEFb is somehow involved in this process (Barboric et al., 2001; Pagans et al., 2005; Yeung et al., 2004).
SIRT1 targets many transcriptional activators in addition to p65, including p53, Forkhead transcription factors (FOXO1, 3a, and 4), as well as transcriptional coactivators PGC-1α, PCAF and p300 (Sauve et al., 2006). The observation that Tat inhibits a central regulator of the function of many transcription factors and transcriptional coactivators provides an explanation of Tat’s reported pleiotropism. However, these observations raise a number of additional questions. One remaining question concerns the role of the second exon of the tat gene in T-cell hyperactivation. We and others have previously reported that only the 101-amino acid Tat protein encoded by both tat exons induces T-cell hyperactivation, while the shorter 72-amino acid one-exon Tat variant does not (Ott et al., 1997; Westendorp et al., 1994). Our new data show that Tat72 effectively blocks the SIRT1 deacetylase activity in vitro and link residues within the transactivating domain of Tat shared by Tat101 and Tat72 with the hyperacetylation of p65 and T-cell hyperactivation. We have observed that Tat72 is markedly less expressed in infected T cells than Tat101 (H-S. Kwon and M. Ott, unpublished data). Future studies will investigate whether this difference in expression may account for the different in vivo functions of the two Tat variants or whether residues in the C terminus of Tat101 are also involved in this process.
T-cell activation is a highly controlled process and involves positive and negative regulatory mechanisms. Our results showing that inhibition of SIRT1 by Tat and nicotinamide lead to T-cell hyperactivation identify SIRT1 as a negative regulator of T-cell activation. This concept is supported by our preliminary findings that SIRT1 protein levels are upregulated after activation of human primary CD4+ T cells (H-S. Kwon and M. Ott, unpublished data). This upregulation occurs rather late after activation and may serve to “terminate” NF-κB function through p65 deacetylation.
T-cell activation is dysregulated during the immune hyperactivation syndrome present in individuals infected with HIV-1. Increased production of proinflammatory cytokines is found in circulating blood lymphocytes as well as in T cells isolated from lymphoid tissue (Fauci et al., 1996). Many of these cytokines are regulated by NF-κB and are dysregulated by Tat (Buonaguro et al., 1994; Scala et al., 1994). Tat-mediated inhibition of the SIRT1 deacetylase and the resulting hyperactivation of NF-κB function may thereby directly participate in the immune dysfunctions in infected patients. While the interaction of Tat with SIRT1 and NF-κB is likely restricted to infected cells, the resulting increased production of proinflammatory cytokines dysregulates neighboring uninfected and infected immune cells. In addition, Tat and Nef open reading frames are transcribed from nonintegrated viral cDNAs after infection of quiescent T cells and cooperate in the induction of T-cell activation and chromosomal integration of the provirus (Wu and Marsh, 2001).
We speculate that the stimulatory role of Tat may be important in the very early stages of infection when few CD4+ T cells in lymph nodes of patients are activated and infected. This stimulatory role is likely to cooperate with pro-enhancing effects of Nef on TCR and CD28-mediated signals (Fenard et al., 2005; Schindler et al., 2006; Wang et al., 2000). Interestingly, elevated levels of IL-2 are present in lymph nodes of patients early after infection (Gray et al., 1996; Graziosi et al., 1996). At this time, the stimulatory effect of Tat may contribute to priming naïve T cells for HIV infection and creating a permissive environment for the unrestricted propagation of the infectious process (Li et al., 1997).
The finding that Tat induces T-cell hyperactivation through inhibition of SIRT1 opens the possibility for therapeutic intervention. The role of yeast Sir2 and possibly human SIRT1 in lifespan extension has fueled the search for sirtuin activators (Baur and Sinclair, 2006). Activation of SIRT1 might counterbalance Tat-induced T-cell hyperactivation thus leading to the possibility of including activators of the SIRT1 deacetylase activity into the therapeutic regimen of HIV-infected individuals.
Experimental Procedures
Cells and plasmids
HeLa, 293, 293T and Jurkat T cells were cultured under standard tissue culture conditions. MEF cells derived from SIRT1−/− cells were grown as described (Pagans et al., 2005). Mutant constructs for SIRT1 HDAC domain were prepared by site-directed mutagenesis using FLAG-tagged SIRT1 as a template. The Sir2α deletion constructs were kindly provided by V. Sartorelli, NIH, the 3X-κB luciferase reporter by N. Chirmule, Merck Inc, the IκBα luciferase reporter by K. Yamamoto, UCSF, the E-selectin luciferase reporter by J. Pober, Yale University, the 6X histidine-tagged SIRT1 plasmid by D. Sinclair, Harvard University.
Infection with lentiviral vectors
LTR-GFP and LTR-Tat-GFP are HIV-based vectors derived from the pHR′ series, in which GFP alone or Tat (101 aa) and GFP are under the control of the HIV-1 LTR through the use of an internal ribosomal entry site (Jordan et al., 2001). For experiments with mutant Tat, the EF-1α promoter was inserted into the LTR-GFP vector upstream of wild type or mutant Tat (EF1α-Tat-GFP). Details for viral production and infection are described in the Supplemental Experimental Procedures. Flow cytometry analysis of GFP expression (FACSCalibur, BD Bioscience) showed that infection efficiencies ranged from 70–98% GFP+ cells in individual experiments.
Generation of SIRT1- and Tat-expressing MEF cell lines
Open reading frames corresponding to Myc-tagged human SIRT1 and T7-tagged HIV-1 Tat (101 aa) were inserted into the murine stem cell virus (MSCV)-based retroviral vectors MSCV-puromycin and MSCV-zeocin, respectively (Clontech). Details for viral production and infection are described in the Supplemental Experimental Procedures.
RNA purification, cDNA synthesis, and real-time RT-PCR
Total RNA was extracted using RNA STAT-60 reagent (Tel-Test) according to the manufacturer’s instruction. The first strand cDNA was generated using 2 μg of total RNA and SuperScript reverse transcriptase (Invitrogen). Relative gene expression ratios between stimulated and nonstimulated conditions were calculated by the equation, ratio = 2−ΔΔCt, comparative Ct method (Applied Biosystems). Details are described in the Supplemental Experimental Procedures.
Transfections and coimmunoprecipitations
Luciferase reporter constructs and protein expression vectors were transiently cotransfected into HeLa cells using Lipofectamine (Invitrogen). 24 h after transfection, cells were lysed and processed for luciferase assays (Promega). P values (paired t-test) were used for statistical analysis.
293 cells grown in 6-well plates at 70% confluence were cotransfected with expression vectors indicated in the Figure legends. Transfection, cell lysis, immunoprecipitation, and western blotting were previously described (Pagans et al., 2005). The acetylated p65 protein was quantified using the ImageJ software available at http://rsb.info.nih.gov/ij.
Fluorescent deacetylase assay
Full-length human 6X histidine-tagged SIRT1 was purified using Ni-NTA agarose (Qiagen) and Superdex 200 gel filtration. The rate of 1 μM SIRT1 was calculated for 200 μM fluorogenic deacetylase substrate (BioMol) in the presence of twelve concentrations (1, 2, 3, 4, 5, 6, 10, 15, 25, 50, 126, 252 μM) of synthetic Tat (72 aa; Peptide Specialty Laboratory, Heidelberg, Germany). Details of IC50 calculations are described in the Supplemental Experimental Procedures.
Radioactive HDAC assays
Expression plasmids for FLAG-tagged wild type and mutants SIRT1 (1 μg) were transfected in 293 cells with lipofectamine reagent. Equal amounts of whole cell extracts (2 mg) were immunoprecipitated and in vitro HDAC assays were performed as previously described (Pagans et al., 2005).
Recombinant p65 protein was prepared as previously described (Chen et al., 2005) and used for HDAC assay with recombinant SIRT1 and synthetic Tat proteins. Details are described in the Supplemental Experimental Procedures.
Supplementary Material
Acknowledgments
We thank Vittorio Sartorelli, Narendra Chirmule, Keith Yamamoto, David Sinclair and Jordan Pober for sharing their reagents, Angelika Pedal and Thomas Conrad for technical assistance, Brandi Sanders and Eric Verdin for helpful discussions, John Carroll and Alisha Wilson for graphics, Gary Howard for editing the manuscript and Veronica Fonseca for administrative assistance. H-S. Kwon is supported by a fellowship from the University-wide AIDS Research Program (UARP), L-F. Chen by an investigator award from the Arthritis Foundation, and M. Ott by grants from the Gladstone Institute, the NIH (1R56AI067118-01A2) and the UARP (ID07-GI103).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agbottah E, Deng L, Dannenberg LO, Pumfery A, Kashanchi F. Effect of SWI/SNF chromatin remodeling complex on HIV-1 Tat activated transcription. Retrovirology. 2006;3:48. doi: 10.1186/1742-4690-3-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avalos JL, Celic I, Muhammad S, Cosgrove MS, Boeke JD, Wolberger C. Structure of a Sir2 enzyme bound to an acetylated p53 peptide. Mol Cell. 2002;10:523–535. doi: 10.1016/s1097-2765(02)00628-7. [DOI] [PubMed] [Google Scholar]
- Barboric M, Nissen RM, Kanazawa S, Jabrane-Ferrat N, Peterlin BM. NF-kappaB binds P-TEFb to stimulate transcriptional elongation by RNA polymerase II. Mol Cell. 2001;8:327–337. doi: 10.1016/s1097-2765(01)00314-8. [DOI] [PubMed] [Google Scholar]
- Barboric M, Peterlin BM. A New Paradigm in Eukaryotic Biology: HIV Tat and the Control of Transcriptional Elongation. PLoS Biol. 2005;3:e76. doi: 10.1371/journal.pbio.0030076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baur JA, Sinclair DA. Therapeutic potential of resveratrol: the in vivo evidence. Nat Rev Drug Discov. 2006;5:493–506. doi: 10.1038/nrd2060. [DOI] [PubMed] [Google Scholar]
- Bieniasz PD, Grdina TA, Bogerd HP, Cullen BR. Recruitment of a protein complex containing Tat and cyclin T1 to TAR governs the species specificity of HIV-1 Tat. EMBO J. 1998;17:7056–7065. doi: 10.1093/emboj/17.23.7056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blander G, Guarente L. The Sir2 family of protein deacetylases. Annu Rev Biochem. 2004;73:417–435. doi: 10.1146/annurev.biochem.73.011303.073651. [DOI] [PubMed] [Google Scholar]
- Borgstrom B. On the interactions between pancreatic lipase and colipase and the substrate, and the importance of bile salts. J Lipid Res. 1975;16:411–417. [PubMed] [Google Scholar]
- Buonaguro L, Buonaguro FM, Giraldo G, Ensoli B. The human immunodeficiency virus type 1 Tat protein transactivates tumor necrosis factor beta gene expression through a TAR-like structure. J Virol. 1994;68:2677–2682. doi: 10.1128/jvi.68.4.2677-2682.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Fischle W, Verdin E, Greene WC. Duration of nuclear NF-kappaB action regulated by reversible acetylation. Science. 2001;293:1653–1657. doi: 10.1126/science.1062374. [DOI] [PubMed] [Google Scholar]
- Chen LF, Mu Y, Greene WC. Acetylation of RelA at discrete sites regulates distinct nuclear functions of NF-kappaB. EMBO J. 2002;21:6539–6548. doi: 10.1093/emboj/cdf660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LF, Williams SA, Mu Y, Nakano H, Duerr JM, Buckbinder L, Greene WC. NF-kappaB RelA phosphorylation regulates RelA acetylation. Mol Cell Biol. 2005;25:7966–7975. doi: 10.1128/MCB.25.18.7966-7975.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chirmule N, Pahwa S. Envelope glycoproteins of human immunodeficiency virus type 1: profound influences on immune functions. Microbiol Rev. 1996;60:386–406. doi: 10.1128/mr.60.2.386-406.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu YL, Soros VB, Kreisberg JF, Stopak K, Yonemoto W, Greene WC. Cellular APOBEC3G restricts HIV-1 infection in resting CD4+ T cells. Nature. 2005;435:108–114. doi: 10.1038/nature03493. [DOI] [PubMed] [Google Scholar]
- Col E, Caron C, Seigneurin-Berny D, Gracia J, Favier A, Khochbin S. The histone acetyltransferase, hGCN5, interacts with and acetylates the HIV transactivator, Tat. J Biol Chem. 2001;276:28179–28184. doi: 10.1074/jbc.M101385200. [DOI] [PubMed] [Google Scholar]
- Cota-Gomez A, Flores NC, Cruz C, Casullo A, Aw TY, Ichikawa H, Schaack J, Scheinman R, Flores SC. The human immunodeficiency virus-1 Tat protein activates human umbilical vein endothelial cell E-selectin expression via an NF-kappa B-dependent mechanism. J Biol Chem. 2002;277:14390–14399. doi: 10.1074/jbc.M108591200. [DOI] [PubMed] [Google Scholar]
- Demarchi F, d’Adda di Fagagna F, Falaschi A, Giacca M. Activation of transcription factor NF-kappaB by the Tat protein of human immunodeficiency virus type 1. J Virol. 1996;70:4427–4437. doi: 10.1128/jvi.70.7.4427-4437.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorr A, Kiermer V, Pedal A, Rackwitz HR, Henklein P, Schubert U, Zhou MM, Verdin E, Ott M. Transcriptional synergy between Tat and PCAF is dependent on the binding of acetylated Tat to the PCAF bromodomain. EMBO J. 2002;21:2715–2723. doi: 10.1093/emboj/21.11.2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douek DC, Picker LJ, Koup RA. T cell dynamics in HIV-1 infection. Annu Rev Immunol. 2003;21:265–304. doi: 10.1146/annurev.immunol.21.120601.141053. [DOI] [PubMed] [Google Scholar]
- Edelstein LC, Pan A, Collins T. Chromatin modification and the endothelial-specific activation of the E-selectin gene. J Biol Chem. 2005;280:11192–11202. doi: 10.1074/jbc.M412997200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emiliani S, Fischle W, Ott M, Van Lint C, Amella CA, Verdin E. Mutations in the tat gene are responsible for human immunodeficiency virus type 1 postintegration latency in the U1 cell line. J Virol. 1998;72:1666–1670. doi: 10.1128/jvi.72.2.1666-1670.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fackler OT, Baur AS. Live and let die: Nef functions beyond HIV replication. Immunity. 2002;16:493–497. doi: 10.1016/s1074-7613(02)00307-2. [DOI] [PubMed] [Google Scholar]
- Fauci AS, Pantaleo G, Stanley S, Weissman D. Immunopathogenic mechanisms of HIV infection. Ann Intern Med. 1996;124:654–663. doi: 10.7326/0003-4819-124-7-199604010-00006. [DOI] [PubMed] [Google Scholar]
- Fenard D, Yonemoto W, de Noronha C, Cavrois M, Williams SA, Greene WC. Nef is physically recruited into the immunological synapse and potentiates T cell activation early after TCR engagement. J Immunol. 2005;175:6050–6057. doi: 10.4049/jimmunol.175.9.6050. [DOI] [PubMed] [Google Scholar]
- Fortin JF, Barat C, Beausejour Y, Barbeau B, Tremblay MJ. Hyper-responsiveness to stimulation of human immunodeficiency virus-infected CD4+ T cells requires Nef and Tat virus gene products and results from higher NFAT, NF-kappaB, and AP-1 induction. J Biol Chem. 2004;279:39520–39531. doi: 10.1074/jbc.M407477200. [DOI] [PubMed] [Google Scholar]
- Fulco M, Schiltz RL, Iezzi S, King MT, Zhao P, Kashiwaya Y, Hoffman E, Veech RL, Sartorelli V. Sir2 regulates skeletal muscle differentiation as a potential sensor of the redox state. Mol Cell. 2003;12:51–62. doi: 10.1016/s1097-2765(03)00226-0. [DOI] [PubMed] [Google Scholar]
- Garber ME, Wei P, KewalRamani VN, Mayall TP, Herrmann CH, Rice AP, Littman DR, Jones KA. The interaction between HIV-1 Tat and human cyclin T1 requires zinc and a critical cysteine residue that is not conserved in the murine CycT1 protein. Genes Dev. 1998;12:3512–3527. doi: 10.1101/gad.12.22.3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray CM, Morris L, Murray J, Keeton J, Shalekoff S, Lyons SF, Sonnenberg P, Martin DJ. Identification of cell subsets expressing intracytoplasmic cytokines within HIV-1-infected lymph nodes. AIDS. 1996;10:1467–1475. doi: 10.1097/00002030-199611000-00003. [DOI] [PubMed] [Google Scholar]
- Graziosi C, Gantt KR, Vaccarezza M, Demarest JF, Daucher M, Saag MS, Shaw GM, Quinn TC, Cohen OJ, Welbon CC, et al. Kinetics of cytokine expression during primary human immunodeficiency virus type 1 infection. Proc Natl Acad Sci USA. 1996;93:4386–4391. doi: 10.1073/pnas.93.9.4386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan A, Defechereux P, Verdin E. The site of HIV-1 integration in the human genome determines basal transcriptional activity and response to Tat transactivation. EMBO J. 2001;20:1726–1738. doi: 10.1093/emboj/20.7.1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiernan R, Bres V, Ng RW, Coudart MP, El Messaoudi S, Sardet C, Jin DY, Emiliani S, Benkirane M. Post-activation turn-off of NF-kappa B-dependent transcription is regulated by acetylation of p65. J Biol Chem. 2003;278:2758–2766. doi: 10.1074/jbc.M209572200. [DOI] [PubMed] [Google Scholar]
- Kiernan RE, Vanhulle C, Schiltz L, Adam E, Xiao H, Maudoux F, Calomme C, Burny A, Nakatani Y, Jeang KT, et al. HIV-1 tat transcriptional activity is regulated by acetylation. EMBO J. 1999;18:6106–6118. doi: 10.1093/emboj/18.21.6106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinter AL, Poli G, Fox L, Hardy E, Fauci AS. HIV replication in IL-2-stimulated peripheral blood mononuclear cells is driven in an autocrine/paracrine manner by endogenous cytokines. J Immunol. 1995;154:2448–2459. [PubMed] [Google Scholar]
- Kreisberg JF, Yonemoto W, Greene WC. Endogenous factors enhance HIV infection of tissue naive CD4 T cells by stimulating high molecular mass APOBEC3G complex formation. J Exp Med. 2006;203:865–870. doi: 10.1084/jem.20051856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landry J, Slama JT, Sternglanz R. Role of NAD(+) in the deacetylase activity of the SIR2-like proteins. Biochem Biophys Res Commun. 2000;278:685–690. doi: 10.1006/bbrc.2000.3854. [DOI] [PubMed] [Google Scholar]
- Lawn SD, Butera ST, Folks TM. Contribution of immune activation to the pathogenesis and transmission of human immunodeficiency virus type 1 infection. Clin Microbiol Rev. 2001;14:753–777. doi: 10.1128/CMR.14.4.753-777.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YW, Eum SY, Nath A, Toborek M. Estrogen-mediated protection against HIV Tat protein-induced inflammatory pathways in human vascular endothelial cells. Cardiovasc Res. 2004;63:139–148. doi: 10.1016/j.cardiores.2004.03.006. [DOI] [PubMed] [Google Scholar]
- Li CJ, Ueda Y, Shi B, Borodyansky L, Huang L, Li YZ, Pardee AB. Tat protein induces self-perpetuating permissivity for productive HIV-1 infection. Proc Natl Acad Sci USA. 1997;94:8116–8120. doi: 10.1073/pnas.94.15.8116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmoudi T, Parra M, Vries RG, Kauder SE, Verrijzer CP, Ott M, Verdin E. The SWI/SNF Chromatin-remodeling Complex Is a Cofactor for Tat Transactivation of the HIV Promoter. J Biol Chem. 2006;281:19960–19968. doi: 10.1074/jbc.M603336200. [DOI] [PubMed] [Google Scholar]
- Min J, Landry J, Sternglanz R, Xu RM. Crystal structure of a SIR2 homolog-NAD complex. Cell. 2001;105:269–279. doi: 10.1016/s0092-8674(01)00317-8. [DOI] [PubMed] [Google Scholar]
- Mujtaba S, He Y, Zeng L, Farooq A, Carlson JE, Ott M, Verdin E, Zhou MM. Structural basis of lysine-acetylated HIV-1 Tat recognition by PCAF bromodomain. Mol Cell. 2002;9:575–586. doi: 10.1016/s1097-2765(02)00483-5. [DOI] [PubMed] [Google Scholar]
- Nabel G, Baltimore D. An inducible transcription factor activates expression of human immunodeficiency virus in T cells. Nature. 1987;326:711–713. doi: 10.1038/326711a0. [DOI] [PubMed] [Google Scholar]
- Ott M, Emiliani S, Van Lint C, Herbein G, Lovett J, Chirmule N, McCloskey T, Pahwa S, Verdin E. Immune hyperactivation of HIV-1-infected T cells mediated by Tat and the CD28 pathway. Science. 1997;275:1481–1485. doi: 10.1126/science.275.5305.1481. [DOI] [PubMed] [Google Scholar]
- Ott M, Lovett JL, Mueller L, Verdin E. Superinduction of IL-8 in T cells by HIV-1 Tat protein is mediated through NF-kappaB factors. J Immunol. 1998;160:2872–2880. [PubMed] [Google Scholar]
- Ott M, Schnolzer M, Garnica J, Fischle W, Emiliani S, Rackwitz HR, Verdin E. Acetylation of the HIV-1 Tat protein by p300 is important for its transcriptional activity. Curr Biol. 1999;9:1489–1492. doi: 10.1016/s0960-9822(00)80120-7. [DOI] [PubMed] [Google Scholar]
- Pagans S, Pedal A, North BJ, Kaehlcke K, Marshall BL, Dorr A, Hetzer-Egger C, Henklein P, Frye R, McBurney MW, et al. SIRT1 Regulates HIV Transcription via Tat Deacetylation. PLoS Biol. 2005;3:e41. doi: 10.1371/journal.pbio.0030041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peruzzi F. The multiple functions of HIV-1 Tat: proliferation versus apoptosis. Front Biosci. 2006;11:708–717. doi: 10.2741/1829. [DOI] [PubMed] [Google Scholar]
- Rice AP, Carlotti F. Mutational analysis of the conserved cysteine-rich region of the human immunodeficiency virus type 1 Tat protein. J Virol. 1990;64:1864–1868. doi: 10.1128/jvi.64.4.1864-1868.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauve AA, Wolberger C, Schramm VL, Boeke JD. The Biochemistry of Sirtuins. Annu Rev Biochem. 2006;75:435–465. doi: 10.1146/annurev.biochem.74.082803.133500. [DOI] [PubMed] [Google Scholar]
- Scala G, Ruocco MR, Ambrosino C, Mallardo M, Giordano V, Baldassarre F, Dragonetti E, Quinto I, Venuta S. The expression of the interleukin 6 gene is induced by the human immunodeficiency virus 1 TAT protein. J Exp Med. 1994;179:961–971. doi: 10.1084/jem.179.3.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindler M, Munch J, Kutsch O, Li H, Santiago ML, Bibollet-Ruche F, Muller-Trutwin MC, Novembre FJ, Peeters M, Courgnaud V, et al. Nef-mediated suppression of T cell activation was lost in a lentiviral lineage that gave rise to HIV-1. Cell. 2006;125:1055–1067. doi: 10.1016/j.cell.2006.04.033. [DOI] [PubMed] [Google Scholar]
- Stevenson M, Stanwick TL, Dempsey MP, Lamonica CA. HIV-1 replication is controlled at the level of T cell activation and proviral integration. EMBO J. 1990;9:1551–1560. doi: 10.1002/j.1460-2075.1990.tb08274.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun SC, Ganchi PA, Ballard DW, Greene WC. NF-kappa B controls expression of inhibitor I kappa B alpha: evidence for an inducible autoregulatory pathway. Science. 1993;259:1912–1915. doi: 10.1126/science.8096091. [DOI] [PubMed] [Google Scholar]
- Unutmaz D, Pileri P, Abrignani S. Antigen-independent activation of naive and memory resting T cells by a cytokine combination. J Exp Med. 1994;180:1159–1164. doi: 10.1084/jem.180.3.1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldmann TA. The biology of interleukin-2 and interleukin-15: implications for cancer therapy and vaccine design. Nat Rev Immunol. 2006;6:595–601. doi: 10.1038/nri1901. [DOI] [PubMed] [Google Scholar]
- Wang JK, Kiyokawa E, Verdin E, Trono D. The Nef protein of HIV-1 associates with rafts and primes T cells for activation. Proc Natl Acad Sci USA. 2000;97:394–399. doi: 10.1073/pnas.97.1.394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei P, Garber ME, Fang SM, Fischer WH, Jones KA. A novel CDK9-associated C-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell. 1998;92:451–462. doi: 10.1016/s0092-8674(00)80939-3. [DOI] [PubMed] [Google Scholar]
- Westendorp MO, Li-Weber M, Frank RW, Krammer PH. Human immunodeficiency virus type 1 Tat upregulates interleukin-2 secretion in activated T cells. J Virol. 1994;68:4177–4185. doi: 10.1128/jvi.68.7.4177-4185.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westendorp MO, Shatrov VA, Schulze-Osthoff K, Frank R, Kraft M, Los M, Krammer PH, Droge W, Lehmann V. HIV-1 Tat potentiates TNF-induced NF-kappa B activation and cytotoxicity by altering the cellular redox state. EMBO J. 1995;14:546–554. doi: 10.1002/j.1460-2075.1995.tb07030.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Marsh JW. Selective transcription and modulation of resting T cell activity by preintegrated HIV DNA. Science. 2001;293:1503–1506. doi: 10.1126/science.1061548. [DOI] [PubMed] [Google Scholar]
- Yeung F, Hoberg JE, Ramsey CS, Keller MD, Jones DR, Frye RA, Mayo MW. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004;23:2369–2380. doi: 10.1038/sj.emboj.7600244. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.