

Abstract
Human beta defensin-1 (hBD1) is a component of the immune system which links the innate and adaptive immune responses. We have demonstrated that hBD1 induces rapid cytolysis of prostate cancer cells and that it may also possess tumor suppressive abilities. In addition, there is a high frequency of cancer-specific loss of hBD1 expression which further suggests its potential role in tumor progression. However, the factors responsible for the loss of hBD1 expression are not known. PAX2, a transcriptional regulator normally expressed during early development, has been implicated as an oncogene in carcinomas of the kidney, prostate, breast and ovary. It is known that expression of PAX2 in these tumor cells mediates the evasion of cell death through the suppression of cell death pathways involving the p53 tumor suppressor. However, we have demonstrated that knock-down of PAX2 expression results in cell death independent of p53 status, thus suggesting that additional cell death pathways are negatively regulated by PAX2. Here we describe a novel pathway in which PAX2 represses hBD1 expression through binding of the PAX2 homeodomain to the hBD1 promoter. Furthermore, knock-down of PAX2 expression results in the re-expression of hBD1, and subsequently prostate cancer cell death. These findings are the first to demonstrate that the PAX2 oncogene suppresses hBD1 expression in cancer and further implicate PAX2 as a novel therapeutic target for prostate cancer treatment.
Keywords: PAX2, Oncogene, Human Beta Defensin-1, Tumor Suppressor, Prostate Cancer
1. Introduction
A new etiology for prostate cancer is beginning to emerge in which inflammation and loss of host immune responses may be a possible cause for the pathogenesis of this disease (Nelson et al., 2004). Genes involved in the immune system which protect against infection and inflammation are down-regulated or mutated in cancer, thus leading to a failure of the immune system to recognize inflammation and malignant cells (Nelson et al., 2004; Palapattu et al., 2005). The innate immune system is one of the first lines of defense against inflammation by secreting antimicrobial peptides such as beta defensins to counteract this process (Yang et al., 2004). Defensins can act as direct effectors of the innate immune system through their ability to disrupt the cellular membrane integrity kill via electrostatic interaction which leads to pore formation (Papo and Shai, 2005). Our group and others have shown that human beta defensin-1 (hBD1) expression is frequently lost in prostate and renal cancers, and it is cytotoxic to cancer cells through the disruption of the cell membrane followed by the activation of caspases (Bullard et al., 2008; Donald et al., 2003; Sun et al., 2006).
In addition to its role in innate immunity, hBD1 contributes to adaptive immunity by possessing chemotactic activity to recruit immature dendritic cells (DC) and memory T-cells through the CCR6 chemokine receptor (Yang et al., 1999). The DC are the most powerful antigen-presenting cells and are able to prime naïve T-cells inducing tumor immune responses (Thomas-Kaskel et al., 2007). However, tumors have developed mechanisms to escape recognition and elimination by the immune system which include impairing or decreasing the number of functional DC. Given its cytotoxic activity against cancer cells and the ability to recruit DC to tumors, hBD1 may play an important role in tumor suppression via an anti-tumor response involving the immune system.
PAX2 is a developmental control gene whose expression is normally turned off after terminal differentiation in most tissues (Eccles et al., 2002). However, PAX2 expression is aberrantly turned on in several cancers such as Wilm’s Tumor, breast, ovarian, bladder and prostate (Khoubehi et al., 2001; Muratovska et al., 2003; Silberstein et al., 2002; Tagge et al., 1994). PAX2 has also been identified as an oncogene through its ability to transform cells in culture and form tumors in nude mice (Maulbecker and Gruss, 1993). In addition, it has been shown to regulate downstream target genes such as tumor suppressors. For example, PAX2 has been reported to act as a transcriptional activator of WT1 and a transcriptional repressor of p53 (Dehbi et al., 1996). In our earlier studies, we found that inhibition of PAX2 can promote cell death independent of p53 status suggesting that PAX2 may suppress an additional cell death pathway (Gibson et al., 2007).
In this study, we describe the cellular factors responsible for hBD1 loss in prostate cancer. We have identified an additional mechanism by which PAX2 promotes cancer cell survival via transcriptional repression of the putative tumor suppressor hBD1. Furthermore, knock-down of PAX2 expression resulted in the re-expression of hBD1, and subsequently hBD1-mediated cell death. Given that PAX2 may contribute to prostate cancer progression through the suppression of this unique component of the immune system involved in cancer cell recognition and destruction, targeting PAX2 for the induction of hBD1 expression may serve as a novel therapeutic approach to treat prostate cancer.
2. Material and methods
2.1. Maintenance of Cell Lines
The prostate cancer cell lines were obtained from the American Type Culture Collection (Manassas, VA). DU145 cells were cultured in DMEM medium and PC3 were grown in F12 medium. The hPrEC cells were cultured in prostate epithelium basal media (Cambrex Bio Science Inc., Walkersville, MD). Cells were maintained at 37°C in 5% CO2.
2.2. Reporter Constructs
The pGL3-hBD1 construct was created by cloning in a 160bp region representing the hBD1 minimal promoter (−226 to −67), which contained the PAX2 consensus recognition sequence. The fragment was generated by PCR amplification with the following primers designed to include 5′ KpnI and 3′ NheI restriction sites: forward, 5′-TAGGGGTACCCCCATGTGACTGCTGACTGC-3′ and reverse, 5′- CTAGCTAGCAGGCT-GGAGCGTCACTGTA-3. The PCR product was then restriction digested with KpnI and NheI and ligated into pGL3 basic vector digested with the same restriction enzymes. The pGL3-hBD1 vector was transformed in XL-1 Blue competent cells and the plasmids were verified by restriction digestion and automated sequence analysis.
2.3. Site-directed Mutagenesis and Luciferase Assay
Site-directed mutagenesis was performed with the QuickChange II XL Site-Directed Mutagenesis Kit according to the manufacture’s instruction (Stratagene, La Jolla, CA, USA). Briefly, using the pGL3-hBD1 wild-type construct as a template, the PAX2 recognition sequence was disrupted by altering three bases at positions −169, −165, −162 (underlined) from the transcriptional start site by PCR with the following primers: forward, 5′-GCGATTAGAAGTTAACCATTAACTGTGGCACCTCCC-3′ and reverse, 5′-GGGAGGTGCCACAGTTAATGGTTAACTTCTAATCGC-3′. Approximately 5 × 104 cells were seeded on a 96 well plate and grown overnight. Next, the cells were transfected with either 0.01 μg of PGL3-hBD1 wild-type vector or pGL3-hBD1 mutant vector using lipofectamine 2000 (Invitrogen). Twenty-four hours after transfection, the cells were lysed in 200μl of Bright-Glo luciferase reagent (Promega), and luciferase activity was quantified by a microplate luminometer.
2.4. SiRNA Silencing of PAX2 and hBD1
SiRNA knockdown of PAX2 was performed as previously described (Gibson et al., 2007). In addition to siRNA against PAX2, prostate cancer cells were treated for 48 hours with 5 pg/cell of hBD1 siRNA (Ambion, Inc) alone, or with PAX2 and hBD1 siRNA using the Codebreaker lipofectamine reagent (Promega). As a negative control, cells were treated for 48 hours with 5 pg/cell scramble siRNA.
2.5. Immunocytochemistry
Immunocytochemistry was performed as previously described (Bullard et al., 2008) with some modifications. Briefly, DU145 (1 × 104) cells were plated onto polysine coated slides and fixed in 4 % paraformaldehyde for 1hr at room temperature. The cells were washed six times and placed in blocking solution (1 × PBS, 2 % BSA, 0.8 % normal goat serum, 0.4 % TritonX-100) for 1 hour at room temperature. Following blocking, the cells were incubated with 1:1000 dilution of hBD-1 polyclonal antibody (Peprotech, Inc.) in blocking solution overnight at 4 °C. Next the cells were washed six times in blocking solution and incubated with Alex Fluor 488 goat anti rabbit IgG (H+L) secondary antibody in a 1:1000 dilution of blocking buffer for 1 hour at room temperature. Lastly, a cover slip was placed over the cells with mounting medium and visualized using a fluorescence microscope.
2.6. Preparation of Nuclear Extract
Briefly, nuclear extracts were prepared as previously described (Andrews and Faller, 1991). Here, cells were harvested with trypsin-EDTA, washed with ice cold 1× PBS containing 0.1 % protease inhibitor cocktail and the pellet was re-suspended in 400 μl of low salt buffer containing 10mM HEPES-KOH(pH7.9),1.5mM MgCl2, 10mM KCl, 1mM DTT and 0.1% protease inhibitor cocktail (Sigma). After incubation on ice for 10 min, suspensions were centrifuged at 12000 rpm for 2 min at 4 °C. The pellet obtained was re-suspended in high salt buffer containing 20mM HEPES-KOH (pH7.9), 10% glycerol, 420mM NaCl, 1.5mM MgCl2, 0.2mM EDTA, 1mM DTT and 0.1% protease inhibitor cocktail (Sigma), incubated on ice for 20 min and centrifuged at 12000g for 2 min. The supernatants, which contained nuclear fractions, were used for the EMSA experiment.
2.7 Electromobility Shift Assay
A 160 base pair region of the hBD1 promoter containing the PAX2 recognition sequence was PCR amplified as described above, and was used as a probe for the shift assay. Here, the resulting PCR product was purified by gel extraction (Qiagen) and was end-labeled with T4 polynucleotide kinase and [32P] ATP (6000 Ci/mmol). The labeled probe was purified with a gel filtration spin column. Binding reactions were performed in a total volume of 20μl at room temperature for 30 min. The reactions contained approximately 12μg of nuclear extract, 100ng of poly (dI-dC), 32P-labeled probe (20,000 cpm) and 0.5 mg/ml BSA in binding buffer (15 mM Tris-HCl pH 8.0, 100mM KCl, 5 mM MgCl2, 1 mM EDTA, 6.5 % glycerol, 0.5 mM DTT and protease inhibitor cocktail). For competition experiments, the nuclear extract was pre-incubated with a 10- to 75-fold molar excess of unlabeled DNA or 75-fold molar excess of poly (dI-dC) for 10 min before addition of the 32P-labeled probe. For antibody shift assays, 1.0 μg of protein A purified rabbit anti-PAX2 IgG or normal rabbit IgG were incubated with nuclear extract for 30–45 min at room temperature prior to the addition of 32P labeled probe. Free DNA and DNA-protein complexes were resolved at 4 °C on 5% neutral polyacrylamide gels in 0.5X Tris-Borate-EDTA (TBE) buffer at 150 V for 4 hours, dried on nitran membrane and analyzed by phosphorimaging using the STORM™ phosphoimager (Molecular Dynamics).
2.8. Chromatin Immunoprecipitation (ChIP)
The ChIP assay was performed using the Millipore ChIP assay kit (catalog # 17-295) according to the manufacturer’s protocol (Billerica, MA). Briefly, DU145 and PC3 prostate cancer cells (7–8 × 106) were treated with media containing 1% formaldehyde (Sigma) for 10 min to crosslink chromatin. The cells were harvested, centrifuged, and washed twice with 10 ml of ice-cold PBS containing 0.1% protease inhibitor cocktail. The cell pellets were resuspended in 500 μl of SDS Lysis Buffer and incubated on ice for 10min followed by sonication to shear the DNA. Sonication was optimized to generate DNA fragments between 200–1000 base pairs in size. Following centrifugation at 12000×g at 4°C, cell lysates were diluted 10-fold with ChIP dilution buffer containing 0.1% protease inhibitors. Samples were then pre-cleared by incubation with 60 μl of Salmon Sperm DNA/Protein A Agarose-50% slurry for 2 hours at 4 °C. The supernatant obtained by centrifugation was immunoprecipitated either with PAX2 specific rabbit polyclonal antibody or with rabbit IgG overnight at 4 °C with rotation at 1000 rpm for 5 mins followed by washing with wash buffers. Next, the chromatin was eluted from the antibody-protein complex by a freshly prepared elution buffer (1% SDS, 0.1 M NaHCO3). To reverse the cross-linking, samples were treated with 10μl of 5M NaCl at 65 °C overnight followed by treatment with proteinase K. The DNA was recovered using Qiagen PCR purification Kit. PCR amplification was conducted for the detection of the 160-bp fragment of the hBD1 promoter containing the PAX2 consensus sequence under standard conditions with the following primers: ChIP Primer A, 5′- TAGGGGTACCCCCATGTGACTGCTGACT-3′, and ChIP Primer B, 5′-TCTGCATAAGGGGAGAGATGAGA-3′. As a negative control, a randomly selected region of the hBD1 promoter which was null for the PAX2 recognition sequence was also amplified with the following primers: ChIP Primer C, 5′- CGCTCGGCTCTAAGCTGG-3′ and ChIP Primer D, 5′ TCTGCATAAGGGGAGAGATGAGA-3′. Amplification products were analyzed by 2% agarose gel electrophoresis.
2.9. Acridine Orange/Ethidium Bromide Assay
Membrane integrity was examined as previously described with slight modification (Bullard et al., 2008). Acridine orange (AO) stains viable cells, whereas ethidium bromide (EtBr) stains dying cells that have compromised membranes. Briefly, cells were seeded into 2-chamber culture slides (BD Falcon). Cells were then treated with negative control siRNA, PAX2 siRNA or PAX2 and hBD-1 siRNA simultaneously. Next, cells were washed with PBS and stained with 2 ml of a mixture (1:1) of AO (Sigma, St. Louis, MO) and EtBr (Promega) (5 μg/ml) solution for 5 mins, washed with 1×PBS and viewed by Confocal microscopy.
Fluorescence was viewed with a Zeiss LSM 5 Pascal Vario 2 LASER scanning Confocal microscope (Carl Zeiss). The emission filter color wheel contained BS505–530 (green) and LP560 (red) filter blocks to allow for the separation of emitted green light from AO into the green channel and red light from EtBr into the red channel. The excitation was provided by a Kr/Ar mixed gas laser at wavelengths of 543 nm for AO and 488 nm for EtBr. Slides were analyzed under 40× magnification and digital images were stored as uncompressed TIFF files and exported into Photoshop CS software (Adobe Systems) for image processing and hard copy presentation.
2.10. MTT Cytotoxicity Assay
Cell viability was measured by MTT assay as previously described with modification (21). Briefly, DU145 cell suspensions were diluted and seeded onto a 96-well plate at 1–5 ×103 cells per well. The next day they were transfected with 5pg/cell of the PAX2 siRNA pool or negative control siRNA pool using Codebreaker transfection reagent (Promega) and allowed to grow for 2-, 4- or 6 days after treatment. Cell viability was determined by measuring the conversion of 3-[4,5-dimethylthiazol-2yl]-2,5 diphenyl tetrazolium bromide, MTT (Promega), to a colored formazan product. Absorbance was read at 540 nm on a scanning multi-well spectrophotometer. Percent viability was normalized to cells treated with non-specific negative control siRNA and data is presented as relative decrease in viability.
2.11. Statistical Analysis
Statistical differences were evaluated using the Student’s t-test for unpaired values. P values were determined by a two-sided calculation, and a P value of less than 0.05 was considered statistically significant.
3. Results
3.1. HBD1 Promoter Contains PAX2 Consensus Sequence
It has been reported that PAX2 has both activation and inhibition domains and possesses specific DNA binding activity in both its homeodomain and paired domain regions (Fickenscher et al., 1993). The hBD1 promoter was examined for potential PAX2 binding sites using the TFSearch program (http://www,cbrc.jp/research/db/TFSEARCH.html). The PAX2 consensus recognition sequence (Brophy et al., 2001) was located upstream of the hBD1 TATA Box and transcriptional start site at position −170 to −159 (Fig. 1A). Furthermore, this region shared significant homology to the published paired recognition sequences for PAX2 (Dziarmaga et al., 2006; Epstein et al., 1994; Phelps and Dressler, 1996) and the published consensus sequence (Brophy et al., 2001) which spans 11 core nucleotides in the hBD1 gene promoter (Fig. 1B). The PAX2 recognition sequence in the hBD1 promoter contained 7 of the 11 core nucleotides of the published PAX2 consensus sequence.
Fig. 1. Computational Analysis of hBD1 promoter and comparison of PAX2 consensus sequence to hBD1 sequences.


A, TF Search promoter analysis computer program revealed the PAX2 consensus binding motif containing the core CCTTG sequence located upstream of the hBD1 TATA BOX. B, PAX2 homology to other published PAX2 binding sites. The PAX2 consensus sequence in the hBD1 promoter has a high degree of similarity with published PAX2 binding domains.
3.2. Knock-down of PAX2 Results in the Re-expression of hBD1
To investigate whether PAX2 suppresses hBD1 protein expression in prostate cancer, we examined hBD1 expression following PAX2 knock-down with siRNA. Previously we demonstrated that inhibition of PAX2 in prostate cancer cells resulted in an upregulation of hBD1 mRNA compared to cells treated with negative control siRNA (Bullard et al., 2008). To verify hBD1 protein expression, immunocytochemistry was performed in cells following PAX2 knock-down. Protein expression was monitored by staining with primary antibody against hBD1 and a green fluorescing secondary antibody (Fig. 2). Cells were analyzed by confocal laser microscopy at 488 nm as previously described (Bullard et al., 2008). HPrEC prostate epithelial cells which endogenously express hBD1 were examined as a positive control (data not shown). Analysis under DIC verified the presence of DU145 cells treated with media only (Fig. 2A), negative control siRNA (Fig. 2C) or PAX2 siRNA (Fig. 2E) cells. Confocal analysis revealed no detectable green fluorescence in DU145 following 6 days of treatment with media only (Fig. 2B), nor with negative control siRNA (Fig. 2D). However, cells treated with PAX2 siRNA emitted green fluorescence indicating the presence of hBD1 protein (Fig. 2F). These results further suggest that PAX2 may be a transcriptional repressor of hBD1.
Fig. 2. Expression of hBD1 after PAX2 knockdown in DU145 cells.
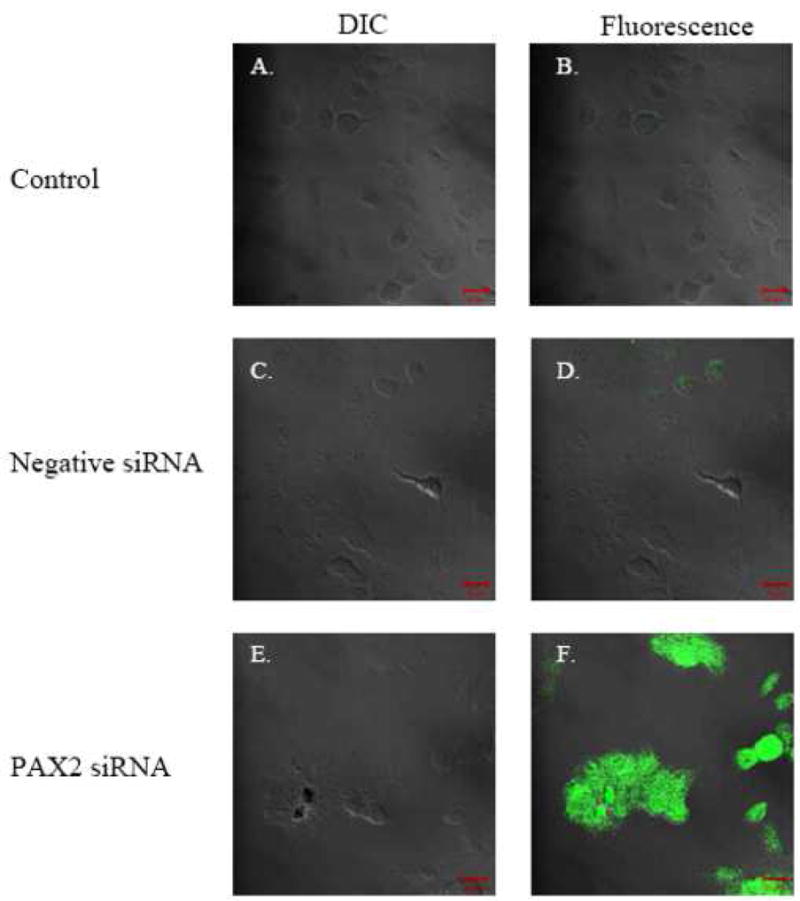
The presence of cells were verified under DIC for control (A), negative siRNA (C) and PAX2 siRNA (E). Under fluorescence, no hBD1 expression was detected in DU145 cells treated with media (B) or negative siRNA (D). However, cells treated with PAX2 siRNA for 6 days exhibited hBD1 protein expression (F).
3.3. PAX2 Protein Binds Recognition Sequence in the hBD1 Promoter
To examine PAX2 binding affinity for the hBD1 promoter, DNA gel mobility shift assays were performed (Fig. 3A). Compared to the free probe (lane1), migration of the radiolabeled probe was retarded in the gel showing the formation of three major bands when incubated with nuclear extracts from DU145 prostate cancer cells which suggests the formation of DNA-protein complexes (lane 2). To test whether these formed complexes contained the PAX2 protein, PAX2 antibody was added to the nuclear extracts prior to addition of the radiolabeled probe. This resulted in a significant decrease in intensity of two of the retarded protein-DNA complexes (lane 4) containing the PAX2 protein (Complex 1 and 2). However, the reaction performed in the presence of anti-rabbit IgG resulted in no change in the intensity of the shifted band which served as a negative control (lane 3).
Fig. 3. PAX2 binds to the hBD1 promoter in vitro.
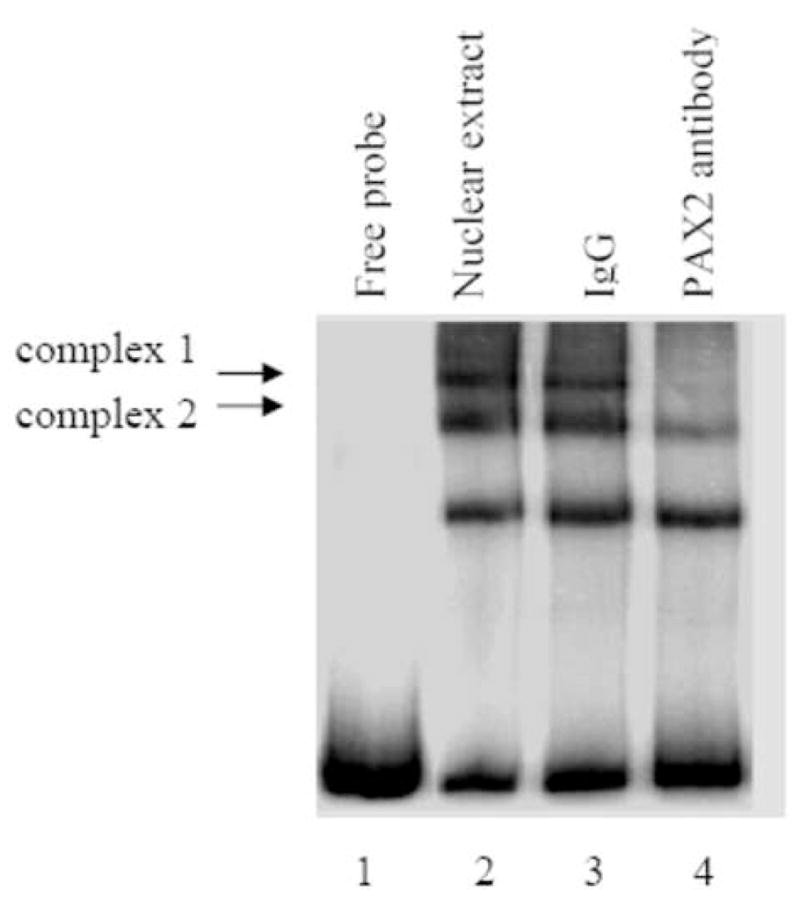
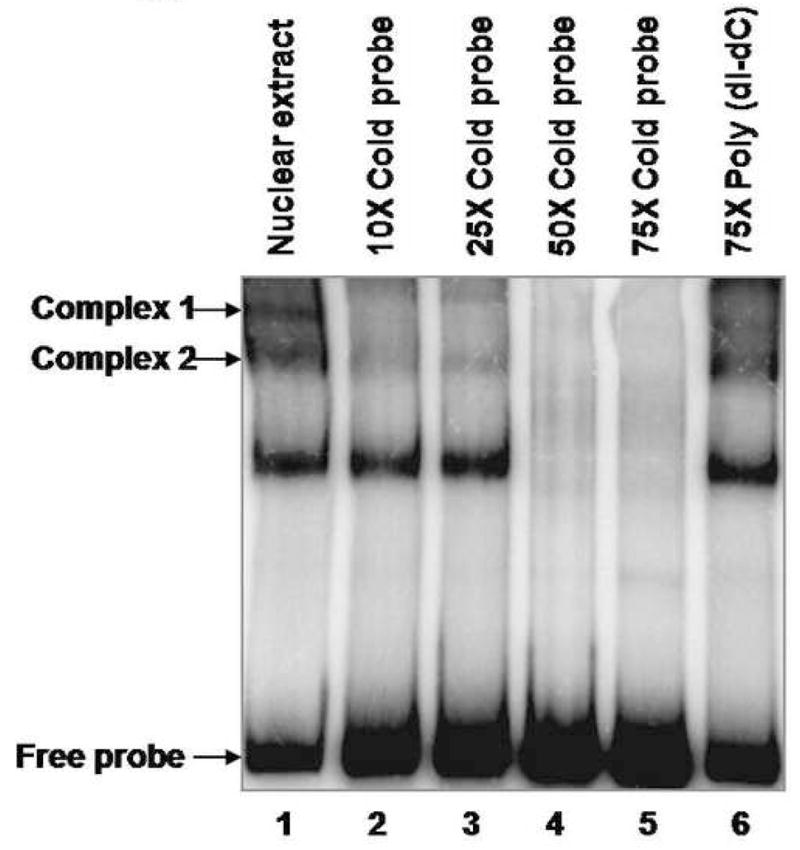

A, Electrophoretic mobility shift assay of the 32P-hBD1 DNA probe without nuclear extract sample (lane 1), or in the presence of 12 μg of nuclear extract from DU145 cells (lane 2). The presence of PAX2 in protein-DNA complexes was tested by adding anti-PAX2 antibody to the reaction mixture (lane 4) which showed decrease in intensity of two complexes. However, incubations of nuclear extract with normal rabbit IgG, no change in the intensities of the complexes was observed. B, Gel mobility shift competition assay was performed to test the specificity of PAX2 binding to hBD1-promoter. Electrophoresis of the 32P-labeled hBD1-DNA with DU145 nuclear extract showed bands of DNA-protein complexes (lane 1). To examine the specificity of DNA binding, nuclear extract was incubated with 32P-hBD1 DNA in the presence of 10X (lane 2), 25X (lane 3), 50X (lane 4) and 75X (lane 5) molar excess of unlabeled competitor homologous hBD1-DNA or 75X molar excess of poly-dI-dC as a negative control (lane 6). C, SiRNA knock-down of PAX2 was performed to determine the effect of PAX2 suppression on protein-DNA complex expression. Electrophoresis of the 32P-labeled hBD1-DNA without nuclear extract (lane 1) or incubated with nuclear extract from DU145 cells treated with scramble siRNA (lane 2) or PAX2 siRNA (lane 3). After incubation in binding buffer, samples were separated on a 5% acrylamide gel which was dried and analyzed by phosphor-imaging.
To examine the specificity of PAX2 binding to hBD1 promoter, competition DNA gel-shift assays were performed (Fig. 3B). Probe incubated with extracts showed mobility shift of the DNA-protein complexes compared to the migration patterns of lower band which represents the free probe (lane 1). However, the radiolabeled complexes were greatly reduced in the presence of 10X (lane 2), 25× (lane 3), 50X (lane 4) and 75X (lane 5) molar excess of the unlabelled homologous competitor. Conversely, 75-fold molar excess of poly (dI-dC) did not inhibit the formation of PAX2- hBD1 complexes (lane 6).
To further examine whether the protein-DNA complexes were being formed specifically by PAX2 protein, DU145 prostate cancer cells were treated with PAX2 siRNA or scramble siRNA prior to collecting nuclear extracts (Fig. 3C). Compared to the negative control lane in which no protein extract was added to the radiolabeled probe (lane 1), protein-DNA complexes were formed with nuclear extracts from cells treated with scramble siRNA (lane 2) similar to untreated cells as observed in Fig 3B. However, nuclear extracts from DU145 cells treated with PAX2 siRNA failed to form Complex 1 and Complex 2 (lane 3). These data demonstrate that PAX2 binds to the hBD1 promoter in prostate cancer cells.
3.4. PAX2 Actively Binds to the hBD1 Promoter in Prostate Cancer Cells in Vivo
We examined PAX2 interaction with the endogenous hBD1 promoter in DU145 and PC3 cancer cells by chromatin- immunoprecipitation assay. PAX2 protein-DNA complexes were immunoprecipitated from formaldehyde cross-linked DU145 and PC3 prostate cancer cells with PAX2 antibody and the DNA collected from the reactions were subjected to PCR amplification. Primer sets used to amplify the DNA from the immunoprecipitated samples are indicated in Fig. 4A. Reaction with primers A and B generated a 160bp fragment confirming that PAX2 protein was bound to the region of the hBD1 promoter containing PAX2 recognition sequence as shown in Fig. 4B (lane 3 for DU145 and lanes 3–4 for PC3). However, the reaction with normal rabbit IgG or without antibody did not amplify the 160 bp fragment (lanes 1–2). Total input control DNA (Input C) was used as a positive control (lane 4 for DU145 and lane 5 for PC3). Furthermore, no product was generated from any of the samples amplified with the primers C and D which flanked a randomly chosen region in the hBD1 promoter null for the PAX2 consensus sequence (data not shown). These data demonstrated that PAX2 is bound to the hBD1 promoter specifically at the PAX2 consensus recognition sequence under in vivo conditions.
Fig. 4. ChIP analysis of PAX2 binding to the hBD1 promoter, in vivo.
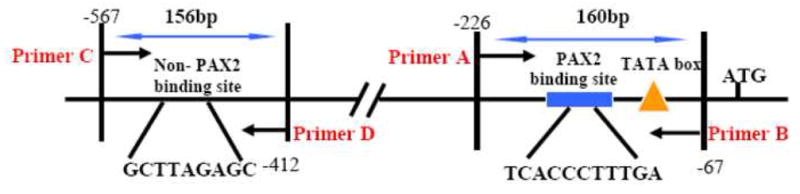

Chromatin immunoprecipitation- assays were performed to examine PAX2 binding activity in situ using DU145 and PC3 cells. A, PCR amplification of precipitated DNA was performed with primers flanking the PAX2 binding motif (A and B) or a region null for the PAX2 recognition sequence (C and D). B, Whole cell lysates from formaldehyde cross-linked DU145 or PC3 cells were incubated with antibodies and DNA-protein-antibody complexes were recovered by incubation with Protein A sepharose beads. The presence of hBD1-DNA in immunoprecipitated complexes was detected by PCR using primer sets A and B. PCR products were analyzed on a 2% agarose gel, which was stained with ethidium bromide. Precipitation without antibody (lane 1) or with normal IgG (lane 2) resulted in no amplification product. However, amplification of DNA precipitated with anti-PAX2 antibody generated 160 bp products in DU145 cells (lane3) and PC3 cells (lanes 3–4).
3.5. PAX2 Consensus Sequence is required for Transcriptional Repression of the hBD1 Promoter
We cloned the hBD1 minimal promoter region containing the PAX2 consensus sequence into a pGL3 luciferase vector (pGL3-hBD1-wildtype) and compared hBD1 promoter activity to a mutant construct of the PAX2 recognition motif (Fig. 1B). Cells transfected with empty vector or pGL3-hBD1 wild-type construct demonstrated low levels hBD1 transcriptional activity in DU145 and PC3 cell lines that constitutively expresses PAX2 (Fig. 5). Furthermore, we observed that promoter activity in DU145 prostate cancer cells was significantly less than the PAX2-null hPrEC cells transfected with the pGL3-hBD1 wild-type construct (unpublished data). However, cells transfected with the pGL3-hBD1 mutant construct exhibited hBD1 promoter activity that was 3-fold higher in DU145 cells and 4-fold higher in PC3 cells compared to DU145 and PC3 cells containing pGL3-hBD1 wild-type, respectively. These findings indicate that the PAX2 recognition motif is required for PAX2 mediated suppression of hBD1 promoter activity.
Fig. 5. Effect of sequence integrity of the PAX2 binding region on hBD1 promoter activity.
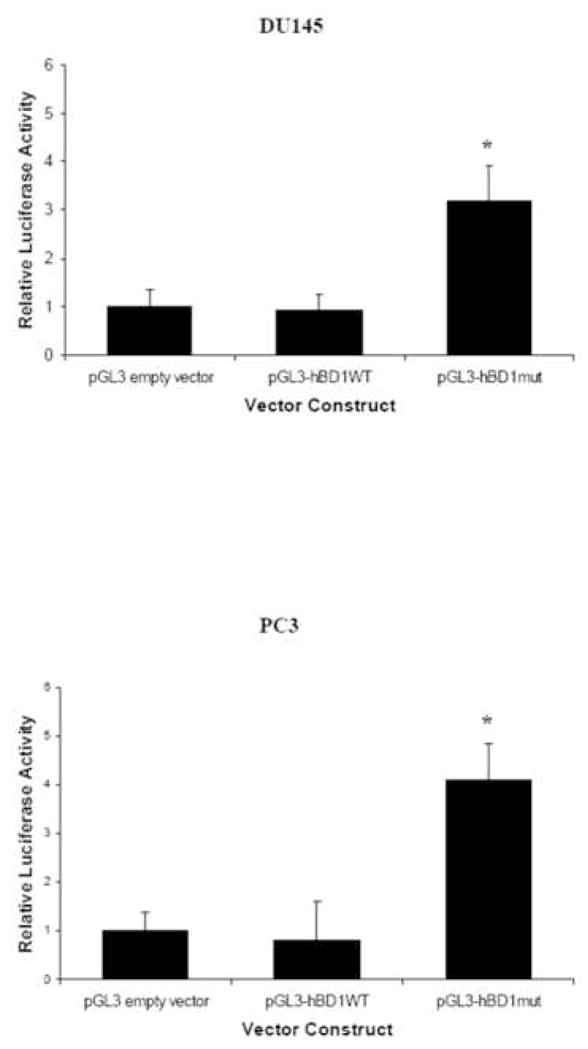
HBD1 promoter activity is reported as luciferase activity relative to pGL3 empty vector. The asterisk represents statistically significant levels of expression compared to empty vector. Each experiment was performed three times in triplicate (p<0.05). Asterisks represent statistical differences.
3.6. Knock-down of PAX2 Leads to hBD1 Mediated Cell Death
To investigate whether PAX2 supports cancer cell survival by suppressing hBD1-mediated cell death, we examined membrane integrity following PAX2 inhibition. We previously demonstrated that hBD1 mediated cell death involves disruption of membrane integrity (Bullard et al., 2008). Here, intact cells fluoresce green due to the incorporation of AO. Cells with compromised plasma membranes fluoresce red when membrane impermeable EtBr leaks into the cytoplasm and become yellow or orange due to co-localization of AO and EtBr in the nuclei (Fig. 6A). Untreated DU145 and PC3 cells stained positively with AO and emitted green fluorescence, but did not stain with EtBr indicating that the cells had intact membranes. Following PAX2 knockdown, both DU145 and PC3 cells exhibited condensed nuclei that appeared yellow due to the co-localization of green and red staining from AO and EtBr, respectively. These results suggest that DU145 and PC3 cells are undergoing cell death pathway involving necrosis. However, co-treatment of cells with PAX2 and hBD1 siRNA resulted in only green fluorescence indicative of intact cell membranes. Collectively, these results suggest that PAX2 expression in prostate cancer suppresses hBD1-mediated cell death.
Fig. 6. PAX2 suppresses hBD1 mediated cell death.
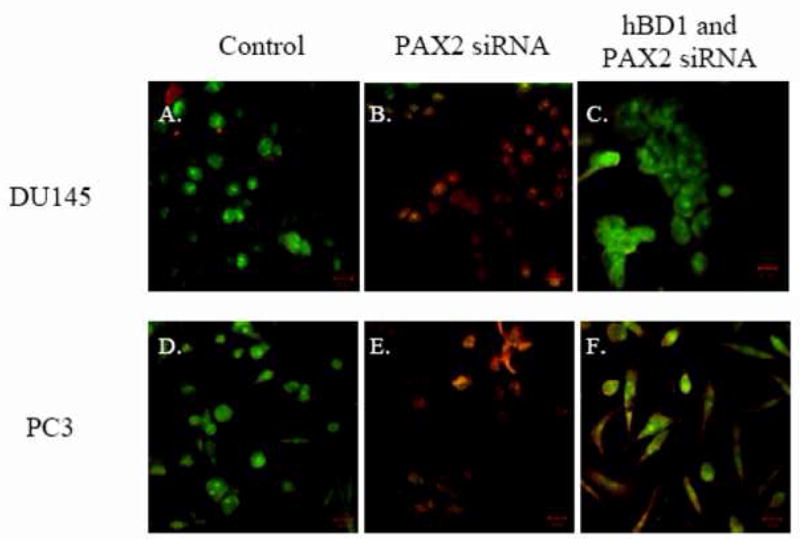
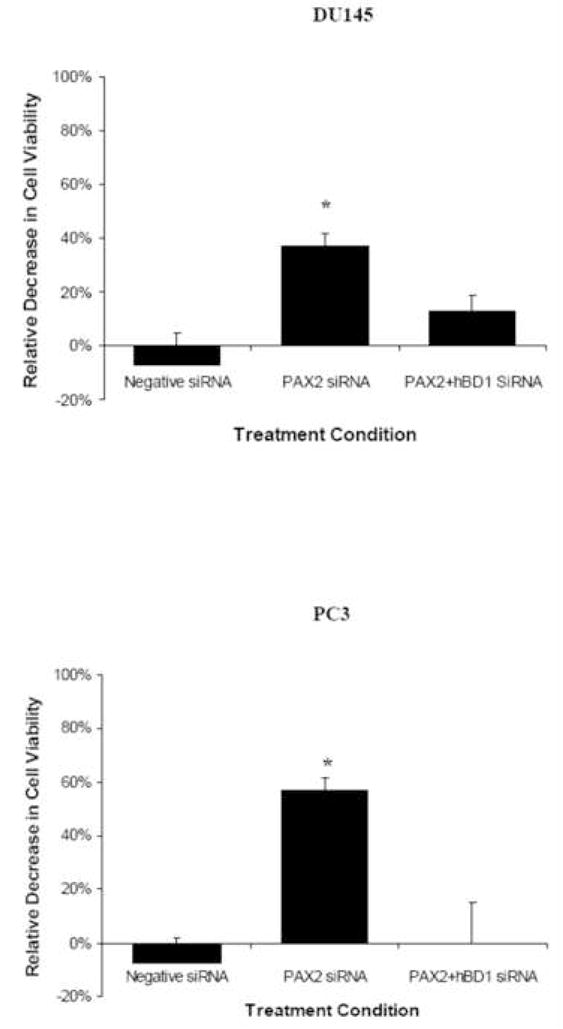
A, AO/EtBr bromide assay was used to monitor membrane permeability. DU145 (A) and PC3 (D) cells treated with media only fluoresced green indicating viable cells with intact membranes. Treatment with PAX2 siRNA resulted in cells staining with both AO and EtBr generating yellow nuclei (B and E). However, treatment of cells with PAX2 and hBD1 siRNA simultaneously resulted in green staining indicative of viable cells with intact membranes (lanes C and F). B, Cellular viability was measured by MTT assay after 6 days of treatment with negative control siRNA, PAX2 siRNA or a double knockdown with PAX2 siRNA and hBD1 siRNA, respectively. Each experiment was performed three times in triplicate. Asterisks represent statistical differences (p<0.05).
We previously demonstrated that hBD1 mediated cell death in prostate cancer cells involves an increase in cell permeability and the activation of caspases (Bullard et al., 2008). Treatment of prostate cancer cells with siRNA to knock-down PAX2 expression resulted in a similar finding with an increase in membrane permeability and activation of caspases (Gibson et al., 2007). To further examine this, cell viability was examined which revealed that knockdown of PAX2 resulted in a 37% decrease in DU145 (Fig. 6B). However, simultaneous knockdown of PAX2 and hBD1 resulted in an 11% decrease in viability. In PC3 cells, inhibition of PAX2 resulted in a 57% decrease in cell viability. However, simultaneous treatment of the cells with PAX2 siRNA and hBD1 siRNA, resulted in no significant decrease in cell viability compared to cells treated with media only. These results demonstrate that PAX2 may promote cancer cell survival by negatively regulating hBD1-mediated cell death.
4. Discussion
Currently, little is known about the specific components of the innate immune system that play a role in tumor suppression. It has been suggested that there is an imbalance between immune activation and suppression during tumor growth, thus allowing the tumor cells to escape immune destruction (Selsted and Ouellette, 2005). Furthermore, tumor progression has been linked to an increase in immune suppression in prostate cancer patients (Palapattu et al., 2005). We have shown that hBD1 may be a putative tumor suppressor gene given its strong anti-cancer activity and that its loss of expression may contribute to prostate cancer progression (Bullard et al., 2008). There are also reports of altered expression of human beta defensins in other cancers such as lung and oral squamous cell carcinomas, as well as basal cell carcinoma (Abiko et al., 1999; Arimura et al., 2004; Gambichler et al., 2006). However, the molecular mechanism of hBD1 expression loss is not clear.
Sun et al. reported that a loss or reduction of hBD1 expression was due to the presence of 5′ UTR single nucleotide polymorphisms (SNPs) in the hBD1 promoter. Other reports contradict these findings in that the SNPs did not demonstrate a statistical difference in reduction (Braida et al., 2004; Milanese et al., 2006) suggesting that hBD1 loss may be related to other factors (Milanese et al., 2007). Furthermore, the SNPs identified in the report by Sun et al. did not correlate with the previous observation that loss of hBD1 expression occurred in malignant prostate tissue, but was retained in the adjacent normal tissue from patients (Donald et al., 2003). Here we demonstrate that cancer specific loss of hBD1 is due to transcriptional repression by the PAX2 oncogene. It is known that PAX2 is a transcriptional regulator and that its expression is aberrantly turned on in a number of cancers including prostate carcinomas. It is also known that PAX2 promotes cell survival through the alteration of the expression of the p53 tumor suppressor genes. However, we previously demonstrated that PAX2 negatively regulates cell death pathways independent of p53 (Gibson et al., 2007). In this study, we show that PAX2 suppresses transcriptional activity of the hBD1 promoter by binding to a PAX2 recognition sequence located within the hBD1 promoter. Gel mobility shift analysis demonstrated that PAX2 possesses DNA binding affinity to the region of the hBD1 promoter containing the PAX2 consensus recognition sequence. Furthermore, chromatin immunoprecipitation analysis demonstrated that the endogenous hBD1 promoter sequence in prostate cancer cells is specifically associated with the PAX2 transcription factor.
Our data further demonstrated that PAX2 negatively regulates hBD1 expression and its ability to suppress hBD1-mediated cell death in prostate cancer cells. We have shown previously that there is an inverse relationship between PAX2 and hBD1 expression in prostate tissue and that knockdown of PAX2 by siRNA in prostate cancer cells result in an increase of hBD1 expression (Bullard et al., 2008). This was further supported by our functional analysis of the effect of PAX2 knockdown on cell membrane integrity. HBD1 exerts its cytotoxic effect on cancer cells by increasing membrane permeability resulting in necrotic death (Bullard et al., 2008; Papo et al., 2004). In the AO/EtBr studies presented here, we found that inhibition of PAX2 also resulted in loss of membrane integrity and cell death in prostate cancer cells. Furthermore, when PAX2 and hBD1 were knocked-down simultaneously by siRNA, cells maintained intact membranes and necrotic cell death did not occur. Interestingly, the production of necrotic cells is important to the immune system because they are important activators of DC, powerful antigen presenting cells to T cells (Gabrilovich, 2007). The ability of hBD1 to recruit DC and promote necrosis in prostate cancer cells suggests that hBD1 may have an important role in host anti-tumor immunity. Therefore, PAX2 may subvert this important cascade of events for cancer cell recognition and destruction through hBD1 suppression. PAX2 has also been shown to influence additional components of the immune system via the up-regulation of PAX5 expression which interferes with T cell development by activating B cell-specific genes and repressing T cell-linage genes (Souabni et al., 2007). Our data is the first to show that the PAX2 oncogene promotes cancer cell survival through the suppression of hBD1 expression, thus inhibiting its role as an important component of the innate immune system which may include anti-tumor activity. Collectively, these finding further implicate PAX2 in cancer cell survival and tumor progression specifically through its ability to down-regulate components involved in tumor immunity.
The ability of the human immune system to recognize and destroy clones of cancer cells has become of great interest in cancer therapy research. However, cancer cells usually evade immune surveillance, despite evidence that immune effectors can play a significant role in controlling tumor growth under natural conditions or in response to therapeutic manipulation. Innate immunity peptides can overcome these limitations via a unique mechanism of cancer cell killing that involves membrane lysis and necrotic cell death. Therefore, suppression of these peptides may be another mechanism in which cancer cells elude destruction and promote tumorigenesis.
Overall, our data demonstrated that PAX2 acts as a transcriptional repressor of hBD1 by binding to the PAX2 consensus recognition sequence located in the hBD1 promoter. In addition, the knock-down of PAX2 expression resulted in the re-expression of hBD1, and subsequently hBD1-mediated cell death in prostate cancer cells. These findings may offer a novel approach to prostate cancer therapy through the modulation of the patient’s innate immune system. Therefore, further consideration of PAX2 as a molecular target for cancer therapy may be warranted.
Acknowledgments
The authors would like to thank Dr. Sashidhar Nakarakanti and Mr. Arindam Saha for their technical assistance with our ChIP and mobility shift assays, and the Biotechnology Resource Laboratory of the Medical University of South Carolina. This work was supported in part by Grant C06RR14516 from the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abiko Y, Mitamura J, Nishimura M, Muramatsu T, Inoue T, Shimono M, Kaku T. Pattern of expression of beta-defensins in oral squamous cell carcinoma. Cancer Lett. 1999;143:37–43. doi: 10.1016/s0304-3835(99)00171-8. [DOI] [PubMed] [Google Scholar]
- Andrews NC, Faller DV. A rapid micropreparation technique for extraction of DNA-binding proteins from limiting numbers of mammalian cells. Nucleic Acids Res. 1991;19:2499. doi: 10.1093/nar/19.9.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arimura Y, Ashitani J, Yanagi S, Tokojima M, Abe K, Mukae H, Nakazato M. Elevated serum beta-defensins concentrations in patients with lung cancer. Anticancer Res. 2004;24:4051–7. [PubMed] [Google Scholar]
- Braida L, Boniotto M, Pontillo A, Tovo PA, Amoroso A, Crovella S. A single-nucleotide polymorphism in the human beta-defensin 1 gene is associated with HIV-1 infection in Italian children. Aids. 2004;18:1598–600. doi: 10.1097/01.aids.0000131363.82951.fb. [DOI] [PubMed] [Google Scholar]
- Brophy PD, Ostrom L, Lang KM, Dressler GR. Regulation of ureteric bud outgrowth by Pax2-dependent activation of the glial derived neurotrophic factor gene. Development. 2001;128:4747–56. doi: 10.1242/dev.128.23.4747. [DOI] [PubMed] [Google Scholar]
- Bullard RS, Gibson W, Bose SK, Belgrave JK, Eaddy AC, Wright CJ, Hazen-Martin DJ, Lage JM, Keane TE, Ganz TA, Donald CD. Functional analysis of the host defense peptide Human Beta Defensin-1: new insight into its potential role in cancer. Mol Immunol. 2008;45:839–48. doi: 10.1016/j.molimm.2006.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehbi M, Ghahremani M, Lechner M, Dressler G, Pelletier J. The paired-box transcription factor, PAX2, positively modulates expression of the Wilms’ tumor suppressor gene (WT1) Oncogene. 1996;13:447–53. [PubMed] [Google Scholar]
- Donald CD, Sun CQ, Lim SD, Macoska J, Cohen C, Amin MB, Young AN, Ganz TA, Marshall FF, Petros JA. Cancer-specific loss of beta-defensin 1 in renal and prostatic carcinomas. Lab Invest. 2003;83:501–5. doi: 10.1097/01.lab.0000063929.61760.f6. [DOI] [PubMed] [Google Scholar]
- Dziarmaga A, Hueber PA, Iglesias D, Hache N, Jeffs A, Gendron N, Mackenzie A, Eccles M, Goodyer P. Neuronal apoptosis inhibitory protein is expressed in developing kidney and is regulated by PAX2. Am J Physiol Renal Physiol. 2006;291:F913–20. doi: 10.1152/ajprenal.00004.2006. [DOI] [PubMed] [Google Scholar]
- Eccles MR, He S, Legge M, Kumar R, Fox J, Zhou C, French M, Tsai RW. PAX genes in development and disease: the role of PAX2 in urogenital tract development. Int J Dev Biol. 2002;46:535–44. [PubMed] [Google Scholar]
- Epstein J, Cai J, Glaser T, Jepeal L, Maas R. Identification of a Pax paired domain recognition sequence and evidence for DNA-dependent conformational changes. J Biol Chem. 1994;269:8355–61. [PubMed] [Google Scholar]
- Fickenscher HR, Chalepakis G, Gruss P. Murine Pax-2 protein is a sequence-specific trans-activator with expression in the genital system. DNA Cell Biol. 1993;12:381–91. doi: 10.1089/dna.1993.12.381. [DOI] [PubMed] [Google Scholar]
- Gabrilovich DI. Molecular mechanisms and therapeutic reversal of immune suppression in cancer. Curr Cancer Drug Targets. 2007;7:1. [PubMed] [Google Scholar]
- Gambichler T, Skrygan M, Huyn J, Bechara FG, Sand M, Altmeyer P, Kreuter A. Pattern of mRNA expression of beta-defensins in basal cell carcinoma. BMC Cancer. 2006;6:163. doi: 10.1186/1471-2407-6-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson W, Green A, Bullard RS, Eaddy AC, Donald CD. Inhibition of PAX2 expression results in alternate cell death pathways in prostate cancer cells differing in p53 status. Cancer Lett. 2007;248:251–61. doi: 10.1016/j.canlet.2006.08.007. [DOI] [PubMed] [Google Scholar]
- Khoubehi B, Kessling AM, Adshead JM, Smith GL, Smith RD, Ogden CW. Expression of the developmental and oncogenic PAX2 gene in human prostate cancer. J Urol. 2001;165:2115–20. doi: 10.1097/00005392-200106000-00080. [DOI] [PubMed] [Google Scholar]
- Maulbecker CC, Gruss P. The oncogenic potential of Pax genes. Embo J. 1993;12:2361–7. doi: 10.1002/j.1460-2075.1993.tb05890.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milanese M, Segat L, Crovella S. Transcriptional effect of DEFB1 gene 5′ untranslated region polymorphisms. Cancer Res. 2007;67:5997. doi: 10.1158/0008-5472.CAN-06-3544. author reply 5997. [DOI] [PubMed] [Google Scholar]
- Milanese M, Segat L, Pontillo A, Arraes LC, de Lima Filho JL, Crovella S. DEFB1 gene polymorphisms and increased risk of HIV-1 infection in Brazilian children. Aids. 2006;20:1673–5. doi: 10.1097/01.aids.0000238417.05819.40. [DOI] [PubMed] [Google Scholar]
- Muratovska A, Zhou C, He S, Goodyer P, Eccles MR. Paired-Box genes are frequently expressed in cancer and often required for cancer cell survival. Oncogene. 2003;22:7989–97. doi: 10.1038/sj.onc.1206766. [DOI] [PubMed] [Google Scholar]
- Nelson WG, De Marzo AM, DeWeese TL, Isaacs WB. The role of inflammation in the pathogenesis of prostate cancer. J Urol. 2004;172:S6–11. doi: 10.1097/01.ju.0000142058.99614.ff. discussion S11–2. [DOI] [PubMed] [Google Scholar]
- Palapattu GS, Sutcliffe S, Bastian PJ, Platz EA, De Marzo AM, Isaacs WB, Nelson WG. Prostate carcinogenesis and inflammation: emerging insights. Carcinogenesis. 2005;26:1170–81. doi: 10.1093/carcin/bgh317. [DOI] [PubMed] [Google Scholar]
- Papo N, Braunstein A, Eshhar Z, Shai Y. Suppression of human prostate tumor growth in mice by a cytolytic D-, L-amino Acid Peptide: membrane lysis, increased necrosis, and inhibition of prostate-specific antigen secretion. Cancer Res. 2004;64:5779–86. doi: 10.1158/0008-5472.CAN-04-1438. [DOI] [PubMed] [Google Scholar]
- Papo N, Shai Y. Host defense peptides as new weapons in cancer treatment. Cell Mol Life Sci. 2005;62:784–90. doi: 10.1007/s00018-005-4560-2. [DOI] [PubMed] [Google Scholar]
- Phelps DE, Dressler GR. Identification of novel Pax-2 binding sites by chromatin precipitation. J Biol Chem. 1996;271:7978–85. doi: 10.1074/jbc.271.14.7978. [DOI] [PubMed] [Google Scholar]
- Selsted ME, Ouellette AJ. Mammalian defensins in the antimicrobial immune response. Nat Immunol. 2005;6:551–7. doi: 10.1038/ni1206. [DOI] [PubMed] [Google Scholar]
- Silberstein GB, Dressler GR, Van Horn K. Expression of the PAX2 oncogene in human breast cancer and its role in progesterone-dependent mammary growth. Oncogene. 2002;21:1009–16. doi: 10.1038/sj.onc.1205172. [DOI] [PubMed] [Google Scholar]
- Souabni A, Jochum W, Busslinger M. Oncogenic role of Pax5 in the T-lymphoid lineage upon ectopic expression from the immunoglobulin heavy-chain locus. Blood. 2007;109:281–9. doi: 10.1182/blood-2006-03-009670. [DOI] [PubMed] [Google Scholar]
- Sun CQ, Arnold R, Fernandez-Golarz C, Parrish AB, Almekinder T, He J, Ho SM, Svoboda P, Pohl J, Marshall FF, Petros JA. Human beta-defensin-1, a potential chromosome 8p tumor suppressor: control of transcription and induction of apoptosis in renal cell carcinoma. Cancer Res. 2006;66:8542–9. doi: 10.1158/0008-5472.CAN-06-0294. [DOI] [PubMed] [Google Scholar]
- Tagge EP, Hanson P, Re GG, Othersen HB, Jr, Smith CD, Garvin AJ. Paired box gene expression in Wilms’ tumor. J Pediatr Surg. 1994;29:134–41. doi: 10.1016/0022-3468(94)90308-5. [DOI] [PubMed] [Google Scholar]
- Thomas-Kaskel AK, Waller CF, Schultze-Seemann W, Veelken H. Immunotherapy with dendritic cells for prostate cancer. Int J Cancer. 2007;121:467–73. doi: 10.1002/ijc.22859. [DOI] [PubMed] [Google Scholar]
- Yang D, Biragyn A, Hoover DM, Lubkowski J, Oppenheim JJ. Multiple roles of antimicrobial defensins, cathelicidins, and eosinophil-derived neurotoxin in host defense. Annu Rev Immunol. 2004;22:181–215. doi: 10.1146/annurev.immunol.22.012703.104603. [DOI] [PubMed] [Google Scholar]
- Yang D, Chertov O, Bykovskaia SN, Chen Q, Buffo MJ, Shogan J, Anderson M, Schroder JM, Wang JM, Howard OM, Oppenheim JJ. Beta-defensins: linking innate and adaptive immunity through dendritic and T cell CCR6. Science. 1999;286:525–8. doi: 10.1126/science.286.5439.525. [DOI] [PubMed] [Google Scholar]