

Abstract
The association of cargoes to kinesins is thought to promote kinesin activation, yet the validation of such a model with native cargoes is lacking because none is known to activate kinesins directly in an in vitro system of purified components. The RAN-binding protein 2 (RANBP2), through its kinesin-binding domain (KBD), associates in vivo with kinesin-1, KIF5B/KIF5C. Here, we show that KBD and its flanking domains, RAN GTPase-binding domains 2 and 3 (RBD2/RBD3), activate the ATPase activity of KIF5B approximately 30-fold in the presence of microtubules and ATP. The activation kinetics of KIF5B by RANBP2 is biphasic and highly cooperative. Deletion of one of its RBDs lowers the activation of KIF5B threefold and abolishes cooperativity. Remarkably, RBD2–KBD–RBD3 induces unfolding and modest activation of KIF5B in the absence of microtubules. Hence, RANBP2 is the first native and positive allosteric activator known to jump-start and boost directly the activity of a kinesin.
Keywords: ATPase, kinetics, mechanotransduction, kinesin, RAN-binding protein 2
Introduction
Kinesins are microtubule-based motor proteins that mediate generally the intracellular transport of cargoes towards the plus end of microtubules (cell periphery; Brady, 1985; Vale et al, 1985). The conventional kinesin heavy chains, kinesin-1, which include the vertebrate kinesin superfamily protein 5 (KIF5) isoforms KIF5A, KIF5B and KIF5C, exist as homodimers (DeBoer et al, 2008). The amino-terminal domain of kinesin-1 comprises a motor head domain with ATPase and microtubule-binding activities, whereas the carboxy-terminal end comprises coiled-coil and tail domains with binding activity, singly or in combination with accessory proteins, towards a wide variety of cargoes (Adio et al, 2006; Gindhart, 2006).
Cargoless kinesin exists in a folded and compact state in which the tail folds over the motor domains, leading to inactivation of the ATPase activity of the motor domains (Coy et al, 1999a). This is thought to prevent, among other things, the futile ATP depletion from the cell and the amassing of kinesin at the plus ends of microtubules. Cargo binding to the tail domain of kinesin relieves the inhibitory constraint of the tail over the motor domain, and switches kinesin to an extended and microtubule-based ATPase active state that propels kinesin along the microtubules Coy et al, 1999a). Hence, the conversion of kinesin between these two states reflects a crucial regulatory mechanism of kinesin activity. Support for this model is based mainly on the facts that (i) the tail of Drosophila kinesin-1 inhibits the constitutive ATPase activity of its motor domain (Coy et al, 1999a; Hackney & Stock, 2000), and (ii) an artificial cargo adsorbed to kinesin-1 stimulates kinesin activity (Coy et al, 1999a). Yet, no native cargo identified so far has been shown to activate kinesin directly. Identification of native cargoes that are able to activate kinesin directly is of crucial significance to validate functionally the two-state conformational model and to establish such cargo as a specific and authentic activator of kinesin motor (transport) activity. It will also allow the interplay of a host of regulatory and pathological processes between kinesin and its cargo to be probed mechanistically in a well-defined system.
The RAN-binding protein 2 (RANBP2), through its kinesin-binding domain (KBD), specifically associates in vivo, in vitro and directly with an approximately 100-residue segment, which spans the coiled-coil and tail domains of the highly homologous KIF5B and KIF5C, but not KIF5A (Cai et al, 2001; Cho et al, 2007). Ectopic expression of KBD of RANBP2 in cell cultures prevents the outward dispersion of mitochondria, whereas a mutant KBD incapable of binding to KIF5B/KIF5C renders KBD biologically inactive (Cho et al, 2007). Here, we report that RANBP2 jump-starts and boosts the activity of KIF5B in a biochemically defined cell-free system, and the discovery of unusual properties that are linked to the activation kinetics of KIF5B by RANBP2.
Results
Full-length KIF5B has low intrinsic ATPase activity
To probe whether the mode of autoregulation of KIF5B motor activity mirrors that observed for the Drosophila kinesin-1 (Coy et al, 1999a), we carried out end-point ATPase activity assays for three recombinant KIF5B constructs in the presence or absence of microtubules: full-length KIF5B (KIF5B), the N-terminal motor (N-KIF5B) and the C-terminal coiled-coil tail domains (KIF5B-C) (Fig 1A). As shown in Fig 1B, the ATPase activity of N-KIF5B is stimulated by the presence of microtubules, but full-length KIF5B does not present significant ATPase activity in the absence or presence of microtubules. However, KIF5B-C suppresses the ATPase activity of N-KIF5B (Fig 1B) in a concentration-dependent manner (Fig 1C). Hence, KIF5B and the kinesin-1 of Drosophila share similar autoinhibitory mechanisms of the motor activity by their coiled-coil tail domains.
Figure 1.

Regulation of the motor activity of KIF5B by its tail domain and domains of RANBP2. (A) Schematic diagram of KIF5B domains and constructs used in this study. KIF5B, N-KIF5B and KIF5B-C comprise the full-length, the amino-terminal motor/ATPase and neck domains, and the carboxy terminal coiled-coil tail domains, respectively. (B) The ATPase activity of N-KIF5B (50 nM), but not of KIF5B-C (10 μM) and KIF5B (50 nM), is stimulated by microtubules (MT; 300 nM) in end-point assays. KIF5B-C (10 μM) inhibits the activity of N-KIF5B (50 nM). The microtubule-stimulated ATPase activity of N-KIF5B is used as a reference value (100%) for subsequent end-point assays under the same experimental conditions (C,E,F). (C) The increasing concentrations of KIF5-C inhibit the ATPase activity of N-KIF5B (50 nM) to levels close to that observed with KIF5B alone. (D) Primary structure and functional domains of RANBP2. RBD2, KBD and RBD3 are the subjects of this study. (E) KBD, but not KBDMut, partly suppresses the inhibitory activity in trans of KIF5B-C (2 μM) over N-KIF5B (50 nM). (F) RBD2–KBD–RBD3 (R2KR3) restores the activity of N-KIF5B in the presence of KIF5B-C (2 μM) to levels comparable to that observed with N-KIF5B alone (50 nM), whereas RBD2–KBDMut–RBD3 (R2KMutR3) has no effect. Data are means of three independent experiments±s.d. CLD, cyclophilin-like domain; CY, cyclophilin domain; IR, internal repeats 1 and 2; KBD, kinesin-binding domain; KIF5, kinesin superfamily protein 5; LD, leucine-rich domain; RBD1–4, RAN-binding domains 1–4; RANBP2, RAN-binding protein 2; SDS–PAGE, SDS–polyacrylamide gel electrophoresis; ZnF, zinc-finger-rich domain.
Stimulation of KIF5B by domains of RANBP2
We used the same end-point assays described to test the activity of N-KIF5B in the presence of a fixed inhibitory concentration of KIF5B-C and either KBD alone (Fig 1D,E), KBD with its flanking domains, RAN GTPase-binding domains 2 and 3 (RBD2/RBD3) (Fig 1D,F) or corresponding constructs with point mutations in KBD (Fig 1E,F; Cho et al, 2007). The biologically inactive constructs with mutations in KBD act as crucial controls for the presence of potential contaminants able to influence kinesin activation (Cho et al, 2007). As shown in Fig 1E, increasing concentrations of KBD relieve significantly the trans inhibitory activity of KIF5B-C over the motor activity of N-KIF5B up to approximately 2.5-fold, whereas KBDMut has no effect (Fig 1E). RBD2 and RBD3 flanking the KBD of RANBP2 significantly enhance the association of KIF5B/KIF5C with KBD in tissue extracts (Cai et al, 2001; Y. Cai and P. Ferreira, unpublished data). Hence, RBD2 and RBD3 probably modulate further the activation of KIF5B by KBD. As shown in Fig 1F, the increasing concentrations of the RBD2–KBD–RBD3 (R2KR3) domains of RANBP2 can completely suppress the inhibition in trans of KIF5B-C over N-KIF5B, whereas RBD2–KBDMut–RBD3 (R2KMutR3) has no effect (Fig 1F).
Activation kinetics of KIF5B by R2KR3 and domains thereof
Next, we carried out detailed analyses of the kinetics of KIF5B activation by the R2KR3 and constructs thereof. The linear rates of KIF5B activation were examined in the presence of increasing amounts of each construct, saturating ATP and microtubules. The microtubule-stimulated ATPase rate of KIF5B alone is very low (0.75±0.25 s−1), as is that of the RANBP2 constructs alone, both of which were subtracted from all ATPase measurements in the presence of KIF5B and constructs of RANBP2. KBD activates KIF5B by 10-fold in the presence of microtubules (Fig 2A,D), whereas no significant activity is measurable with KBDMut (Fig 2A,D) or in the absence of microtubules (supplementary Fig 1A online). The presence of RBD3 after KBD does not cause significant change in the ATPase activity of KIF5B in the presence (Fig 2B,D) or absence of microtubules (supplementary Fig 1B online), whereas RBD3 alone has no measurable effect on KIF5B (Fig 2B,D; supplementary Fig 1B online). However, the presence of RBD2 and RBD3 flanking KBD (R2KR3) promotes the activation of KIF5B over 30-fold in the presence of microtubules (Fig 2B,D). The plot of KIF5B activation versus [R2KR3] shows biphasic kinetics comprising an initial hyperbolic phase revealed by a linear double reciprocal plot at concentrations below 0.5 μM, followed by a sigmoid phase above 0.5 μM (Fig 2B). Mutations in KBD of R2KR3 (R2KMutR3) abolish the cooperative phase, but do not affect the hyperbolic phase (Fig 2C). KIF5B acts as a positively cooperative enzyme with a large Hill coefficient (nH) of approximately 3.9 (Fig 2B). Hence, the biphasic kinetics of KIF5B activation supports the finding that R2KR3 is a positive allosteric activator of KIF5B and that there is a tight conformational coupling to four binding sites in the kinesin dimer (two sites in each monomer). Interference with such coupling by mutation of R2KR3 collapses the system to a hyperbolic scheme in which all the cargo sites are equivalent (Fig 2C). For cooperative schemes, 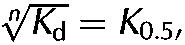 where Kd is the intrinsic binding dissociation constant for ligand and K0.5 is the observed ligand concentration yielding half maximal activity (Segel, 1975), the nth root of the Kd (0.074 μM, nH=3.9) for binding of the cargo to kinesin is 0.51 μM, in good agreement with the empirical value for K0.5 of 0.76 (Fig 2E), suggests that the binding affinity of R2KR3 towards KIF5B remains constant at all loading stoichiometries. The observation that kcat increases with increasing concentration of R2KR3 indicates that cooperativity increases the Vmax with KIF5B reaching a maximal ATPase rate of 30±2.8 ATP s−1 (Fig 2D,E); therefore, the system formally shows V-type positive allosteric behaviour. Finally, R2KR3 also increases significantly, but modestly (fivefold), the activity of KIF5B in the absence of microtubules and independently of the specific-binding activity of KBD, indicating that the binding site in KIF5B is bipartite (supplementary Fig 1B and C online; Fig 2D).
where Kd is the intrinsic binding dissociation constant for ligand and K0.5 is the observed ligand concentration yielding half maximal activity (Segel, 1975), the nth root of the Kd (0.074 μM, nH=3.9) for binding of the cargo to kinesin is 0.51 μM, in good agreement with the empirical value for K0.5 of 0.76 (Fig 2E), suggests that the binding affinity of R2KR3 towards KIF5B remains constant at all loading stoichiometries. The observation that kcat increases with increasing concentration of R2KR3 indicates that cooperativity increases the Vmax with KIF5B reaching a maximal ATPase rate of 30±2.8 ATP s−1 (Fig 2D,E); therefore, the system formally shows V-type positive allosteric behaviour. Finally, R2KR3 also increases significantly, but modestly (fivefold), the activity of KIF5B in the absence of microtubules and independently of the specific-binding activity of KBD, indicating that the binding site in KIF5B is bipartite (supplementary Fig 1B and C online; Fig 2D).
Figure 2.

Analyses of the activation of kinetics of KIF5B by RBD2–KBD–RBD3 and domains thereof in the presence of microtubules (300 nM). (A) The ATPase rate of KIF5B (2.5 nM) is stimulated by KBD, but not by KBDMut. (B) Activation of the ATPase rate of KIF5B (2.5 nM) in the presence of increasing concentrations of R2KR3 comprises an initial hyperbolic phase and a subsequent sigmoidal phase. The sigmoid phase of the Hill reaction has a Hill coefficient (nH) of 3.9. Removal of RBD2 (R2) from R2KR3 (KR3) collapses the biphasic activation of KIF5B to a hyperbolic scheme that is kinetically indistinguishable from that of KBD alone, whereas RBD3 does not activate KIF5B. (C) A mutation in KBD of R2KR3, R2KMutR3, collapses the biphasic activation of KIF5B (2.5 nM) to a hyperbolic scheme. (D,E) The kinetic parameters of Michaelis–Menton (MM rx) and Hill reactions (Hill rx) of KIF5B activation by R2KR3 and constructs thereof. The dissociation constant (Kd) is considered as the concentration of R2KR3 needed for half-maximal velocity of the hyperbolic or the sigmoidal kinetic phase. Data are means of three (n=3) independent experiments±s.d. KBD, kinesin-binding domain; KIF5, kinesin superfamily protein 5; RANBP2, RAN-binding protein 2; R2KR3, RBD2–KBD–RBD3.
R2KR3 promotes binding of KIF5B to microtubules
AMPPNP produces a non-motile rigour state of kinesin tightly bound to microtubules (Vale et al, 1985; Saxton, 1994); hence, the microtubule-binding assay also acts to determine the conformational state of kinesin activation. We used pull-down assays with R2KR3 and R2KMutR3 constructs of KIF5B in the presence of polymerized microtubules and either ATP or AMPPNP. As shown in Fig 3, R2KR3 co-precipitates KIF5B and microtubules in the presence of AMPPNP but not ATP. By contrast, although the R2KMutR3 construct expectedly pulls down less KIF5B, there is no co-precipitation of microtubules. Hence, R2KR3 produces a conformational active state of KIF5B with strong binding capacity towards microtubules.
Figure 3.

GST pulldown indicates that KIF5B binds to taxol-polymerized microtubules in the presence of RBD2–KBD–RBD3 and AMPPNP, but not ATP. KIF5B does not bind to microtubules in the presence of the mutant construct (R2KMutR3) with either AMPPNP or ATP. Top and lower panels are immunoblots (IBs) of KIF5B and α-tubulin, respectively. The input comprises 3% and 5% of KIF5B and microtubules, respectively, used for a pull-down reaction. AMPPNP, adenylylimidodiphosphate; KBD, kinesin-binding domain; KIF5, kinesin superfamily protein 5; MT, microtubule; R2KR3, RBD2–KBD–RBD3; RBD, RAN-binding domain.
R2KR3 domains of RANBP2 unfold KIF5B
Low-angle-rotary shadowing studies on kinesins helped to determine the molecular conformations of various classes of kinesins (Hirokawa, 1998). Studies on kinesin-1 have shown that at high ionic strength conditions, kinesin-1 changes from a folded to an extended conformation (Hirokawa et al, 1989; Hackney et al, 1992). Hence, we took a complementary approach to probe the effect of R2KR3 on the conformation of KIF5B under the same ionic buffer conditions used in the kinetic assays. As shown in Fig 4A,B,M, KIF5B alone exists exclusively as a folded conformer. By contrast, a large number of extended KIF5B molecules are visible on incubation with R2KR3 (Fig 4C–L,N). The association of R2KR3 with KIF5B is observed at the end of the KIF5B tail (Fig 4C–L). Quantitative morphometric analysis of KIF5B in the presence of R2KR3 (Fig 4N) shows that a significant fraction of kinesin molecules seems to form tail-to-tail conformers of approximately 160 nm (see Fig 4K), probably attributable to either the high concentration of KIF5B or the absence of light chains, which might promote the biogenesis of kinesin-1 similar to that purified from the brain (Hirokawa et al, 1989; Hackney et al, 1992).
Figure 4.

Electron micrographs of rotary shadowed KIF5B alone and in the presence of RBD2–KBD–RBD3. (A,B) KIF5B molecules without R2KR3; only compact KIF5Bs were observed. (C–L) Extended KIF5B conformers in the presence of R2KR3. (C,E,G) Molecules with R2KR3 globular structures at one end (arrows), (I) a molecule with two globular structures and (K) one with a globule apparently in the middle (arrow). (D,F,H,J,L) Molecules with the hint of two globular motor structures (arrowheads) at the right end of the molecule. Scale bar, 100 nm. (M,N) Histograms of morphometric analysis of the length distribution of KIF5B in the absence and presence of R2KR3, respectively. KBD, kinesin-binding domain; KIF5, kinesin superfamily protein 5; R2KR3, RBD2–KBD–RBD3; RBD, RAN-binding domain.
Discussion
These studies are the first to show the ability of a native cargo (RANBP2) to activate directly a kinesin (KIF5B), validating a role of the R2KR3 domains of RANBP2 in the function of KIF5B in a biochemically defined cell-free system. This study provides crucial insights into KIF5B activation but differs from previous reports on the kinetic properties of Drosophila kinesin-1 and KIF5B in several respects (Coy et al, 1999a; Friedman & Vale, 1999). First, this method of KIF5B purification yields a protein with substantially lower intrinsic ATPase activity (0.75±0.25 s−1) than the reported kcat values of 28±2 s−1 (Coy et al, 1999a) and 33±4.0 s−1 (Hackney & Stock, 2000) for Drosophila kinesin-1, and 6.5±0.6 s−1 per head of KIF5B (∼13 s−1 per kinesin molecule; Friedman & Vale, 1999) under low ionic strength, allowing much better resolution of new kinetic properties linked to KIF5B. Second, nonspecifically adsorbed casein-coated silica beads induced a three- to sevenfold activation over the basal ATPase rate of Drosophila kinesin-1 (Coy et al, 1999a). By contrast, we obtained a 33-fold activation of KIF5B when fully loaded with the R2KR3 domains of RANBP2 and observed new biphasic kinetics for KIF5B activation (Fig 2). These differences account for crucial variations in the nature of cargoes used, native versus artificial and nonspecific (Coy et al, 1999a). Third, cargoless KIF5B with mutations constraining its conformation lead to an increase in its ATPase rate up to approximately fourfold (Friedman & Vale, 1999). Finally, it is reported that two other kinesin-1 binding partners, the fasciculation and elongation protein ζ and c-Jun N-terminal kinase-interacting protein-1 together are required to activate kinesin (Blasius et al, 2007). In the light of the number of other purported cargoes that are known to associate with kinesin-1 (Adio et al, 2006; Gindhart, 2006), contamination of these components from cell lysates prevents one from distinguishing direct from indirect effects (Hackney, 2007). This, among other factors, does not allow observation of the ‘cooperative' and quantitative effects of the partners on kinesin activation. The salient features of our data support the following model (Fig 5; supplementary Fig 2 online): (i) R2KR3 cargo relieves the intrinsic KIF5B autoinhibition (Figs 1, 2, 3 and 4) and thus is formally an allosteric activator; (ii) there are four cargo-binding sites per kinesin dimer (Fig 2); and (iii) maximum activity is observed only when all four cargo sites are occupied (Fig 2).
Figure 5.

Schematic modelling of the activation of kinesin-1 by the R2KR3 of RANBP2; R2KR3 is depicted as a cargo of KIF5B. Cargoless kinesin-1 is in a compact conformation with the tail folded over the motor domain causing the inhibition of the ATPase activity. At low concentrations, R2KR3 binds to one of the two bipartite binding sites in kinesin-1 and partly reverts the inhibition by the tails over its motor domain, generating an idle kinesin-1 with low ATPase activity (1). At high concentration of R2KR3 (2), the bipartite sites on both chains become occupied (3). This leads to an extended (relaxed) kinesin-1 by tight conformational coupling and boosting of the motor activity of kinesin-1 in the presence of microtubules (3). KIF5, kinesin superfamily protein 5; RANBP2, RAN-binding protein 2; R2KR3, RBD2–KBD–RBD3.
In our minimum kinetic model (supplementary Fig 2 online), KIF5B exists in two conformers: an inactive T state in which the ATPase activity is inhibited by binding of the tail domain(s) to the head domain(s); and an active R state in which dissociation of the tail domains results in full activity. R2KR3 cargo relieves the inhibition exerted by the tail domain(s) and acts as an allosteric activator. By definition, the R2KR3 tripartite domains have greater affinity for the R state than for the T state (Segel, 1975). The observed cooperativity results from a successive mass action shift of the conformational equilibrium from the T state to the R state, with successive binding of the activator. The R state differs from the T state in showing a greater intrinsic kcat for ATPase activity (supplementary Fig 2 online). The initial hyperbolic phase is a consequence of partial removal of tail inhibition (Figs 2B, 5). The Hill coefficient unambiguously indicates the presence of four tightly and conformationally coupled binding sites for the allosteric activator; however, the data do not allow us to distinguish between four sites for the R2KR3 motif or two sites in which independent binding of the R2–R3 motifs constitutes the four sites. Future stoichiometry studies should resolve this question.
Kinesins walk processively from the minus to the plus end of microtubules in 8 nm steps (Howard et al, 1989; Block et al, 1990; Coy et al, 1999b). As there is a one-to-one coupling between the mechanical and chemical (ATP hydrolysis) cycles (Howard et al, 1989; Block et al, 1990; Coy et al, 1999b), the speed of kinesin is equal to ν=kATPaseΔ (Δ, distance travelled by the kinesin per mechanical cycle; kATPase, rate of ATP hydrolysis by kinesin; Hua et al, 1997; Schnitzer & Block, 1997). The maximum ATPase rate obtained with our assays of KIF5B in the presence of R2KR3 (33 ATP s−1 per kinesin molecule) produce a calculated speed of approximately 264 nm s−1. This value is still approximately two- to threefold lower than that observed in vitro with the KIF5C motor domain alone (Dunn et al, 2008; Ross et al, 2008), the bovine brain, the squid kinesin and recombinant Drosophila kinesin-1 (Howard et al, 1989; Schnitzer & Block, 1997; Coy et al, 1999b). Our value is also approximately 3.5- and 6-fold lower, respectively, than the speed of KIF5C in vivo (Dunn et al, 2008) and anterograde transport rate by kinesin in squid axoplasm (Brady et al, 1990). These discrepancies suggest that other factors are still required to stimulate further the motor activity of KIF5B (Reed et al, 2006; Dunn et al, 2008). Factors such as RAN GTPase, which associates with the RBDs of RANBP2, in combination with the minimal in vitro system described herein, will provide new insights on how conventional kinesin undergoes multiple levels of regulation. Finally, mutations in RANBP2 coupled with various febrile infections lead selectively to rampant necrosis of neurons of the central nervous system (Neilson et al, 2009). As TNF-α is a crucial pyrogenic cytokine and induces the phosphorylation of c-Jun N-terminal kinase, dissociation of KIF5B from tubulin in axons, but not cell bodies, and inhibition of mitochondria transport (Stagi et al, 2006), a traffic event also affected by RANBP2 (Cho et al, 2007), it is possible that RANBP2 is important in this pathogenic KIF5B-dependent process, as well as in various other disease states (Hirokawa & Takemura, 2003).
Methods
Purification of KIF5B. Full-length human KIF5B tagged with 6 × histidine at the C-terminal end was expressed in BL21(DE) bacterial cells. A cell pellet was resuspended and lysed in lysis buffer (50 mM sodium phosphate buffer and 300 mM NaCl, pH 8.0) using French press. The pellet of the lysed cells was dissolved and incubated in a chaotropic extraction buffer (8 M urea; 50 mM sodium phosphate, pH 8.0; 300 mM NaCl; and 10 mM 2-mercaptoethanol) containing EDTA-free complete protease inhibitor cocktail tablet (Roche, Indianapolis, IN, USA) for 30 min. After centrifuging at 10,000g for 25 min, the supernatant was incubated with TALON™ resin (BD Biosciences, San Jose, CA, USA) for 20 min at 4°C, washed and eluted with chaotropic extraction buffer containing 500-mM imidazole. The eluate was renatured by dialysis in a stepwise manner by placing the purified protein (approximately 3 ml) in a Slide-A-Lyzer® Dialysis Cassette against 1 l of initial dialysis buffer (6 M urea, 50 mM sodium phosphate, pH 8.0; 300 mM NaCl; and 10 mM 2-mercaptoethanol) at 4°C. After 2 h, 0.5 l buffer was discarded and 0.5 l of storage buffer (50 mM Tris, pH 8.0; 10% glycerol; and 10 mM 2-mercaptoethanol) was added. We repeated this procedure sequentially every 2 h (including an 8 h equilibration overnight) until the concentration of urea reached 0.1 M. Then, the protein was dialysed against storage buffer three more times and stored at −80°C.
A description of additional methods is provided as supplementary information online.
Supplementary information is available at EMBO reports online (http://www.emboreports.org)
Supplementary Material
Supplementary Methods
Acknowledgments
This study was supported by National Institutes of Health grant EY011993 from the Pearle Vision Foundation to P.A.F. and National Institutes of Health 2P30-EY005722-21. P.A.F. is the Jules & Doris Stein Research to Prevent Blindness Professor.
Footnotes
The authors declare that they have no conflict of interest.
References
- Adio S, Reth J, Bathe F, Woehlke G (2006) Review: regulation mechanisms of Kinesin-1. J Muscle Res Cell Motil 27: 153–160 [DOI] [PubMed] [Google Scholar]
- Blasius TL, Cai D, Jih GT, Toret CP, Verhey KJ (2007) Two binding partners cooperate to activate the molecular motor Kinesin-1. J Cell Biol 176: 11–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block SM, Goldstein LS, Schnapp BJ (1990) Bead movement by single kinesin molecules studied with optical tweezers. Nature 348: 348–352 [DOI] [PubMed] [Google Scholar]
- Brady ST (1985) A novel brain ATPase with properties expected for the fast axonal transport motor. Nature 317: 73–75 [DOI] [PubMed] [Google Scholar]
- Brady ST, Pfister KK, Bloom GS (1990) A monoclonal antibody against kinesin inhibits both anterograde and retrograde fast axonal transport in squid axoplasm. Proc Natl Acad Sci USA 87: 1061–1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Y, Singh BB, Aslanukov A, Zhao H, Ferreira PA (2001) The docking of kinesins, KIF5B and KIF5C, to Ran-binding protein 2 (RanBP2) is mediated via a novel RanBP2 domain. J Biol Chem 276: 41594–41602 [DOI] [PubMed] [Google Scholar]
- Cho KI, Cai Y, Yi H, Yeh A, Aslanukov A, Ferreira PA (2007) Association of the kinesin-binding domain of RanBP2 to KIF5B and KIF5C determines mitochondria localization and function. Traffic 8: 1722–1735 [DOI] [PubMed] [Google Scholar]
- Coy DL, Hancock WO, Wagenbach M, Howard J (1999a) Kinesin's tail domain is an inhibitory regulator of the motor domain. Nat Cell Biol 1: 288–292 [DOI] [PubMed] [Google Scholar]
- Coy DL, Wagenbach M, Howard J (1999b) Kinesin takes one 8-nm step for each ATP that it hydrolyzes. J Biol Chem 274: 3667–3671 [DOI] [PubMed] [Google Scholar]
- DeBoer SR et al. (2008) Conventional kinesin holoenzymes are composed of heavy and light chain homodimers. Biochemistry 47: 4535–4543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn S, Morrison EE, Liverpool TB, Molina-Paris C, Cross RA, Alonso MC, Peckham M (2008) Differential trafficking of Kif5c on tyrosinated and detyrosinated microtubules in live cells. J Cell Sci 121: 1085–1095 [DOI] [PubMed] [Google Scholar]
- Friedman DS, Vale RD (1999) Single-molecule analysis of kinesin motility reveals regulation by the cargo-binding tail domain. Nat Cell Biol 1: 293–297 [DOI] [PubMed] [Google Scholar]
- Gindhart JG (2006) Towards an understanding of kinesin-1 dependent transport pathways through the study of protein–protein interactions. Brief Funct Genomic Proteomic 5: 74–86 [DOI] [PubMed] [Google Scholar]
- Hackney DD (2007) Jump-starting kinesin. J Cell Biol 176: 7–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackney DD, Stock MF (2000) Kinesin's IAK tail domain inhibits initial microtubule-stimulated ADP release. Nat Cell Biol 2: 257–260 [DOI] [PubMed] [Google Scholar]
- Hackney DD, Levitt JD, Suhan J (1992) Kinesin undergoes a 9 S to 6 S conformational transition. J Biol Chem 267: 8696–8701 [PubMed] [Google Scholar]
- Hirokawa N (1998) Kinesin and dynein superfamily proteins and the mechanism of organelle transport. Science 279: 519–526 [DOI] [PubMed] [Google Scholar]
- Hirokawa N, Takemura R (2003) Biochemical and molecular characterization of diseases linked to motor proteins. Trends Biochem Sci 28: 558–565 [DOI] [PubMed] [Google Scholar]
- Hirokawa N, Pfister KK, Yorifuji H, Wagner MC, Brady ST, Bloom GS (1989) Submolecular domains of bovine brain kinesin identified by electron microscopy and monoclonal antibody decoration. Cell 56: 867–878 [DOI] [PubMed] [Google Scholar]
- Howard J, Hudspeth AJ, Vale RD (1989) Movement of microtubules by single kinesin molecules. Nature 342: 154–158 [DOI] [PubMed] [Google Scholar]
- Hua W, Young EC, Fleming ML, Gelles J (1997) Coupling of kinesin steps to ATP hydrolysis. Nature 388: 390–393 [DOI] [PubMed] [Google Scholar]
- Neilson DE et al. (2009) Infection-triggered familial or recurrent cases of acute necrotizing encephalopathy caused by mutations in a component of the nuclear pore, RANBP2. Am J Hum Genet 84: 44–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed NA, Cai D, Blasius TL, Jih GT, Meyhofer E, Gaertig J, Verhey KJ (2006) Microtubule acetylation promotes kinesin-1 binding and transport. Curr Biol 16: 2166–2172 [DOI] [PubMed] [Google Scholar]
- Ross JL, Shuman H, Holzbaur EL, Goldman YE (2008) Kinesin and dynein-dynactin at intersecting microtubules: motor density affects dynein function. Biophys J 94: 3115–3125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton WM (1994) Isolation and analysis of microtubule motor proteins. Methods Cell Biol 44: 279–288 [DOI] [PubMed] [Google Scholar]
- Schnitzer MJ, Block SM (1997) Kinesin hydrolyses one ATP per 8-nm step. Nature 388: 386–390 [DOI] [PubMed] [Google Scholar]
- Segel IH (1975) Enzyme Kinetics. New York, NY, USA: Wiley-Interscience [Google Scholar]
- Stagi M, Gorlovoy P, Larionov S, Takahashi K, Neumann H (2006) Unloading kinesin transported cargoes from the tubulin track via the inflammatory c-Jun N-terminal kinase pathway. FASEB J 20: 2573–2575 [DOI] [PubMed] [Google Scholar]
- Vale RD, Reese TS, Sheetz MP (1985) Identification of a novel force-generating protein, kinesin, involved in microtubule-based motility. Cell 42: 39–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Methods