

Abstract
Background
Eukaryotic cells contain numerous compartments, which have different protein constituents. Proteins are typically directed to compartments by short peptide sequences that act as targeting signals. Translocation to the proper compartment allows a protein to form the necessary interactions with its partners and take part in biological networks such as signalling and metabolic pathways. If a protein is not transported to the correct intracellular compartment either the reaction performed or information carried by the protein does not reach the proper site, causing either inactivation of central reactions or misregulation of signalling cascades, or the mislocalized active protein has harmful effects by acting in the wrong place.
Results
Numerous methods have been developed to predict protein subcellular localization with quite high accuracy. We applied bioinformatics methods to investigate the effects of known disease-related mutations on protein targeting and localization by analyzing over 22,000 missense mutations in more than 1,500 proteins with two complementary prediction approaches. Several hundred putative localization affecting mutations were identified and investigated statistically.
Conclusion
Although alterations to localization signals are rare, these effects should be taken into account when analyzing the consequences of disease-related mutations.
Background
Eukaryotic cells contain numerous compartments, such as cytoplasm, mitochondria, Golgi apparatus, and peroxisomes, all of which contain different protein constituents and have different functions. Proteins are typically directed to these compartments by short peptide sequences that act as targeting signals. For example, secretory, chloroplast and mitochondrial targeting peptides are located at the N terminus, whereas signals for other compartments can be within the amino acid sequence. Terminal signal peptides are typically cleaved during the protein translocation process.
Protein function depends on numerous factors. One important but often neglected property is its subcellular localization. Translocation to the proper compartment allows a protein to form the necessary interactions with its partners and take part in biological networks. For example, signalling and metabolic pathways are dependent on the location of the constituent proteins. Failure to be transported to the correct intracellular compartment can have detrimental effects, which appear in different ways. Either the reaction performed or information carried by the protein does not reach the proper site, causing either inactivation of central reactions or misregulation of, eg, signalling cascades, or the mislocalized protein is active, but has harmful effects by acting in the wrong place.
Subcellular localization of proteins and peptides has long been investigated using numerous methods. Recently, high-throughput methods have been developed based either on the use of reporter genes/tags or by purification, fractionation and analysis of cellular compartments [1,2]. Information on protein localization is scattered throughout publications and numerous databases. Fortunately, central resources such as the Human Protein Reference Database (HPRD) [3], UniProt [4] and Gene Ontologies [5] now exist to integrate information from several sources. A problem with these databases, however, is that data quality and experimental methods vary. Further, some databases contain experimentally validated localization information whereas others also contain localization predictions. The picture is further complicated by the fact that a protein can be localized in more than one compartment, often depending on the state of the cell. Thus, databases that contain only experimentally validated data may not provide complete information for all proteins.
Numerous methods have been developed to predict protein subcellular localization (for review, see eg, [6]). The very first methods in the 1970's were developed to identify microbial signal peptides [7,8]. Now, methods and protocols exist for the prediction of over 10 cellular compartments and subcompartments. Although the actual prediction algorithms and methods differ, all are based on sequence signature patterns. Some general predictors are useful for all subcompartments, but the majority of methods are specific for individual compartments and organisms or groups of organisms. The reliability of individual methods is relatively high, close to 90% (see, eg, [9-11])
Disease-causing mutations result in abnormal cellular function through numerous mechanisms. To date, pathological mechanisms have been revealed for only a fraction of all known mutations. Mutation information has been collected and stored in locus-specific (eg, [12,13]) and general (such as Online Mendelian Inheritance in Man (OMIM) and Human Gene Mutation Database (HGMD)) databases. Many experimental methods are tedious, expensive and difficult to use. Disease-causing mutations are identified for diagnostic purposes, and thus most medical centers identify a genetic mutation(s) without acquiring further information about the protein. We and others have applied numerous bioinformatic methods to predict and explain the consequences of mutations. Recently, we discussed the applicability of some 40 analysis and prediction methods [14,15]. The effects and consequences vary depending on the site and type of mutation, with insertions and deletions usually leading to truncated proteins. These cases are easy to explain if a substantial part of the protein is missing. To understand protein structure and function, however, missense mutations are most interesting because they often indicate residues that are critical for, and changes that are deleterious to, structure and/or function. Most mutations reduce protein activity, but increasing numbers of gain-of-function mutations [16,17] are also being identified. Relatively few detailed investigations have described protein mislocalization due to disease-related mutations or introduced genetic alterations. In addition, all such publications report a limited number of mutations in a single protein.
Targeting signals tend to be conserved and thus sensitive to alterations; therefore, we can assume that these methods can be applied to the analysis of point mutations. Here we use bioinformatics to investigate the effects of known disease-related mutations on protein targeting and localization by analyzing 22,416 missense mutations. Several hundred putative localization mutations were identified with two complementary multiprediction approaches. The results indicate that although alterations to localization signals are rare, localization predictors should be added to the methods arsenal of a mutations analyst. Our results also suggest pathological mechanisms for a number of mutations and depict cases for further experimental investigation.
Results and discussion
We investigated the effects of disease-related mutations on protein localization by performing large-scale analysis and prediction with two different but complementary methods. Because we needed unambiguous mapping of DNA mutations to protein sequences, we performed filtering steps. We obtained experimentally identified protein localizations from HPRD [3], which is considered a highly accurate, consistent and reliable source of protein annotations.
Reliability of the individual localization predictors
Before approaching the mutation effect predictions, we wanted to test the applicability of the methods to the dataset. Because HPRD contains experimentally verified data, we compared the localizations to predictions for the wild type proteins. The analysis was made for localizations for which SP and/or WoLF PSORT make predictions.
The compartments with the largest numbers of proteins are plasma membrane, cytoplasm, nucleus, extracellular space, and mitochondria (see Additional file 1). Endoplasmic reticulum (ER) had the highest number of proteins as a secondary classification (see Additional file 2). Proteins were distributed unequally among the different compartments. Although disease-related proteins form a special group, they still reflect the overall properties of all proteins.
Table 1 indicates that results for TMHMM, TargetP predictions for mitochondrial proteins, PeroxiP and PTS1 have high accuracy while Golgipredictor and PredictNLS have only moderate performance. Precision values indicate that except for Golgipredictor and PeroxiP the predictors mainly detect the proteins well. However, when recall is considered, only TargetP and TMHMM are highly reliable. The overall parameter MCC values range from 0.056 to 0.58.
Table 1.
Prediction results for the localization of wild type proteins with the individual predictorsa
| Compartment | tp | rp | tn | fn | Accuracy | Precision | Recall | MCC | Proteins located |
| TargetP (mitochodrial) | 101 | 109 | 1254 | 52 | 0.894 | 0.481 | 0.660 | 0.506 | 210 |
| TMHMM | 377 | 144 | 853 | 142 | 0.811 | 0.724 | 0.726 | 0.581 | 521 |
| Golgipredictor | 23 | 178 | 227 | 20 | 0.558 | 0.114 | 0.535 | 0.056 | 201 |
| PeroxiP | 6 | 24 | 423 | 12 | 0.923 | 0.200 | 0.333 | 0.220 | 30 |
| PTS1 | 3 | 1 | 446 | 15 | 0.966 | 0.750 | 0.167 | 0.343 | 4 |
| PredictNLS | 98 | 23 | 248 | 217 | 0.590 | 0.810 | 0.311 | 0.279 | 121 |
| Summary | 0.790b | 0.513b | 0.455b | 0.331b | 1087 |
atp, the number of positive cases that were correctly predicted; tn, the number of negative cases correctly predicted; fp, the number of positive cases incorrectly predicted; fn, the number of negative cases incorrectly predicted.
baverage value.
One reason for the poor behaviour of certain predictors is likely the fact that they are usually not used alone i.e. other programs are used to sort the data to localization routes before applying these tools. Overall, the methods obtained good precision at the cost of recall (false negatives). In summary, the individual methods can be applied with relatively high accuracy and precision to localization predictions. Methods, which predict the localization at the end of a complex pathway, are less reliable when applied directly to sequences.
Reliability of the combined localization predictors
The SP predicted localization for 12 possible compartments and WoLF PSORT (animal version) predicted ten localizations. The results for the two approaches and comparison to experimental data for the wild type proteins are shown in Tables 2 and 3. Several parameters were calculated to describe the prediction performance. For the SP, altogether 60.9% (966/1586) of predictions were correct. Seventy proteins received two predictions in TargetP and thus two routes in SignalP. In these cases both predictions are included. No mitochondrial periplasmic space proteins were predicted and the false negative rate is very high for these cases. The precision and recall of Golgi, transmembrane is low as well as the values for peroxisomal localization. Accuracy and precision are usually clearly better than the recall values, which is in line with the results for individual predictors (Table 1). The results for gPM and mPM were combined to those for plasma membrane, since these localizations were predicted only for 3 and 6 proteins, respectively.
Table 2.
Prediction results for the localization of wild type proteins with the Scandinavian protocol.
| Compartmenta | tp | fp | tn | fn | Accuracy | Precision | Recall | MCC | Proteins located |
| Mtm | 10 | 29 | 1337 | 140 | 0.889 | 0.256 | 0.067 | 0.086 | 39 |
| Mps | 0 | 6 | 1363 | 147 | 0.899 | 0.000 | 0.000 | -0.021 | 6 |
| Mma | 91 | 74 | 1293 | 58 | 0.913 | 0.552 | 0.611 | 0.532 | 165 |
| Gtm | 23 | 178 | 1240 | 75 | 0.833 | 0.114 | 0.235 | 0.079 | 201 |
| PM | 221 | 35 | 619 | 641 | 0.554 | 0.863 | 0.256 | 0.268 | 256 |
| S | 246 | 84 | 1093 | 93 | 0.883 | 0.745 | 0.726 | 0.661 | 330 |
| ER | 2 | 1 | 1330 | 183 | 0.879 | 0.667 | 0.011 | 0.074 | 3 |
| N | 98 | 23 | 1097 | 298 | 0.788 | 0.810 | 0.247 | 0.368 | 121 |
| P | 6 | 24 | 1462 | 24 | 0.968 | 0.200 | 0.200 | 0.184 | 30 |
| C | 269 | 166 | 852 | 229 | 0.739 | 0.618 | 0.540 | 0.392 | 435 |
| Summary | 966b | 620b | 1888b | 0.835c | 0.483c | 0.289c | 0.262c | 1586b |
Abbreviations for statistical parameters as in Table 1.
a C, cytosol; Gtm, Golgi, transmembrane; Mma, mitochondrial matrix; Mps, mitochondrial, periplasmic space; Mtm, mitochondrial transmembrane; N, nuclear; P, peroxisomal; PM, plasma membrane; S, secreted
btotal number
caverage value.
Table 3.
Prediction results for the localization of wild type proteins with WoLF PSORT.
| Mutant compartment | |||||||||
| Wild type compartmenta | tp | fp | tn | fn | Accuracy | Precision | Recall | MCC | Proteins located |
| CK | 2 | 38 | 1471 | 5 | 0.972 | 0.050 | 0.286 | 0.110 | 40 |
| CK_PM | 0 | 31 | 1485 | 31 | 0.960 | 0.000 | 0.000 | -0.020 | 31 |
| C | 339 | 304 | 713 | 160 | 0.694 | 0.527 | 0.679 | 0.362 | 643 |
| C_G | 0 | 29 | 1450 | 37 | 0.956 | 0.000 | 0.000 | -0.022 | 29 |
| C_M | 8 | 145 | 1328 | 35 | 0.881 | 0.052 | 0.186 | 0.048 | 153 |
| C_N | 102 | 365 | 963 | 86 | 0.703 | 0.218 | 0.543 | 0.191 | 467 |
| C_P | 3 | 77 | 1425 | 11 | 0.942 | 0.038 | 0.214 | 0.070 | 80 |
| ER | 85 | 253 | 1081 | 97 | 0.769 | 0.251 | 0.467 | 0.217 | 338 |
| ER_G | 4 | 70 | 1392 | 50 | 0.921 | 0.054 | 0.074 | 0.023 | 74 |
| ER_M | 2 | 92 | 1413 | 9 | 0.933 | 0.021 | 0.182 | 0.042 | 94 |
| S | 281 | 321 | 856 | 58 | 0.750 | 0.467 | 0.829 | 0.474 | 602 |
| S_PM | 4 | 148 | 1358 | 6 | 0.898 | 0.026 | 0.400 | 0.081 | 152 |
| G | 13 | 74 | 1344 | 85 | 0.895 | 0.149 | 0.133 | 0.085 | 87 |
| L | 33 | 202 | 1258 | 23 | 0.852 | 0.140 | 0.589 | 0.235 | 235 |
| M | 128 | 325 | 1038 | 25 | 0.769 | 0.283 | 0.837 | 0.394 | 453 |
| M_N | 0 | 33 | 1459 | 24 | 0.962 | 0.000 | 0.000 | -0.019 | 33 |
| M_P | 1 | 112 | 1397 | 6 | 0.922 | 0.009 | 0.143 | 0.018 | 113 |
| N | 325 | 330 | 790 | 71 | 0.735 | 0.496 | 0.821 | 0.467 | 655 |
| P | 12 | 302 | 1184 | 18 | 0.789 | 0.038 | 0.400 | 0.068 | 314 |
| PM | 356 | 185 | 841 | 134 | 0.790 | 0.658 | 0.727 | 0.533 | 542 |
| Summary | 1696b | 3398b | 966b | 0.854c | 0.174c | 0.375c | 0.168c | 5095b | |
Abbreviations for statistical parameters as in Table 1.
aC, cytosol; CK, cytoskeleton; G, Golgi compartment; M, mitochondrial; N, nuclear; P, peroxisomal; PM, plasma membrane; S, secreted
btotal number. Underline sign indicates multiple predictions.
caverage value
In the case of WoLF PSORT, 33.7% (1696/5095) gave correct predictions (Table 3). There are a number of dual predictions, eg for proteins, which shuttle between cytosol and nucleus. Results for these predictions were considered as correct only if the protein was found from both compartments. Values for accuracy ranged from 0.69 to 0.98 (average 0.854), whereas recall ranged from 0 to 0.84 (average 0.375). Peroxisomal proteins clearly had the lowest prediction accuracy. The results for WoLF PSORT do not allow a direct comparison with the SP, because WoLF PSORT considers combined predictions to be correct when one of the predictions is correct. Actually, just six classes had a substantial number of predicted proteins. The overall accuracy is almost identical for the two protocols whereas SP has clearly better precision and somewhat higher MCC score. The recall is slightly better for WoLF PSORT.
In conclusion, detailed analysis of the prediction performance indicates that the subcellular localization predictors still have much to improve. However, because the accuracy of individual predictions are rather high, these methods are indeed applicable to systematic analysis of mutations even though the precision, recall and MCC are clearly suboptimal. The more steps there are in the analysis the lower the expected accuracy (and other parameter values). Thus, if the analysis is based on five consecutive steps (as in SP) in which each step has 90% accuracy the final expected accuracy would be 59% (0.95).
Analysis of mutation effects
As the results above indicate, the subcellular localization of individual compartments of the investigated proteins can be predicted with rather high accuracy and also multipredictors provide useful data. The effect of mutations on protein localization was tested for all 22,416 missense mutations. In this analysis we looked for differences in predicted localization compared with that for wild type forms. Even if the prediction of the compartment was incorrect, a change in the predicted localization due to mutation might indicate the mutation mechanism and be useful for further studies. Similar effect has been useful also in some other bioinformatics predictions such as protein secondary structures.
The SP predicted that 203 mutations would alter protein localization. Results in Table 4 and in Additional file 3 show the distribution of mutations in the different subcompartments for the mutations and proteins in which the mutations appear, respectively. The numbers represent correctly predicted proteins and the total number of mutations for each category. The most common original compartments for proteins whose localization changed on account of the mutation were plasma membrane, Golgi transmembrane, nucleus, and cytoplasm. Most common among the mutant sublocalizations were plasma membrane, Golgi transmembrane, and nucleus. The single most common predicted mutation type was from plasma membrane (for wild type localization) to Golgi transmembrane–altogether 47 cases, 17 of which had the correct prediction for the wild type form. Although the number of correct wild type predictions was not directly related to the mutation predictions, the numbers varied widely–as an extreme case C to N prediction, with 19 of 20 having the correct wild type prediction for 11 proteins out of 12. The range of changes to localizations of mutations varied from one to five, the highest being for Gtm proteins. Similarly, the predicted range of mislocalizations from one subcompartment to others varied from one to six, with cytoplasmic proteins being redirected to six different compartments when mutated.
Table 4.
Changes in SP localization prediction due to mutations.
| Mutant compartment | ||||||||||||||||
| Wild type compartmenta | Gtm | Mtm/Gtm | Mma/C | Mma | Mma/PM | Mps/S | C | Mma/P | N | PM | Mtm | S | Mtm/PM | Mma/N | P | Total |
| Mtm/Gtm | 0/3 | 1/1 | 1/4 | |||||||||||||
| Mtm/PM | 0/6 | 0/9 | 1/1 | 1/16 | ||||||||||||
| PM | 17/47 | 0/1 | 4/5 | 21/53 | ||||||||||||
| S | 0/4 | 7/8 | 7/12 | |||||||||||||
| C | 0/1 | 8/8 | 19/20 | 1/1 | 1/1 | 0/2 | 29/33 | |||||||||
| Mtm | 0/3 | 1/1 | 1/4 | |||||||||||||
| Mma/Gtm | 0/2 | 0/2 | ||||||||||||||
| Gtm | 0/5 | 17/21 | 0/1 | 2/4 | 19/31 | |||||||||||
| Mtm/C | 0/1 | 1/4 | 1/5 | |||||||||||||
| Mps/C | 1/1 | 1/1 | ||||||||||||||
| Mma | 6/6 | 1/1 | 2/2 | 1/1 | 10/10 | |||||||||||
| Mma/S | 2/2 | 1/1 | 1/1 | 4/4 | ||||||||||||
| Mma/C | 4/4 | 4/4 | ||||||||||||||
| Mps | 2/2 | 2/2 | ||||||||||||||
| Mma/N | 1/1 | 1/1 | 2/2 | |||||||||||||
| P | 1/1 | 1/1 | ||||||||||||||
| N | 0/18 | 1/1 | 1/19 | |||||||||||||
| Total | 17/61 | 0/19 | 17/18 | 3/6 | 2/2 | 4/4 | 7/8 | 1/1 | 21/22 | 25/48 | 0/1 | 3/5 | 4/5 | 1/1 | 0/2 | 105/203 |
The numbers separated by the slash sign are for how many proteins the wild type localization has been correctly predicted, and the number of analyzed mutations, respectively.
aC, cytosol; Gtm, Golgi, transmembrane; Mma, mitochondrial matrix; Mps, mitochondrial, periplasmic space; Mtm, mitochondrial transmembrane; N, nuclear; P, peroxisomal; PM, plasma membrane; S, secreted. Slash sign indicates alternative predicted localizations.
Results for the mutations and proteins analyzed by WoLF PSORT are shown in Table 5 and Additional file 4, respectively. To avoid excessive partitioning of the results to very small groups, only the results for the highest prediction score are indicated. About 50% of the wild type proteins had the correct localization. Altogether, WoLF PSORT found 183 cases with predicted alteration caused by mutation. The highest number of mutation-based rerouting to other compartments was for proteins whose wild type form was predicted to localize to the cytoplasm. Extracellular, cytoplasm, plasma membrane, nuclear and mitochondria are the most common localizations for mutant proteins. In comparison to SP, WoLF PSORT had somewhat lower numbers in target compartments. The changes with the largest number of mutations were CN to C, and N to C, which are related predictions. WoLF PSORT may suffer from using BLAST as part of its algorithm. In the case of SP, the search for homologues was not implemented, however, that was not possible to do for WoLF PSORT.
Table 5.
Changes in WoLF PSORT localization prediction due to mutations.
| Mutant compartment | ||||||||||||||||||
| Wild type compartmenta | PM | S | S_PM | C | C_N | C_M | N | N/C_N | N/C/ C_N |
ER | ER_ M |
M | M_N | M_N/ C_M |
P | L | CK | Total |
| P | 2/2 | 0/1 | 1/6 | 2/2 | 0/1 | 0/6 | 1/1 | 6/19 | ||||||||||
| S | 0/1 | 0/4 | 0/1 | 4/5 | 1/1 | 4/9 | 0/2 | 9/23 | ||||||||||
| S/S_PM | 0/1 | 0/1 | ||||||||||||||||
| C | 0/6 | 2/3 | 3/9 | 3/3 | 5/6 | 0/1 | 3/6 | 2/2 | 18/36 | |||||||||
| C/C_N | 1/1 | 1/1 | ||||||||||||||||
| C_N | 0/2 | 18/19 | 4/8 | 2/2 | 2/2 | 3/3 | 29/36 | |||||||||||
| C_M | 1/1 | 1/1 | ||||||||||||||||
| N | 0/1 | 0/10 | 6/10 | 7/7 | 2/4 | 15/32 | ||||||||||||
| G/ER_G | 0/1 | 0/1 | ||||||||||||||||
| ER | 0/11 | 0/1 | 1/1 | 0/1 | 2/2 | 3/16 | ||||||||||||
| M | 0/2 | 3/3 | 6/6 | 0/2 | 0/2 | 9/15 | ||||||||||||
| ER_M | 1/1 | 1/1 | ||||||||||||||||
| M/P/M_P | 0/1 | 0/1 | ||||||||||||||||
| Total | 0/21 | 7/21 | 0/1 | 37/53 | 7/11 | 3/3 | 12/20 | 0/3 | 0/1 | 4/7 | 1/1 | 11/28 | 2/2 | 3/3 | 3/3 | 0/3 | 2/2 | 92/183 |
The numbers separated by the slash sign are for how many proteins the wild type localization have been correctly predicted, and the number of analyzed mutations, respectively.
aC, cytosol; CK, cytoskeleton; G, Golgi compartment; M, mitochondrial; N, nuclear; P, peroxisomal; PM, plasma membrane; S, secreted. Slash sign indicates alternative localization predictions and underline sign multiple predicted localizations.
The results for the identified changes in protein localization due to missense mutations are shown in Additional file 5 and Additional file 6. The two prediction approaches, SP and WoLF PSORT, agreed on 17267 (77%) of the total 22,416 mutations when all predictions of WoLF PSORT were taken into account. Of the two approaches, 203 and 183 mutations were predicted to alter the target compartments of mutant proteins, affecting 105 and 92 proteins, respectively. 18 of these proteins were common for the two methods, and in these proteins the protocols agreed on 12 mutations to affect proteins localization. The two methods predicted the same compartment mislocalization in seven cases. This indicates that neither of the methods was able to detect all putative localization mutations. Similar result calling for use of several tools was apparent when splice site prediction tools were tested for mutation analysis [18].
We can estimate the number of expected mutations in localization sites. Our data set contains 1,516 proteins, which consist of 1,054,823 amino acid residues, and which have 2373 localizations based on HPRD. The length of the targeting peptides varies from a few residues to close to 30. If we use an average value of eight residues for the targeting peptide, we should see 403 (2373*8/1054823*22416) mutations in localization signals. This number is almost exactly what was observed.
The distribution of the amino acid changes in the predicted localization alterations is shown in Fig. 1. The amino acid distributions for mutations were compared with information for all human proteins taken from Codon Usage Tabulated from GenBank (CUTG) [19]. The distribution of all the mutations was significantly biased compared to random distribution in all amino acid types except for D and H (see Additional file 7). The results are in line with previous mutation distribution studies for numerous proteins and secondary structural elements within them [20-22], including mutations in the protein kinase family [23] and in immunodeficiencies [12]. These studies indicated highly skewed distribution for disease mutations, which varies also between secondary structural elements. Data for the SP indicated that mutations are most common in R (Fig. 2 and Additional file 8). Arginine is coded by six synonymous codons, four of which contain a CpG dinucleotide, a well known mutational hot spot [24]. Also G, L and M are frequently mutated. The most common mutant residues were R, C, and P, of which arginine is the most common. Arginine was usually replaced by C (14 of 50 cases), making this the single most frequent mutation type. Eight of 9 mutations to W were from R, and 7 of 8 mutations in Q were from arginine. Arginine was mutated altogether to 10 other residues, i.e., all except two (K and M) of the possible substitutions with single nucleotide changes. Arginine was also the most common resulting residue from mutations in other codons, and it was the residue type with the highest number of original residues, 9. Somewhat surprisingly, no localization mutations were identified in Q, which however occurred 8 times as a mutant residue.
Figure 1.

Amino acid distribution for the two prediction schemes. Scandinavian protocol (A, B) and WoLF PSORT (C, D) predictions of (A, C) amino acids in wild type proteins that are predicted to be mutated in localization mutants, and (B, D) mutant amino acids in localization mutants.
Figure 2.

Schematic illustration of the analysis of protein localization with the Scandinavian protocol. The predicted compartments are indicated with corresponding numbers in black circles. The localizations are listed in the middle of the figure. RC, reliability coefficient; SP, signal peptide; TM, transmembrane region.
Of note, only two mutations to A and three mutations to F were predicted to be disease-related. H, Q and E were the least frequently mutated residues. These results follow somewhat the general amino acid distribution with prominent exceptions like arginine.
WoLF PSORT results show some differences from SP, which may have originated from the prediction algorithm. R, G, and L were the most commonly mutated residues. However, arginine did not show the clear overprediction as in the SP data. D, H and K were the least mutated residues (Fig. 2 and Additional file 9). Mutations to G and R both appeared in seven original residue types, whereas S was mutated from eight original residues. These were also the residues that had the highest number of mutant residue types. Only one change to H, two to N, D or I were predicted to be related to diseases.
Comparison to known mislocalization mutations
Our results predicted localization changes that underlie many different types of diseases, including those involving signal transduction, metabolism, immunodeficiencies, eye diseases, developmental disorders and cancers (see Additional file 5 and Additional file 6). Some disease-related mutations, which have been confirmed to affect protein localization, have been described. These cases are usually sporadic in the literature. Because no database is available for such mutations, we performed a literature search and identified a number of cases.
Mutations in SHOX, homeobox-containing gene, cause idiopathic short stature, Leri-Well dyschondrosteosis and Langer mesomelic dysplasia. The substitution R173C prevents the transport of the SHOX-encoded protein to the nucleus and its subsequent function as a transcription activator [25]. Both the SP and WoLF PSORT correctly predicted the mislocalization and the effect of the mutation.
AIRE, autoimmune regulator, is a nuclear protein and transcriptional regulator. Wild type AIRE appears both in nuclear dots, as evenly distributed in the nucleus, and in the cytoplasm. Several mutations have been shown to affect the distribution of AIRE between compartments [26-28]. Mutations R14L, T16M, A21V and Y85C were correctly predicted to affect protein localization by the SP and L28P and L29P by WoLF PSORT predictor. However, the predicted changes were not accurate, because the SP has a change from cytoplasmic and mitochondrial matrix to cytoplasm and WoLF PSORT from secreted to mitochondrial.
Similar results were obtained for BSND mutations. Barttin, encoded by BSND, is involved in Bartter syndrome, a renal tubular salt-wasting disease. Barttin localizes to the plasma membrane, whereas mutant forms are retained in the ER [29]. R8L was predicted by SP to change the localization from Golgi transmembrane to plasma membrane. A milder form, G10S, which appears in both the ER and the plasma membrane, was not predicted to affect localization. We also consider this kind of prediction useful because a localization change is forecast due to the mutation. Thus, the predictions can give a hint of the possible mechanism, even though the final validation must be obtained experimentally.
We did predictions for cases, which according to liture affect the localization in ATP7B mutations in Wilson disease [30], ABCA1 mutations in Scott syndrome [31], RPS19 mutations in Diamond-Blackfan anemia [32], ABCA1 mutations in Tangier disease [33], and laminin A/C mutations in heritable dilated cardiomyopathy [34]. However, the predictions agreed with the experimental data only for the FXYD2 mutation in hereditary primary hypomagnesia [35]. These results show the poor recall of the methods.
Several reasons account for failure of the predictions to detect all the localization changes. As noted above, the predictions are characterized by high accuracy and low recall. Even the experimental information can sometimes be misleading, because the localization effect can be secondary and may not have been investigated in detail. In the androgen receptor C169Y missense mutation, mutant receptor aggregation causes a change in localization [36]. The wild type protein is in nucleoplasm whereas the mutant forms aggregate in both nucleus and cytoplasm. In nine mutations in nonmuscle myosin heavy chain A (MYH9), the mutant proteins aggregate, causing several disorders characterized by giant platelets, thrombocytopenia, and Döhle body-like cytoplasmic inclusions in granulocytes [37].
Sometimes localization-changing mutations appear outside the targeting signals. Forkhead box (FOX) P2 involved in a speech/language disorder has two separate nuclear localization signals. Mutant protein R553H is mainly targeted to the cytoplasm instead of the nucleus [38]. The mutation appears in the region between but not within the two nuclear localization signal sequences. Neutral evolution can generate novel targeting signals. Putative peroxisomal targeting signals were identified from a number of non-peroxisomal proteins and were shown to have a potential to be activated if the original target signal is changed or not accessible [39]. A mutation in the pleckstrin homology domain of AKT1 kinase leads to cancer because of pathological localization to the plasma membrane [40]. AKT1 is normally translocated from nucleus to the plasma membrane in response to growth factor stimulation. A mutant form of E17K, which has increased phosphorylation, is located at the plasma membrane in response to growth factor stimulation.
Cell type-specific alternative splicing can alter the localization of proteins, including myotonic dystrophy protein kinase (DMPK) [41]. Still another mechanism affects the Menkes disease copper ATPase, in which the mutation G1019D interferes with protein folding [42]. Similar effects have been seen for certain breast cancer 1 (BRCA1) mutations [43]. In tafazzin, mutations disrupt the membrane association region [44].
The localization of tyrosine phosphatase SHP-1 is regulated by phosphorylation [45] and alternative start sites [46,47]. The predictions for mutations in any of these proteins indicated no changes to localization.
Conclusion
Applicability of protein localization prediction methods were tested in detecting changes in localization due to point mutations. Altogether 374 mutations were predicted by at least one method to affect protein localization. Because disease mutations are unequally distributed throughout protein sequences, having a higher occurrence in structurally/functionally important sites, we can expect the number of localization mutations to be higher than calculated. The expected number is 403 mutations. Localization mutations are rare events, but they should be taken into account when predicting consequences of mutations. A service for SP predictions will be released in the near future as part of the Pathogenic-Or-Not -Pipeline (PON-P, http://bioinf.uta.fi/PON-P).
Methods
Mutation, localization and sequence data
Missense mutations were obtained from the HGMD http://www.hgmd.cf.ac.uk/ac/index.php[48] (downloaded 17.3.2007) and IDbases [12]. The dataset was filtered to include only genes for which cDNA sequence was available. The experimental localization(s) of each identified protein was collected from the Human Protein Reference Database (HPRD, http://www.hprd.org/) [3] (4.5.2007). We excluded proteins for which the experimental localization was unknown. After these filtering steps, 1,516 proteins remained, which contained altogether 22,416 missense mutations (on average 14.8 per protein). Altogether, we identified 2,373 localizations, indicating that the average per-protein localization was ~1.6 for the 1,516 proteins we investigated. The proteins had 34 primary localizations (Table 1) and altogether 56 localizations (Additional file 2).
We identified both the first (most common) and all localizations for each protein. The wild type protein sequences were translated from cDNA sequences obtained from HGMD. The disease-related mutations were introduced into the protein sequences one by one and analyzed individually. Programs and scripts for the analysis were written in Java or Perl languages.
Localization prediction methods
First, predictions were made separately for certain localizations and then by two strategies for combined predictions. Groups in Stockholm, Sweden, and Lyngby, Denmark, whose long-term efforts have resulted in methods for numerous tasks in subcellular localization prediction, recently published a protocol to combine different predictions developed by them and others into a comprehensive prediction scheme [49]. This Scandinavian protocol (SP) is rather complicated and requires the use of numerous separate prediction tools. To facilitate the analysis, we developed a program that automatically runs all the predictions, parses the results, and provides the outcome of the prediction. The flow chart for the analysis steps and programs is in Fig 2. As a modification, nuclear localization signals were predicted only with the PredictNLS program. Because we analyzed human proteins, it was not necessary to investigate chloroplast localization or prokaryotic predictions. Some of the programs use database searches to identify homologues to strengthen the predictions. We had to omit this step because the wild type sequences in the databases are identical to the mutant sequences, apart from the single missense mutation. Because sequence conservation is indicative of protein colocalization [50,51], database searches would have selected the wild type sequence for prediction and thereby hampered the analysis of mutants. Also the step for β-barrel prediction was omitted.
The programs TargetP [52], SignalP [53] and TMHMM [54,55] were downloaded from http://www.cbs.dtu.dk/services/ and were run locally, whereas programs Big-PI [56-59]http://mendel.imp.ac.at/sat/gpi/gpi_server.html, NMT http://mendel.imp.ac.at/myristate/SUPLpredictor.htm, PeroxiP [60]http://bioinfo.se/PeroxiP/, PredictNLS [11]http://cubic.bioc.columbia.edu/predictNLS/, PTS1 [61,62]http://mendel.imp.ac.at/mendeljsp/sat/pts1/PTS1predictor.jsp, Golgipredictor [63]http://ccb.imb.uq.edu.au/golgi/, Phobius [64]http://phobius.sbc.su.se/ and Prosite [65]http://au.expasy.org/prosite/ were run over the Internet. Altogether, this procedure could predict 12 different localizations (Fig 1).
In the SP protocol, first the TargetP assigns whether the proteins go to mitochondia or secretory pathway or not. The mitochondrial proteins are classified further to transmembrane, periplasmic space or matrix based on the analysis of transmembrane and signal peptide sequences. Transmembrane proteins are predicted via two routes and are then classified to those ending in Golgi transmembrane or plasma membrane. Signal peptide(s) containing proteins are classified to different compartments whether they contain transmembrane region(s), signal peptide, are myristoylated, have GPI anchors or are predicted to endoplasmic reticulum.
The other method we applied, WoLF PSORT, is an integrated program that makes predictions for 10 subcellular compartments [66]. WoLF PSORT was run locally with default parameters. WoLF PSORT program was downloaded from http://wolfpsort.cbrc.jp/ and run locally.
We ran each prediction strategy for both wild type and mutated sequences and determined whether the mutation(s) changed the localization prediction. Both protocols may predict multiple localizations for a protein–for example nucleus and cytosol for a protein that is transported between nucleus and cytosol. Thus, all highest-score predictions provided by the programs were taken into account. In SP, if TargetP had problems to resolve the localization for a protein predicted to mitochondria with poor reliability coefficient (RC) (value 4 or 5) then the protein was predicted also with SignalP and it gets two alternative localizations (Fig 1).
The two methods predict proteins to following compartments. The SP predicted localization for 12 possible compartments, which include the mitochondrial membrane (transmembrane) (Mtm), mitochondrial periplasmic space (Mps), mitochondrial matrix (Mma), Golgi, transmembrane (Gtm), plasma membrane (PM), secreted (S), ER lumen (ER), nucleus (N), peroxisome (P), cytoplasmic C), plasma membrane, GPI anchor (gPM), and plasma membrane, myristoylated (mPM). WoLF PSORT (animal version) predicted ten localizations: cytosol (C), cytoskeleton (CK), ER, extracellular (S), Golgi apparatus (G), lysosome (L), mitochondria (M), nucleus (N), peroxisome (P) and plasma membrane (PM). All these were present in the dataset.
The quality of the predictions was measured by four parameters: accuracy, recall, precision, and the Matthew's correlation coefficient (MCC) as follows:
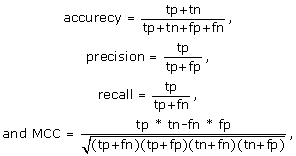 |
where tp is the number of positive cases that were correctly predicted, tn is the number of negative cases correctly predicted, fp is the number of positive cases incorrectly predicted, and the fn is the number of negative cases incorrectly predicted.
Abbreviations
C: cytoplasmic; CK: cytoskeleton; ER: ER lumen; fn: false negative. false positive; G: Golgi apparatus; gPM: plasma membrane, GPI anchor; Gtm: Golgi, transmembrane; HGMD: Human Gene Mutation Database; HPRD: Human Protein Reference Database; L: lysosome; M: mitochondria; Mma: mitochondrial matrix; mPM: plasma membrane, myristoylated; Mps: mitochondrial periplasmic space; Mtm: mitochondrial membrane, transmembrane; N: nucleus; OMIM: Online Mendelian Inheritance in Man; P: peroxisome; PM: plasma membrane; RC: reliability coefficient; S: secreted; SP: Scandinavian Protocol; tn: true negative; tp: true positive.
Authors' contributions
KL collected data, performed the statistical analysis and drafted the manuscript. MV conceived of the study, designed the study, analyzed the data and drafted the manuscript. All authors read and approved the final manuscript.
Supplementary Material
The primary localization of the proteins according to HPRD. Number of proteins localizing according to HPRD (first localization only).
All localizations of the proteins according to HPRD. Information about all the localizations for the studied proteins.
Changes in SP localization prediction due to mutations (data for affected proteins). Information for mutations related to diseases according to Scandinavian protocol.
Changes in WoLF PSORT localization prediction due to mutations (data for affected proteins). Information for mutations related to diseases according to WoLF PSORT.
Mutations predicted by SP to alter protein localization. List of disease-causing mutations predicted to be related to protein localization by SP.
Mutations predicted by WolF PSORT to alter protein localization. List of disease-causing mutations predicted to be related to protein localization by WoLF PSORT.
Comparison of observed and expected numbers of mutations in the dataset. Statistical analysis of numbers of mutations.
Amino acid distribution of localization mutations predicted with SP. Statistics of amino acid changes in the disease-causing mutations predicted to change localization by SP.
Amino acid distribution of localization mutations predicted with WolF PSORT. Statistics of amino acid changes in the disease-causing mutations predicted to change localization by WoLF PSORT.
Acknowledgments
Acknowledgements
Financial support from the Medical Research Fund of Tampere University Hospital, Academy of Finland, Tampere Graduate School in Information Science and Engineering (TISE), Sigrid Juselius Foundation, and the Finnish Foundation for Technology Promotion is gratefully acknowledged.
Contributor Information
Kirsti Laurila, Email: kirsti.laurila@uta.fi.
Mauno Vihinen, Email: mauno.vihinen@uta.fi.
References
- Davis TN. Protein localization in proteomics. Curr Opin Chem Biol. 2004;8:49–53. doi: 10.1016/j.cbpa.2003.11.003. [DOI] [PubMed] [Google Scholar]
- Falk R, Ramström M, Ståhl S, Hober S. Approaches for systematic proteome exploration. Biomol Eng. 2007;24:155–168. doi: 10.1016/j.bioeng.2007.01.001. [DOI] [PubMed] [Google Scholar]
- Mishra GR, Suresh M, Kumaran K, Kannabiran N, Suresh S, Bala P, Shivakumar K, Anuradha N, Reddy R, Raghavan TM, et al. Human protein reference database–2006 update. Nucleic Acids Res. 2006:D411–414. doi: 10.1093/nar/gkj141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The UniProt Consortium The Universal Protein Resource (UniProt) Nucleic Acids Res. 2007:D193–197. doi: 10.1093/nar/gkl929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider G, Fechner U. Advances in the prediction of protein targeting signals. Proteomics. 2004;4:1571–1580. doi: 10.1002/pmic.200300786. [DOI] [PubMed] [Google Scholar]
- Austen BM. Predicted secondary structures of amino-terminal extension sequences of secreted proteins. FEBS Lett. 1979;103:308–313. doi: 10.1016/0014-5793(79)81351-4. [DOI] [PubMed] [Google Scholar]
- von Heijne G. Patterns of amino acids near signal-sequence cleavage sites. Eur J Biochem. 1983;133:17–21. doi: 10.1111/j.1432-1033.1983.tb07424.x. [DOI] [PubMed] [Google Scholar]
- Klee EW, Ellis LB. Evaluating eukaryotic secreted protein prediction. BMC Bioinformatics. 2005;6:256. doi: 10.1186/1471-2105-6-256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprenger J, Fink JL, Teasdale RD. Evaluation and comparison of mammalian subcellular localization prediction methods. BMC Bioinformatics. 2006;7:S3. doi: 10.1186/1471-2105-7-S5-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cokol M, Nair R, Rost B. Finding nuclear localization signals. EMBO Rep. 2000;1:411–415. doi: 10.1093/embo-reports/kvd092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piirilä H, Väliaho J, Vihinen M. Immunodeficiency mutation databases (IDbases) Hum Mutat. 2006;27:1200–1208. doi: 10.1002/humu.20405. [DOI] [PubMed] [Google Scholar]
- Cotton RG, Phillips K, Horaitis O. A survey of locus-specific database curation. Human Genome Variation Society. J Med Genet. 2007;44:e72. doi: 10.1136/jmg.2006.044081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thusberg J, Vihinen M. Bioinformatic analysis of protein structure-function relationships: case study of leukocyte elastase (ELA2) missense mutations. Hum Mutat. 2006;27:1230–1243. doi: 10.1002/humu.20407. [DOI] [PubMed] [Google Scholar]
- Thusberg J, Vihinen M. Pathogenic or not? And if so, then how? Studying the effects of missense mutations using bioinformatics methods. Hum Mutat. 2009 doi: 10.1002/humu.20938. [DOI] [PubMed] [Google Scholar]
- Zhang SJ, Ma LY, Huang QH, Li G, Gu BW, Gao XD, Shi JY, Wang YY, Gao L, Cai X, et al. Gain-of-function mutation of GATA-2 in acute myeloid transformation of chronic myeloid leukemia. Proc Natl Acad Sci USA. 2008;105:2076–2081. doi: 10.1073/pnas.0711824105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percy MJ, Furlow PW, Lucas GS, Li X, Lappin TR, McMullin MF, Lee FS. A gain-of-function mutation in the HIF2A gene in familial erythrocytosis. N Engl J Med. 2008;358:162–168. doi: 10.1056/NEJMoa073123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houdayer C, Dehainault C, Mattler C, Michaux D, Caux-Moncoutier V, Pagès-Berhouet S, d'Enghien CD, Laugé A, Castera L, Gauthier-Villars M, et al. Evaluation of in silico splice tools for decision-making in molecular diagnosis. Hum Mutat. 2008;29:975–982. doi: 10.1002/humu.20765. [DOI] [PubMed] [Google Scholar]
- Nakamura Y, Gojobori T, Ikemura T. Codon usage tabulated from international DNA sequence databases: status for the year 2000. Nucleic Acids Res. 2000;28:292. doi: 10.1093/nar/28.1.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer-Costa C, Orozco M, de la Cruz X. Characterization of disease-associated single amino acid polymorphisms in terms of sequence and structure properties. J Mol Biol. 2002;315:771–786. doi: 10.1006/jmbi.2001.5255. [DOI] [PubMed] [Google Scholar]
- Khan S, Vihinen M. Spectrum of disease-causing mutations in protein secondary structures. BMC Struct Biol. 2007;7:56. doi: 10.1186/1472-6807-7-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitkup D, Sander C, Church GM. The amino-acid mutational spectrum of human genetic disease. Genome Biol. 2003;4:R72. doi: 10.1186/gb-2003-4-11-r72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortutay C, Väliaho J, Stenberg K, Vihinen M. KinMutBase: a registry of disease-causing mutations in protein kinase domains. Hum Mutat. 2005;25:435–442. doi: 10.1002/humu.20166. [DOI] [PubMed] [Google Scholar]
- Ollila J, Lappalainen I, Vihinen M. Sequence specificity in CpG mutation hotspots. FEBS Lett. 1996;396:119–122. doi: 10.1016/0014-5793(96)01075-7. [DOI] [PubMed] [Google Scholar]
- Sabherwal N, Schneider KU, Blaschke RJ, Marchini A, Rappold G. Impairment of SHOX nuclear localization as a cause for Leri-Weill syndrome. J Cell Sci. 2004;117:3041–3048. doi: 10.1242/jcs.01152. [DOI] [PubMed] [Google Scholar]
- Björses P, Halonen M, Palvimo JJ, Kolmer M, Aaltonen J, Ellonen P, Perheentupa J, Ulmanen I, Peltonen L. Mutations in the AIRE gene: effects on subcellular location and transactivation function of the autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy protein. Am J Hum Genet. 2000;66:378–392. doi: 10.1086/302765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halonen M, Kangas H, Rüppell T, Ilmarinen T, Ollila J, Kolmer M, Vihinen M, Palvimo J, Saarela J, Ulmanen I, et al. APECED-causing mutations in AIRE reveal the functional domains of the protein. Hum Mutat. 2004;23:245–257. doi: 10.1002/humu.20003. [DOI] [PubMed] [Google Scholar]
- Ilmarinen T, Eskelin P, Halonen M, Rüppell T, Kilpikari R, Torres GD, Kangas H, Ulmanen I. Functional analysis of SAND mutations in AIRE supports dominant inheritance of the G228W mutation. Hum Mutat. 2005;26:322–331. doi: 10.1002/humu.20224. [DOI] [PubMed] [Google Scholar]
- Hayama A, Rai T, Sasaki S, Uchida S. Molecular mechanisms of Bartter syndrome caused by mutations in the BSND gene. Histochem Cell Biol. 2003;119:485–493. doi: 10.1007/s00418-003-0535-2. [DOI] [PubMed] [Google Scholar]
- Huster D, Hoppert M, Lutsenko S, Zinke J, Lehmann C, Mössner J, Berr F, Caca K. Defective cellular localization of mutant ATP7B in Wilson's disease patients and hepatoma cell lines. Gastroenterology. 2003;124:335–345. doi: 10.1053/gast.2003.50066. [DOI] [PubMed] [Google Scholar]
- Albrecht C, McVey JH, Elliott JI, Sardini A, Kasza I, Mumford AD, Naoumova RP, Tuddenham EG, Szabo K, Higgins CF. A novel missense mutation in ABCA1 results in altered protein trafficking and reduced phosphatidylserine translocation in a patient with Scott syndrome. Blood. 2005;106:542–549. doi: 10.1182/blood-2004-05-2056. [DOI] [PubMed] [Google Scholar]
- Da Costa L, Tchernia G, Gascard P, Lo A, Meerpohl J, Niemeyer C, Chasis JA, Fixler J, Mohandas N. Nucleolar localization of RPS19 protein in normal cells and mislocalization due to mutations in the nucleolar localization signals in 2 Diamond-Blackfan anemia patients: potential insights into pathophysiology. Blood. 2003;101:5039–5045. doi: 10.1182/blood-2002-12-3878. [DOI] [PubMed] [Google Scholar]
- Tanaka AR, Abe-Dohmae S, Ohnishi T, Aoki R, Morinaga G, Okuhira K, Ikeda Y, Kano F, Matsuo M, Kioka N, et al. Effects of mutations of ABCA1 in the first extracellular domain on subcellular trafficking and ATP binding/hydrolysis. J Biol Chem. 2003;278:8815–8819. doi: 10.1074/jbc.M206885200. [DOI] [PubMed] [Google Scholar]
- Charniot JC, Pascal C, Bouchier C, Sebillon P, Salama J, Duboscq-Bidot L, Peuchmaurd M, Desnos M, Artigou JY, Komajda M. Functional consequences of an LMNA mutation associated with a new cardiac and non-cardiac phenotype. Hum Mutat. 2003;21:473–481. doi: 10.1002/humu.10170. [DOI] [PubMed] [Google Scholar]
- Meij IC, Koenderink JB, De Jong JC, De Pont JJ, Monnens LA, Heuvel LP Van Den, Knoers NV. Dominant isolated renal magnesium loss is caused by misrouting of the Na+, K+-ATPase gamma-subunit. Ann N Y Acad Sci. 2003;986:437–443. doi: 10.1111/j.1749-6632.2003.tb07226.x. [DOI] [PubMed] [Google Scholar]
- Nazareth LV, Stenoien DL, Bingman WE, 3rd, James AJ, Wu C, Zhang Y, Edwards DP, Mancini M, Marcelli M, Lamb DJ, et al. A C619Y mutation in the human androgen receptor causes inactivation and mislocalization of the receptor with concomitant sequestration of SRC-1 (steroid receptor coactivator 1) Mol Endocrinol. 1999;13:2065–2075. doi: 10.1210/me.13.12.2065. [DOI] [PubMed] [Google Scholar]
- Kunishima S, Matsushita T, Kojima T, Sako M, Kimura F, Jo EK, Inoue C, Kamiya T, Saito H. Immunofluorescence analysis of neutrophil nonmuscle myosin heavy chain-A in MYH9 disorders: association of subcellular localization with MYH9 mutations. Lab Invest. 2003;83:115–122. doi: 10.1097/01.lab.0000050960.48774.17. [DOI] [PubMed] [Google Scholar]
- Mizutani A, Matsuzaki A, Momoi MY, Fujita E, Tanabe Y, Momoi T. Intracellular distribution of a speech/language disorder associated FOXP2 mutant. Biochem Biophys Res Commun. 2007;353:869–874. doi: 10.1016/j.bbrc.2006.12.130. [DOI] [PubMed] [Google Scholar]
- Neuberger G, Kunze M, Eisenhaber F, Berger J, Hartig A, Brocard C. Hidden localization motifs: naturally occurring peroxisomal targeting signals in non-peroxisomal proteins. Genome Biol. 2004;5:R97. doi: 10.1186/gb-2004-5-12-r97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpten JD, Faber AL, Horn C, Donoho GP, Briggs SL, Robbins CM, Hostetter G, Boguslawski S, Moses TY, Savage S, et al. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature. 2007;448:439–444. doi: 10.1038/nature05933. [DOI] [PubMed] [Google Scholar]
- Wansink DG, van Herpen RE, Coerwinkel-Driessen MM, Groenen PJ, Hemmings BA, Wieringa B. Alternative splicing controls myotonic dystrophy protein kinase structure, enzymatic activity, and subcellular localization. Mol Cell Biol. 2003;23:5489–5501. doi: 10.1128/MCB.23.16.5489-5501.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim BE, Smith K, Meagher CK, Petris MJ. A conditional mutation affecting localization of the Menkes disease copper ATPase. Suppression by copper supplementation. J Biol Chem. 2002;277:44079–44084. doi: 10.1074/jbc.M208737200. [DOI] [PubMed] [Google Scholar]
- Rodriguez JA, Au WW, Henderson BR. Cytoplasmic mislocalization of BRCA1 caused by cancer-associated mutations in the BRCT domain. Exp Cell Res. 2004;293:14–21. doi: 10.1016/j.yexcr.2003.09.027. [DOI] [PubMed] [Google Scholar]
- Claypool SM, McCaffery JM, Koehler CM. Mitochondrial mislocalization and altered assembly of a cluster of Barth syndrome mutant tafazzins. J Cell Biol. 2006;174:379–390. doi: 10.1083/jcb.200605043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Kruhlak MJ, Hao JJ, Shaw S. Rapid T cell receptor-mediated SHP-1 S591 phosphorylation regulates SHP-1 cellular localization and phosphatase activity. J Leukoc Biol. 2007;82:742–751. doi: 10.1189/jlb.1206736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozisik G, Mantovani G, Achermann JC, Persani L, Spada A, Weiss J, Beck-Peccoz P, Jameson JL. An alternate translation initiation site circumvents an amino-terminal DAX1 nonsense mutation leading to a mild form of X-linked adrenal hypoplasia congenita. J Clin Endocrinol Metab. 2003;88:417–423. doi: 10.1210/jc.2002-021034. [DOI] [PubMed] [Google Scholar]
- Anney RJ, Rees MI, Bryan E, Spurlock G, Williams N, Norton N, Williams H, Cardno A, Zammit S, Jones S, et al. Characterisation, mutation detection, and association analysis of alternative promoters and 5' UTRs of the human dopamine D3 receptor gene in schizophrenia. Mol Psychiatry. 2002;7:493–502. doi: 10.1038/sj.mp.4001003. [DOI] [PubMed] [Google Scholar]
- Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, Thomas NS, Abeysinghe S, Krawczak M, Cooper DN. Human Gene Mutation Database (HGMD): 2003 update. Hum Mutat. 2003;21:577–581. doi: 10.1002/humu.10212. [DOI] [PubMed] [Google Scholar]
- Emanuelsson O, Brunak S, von Heijne G, Nielsen H. Locating proteins in the cell using TargetP, SignalP and related tools. Nat Protoc. 2007;2:953–971. doi: 10.1038/nprot.2007.131. [DOI] [PubMed] [Google Scholar]
- Yu CS, Chen YC, Lu CH, Hwang JK. Prediction of protein subcellular localization. Proteins. 2006;64:643–651. doi: 10.1002/prot.21018. [DOI] [PubMed] [Google Scholar]
- Nair R, Rost B. Sequence conserved for subcellular localization. Protein Sci. 2002;11:2836–2847. doi: 10.1110/ps.0207402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emanuelsson O, Nielsen H, Brunak S, von Heijne G. Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J Mol Biol. 2000;300:1005–1016. doi: 10.1006/jmbi.2000.3903. [DOI] [PubMed] [Google Scholar]
- Bendtsen JD, Nielsen H, von Heijne G, Brunak S. Improved prediction of signal peptides: SignalP 3.0. J Mol Biol. 2004;340:783–795. doi: 10.1016/j.jmb.2004.05.028. [DOI] [PubMed] [Google Scholar]
- Sonnhammer EL, von Heijne G, Krogh A. A hidden Markov model for predicting transmembrane helices in protein sequences. In: Glasgow J, Lathrop R, Littlejohn T, Major F, Peitsch M, Sankoff D, Sensen C, editor. Proc Sixth Int Conf on Intelligent Systems for Molecular Biology. Vol. 6. AAAI Press; 1998. pp. 175–82. [PubMed] [Google Scholar]
- Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- Eisenhaber B, Bork P, Eisenhaber F. Sequence properties of GPI-anchored proteins near the omega-site: constraints for the polypeptide binding site of the putative transamidase. Protein Eng. 1998;11:1155–1161. doi: 10.1093/protein/11.12.1155. [DOI] [PubMed] [Google Scholar]
- Sunyaev SR, Eisenhaber F, Rodchenkov IV, Eisenhaber B, Tumanyan VG, Kuznetsov EN. PSIC: profile extraction from sequence alignments with position-specific counts of independent observations. Protein Eng. 1999;12:387–394. doi: 10.1093/protein/12.5.387. [DOI] [PubMed] [Google Scholar]
- Eisenhaber B, Bork P, Eisenhaber F. Prediction of potential GPI-modification sites in proprotein sequences. J Mol Biol. 1999;292:741–758. doi: 10.1006/jmbi.1999.3069. [DOI] [PubMed] [Google Scholar]
- Eisenhaber B, Bork P, Yuan Y, Löffler G, Eisenhaber F. Automated annotation of GPI anchor sites: case study C. elegans. Trends Biochem Sci. 2000;25:340–341. doi: 10.1016/S0968-0004(00)01601-7. [DOI] [PubMed] [Google Scholar]
- Emanuelsson O, Elofsson A, von Heijne G, Cristobal S. In silico prediction of the peroxisomal proteome in fungi, plants and animals. J Mol Biol. 2003;330:443–456. doi: 10.1016/S0022-2836(03)00553-9. [DOI] [PubMed] [Google Scholar]
- Neuberger G, Maurer-Stroh S, Eisenhaber B, Hartig A, Eisenhaber F. Motif refinement of the peroxisomal targeting signal 1 and evaluation of taxon-specific differences. J Mol Biol. 2003;328:567–579. doi: 10.1016/S0022-2836(03)00318-8. [DOI] [PubMed] [Google Scholar]
- Neuberger G, Maurer-Stroh S, Eisenhaber B, Hartig A, Eisenhaber F. Prediction of peroxisomal targeting signal 1 containing proteins from amino acid sequence. J Mol Biol. 2003;328:581–592. doi: 10.1016/S0022-2836(03)00319-X. [DOI] [PubMed] [Google Scholar]
- Yuan Z, Teasdale RD. Prediction of Golgi Type II membrane proteins based on their transmembrane domains. Bioinformatics. 2002;18:1109–1115. doi: 10.1093/bioinformatics/18.8.1109. [DOI] [PubMed] [Google Scholar]
- Käll L, Krogh A, Sonnhammer EL. A combined transmembrane topology and signal peptide prediction method. J Mol Biol. 2004;338:1027–1036. doi: 10.1016/j.jmb.2004.03.016. [DOI] [PubMed] [Google Scholar]
- de Castro E, Sigrist CJ, Gattiker A, Bulliard V, Langendijk-Genevaux PS, Gasteiger E, Bairoch A, Hulo N. ScanProsite: detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res. 2006:W362–365. doi: 10.1093/nar/gkl124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton P, Park KJ, Obayashi T, Nakai K. Protein Subcellular Localization Prediction with WoLF PSORT. Proceedings of the 4th Annual Asia Pacific Bioinformatics Conference APBC06. Taipei, Taiwan. 2006.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The primary localization of the proteins according to HPRD. Number of proteins localizing according to HPRD (first localization only).
All localizations of the proteins according to HPRD. Information about all the localizations for the studied proteins.
Changes in SP localization prediction due to mutations (data for affected proteins). Information for mutations related to diseases according to Scandinavian protocol.
Changes in WoLF PSORT localization prediction due to mutations (data for affected proteins). Information for mutations related to diseases according to WoLF PSORT.
Mutations predicted by SP to alter protein localization. List of disease-causing mutations predicted to be related to protein localization by SP.
Mutations predicted by WolF PSORT to alter protein localization. List of disease-causing mutations predicted to be related to protein localization by WoLF PSORT.
Comparison of observed and expected numbers of mutations in the dataset. Statistical analysis of numbers of mutations.
Amino acid distribution of localization mutations predicted with SP. Statistics of amino acid changes in the disease-causing mutations predicted to change localization by SP.
Amino acid distribution of localization mutations predicted with WolF PSORT. Statistics of amino acid changes in the disease-causing mutations predicted to change localization by WoLF PSORT.