

Abstract
Drugs of abuse usurp the mechanisms underlying synaptic plasticity in areas of the brain, a process that may contribute to the development of addiction (Kauer & Malenka, 2007). We previously reported that GABAergic synapses onto dopaminergic neurons in the ventral tegmental area (VTA) exhibit long term potentiation (LTPGABA) blocked by in vivo exposure to morphine. The presynaptically-maintained LTP requires that retrogradely released nitric oxide (NO) activates a presynaptic cGMP signaling cascade. Previous work reported that inhibitory GABAA receptor synapses in the VTA are also potentiated by cAMP (Bonci & Williams, 1997; Melis et al., 2002). Here we explored the interactions between cGMP-dependent (PKG) and cAMP- dependent (PKA) protein kinases in the regulation of these GABAergic synapses and LTPGABA. Activation of PKG was required for NO-cGMP signaling and was also essential for the induction of synaptically-elicited LTPGABA, but not for its maintenance. Whereas synapses containing GABAA receptors were potentiated by NO-cGMP signaling, synapses containing GABAB receptors on the same cells were not potentiated. Moreover, although the cAMP-PKA system potentiated GABAA synapses, synaptically-induced LTPGABA was independent of PKA activation. Surprisingly, however, raising cGMP levels saturated potentiation of these synapses, precluding further potentiation by cAMP and suggesting a convergent endpoint for both signaling pathways in the regulation of GABAergic release. We further found that persistent GABAergic synaptic modifications observed with in vivo morphine did not involve the presynaptic cAMP-PKA cascade. Taken together, our data suggest a synapse-specific role for NO-cGMP-PKG signaling pathway in opioid-induced plasticity of VTA GABAA synapses.
Keywords: VTA, LTP, PKA, PKG, GABA, Nitric oxide
Introduction
LTP and LTD are long-lasting synaptic modifications proposed to underlie many examples of experience-dependent plasticity (Malenka & Bear, 2004). Over the last decade, rapid, drug-induced synaptic plasticity has been reported at excitatory glutamatergic synapses in addiction-related brain circuits, suggesting that LTP- and LTD-like changes may also contribute to the development of addiction (Ungless et al., 2001; Hyman et al., 2006; Kauer & Malenka, 2007). Recent evidence suggests that drug-induced plasticity of VTA GABAergic synapses may also contribute to the development of addictive behaviors (Mansvelder et al., 2002; Melis et al., 2002; Liu et al., 2005; Nugent et al., 2007; Nugent & Kauer, 2008; Pan et al., 2008).
Opioids rapidly increase VTA dopamine (DA) cell firing and output through disinhibition, i.e. by reducing the tonic inhibition provided by local interneurons (Johnson & North, 1992). Recently we reported that 24 hours after in vivo morphine exposure NO-dependent LTPGABA is blocked, providing a long-lasting mechanism by which opioids can enhance the excitability of DA neurons and may contribute to the reinforcing effects of opioids. LTPGABA is heterosynaptic, initiated by glutamate release onto NMDA receptors on the postsynaptic DA neuron. Activation of nitric oxide synthase by intracellular Ca2+ generates NO, which then travels retrogradely to activate soluble guanylate cyclase (sGC) in neighboring presynaptic GABAergic nerve terminals. Increased levels of cGMP, presumably acting in presynaptic terminals, promote long-lasting potentiation of GABA release at these synapses (Nugent et al., 2007). In the context of opiate addiction, it is important to further investigate the precise cellular mechanisms underlying LTPGABA.
Cyclic GMP-dependent protein kinase (PKG) is present in neurons throughout the brain, and is a major target of NO-cGMP signaling (el-Husseini et al., 1995; Wang & Robinson, 1997). PKG has previously been implicated in induction and maintenance of synaptic plasticity (Zhuo et al., 1994; Wu et al., 1998; Lu et al., 1999; Santschi et al., 1999; Lu & Hawkins, 2002; Monfort et al., 2002; Chien et al., 2003; Liu et al., 2003; Monfort et al., 2004). The cAMP-PKA signaling pathway also regulates synaptic plasticity in many brain regions (Huang & Kandel, 1994; Weisskopf et al., 1994; Salin et al., 1996a; Salin et al., 1996b; Huang & Kandel, 1998; Castro-Alamancos & Calcagnotto, 1999; Linden & Ahn, 1999; Mellor et al., 2002). Several studies have implicated cAMP-PKA signaling in responses to drugs of abuse. Following acute withdrawal from chronic morphine, cyclic AMP-dependent increases in GABA release in different regions including the VTA have been reported (Bonci & Williams, 1997; Chieng & Williams, 1998; Ingram et al., 1998). Furthermore, Melis et al. (2002) reported that cAMP-PKA signaling is required for induction of a long-lasting potentiation of VTA GABAergic synapses after a single exposure to ethanol. Given that opioids can modulate the release of GABA through an interaction with the presynaptic cAMP cascade (Williams et al., 2001), here we have investigated the roles of PKG and PKA as likely downstream targets for cGMP and cAMP in LTPGABA.
Materials and Methods
Preparation of brain slices
Preparation of slices was as described previously (Jones et al., 2000; Nugent et al., 2007). Sprague-Dawley rats (15-21 days old) were deeply anesthetized using isoflurane and quickly decapitated in accordance with the Brown University Institutional Animal Care and Use Committee guidelines. The brain was rapidly removed into ice-cold artificial cerebrospinal fluid (ACSF) containing (in mM): 126 NaCl, 21.4 NaHCO3, 2.5 KCl, 1.2 NaH2PO4, 2.4 CaCl2, 1.2 MgSO4, 11.1 glucose, 0.4 ascorbic acid, saturated with 95% O2/5% CO2 (pH 7.4). Horizontal midbrain slices containing the VTA (250 μm thick) were cut using a vibratome, stored for at least one hour at 35°C, and transferred to a recording chamber where the slice was submerged in warmed ACSF.
Electrophysiology
Midbrain slices were continuously perfused with ACSF (no ascorbic acid) at 28-32°C at 2-4 ml/min. To study GABAA receptor-mediated synaptic transmission, 6,7-dinitroquinoxaline-2,3-dione (DNQX;10μM), strychnine (1μM), and 1, 3-dipropyl-8-cyclopentylxanthine (DPCPX; 1μM) were added to block AMPA-, glycine-, and A1 adenosine receptor-mediated synaptic currents, respectively. To isolate GABAB receptor-mediated IPSCs, the superfusion medium contained 2-amino-5-phosphonopentanoic acid (AP-5; 100 μM), DNQX (10 μM), picrotoxin (100 μM), strychnine (1 μM), eticlopride (1 μM) and 7-hydroxyiminocyclopropan [b] chromen-1acarboxylic acid ethyl ester (CPCCOEt; 50μM) to block NMDA, AMPA, GABAA, glycine, dopamine D2- and mGluR1-mediated synaptic currents, respectively. The GABABR IPSCs were entirely blocked by the GABAB receptor antagonist CGP55845 (10 μM). Patch pipettes were filled with (in mM): 125 KCl, 2.8 NaCl, 2 MgCl2, 2 ATP-Na+, 0.3 GTP-Li+, 0.6 EGTA, and 10 HEPES. To record GABAAR-mediated IPSCs, cells were voltage-clamped at -70mV except during HFS, and the cell input resistance and series resistance were monitored throughout the experiment; experiments were discarded if these values changed by more than 10% during the experiment. GABABR IPSCs were recorded from cells voltage-clamped at –50mV (see below).
If the steady-state h-current was greater than 60 pA during a step from -50 mV to -100 mV, the neuron was considered a DA neuron. A recent study showed that expression of Ih alone is not sufficient to identify DA cells unequivocally (Margolis et al., 2006), but see (Chen et al., 2008). Therefore in each set of our experiments, a subset of the neurons recorded from and reported here are possibly non-dopaminergic neurons.
GABAAR-mediated IPSCs were stimulated at 0.1 Hz (100 μsec) using a bipolar stainless steel stimulating electrode placed 200-500 μm rostral to the recording site in the VTA. GABABR-mediated IPSCs were stimulated using a train of stimuli; 10 pulses of 250μsec at 66 Hz, repeated once every 60 sec (Bonci & Williams, 1996; Fiorillo & Williams, 2000). LTPGABA was induced by stimulating afferents at 100 Hz for 1 second, the train repeated twice 20 seconds apart (high-frequency stimulation; HFS). Just before HFS, the recorded neuron was taken from voltage-clamp and into bridge mode, so that the HFS trains were delivered with the membrane potential free to vary.
Statistics
Data are presented as means ± s.e.m. Significance was determined using a Student's unpaired t-test with significance level of p<0.05. Levels of LTP are reported as averaged IPSC amplitudes for 5 min just before LTP induction compared with averaged IPSC amplitudes during the 5 min period from 15–20 min after HFS using a Student's paired t-test. Paired-pulse ratios (50 msec interstimulus interval) were measured over five minute epochs of 30 IPSCs each as previously described (Nugent et al., 2007) .
Drug application
Drugs were bath-applied at known concentrations for at least 15 minutes before HFS. Control experiments were interleaved with experiments in which drugs were bath-applied. To assess drug effects, IPSC amplitudes were averaged for 5 minutes during the peak response and were compared with 5 minutes of averaged data prior to drug application. Salts and all other drugs were obtained from Sigma-Research Biochemicals International or Tocris Bioscience, except for KT5823, obtained from Calbiochem.
Treatment with morphine
Rats (15-21 days old) were maintained on a 12-hour light/dark cycle and provided food and water ad libitum. They were injected intraperitoneally with either 10 mg/kg morphine or a comparable volume of saline, placed in a new cage for 2 hours, and then returned to the home cage. They were sacrificed for brain slice preparation 24 hours after injection.
Results
As we previously reported (Nugent et al., 2007; Nugent et al., 2008), GABAergic synapses on VTA DA neurons undergo LTP in response to patterned local electrical stimulation (LTPGABA, Figure 1a). LTPGABA appears to be expressed by an increase in presynaptically released GABA, as the paired pulse ratio and coefficient of variation change after induction (Nugent et al., 2007).
Figure 1. NO is not necessary for the maintenance of LTPGABA.
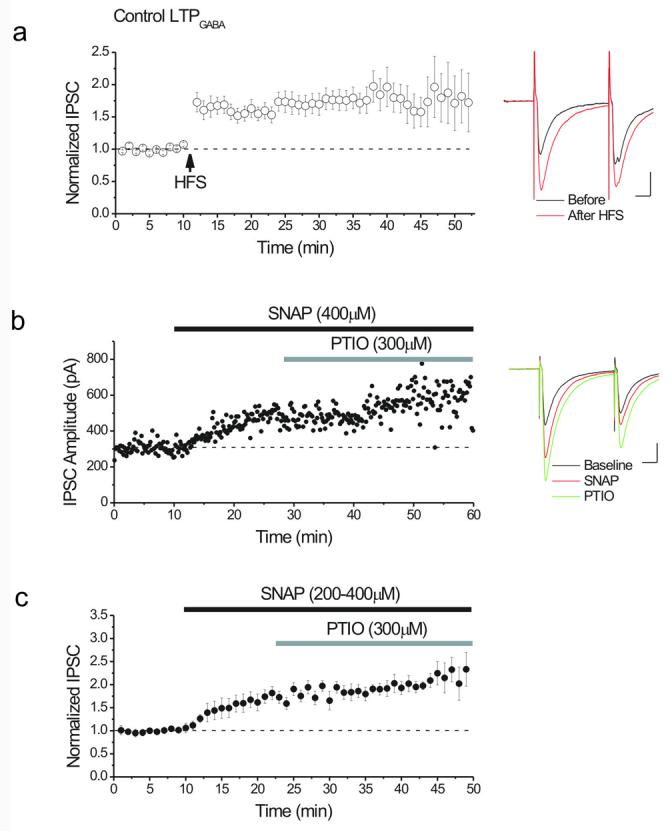
(a) Average of 26 experiments showing LTPGABA recorded from dopamine cells. HFS was delivered at the arrow. LTPGABA: 181 ± 1 % of pre-HFS values, n=26. Inset: averaged IPSCs before (black) and 25 minutes after HFS (red) from single experiment. In this and all figures, ten consecutive IPSCs from each condition were averaged for illustration. Calibration for insets: 10 ms, 100 pA.
(b) Single experiment illustrating the lack of effect of the NO scavenger, PTIO, on SNAP-induced potentiation of GABAergic IPSCs. SNAP (400 μM), an NO donor, potentiated IPSCs. After a new stable level was reached, PTIO (300μM) was bath-applied, but did not reverse the potentiation induced by SNAP. Inset: averaged IPSCs recorded before (black), after 10 minutes in SNAP (red) and after 10 minutes in PTIO (green).
(c) Average of experiments using the protocol outlined in (b) showing that after SNAP potentiated the IPSCs (150 ± 4% of pre-drug values, n=11), PTIO had no effect on SNAP-induced potentiation (121 ± 8% of pre-PTIO values, n=11).
NO is not needed to sustain LTPGABA
Sustained activity of protein kinases such as protein kinase C (PKC) and calcium calmodulin kinase type II (CaMKII) have been proposed to be involved in maintenance and expression of LTP (Lisman, 1985; Lisman & Goldring, 1988; Malinow et al., 1988; Chen et al., 2001; Yang et al., 2004). In VTA dopamine cells, the production of presynaptic cGMP in response to NO release triggers LTPGABA. We first asked whether constitutive release of NO is necessary to sustain LTPGABA, or whether instead, a brief exposure is sufficient to persistently enhance GABA release. Consistent with our previous results the NO donor, SNAP (S-nitroso-N-acetylpenicillamine; 200-400μM), potentiated GABAAR IPSCs, resembling LTPGABA (Figure 1b, c). Yet we observed that when the NO scavenger, PTIO (2-phenyl-4,4,5,5-tetramethylimidazoline-1-oxyl 3-oxide, 300μM) was added after the NO donor elicited synaptic potentiation, the SNAP-induced potentiation did not decay back to control values (Figure 1b,c). The inability of PTIO to reverse the potentiation strongly suggests that the maintenance of LTPGABA does not require the persistent presence of NO.
The NO-cGMP signaling cascade activates PKG to potentiate GABAergic synapses
Consistent with our previous findings, the cGMP analogue, pCPT-cGMP (8-(pchlorophenylthio)-cGMP; 100 μM), potentiated GABAergic IPSCs (Figure 2a) suggesting that NO-mediated activation of guanylate cyclase is required for NO to enhance GABA release. Furthermore, pCPT-cGMP is a selective activator of PKG, with little effect on cyclic nucleotide-gated ion channels or phosphodiesterases (Wang & Robinson, 1997). If NO-cGMP signaling must activate PKG to potentiate GABAergic synapses, then a PKG inhibitor should prevent the potentiation induced either by SNAP or pCPT-cGMP. As predicted, KT5823, a selective PKG inhibitor which interferes at the level of the ATP binding site of the PKG catalytic domain (Hidaka & Kobayashi, 1992), prevented the potentiation induced by either the NO donor (Figure 2b) or the cGMP analogue (Figure 2c). These data support the idea that the sequential activation of the presynaptic sGC and PKG downstream from NO promotes GABA release in these synapses. In addition, KT5823 did not reduce basal synaptic transmission (Figure 2d), implying that PKG activity is not required to maintain basal GABA release from these terminals.
Figure 2. NO-cGMP signaling requires PKG to enhance GABAergic IPSCs.
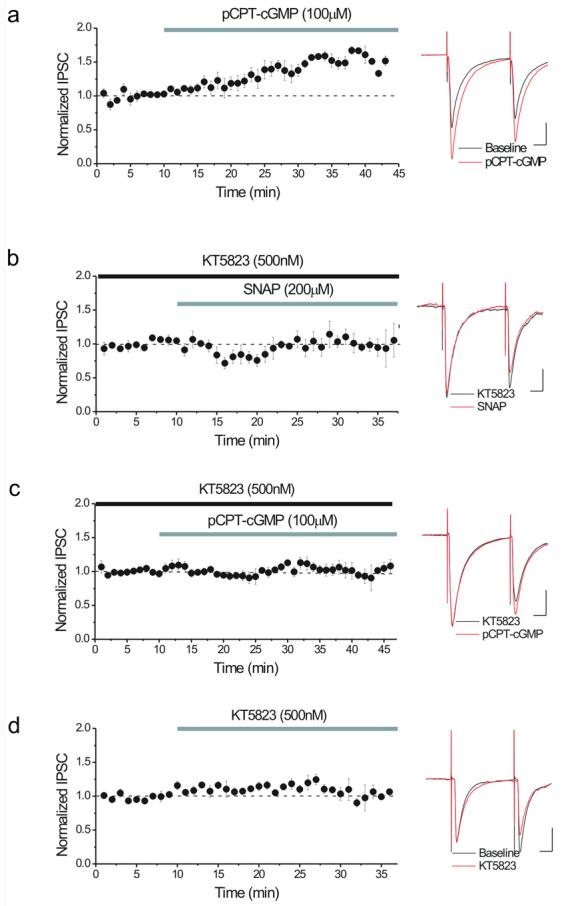
(a) A cGMP analogue, pCPT-cGMP (100 μM), potentiates IPSCs (146 ± 9% of pre-drug values, n=4). Inset: averaged IPSCs recorded during a single such experiment before (black) and after 15 minutes in pCPT-cGMP (red). Calibration for all insets: 10ms, 100 pA.
(b) The PKG inhibitor, KT5823 (500 nM), blocks the enhancement of IPSCs by 200 μM SNAP (101 ± 6% of pre-SNAP values, n=4). KT5823 was applied at least 15 minutes prior to the addition of SNAP. Inset: averaged IPSCs recorded during single experiment in KT5823 (black) and after 15 minutes in SNAP (red).
(c) KT5823 (500 nM) also prevents the potentiation of IPSCs by 100 μM pCPT-cGMP (104 ± 6% of pre-SNAP values, n=6). KT5823 was applied at least 15 minutes prior to the addition of pCPT-cGMP. Inset: averaged IPSCs recorded during single experiment in KT5823 (black) and after 15 minutes in pCPT-cGMP (red).
(d) KT5823 (500 nM) has no effect on basal GABAergic transmission (113 ± 6% of pre-drug values, n=5). Inset: averaged IPSCs recorded during single experiment before (black) and after 10 minutes in KT5823 (red).
Sequential activation of GC, and then PKG is necessary for the induction of LTPGABA but not for its maintenance
We further explored the role of PKG in the induction and maintenance of LTPGABA in response to HFS. Application of KT5823 entirely blocked the induction of LTPGABA (Figure 3a). If persistent PKG activity is also necessary for the maintenance of LTPGABA, synaptic potentiation should be reversed if PKG activity is inhibited after induction. To test this hypothesis, we bath applied KT5823 10 minutes after induction of LTPGABA using synaptic stimulation (HFS). KT5823 had no significant effect on the maintenance of LTPGABA (Figure 3b). Furthermore, after pCPT-cGMP washout and addition of KT5823, the IPSC amplitude remained potentiated, confirming that the maintenance of the potentiation did not require persistent activity of PKG once the LTP was induced (178 ± 4% of pre-drug values, n=3). Taken together, these findings suggest that the induction of LTPGABA requires transient activation of PKG, but the expression of LTPGABA does not require persistent activity of this kinase.
Figure 3. PKG is required for the induction, but not the maintenance of LTPGABA.

(a) Bath application of KT5823 (gray bar, 500 nM) prevents LTPGABA (control LTP, open circles: 212 ± 10% of pre-HFS values, n=6; cells treated for at least 15 minutes prior to HFS with KT5823, closed circles: 100 ± 4% of pre-HFS values, n=6). Insets: averaged IPSCs recorded during single experiment in KT5823 (black), 20 minutes after HFS in KT5823 (red). Bar chart illustrating the magnitude of LTP 25 minutes after HFS in control vs. cells treated with KT5823 for at least 15 minutes prior to HFS, p<0.05.
(b) Bath application of KT5823 (500 nM) 10 minutes after HFS (gray bar) had no effect on the expression of LTPGABA (control LTP, open circles: 174 ± 11% of pre-HFS values, n=10; KT5823 cells, filled circles: 151 ± 5% of pre-HFS values, n=10). Insets: averaged IPSCs recorded during a single experiment before KT5823 (black), and 25 minutes after HFS in KT5823 (red). Bar chart illustrating the magnitude of LTP 25 minutes after HFS in control vs. cells treated with KT5823 after LTPGABA induction, p>0.05. Calibration for insets: 10ms, 100 pA.
GABAB synapses are not potentiated in response to cGMP
Anatomically and functionally distinct sets of GABAergic afferents innervate VTA DA neurons at inhibitory synapses containing either GABAB or GABAA receptors. For example, GABAergic axons from outside the VTA, such as the nucleus accumbens or ventral pallidum, target GABABR-containing synapses, while GABAAR-containing synapses most likely receive their main input from the axons of local GABAergic interneurons in the VTA (Johnson et al., 1992; Sugita et al., 1992; Cameron & Williams, 1993). Since GABAAR LTP is altered after morphine exposure in vivo, and drugs of abuse can also influence GABAB receptor synapses, we next asked whether the NO-cGMP signaling cascade could modulate GABAB synapses. We evaluated the effects of SNAP and pCPT-cGMP on GABAB IPSCs recorded from VTA DA neurons. Unlike GABAA synapses, after bath application of either the NO donor or the cGMP analogue, GABAB synapses were not potentiated (Figure 4). These findings reveal that the molecular machinery for NO-cGMP signaling does not potentiate all GABA-releasing axons in the VTA, but is selective for GABAAR-associated synapses, most likely arising from local VTA GABAergic neurons. The NO-cGMP potentiating mechanism is either absent or non-functional in the VTA GABAB nerve terminals originating from GABAergic neurons outside the VTA.
Figure 4. Neither nitric oxide nor cGMP potentiate GABABR-mediated IPSCs on dopamine neurons.

(a) Single example experiment illustrating that SNAP (400 μM) did not potentiate GABAB IPSCs. Inset: averaged GABAB IPSCs elicited by a train of synaptic stimuli (see methods) recorded before (black) and after 20 minutes in SNAP (red). Calibration: 500ms, 40 pA.
(b) Average of 6 similar experiments using SNAP (400 μM) (87 ± 3% of pre-drug values, n=6).
(c) Single example experiment illustrating that pCPT-cGMP (100 μM) did not potentiate GABAB IPSCs. Inset: averaged GABAB IPSCs recorded before (black) and after 20 minutes in pCPT-cGMP (red). Calibration: 500ms, 40 pA.
(d) Average of 6 similar experiments using pCPT-cGMP (100 μM) (86 ± 7% of pre-drug values, n=6).
Activation of adenylyl cyclase potentiates GABAA synapses and occludes further potentiation by HFS
A rise in presynaptic cAMP following activation of adenylyl cyclase facilitates neurotransmitter release at many synapses, and is involved in the induction and expression of LTP at many excitatory and inhibitory synapses (Briggs et al., 1988; Greengard et al., 1991; Cameron & Williams, 1993; Chavez-Noriega & Stevens, 1994; Huang & Kandel, 1994; Weisskopf et al., 1994; Bonci & Williams, 1996; Salin et al., 1996a; Bonci & Williams, 1997; Huang & Kandel, 1998; Castro-Alamancos & Calcagnotto, 1999; Linden & Ahn, 1999; Mellor et al., 2002). Furthermore, PKA activation has previously been demonstrated to potentiate GABAAR synapses on VTA dopamine neurons (Melis et al., 2002). Therefore, we explored the interactions of the cAMP cascade with LTPGABA using forskolin (10μM) to activate adenylyl cyclase. We confirmed that forskolin enhanced GABAergic responses (Figure 5a,b) and this enhancement was associated with a decrease in the paired-pulse ratio, suggesting that it is likely due to enhanced GABA release, also seen during LTPGABA (Melis et al., 2002; Nugent et al., 2007). Dideoxyforskolin, a biologically inactive analogue that does not stimulate adenylyl cyclase, had no effect on GABAA–mediated responses (Figure 5b). Furthermore, once the potentiation by forskolin had plateaued, HFS failed to produce further synaptic potentiation (Figure 5a,c). Thus, forskolin mimicked and occluded LTPGABA through the activation of adenylyl cyclase and the subsequent rise in cAMP. PKA is the major downstream target for cAMP, and if the activation of PKA mediates synaptic enhancement, Sp-cAMPS (a cAMP mimic and specific activator of PKA) should also enhance GABA release. Consistent with this hypothesis, we found that Sp-cAMPS (20μM) also potentiated GABAA IPSCs and occluded further potentiation induced by HFS (Figure 6 a-c).
Figure 5. Activation of adenylyl cyclase potentiates GABAA IPSCs and occludes further potentiation by HFS.

(a) Single experiment showing occlusion of the HFS-induced potentiation with forskolin (10μM)-induced potentiation. Inset: Averaged GABAA IPSCs recorded during a single experiment before (black), after 20 minutes in forskolin (red) and 20 minutes after HFS (green). Calibration: 10 ms, 100 pA.
(b) Forskolin activated adenylyl cyclase to increase GABAergic transmission, while dideoxyforskolin had no effect on GABAA–mediated responses (forskolin cells, filled circles: 191 ± 22% of pre-drug values, n=10; dideoxyforskolin cells, open circles: 98 ± 6% of pre-drug values, n=3). Inset: Forskolin-induced potentiation was accompanied by a decrease in the paired pulse ratio (PPR), suggesting an increase in presynaptic GABA release after forskolin. Five-minute blocks of data are shown (PPR before forskolin: 1.02±0.13; in forskolin: 0.78±0.05; p<0.05).
(c) After the IPSCs in forskolin reached a stable potentiated level, HFS was delivered. Forskolin-induced potentiation occluded further potentiation of IPSCs by HFS (85 ± 0.6% of pre-HFS values, n=7). Only the portion of the experiment after forskolin-potentiation has plateaued is shown.
Figure 6. PKA is not involved in the induction or expression of LTPGABA.

(a) Single experiment showing occlusion of LTPGABA with Sp-cAMPS (10μM)-induced potentiation. Sp-cAMPS, a cAMP analog, potentiated GABAA IPSCs and occluded the potentiation induced by HFS. Inset: Averaged GABAA IPSCs recorded before (black) and after 25 minutes in Sp-cAMPS (red) and 20 minutes after HFS (green). Calibration: 10ms, 100 pA.
(b) Averaged experiments showing that Sp-cAMPS mimics LTPGABA (152 ± 10% of pre-drug values, n=5).
(c) After the IPSCs in Sp-cAMPS reached a stable potentiated level, HFS was delivered. Only the portion of the experiment after Sp-cAMPS-induced potentiation is shown. Sp-cAMPS-induced potentiation occluded further potentiation of IPSCs by HFS (100 ± 0.5% of pre-HFS values, n=6).
(d) Bath application of Rp-cAMPS (20 μM), a PKA inhibitor, had no effect on the induction or expression of LTPGABA (control LTP, filled circles: 144 ± 14% of pre-HFS values, n=10; Rp-cAMPS cells, open circles: 128 ± 10% of pre-HFS values, n=6). Rp-cAMPS was present in the bath for at least 15 minutes prior to HFS.
Activation of PKA through the cAMP signaling pathway is not necessary for the induction or expression of LTPGABA
Together, these findings indicate that elevation of cAMP or PKA activation enhances synaptic strength through a presynaptic mechanism shared by LTPGABA. However, these experiments do not address whether cAMP/PKA signaling are required for LTPGABA. To test this idea, a specific PKA inhibitor, Rp-cAMPS (20μM) was bath applied prior to HFS and remained throughout the experiment. An even lower concentration of Rp-cAMPS was sufficient to block PKA in an earlier study (Gutlerner et al., 2002). The induction and expression of LTPGABA was entirely unaffected by bath application of Rp-cAMPS (Figure 6d). These data suggest that the cAMP-PKA signaling pathway is not required for LTPGABA but apparently shares downstream mechanisms with LTPGABA that underlie the long-lasting enhancement of GABA release from these terminals.
PKG and PKA signaling pathways converge onto common downstream mechanisms to sustain the potentiation of GABAergic synapses
Our data thus far indicate that elevation of either cGMP or cAMP levels enhances GABA release through the activation of PKG and PKA, respectively, as shown schematically in Figure 7a. PKA and PKG share common substrates that could serve as a mechanism for convergence. If these two pathways share a common target that promotes persistently enhanced GABA release, saturation of potentiation induced by one signaling pathway should preclude further potentiation via the other. To test this idea, we first bath-applied SNAP to potentiate GABAergic synapses through the NO-cGMP-PKG pathway. Once the potentiation by SNAP had plateaued, application of forskolin did not cause further synaptic potentiation (Figure 7b, c). This finding points to a convergence point for PKG and PKA in expressing and sustaining the increased GABA release. However, a trivial explanation might be that when intracellular levels of cGMP or cAMP are sufficiently high, there is cross activation of kinases by the cyclic nucleotides (Wang & Robinson, 1997). To rule out this possibility, we examined the effects of forskolin on GABAergic synapses in the persistent presence of PKG inhibitor, KT5823. If the increased levels of cAMP cross-activate PKG (which would subsequently potentiate these synapses), inhibition of PKG should reduce this potentiation. In contrast, in the presence of the PKG inhibitor, forskolin was still able to induce potentiation comparable to that seen with forskolin alone suggesting that cross-talk between these two pathways cannot explain our results. Instead, the simplest explanation of our data is that the two signaling cascades act on a common target to promote a sustained enhancement of GABA release. Further confirmation of this interpretation comes from bath-application of forskolin for only 10 minutes. The potentiation induced by brief application of forskolin did not require the persistent activation of PKA and still occluded the further potentiation by HFS, suggesting that both kinases converge on a downstream mechanism that is necessary for LTPGABA (99.5 ± 1% of pre-HFS values, n=4).
Figure 7. Convergence of the presynaptic NO-cGMP-PKG and cAMP-PKA signaling pathways on GABA release.

(a) Proposed schematic of signaling molecules mediating the effects of SNAP and forskolin in the presynaptic GABAergic terminal.
(b) Single experiment illustrating the effect of forskolin added after SNAP potentiated GABAergic IPSCs. After the IPSCs in SNAP (400 μM) reached a stable potentiated level, forskolin (10μM), was bath-applied. SNAP occluded the potentiation induced by forskolin. Inset: averaged IPSCs recorded during a single experiment before (black), after 10 minutes in SNAP (red) and after 10 minutes in forskolin (green). Calibration: 10 ms, 100 pA.
(c) Average of 6 experiments using the protocol outlined in (b). Only the portion of the experiment showing the effect of forskolin on SNAP-induced potentiation is shown (108 ± 7% of pre-forskolin values, n=6).
(d) The PKG inhibitor, KT5823 (500 nM), does not prevent the enhancement of IPSCs by 10 μM forskolin (187 ± 8% of pre-forskolin values, n=6), supporting the idea that the effect of forskolin on IPSCs is not mediated by cross-activation of PKG. KT5823 was applied at least 15 minutes prior to the addition of forskolin.
A single in vivo morphine exposure has no effect on the presynaptic cAMP-PKA signaling pathway
Our recent work has shown that in vivo treatment with morphine persistently modulates GABAergic synaptic plasticity as a result of interference with presynaptic NO-cGMP signaling (Nugent et al., 2007). The cAMP-PKA dependent- potentiation of the same GABAergic synapses is also reportedly altered 24 hours following ethanol exposure (Melis et al., 2002). μ-opioid receptors are negatively coupled to adenylyl cyclase via Go, and in the VTA, μ-opioid drugs acutely depress GABAergic synaptic transmission (Johnson & North, 1992; Williams et al., 2001; Nugent et al., 2007). In fact, one of the effectors of opioid receptor activation to decrease GABA release is also the inhibition of adenylyl cyclase. Based on our present results, which suggest that cGMP and cAMP signaling cascades coexist in VTA GABAergic synapses, we tested whether the interaction of in vivo morphine with cAMP signaling in presynaptic terminals has the potential to interfere with synaptic potentiation by the cAMP-PKA pathway. To address this question, rats were treated either with morphine (10 mg/kg i.p.) or with saline and 24 hours after treatment, the effects of forskolin (10μM) were tested on GABAergic synapses. Synapses from both saline- and morphine-treated animals were potentiated after exposure to forskolin (Figure 8a-c) suggesting that the presynaptic cAMP-PKA pathway is unaltered after morphine exposure, in contrast to morphine's effect on the NO-PKG signaling cascade involved in LTPGABA. This result also confirms that the site of disruption of the NO signaling by morphine is upstream to the unidentified converging mechanism for both PKG and PKA.
Figure 8. In vivo morphine does not modulate the presynaptic cAMP signaling cascade.
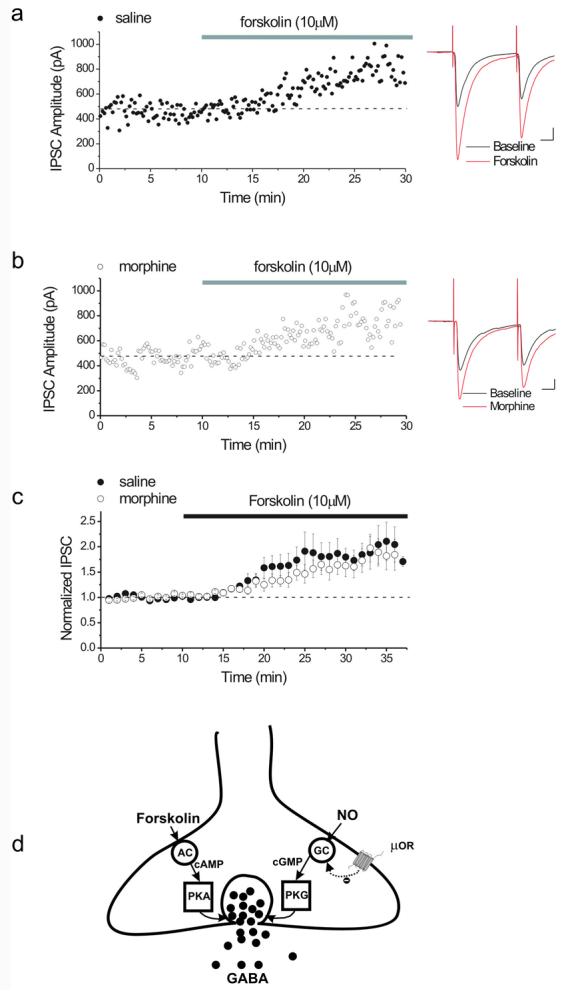
Rats were treated with either 10 mg/kg morphine or saline. 24 hours later, slices were prepared, and IPSCs were recorded from dopamine cells while 10 μM forskolin was bath-applied.
(a) Single experiment illustrating the potentiating effect of forskolin (10μM) on IPSCs from a saline-treated animal. Inset: Averaged GABAA IPSCs recorded before (black) and after 20 minutes in forskolin (red). Calibration for insets: 10ms, 100 pA.
(b) Single experiment illustrating the effect of forskolin (10μM) on IPSCs from a morphine-treated animal. Forskolin still potentiates GABAA-mediated IPSCs. Inset: Averaged GABAA IPSCs recorded before (black) and after 20 minutes in forskolin (red).
(c) Averaged experiments showing the enhancing effect of forskolin on IPSCs in slices from both saline- and morphine-treated animals, demonstrating that morphine in vivo does not alter the effect of forskolin (saline, filled circles, 188 ± 11% of pre-drug values, n=8; morphine, open circles, 167 ± 15% of pre-drug values, n=10).
(d) Proposed model of signaling molecules involved in opioid-induced plasticity at VTA GABAergic synapses. An in vivo injection of morphine alters GABAergic plasticity through modulation of the NO signaling pathway, probably at the level of sGC, without affecting the cAMP signaling cascade.
Discussion
Here we have investigated the involvement of PKG and PKA in the induction and expression of LTPGABA. Furthermore, we provide evidence for the synapse-specificity of NO signaling at VTA GABAA synapses and confirm that in vivo morphine persistently and specifically modulates the plasticity of these synapses through an interaction with the NO signaling pathway without an associated change in the coexistent cAMP signaling cascade.
The NO-cGMP-PKG and cAMP-PKA signaling cascades both potentiate GABAergic synapses
Increasing levels of NO exogenously using SNAP, or application of a cGMP analog, pCPT-cGMP, potentiates GABAergic synapses onto VTA DA neurons. Inhibition of PKG prevented the potentiation induced by NO or cGMP, supporting the role of PKG as the downstream effector from NO-cGMP. However, inhibition of PKG had no effect on basal GABAergic tone suggesting that constitutive PKG activity is not necessary to maintain basal levels of GABA release.
PKG is a serine–threonine kinase that mediates most of the effects of cGMP. Two different classes of PKG have been reported, PKG I and PKG II. While PKG I is highly localized in cerebellar Purkinje cells and a few other sites in brain, the ubiquitous distribution of PKG II and its major localization in neuronal processes make it a major target in mediating cGMP effects in the brain (Wang & Robinson, 1997; de Vente et al., 2001; Williams et al., 2001; Jouvert et al., 2004). Given that pCPT-cGMP is also a specific PKG II activator, PKG II rather than PKG I is the most likely kinase mediating the potentiation of VTA GABA release.
Several studies have shown that stimulation of AC by forskolin increases the release of GABA at VTA GABAergic synapses (Cameron & Williams, 1993; Bonci & Williams, 1996, 1997). We also found that either forskolin treatment or application of a cAMP analog/PKA activator potentiates the GABAergic synapses. We next asked whether the PKG and PKA signaling pathways interact with one another to increase GABA release from these GABAergic terminals. Potentiation of the synapses by using an NO donor prevented subsequent potentiation by forskolin, most likely because these synapses possess the molecular machinery for both NO-cGMP-PKG and cAMP-PKA signaling pathways, with both pathways converging on common downstream effectors to potentiate the GABAergic synapses. PKA and PKG share common phosphorylation substrates and identification of this unknown converging mechanism in GABAergic release machinery deserves further study. Alternatively, it is formally possible that PKG might phosphorylate an unknown cellular target that could in turn inhibit activation of either AC or PKA.
Synapse-specificity of the NO-cGMP-PKG signaling to GABAA synapses
The NO-cGMP signaling pathway can control GABAergic synaptic transmission and plasticity at GABAAR synapses (Stern & Ludwig, 2001; Li et al., 2002; Yu & Eldred, 2005), but many studies also suggest the involvement of GABABRs in drug addiction-related behaviors (Humeniuk et al., 1993; Cameron & Williams, 1994; Bonci & Williams, 1996; Shoji et al., 1997; Shoji et al., 1999; Boehm et al., 2002; Leite-Morris et al., 2004; Ong & Kerr, 2005). Chronic exposure to either morphine or cocaine modulates GABAB receptor function (Bonci & Williams, 1996). Moreover, intra-VTA application of baclofen, a GABAB receptor agonist, interferes with the rewarding properties of intra-cranial self-stimulation (Willick & Kokkinidis, 1995), with self-administration of several addictive drugs including heroin (Xi & Stein, 1999), with morphine-induced place preference (Tsuji et al., 1996), and with opioid-induced motor sensitization (Leite-Morris et al., 2002; Leite-Morris et al., 2004). We therefore next explored the potential presynaptic effects of NO on synaptic transmission mediated by GABAB receptors in the VTA. We found, however, that the NO donor or cGMP analog has no effect on GABAB IPSCs, indicating that the NO signaling pathway selectively potentiates GABAA synapses in the VTA. Although the NO-cGMP signaling pathway did not potentiate GABAB synapses, forskolin activation of the cAMP-PKA pathway has previously been shown to increase GABAB IPSPs (Shoji et al., 1999). This functional selectivity is not entirely surprising given that distinct sets of GABAergic inputs with distinct characteristics appear to innervate GABAA and GABAB synapses in the VTA. Extrinsic GABAergic afferents arising from forebrain selectively provide synaptic inputs to GABAB receptors, whereas GABAA responses are thought to arise from GABA release from local VTA interneurons (Johnson et al., 1992; Sugita et al., 1992; Shoji et al., 1999). In addition to the anatomical differences, D1 and 5-HT1A receptors acting through cAMP-PKA machinery are only expressed on presynaptic GABAergic terminals synapsing on GABABRs on DA neurons (Sugita et al., 1992; Cameron & Williams, 1993, 1994). The synapse-specificity of the NO signaling for GABAA synapses we have observed here emphasizes the fact that the two GABAergic inputs to these important dopamine neurons are quite independent, and modulation or alteration in one will likely spare the other. The distinct machinery available to modulate GABA release in distinct cell populations also potentially provides selective targets for drugs of abuse to exert their modulatory effects on GABAergic neurotransmission. These differences may also be exploited by therapeutic agents targeting only a single type of GABAergic synapse.
PKG but not PKA is involved in LTPGABA
Raising the levels of either cGMP or cAMP increases GABAergic transmission at GABAA synapses, which mimics LTPGABA. Our earlier work demonstrated the role of cGMP in LTPGABA by “occlusion” experiments in which prior potentiation induced by a cGMP analog prevented further HFS-induced LTPGABA, presumably by maximally activating the release potentiating machinery. Comparable sets of occlusion experiments were performed here with forskolin and Sp-cAMPS and we found no further LTPGABA after synaptic HFS, again suggesting an interaction of the cAMP cascade with mechanisms used in LTPGABA. To further clarify the involvement of PKG and PKA in GABAergic plasticity, we used compounds that specifically inhibit protein kinase activity. While inhibition of PKG completely blocked the induction of LTPGABA, the maintenance of LTPGABA was unaffected. These results demonstrate that the induction of NO-dependent LTPGABA is dependent on a rapid activation of PKG; however, the expression and maintenance of LTPGABA does not require persistent PKG activity. On the other hand, inhibition of PKA activity had no effect on the induction or the expression of LTPGABA. The occlusion between SNAP-induced potentiation and forskolin-induced potentiation indicates that LTPGABA requires the NO-cGMP-PKG pathway, and that cAMP-PKA can potentiate GABAA release via a shared cellular mechanism. Phosphorylation of presynaptic proteins provides a potential molecular mechanism to control transmitter release in a nerve terminal, especially in long-term processes such as presynaptic plasticity (Ghijsen & Leenders, 2005). It is also possible that the cAMP-PKA signaling pathway acts in parallel with PKG to increase phosphorylation of an unknown downstream target whose activation is necessary for the expression of LTPGABA. One possible converging downstream mechanism for both kinases is RIM1α, an active zone protein and PKA substrate that is involved in long-term changes in neurotransmitter release (Castillo et al., 2002; Schoch et al., 2002; Chevaleyre et al., 2006; Chevaleyre et al., 2007). However, it is not yet known whether RIM1a is a PKG substrate.
The presynaptic cAMP-PKA cascade is not modulated by a single in vivo morphine exposure
We showed previously that a single in vivo exposure to morphine acts on the NO-cGMP signaling to block LTPGABA at VTA synapses (Nugent et al., 2007). The μ-opioid receptors are coupled through Go to adenylyl cyclase, which in theory could represent an additional morphine target modulated in parallel with the NO-cGMP-PKG signaling cascade. Potentiation of GABA release after withdrawal from chronic morphine resulted from an upregulation of the cAMP-PKA cascade that is sensitive to inhibition by opioids (Chieng & Williams, 1998; Ingram et al., 1998; Shoji et al., 1999; Williams et al., 2001). Moreover, GABAA-mediated synaptic transmission is altered in the VTA by the cAMP-PKA-cascade after a single in vivo exposure to ethanol, and this alteration is proposed to provide an initial maladaptive change at the synaptic level (Melis et al., 2002). However, we found here that increasing cAMP levels in morphine-treated animals still potentiated the GABAA synapses. While in vivo morphine is able to block an increase in GABA release via the NO-cGMP pathway, GABA transmission by the cAMP-PKA pathway is still able to be potentiated 24 hours after morphine. These data also indicate a significant difference between the effects of these two addictive drugs. 24 hours after ethanol exposure, GABAA synapses are potentiated and cAMP-PKA cascades elicit no further potentiation (Melis et al., 2002), while 24 hours after morphine, the synapses are responsive to cAMP-PKA. It is possible that in response to the two drugs synaptic changes occur on different time scales, so that an examination of GABAA synapses at different time points following ethanol or morphine may reveal convergence over time.
One day after morphine exposure LTPGABA is inhibited, presumably removing a normal mechanism limiting dopamine cell firing rate. This inhibition can be bypassed either by cGMP analogues or activation of PKG, or alternatively by activation of the unaffected cAMP-PKA signaling pathway. Our data therefore suggest that raising cAMP or cGMP levels in GABAergic terminals may represent a useful therapeutic strategy to counteract opioid-induced maladaptive changes at GABAergic synapses. Taken together these data indicate the synapse-specific role of NO-cGMP-PKG signaling in opioid-induced plasticity of GABAergic synapses. Understanding the effects of chronic exposure to morphine on the NO-cGMP-PKG signaling pathway would also provide insight into how drugs of abuse reshape the reward circuitry. It is possible that repeated exposure to morphine would upregulate the cAMP-PKA pathway while impairing the NO- cGMP-PKG pathway. It will be of interest to ask whether this modulation by chronic morphine provides a form of homeostatic regulation of inhibitory plasticity in the VTA circuitry during establishment of opiate addiction.
Acknowledgements
This work was supported by NIH grants DA11289 to J.A.K., DA021973 to F.S.N and DA024527 to J.L.N. The authors would like to thank lab members for constructive suggestions and Jeannette Downing-Park for technical assistance.
Footnotes
Disclosure/Conflicts of Interest:
The authors declare no conflicts of interest.
References
- Boehm SL, 2nd, Piercy MM, Bergstrom HC, Phillips TJ. Ventral tegmental area region governs GABA(B) receptor modulation of ethanol-stimulated activity in mice. Neuroscience. 2002;115:185–200. doi: 10.1016/s0306-4522(02)00378-0. [DOI] [PubMed] [Google Scholar]
- Bonci A, Williams JT. A common mechanism mediates long-term changes in synaptic transmission after chronic cocaine and morphine. Neuron. 1996;16:631–639. doi: 10.1016/s0896-6273(00)80082-3. [DOI] [PubMed] [Google Scholar]
- Bonci A, Williams JT. Increased probability of GABA release during withdrawal from morphine. J Neurosci. 1997;17:796–803. doi: 10.1523/JNEUROSCI.17-02-00796.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs CA, McAfee DA, McCaman RE. Long-term regulation of synaptic acetylcholine release and nicotinic transmission: the role of cyclic AMP. Br J Pharmacol. 1988;93:399–411. doi: 10.1111/j.1476-5381.1988.tb11447.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron DL, Williams JT. Dopamine D1 receptors facilitate transmitter release. Nature. 1993;366:344–347. doi: 10.1038/366344a0. [DOI] [PubMed] [Google Scholar]
- Cameron DL, Williams JT. Cocaine inhibits GABA release in the VTA through endogenous 5-HT. J Neurosci. 1994;14:6763–6767. doi: 10.1523/JNEUROSCI.14-11-06763.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo PE, Schoch S, Schmitz F, Sudhof TC, Malenka RC. RIM1alpha is required for presynaptic long-term potentiation. Nature. 2002;415:327–330. doi: 10.1038/415327a. [DOI] [PubMed] [Google Scholar]
- Castro-Alamancos MA, Calcagnotto ME. Presynaptic long-term potentiation in corticothalamic synapses. J Neurosci. 1999;19:9090–9097. doi: 10.1523/JNEUROSCI.19-20-09090.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez-Noriega LE, Stevens CF. Increased transmitter release at excitatory synapses produced by direct activation of adenylate cyclase in rat hippocampal slices. J Neurosci. 1994;14:310–317. doi: 10.1523/JNEUROSCI.14-01-00310.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen BT, Bowers MS, Martin M, Hopf FW, Guillory AM, Carelli RM, Chou JK, Bonci A. Cocaine but not natural reward self-administration nor passive cocaine infusion produces persistent LTP in the VTA. Neuron. 2008;59:288–297. doi: 10.1016/j.neuron.2008.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HX, Otmakhov N, Strack S, Colbran RJ, Lisman JE. Is persistent activity of calcium/calmodulin-dependent kinase required for the maintenance of LTP? J Neurophysiol. 2001;85:1368–1376. doi: 10.1152/jn.2001.85.4.1368. [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Heifets BD, Kaeser PS, Sudhof TC, Castillo PE. Endocannabinoid-mediated long-term plasticity requires cAMP/PKA signaling and RIM1alpha. Neuron. 2007;54:801–812. doi: 10.1016/j.neuron.2007.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevaleyre V, Takahashi KA, Castillo PE. Endocannabinoid-mediated synaptic plasticity in the CNS. Annu Rev Neurosci. 2006;29:37–76. doi: 10.1146/annurev.neuro.29.051605.112834. [DOI] [PubMed] [Google Scholar]
- Chien WL, Liang KC, Teng CM, Kuo SC, Lee FY, Fu WM. Enhancement of long-term potentiation by a potent nitric oxide-guanylyl cyclase activator, 3-(5-hydroxymethyl-2-furyl)-1-benzyl-indazole. Mol Pharmacol. 2003;63:1322–1328. doi: 10.1124/mol.63.6.1322. [DOI] [PubMed] [Google Scholar]
- Chieng B, Williams JT. Increased opioid inhibition of GABA release in nucleus accumbens during morphine withdrawal. J Neurosci. 1998;18:7033–7039. doi: 10.1523/JNEUROSCI.18-17-07033.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vente J, Asan E, Gambaryan S, Markerink-van Ittersum M, Axer H, Gallatz K, Lohmann SM, Palkovits M. Localization of cGMP-dependent protein kinase type II in rat brain. Neuroscience. 2001;108:27–49. doi: 10.1016/s0306-4522(01)00401-8. [DOI] [PubMed] [Google Scholar]
- el-Husseini AE, Bladen C, Vincent SR. Molecular characterization of a type II cyclic GMP-dependent protein kinase expressed in the rat brain. J Neurochem. 1995;64:2814–2817. doi: 10.1046/j.1471-4159.1995.64062814.x. [DOI] [PubMed] [Google Scholar]
- Fiorillo CD, Williams JT. Selective inhibition by adenosine of mGluR IPSPs in dopamine neurons after cocaine treatment. Journal of neurophysiology. 2000;83:1307–1314. doi: 10.1152/jn.2000.83.3.1307. [DOI] [PubMed] [Google Scholar]
- Ghijsen WE, Leenders AG. Differential signaling in presynaptic neurotransmitter release. Cell Mol Life Sci. 2005;62:937–954. doi: 10.1007/s00018-004-4525-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greengard P, Jen J, Nairn AC, Stevens CF. Enhancement of the glutamate response by cAMP-dependent protein kinase in hippocampal neurons. Science. 1991;253:1135–1138. doi: 10.1126/science.1716001. [DOI] [PubMed] [Google Scholar]
- Gutlerner JL, Penick EC, Snyder EM, Kauer JA. Novel protein kinase A-dependent long-term depression of excitatory synapses. Neuron. 2002;36:921–931. doi: 10.1016/s0896-6273(02)01051-6. [DOI] [PubMed] [Google Scholar]
- Hidaka H, Kobayashi R. Pharmacology of protein kinase inhibitors. Annu Rev Pharmacol Toxicol. 1992;32:377–397. doi: 10.1146/annurev.pa.32.040192.002113. [DOI] [PubMed] [Google Scholar]
- Huang YY, Kandel ER. Recruitment of long-lasting and protein kinase A-dependent long-term potentiation in the CA1 region of hippocampus requires repeated tetanization. Learning & memory (Cold Spring Harbor, NY. 1994;1:74–82. [PubMed] [Google Scholar]
- Huang YY, Kandel ER. Postsynaptic induction and PKA-dependent expression of LTP in the lateral amygdala. Neuron. 1998;21:169–178. doi: 10.1016/s0896-6273(00)80524-3. [DOI] [PubMed] [Google Scholar]
- Humeniuk RE, White JM, Ong J. The role of GABAB receptors in mediating the stimulatory effects of ethanol in mice. Psychopharmacology. 1993;111:219–224. doi: 10.1007/BF02245527. [DOI] [PubMed] [Google Scholar]
- Hyman SE, Malenka RC, Nestler EJ. Neural mechanisms of addiction: the role of reward-related learning and memory. Annu Rev Neurosci. 2006;29:565–598. doi: 10.1146/annurev.neuro.29.051605.113009. [DOI] [PubMed] [Google Scholar]
- Ingram SL, Vaughan CW, Bagley EE, Connor M, Christie MJ. Enhanced opioid efficacy in opioid dependence is caused by an altered signal transduction pathway. J Neurosci. 1998;18:10269–10276. doi: 10.1523/JNEUROSCI.18-24-10269.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SW, Mercuri NB, North RA. 5-hydroxytryptamine1B receptors block the GABAB synaptic potential in rat dopamine neurons. J Neurosci. 1992;12:2000–2006. doi: 10.1523/JNEUROSCI.12-05-02000.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SW, North RA. Opioids excite dopamine neurons by hyperpolarization of local interneurons. J Neurosci. 1992;12:483–488. doi: 10.1523/JNEUROSCI.12-02-00483.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S, Kornblum JL, Kauer JA. Amphetamine blocks long-term synaptic depression in the ventral tegmental area. J Neurosci. 2000;20:5575–5580. doi: 10.1523/JNEUROSCI.20-15-05575.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouvert P, Revel MO, Lazaris A, Aunis D, Langley K, Zwiller J. Activation of the cGMP pathway in dopaminergic structures reduces cocaine-induced EGR-1 expression and locomotor activity. J Neurosci. 2004;24:10716–10725. doi: 10.1523/JNEUROSCI.1398-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauer JA, Malenka RC. Synaptic plasticity and addiction. Nat Rev Neurosci. 2007;8:844–858. doi: 10.1038/nrn2234. [DOI] [PubMed] [Google Scholar]
- Leite-Morris KA, Fukudome EY, Kaplan GB. Opiate-induced motor stimulation is regulated by gamma-aminobutyric acid type B receptors found in the ventral tegmental area in mice. Neurosci Lett. 2002;317:119–122. doi: 10.1016/s0304-3940(01)02457-0. [DOI] [PubMed] [Google Scholar]
- Leite-Morris KA, Fukudome EY, Shoeb MH, Kaplan GB. GABA(B) receptor activation in the ventral tegmental area inhibits the acquisition and expression of opiate-induced motor sensitization. J Pharmacol Exp Ther. 2004;308:667–678. doi: 10.1124/jpet.103.058412. [DOI] [PubMed] [Google Scholar]
- Li DP, Chen SR, Pan HL. Nitric oxide inhibits spinally projecting paraventricular neurons through potentiation of presynaptic GABA release. Journal of neurophysiology. 2002;88:2664–2674. doi: 10.1152/jn.00540.2002. [DOI] [PubMed] [Google Scholar]
- Linden DJ, Ahn S. Activation of presynaptic cAMP-dependent protein kinase is required for induction of cerebellar long-term potentiation. J Neurosci. 1999;19:10221–10227. doi: 10.1523/JNEUROSCI.19-23-10221.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman J, Goldring M. Evaluation of a model of long-term memory based on the properties of the Ca2+/calmodulin-dependent protein kinase. J Physiol (Paris) 1988;83:187–197. [PubMed] [Google Scholar]
- Lisman JE. A mechanism for memory storage insensitive to molecular turnover: a bistable autophosphorylating kinase. Proc Natl Acad Sci U S A. 1985;82:3055–3057. doi: 10.1073/pnas.82.9.3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu QS, Pu L, Poo MM. Repeated cocaine exposure in vivo facilitates LTP induction in midbrain dopamine neurons. Nature. 2005;437:1027–1031. doi: 10.1038/nature04050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Rao Y, Daw N. Roles of protein kinase A and protein kinase G in synaptic plasticity in the visual cortex. Cereb Cortex. 2003;13:864–869. doi: 10.1093/cercor/13.8.864. [DOI] [PubMed] [Google Scholar]
- Lu YF, Hawkins RD. Ryanodine receptors contribute to cGMP-induced late-phase LTP and CREB phosphorylation in the hippocampus. Journal of neurophysiology. 2002;88:1270–1278. doi: 10.1152/jn.2002.88.3.1270. [DOI] [PubMed] [Google Scholar]
- Lu YF, Kandel ER, Hawkins RD. Nitric oxide signaling contributes to late-phase LTP and CREB phosphorylation in the hippocampus. J Neurosci. 1999;19:10250–10261. doi: 10.1523/JNEUROSCI.19-23-10250.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Malinow R, Madison DV, Tsien RW. Persistent protein kinase activity underlying long-term potentiation. Nature. 1988;335:820–824. doi: 10.1038/335820a0. [DOI] [PubMed] [Google Scholar]
- Mansvelder HD, Keath JR, McGehee DS. Synaptic mechanisms underlie nicotine-induced excitability of brain reward areas. Neuron. 2002;33:905–919. doi: 10.1016/s0896-6273(02)00625-6. [DOI] [PubMed] [Google Scholar]
- Margolis EB, Lock H, Hjelmstad GO, Fields HL. The ventral tegmental area revisited: is there an electrophysiological marker for dopaminergic neurons? The Journal of physiology. 2006;577:907–924. doi: 10.1113/jphysiol.2006.117069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melis M, Camarini R, Ungless MA, Bonci A. Long-lasting potentiation of GABAergic synapses in dopamine neurons after a single in vivo ethanol exposure. J Neurosci. 2002;22:2074–2082. doi: 10.1523/JNEUROSCI.22-06-02074.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellor J, Nicoll RA, Schmitz D. Mediation of hippocampal mossy fiber long-term potentiation by presynaptic Ih channels. Science (New York, NY. 2002;295:143–147. doi: 10.1126/science.1064285. [DOI] [PubMed] [Google Scholar]
- Monfort P, Munoz MD, Kosenko E, Felipo V. Long-term potentiation in hippocampus involves sequential activation of soluble guanylate cyclase, cGMP-dependent protein kinase, and cGMP-degrading phosphodiesterase. J Neurosci. 2002;22:10116–10122. doi: 10.1523/JNEUROSCI.22-23-10116.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monfort P, Munoz MD, Kosenko E, Llansola M, Sanchez-Perez A, Cauli O, Felipo V. Sequential activation of soluble guanylate cyclase, protein kinase G and cGMP-degrading phosphodiesterase is necessary for proper induction of long-term potentiation in CA1 of hippocampus. Alterations in hyperammonemia. Neurochem Int. 2004;45:895–901. doi: 10.1016/j.neuint.2004.03.020. [DOI] [PubMed] [Google Scholar]
- Nugent FS, Hwong AR, Udaka Y, Kauer JA. High-frequency afferent stimulation induces long-term potentiation of field potentials in the ventral tegmental area. Neuropsychopharmacology. 2008;33:1704–1712. doi: 10.1038/sj.npp.1301561. [DOI] [PubMed] [Google Scholar]
- Nugent FS, Kauer JA. LTP of GABAergic synapses in the ventral tegmental area and beyond. J Physiol. 2008;586:1487–1493. doi: 10.1113/jphysiol.2007.148098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nugent FS, Penick EC, Kauer JA. Opioids block long-term potentiation of inhibitory synapses. Nature. 2007;446:1086–1090. doi: 10.1038/nature05726. [DOI] [PubMed] [Google Scholar]
- Ong J, Kerr DI. Clinical potential of GABAB receptor modulators. CNS Drug Rev. 2005;11:317–334. doi: 10.1111/j.1527-3458.2005.tb00049.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan B, Hillard CJ, Liu QS. Endocannabinoid signaling mediates cocaine-induced inhibitory synaptic plasticity in midbrain dopamine neurons. J Neurosci. 2008;28:1385–1397. doi: 10.1523/JNEUROSCI.4033-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salin PA, Malenka RC, Nicoll RA. Cyclic AMP mediates a presynaptic form of LTP at cerebellar parallel fiber synapses. Neuron. 1996a;16:797–803. doi: 10.1016/s0896-6273(00)80099-9. [DOI] [PubMed] [Google Scholar]
- Salin PA, Scanziani M, Malenka RC, Nicoll RA. Distinct short-term plasticity at two excitatory synapses in the hippocampus. Proc Natl Acad Sci U S A. 1996b;93:13304–13309. doi: 10.1073/pnas.93.23.13304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santschi L, Reyes-Harde M, Stanton PK. Chemically induced, activity-independent LTD elicited by simultaneous activation of PKG and inhibition of PKA. J Neurophysiol. 1999;82:1577–1589. doi: 10.1152/jn.1999.82.3.1577. [DOI] [PubMed] [Google Scholar]
- Schoch S, Castillo PE, Jo T, Mukherjee K, Geppert M, Wang Y, Schmitz F, Malenka RC, Sudhof TC. RIM1alpha forms a protein scaffold for regulating neurotransmitter release at the active zone. Nature. 2002;415:321–326. doi: 10.1038/415321a. [DOI] [PubMed] [Google Scholar]
- Shoji S, Simms D, McDaniel WC, Gallagher JP. Chronic cocaine enhances gamma-aminobutyric acid and glutamate release by altering presynaptic and not postsynaptic gamma-aminobutyric acidB receptors within the rat dorsolateral septal nucleus. J Pharmacol Exp Ther. 1997;280:129–137. [PubMed] [Google Scholar]
- Shoji Y, Delfs J, Williams JT. Presynaptic inhibition of GABA(B)-mediated synaptic potentials in the ventral tegmental area during morphine withdrawal. J Neurosci. 1999;19:2347–2355. doi: 10.1523/JNEUROSCI.19-06-02347.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern JE, Ludwig M. NO inhibits supraoptic oxytocin and vasopressin neurons via activation of GABAergic synaptic inputs. Am J Physiol Regul Integr Comp Physiol. 2001;280:R1815–1822. doi: 10.1152/ajpregu.2001.280.6.R1815. [DOI] [PubMed] [Google Scholar]
- Sugita S, Johnson SW, North RA. Synaptic inputs to GABAA and GABAB receptors originate from discrete afferent neurons. Neurosci Lett. 1992;134:207–211. doi: 10.1016/0304-3940(92)90518-c. [DOI] [PubMed] [Google Scholar]
- Tsuji M, Nakagawa Y, Ishibashi Y, Yoshii T, Takashima T, Shimada M, Suzuki T. Activation of ventral tegmental GABAB receptors inhibits morphine-induced place preference in rats. Eur J Pharmacol. 1996;313:169–173. doi: 10.1016/0014-2999(96)00642-5. [DOI] [PubMed] [Google Scholar]
- Ungless MA, Whistler JL, Malenka RC, Bonci A. Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature. 2001;411:583–587. doi: 10.1038/35079077. [DOI] [PubMed] [Google Scholar]
- Wang X, Robinson PJ. Cyclic GMP-dependent protein kinase and cellular signaling in the nervous system. Journal of neurochemistry. 1997;68:443–456. doi: 10.1046/j.1471-4159.1997.68020443.x. [DOI] [PubMed] [Google Scholar]
- Weisskopf MG, Castillo PE, Zalutsky RA, Nicoll RA. Mediation of hippocampal mossy fiber long-term potentiation by cyclic AMP. Science. 1994;265:1878–1882. doi: 10.1126/science.7916482. [DOI] [PubMed] [Google Scholar]
- Williams JT, Christie MJ, Manzoni O. Cellular and synaptic adaptations mediating opioid dependence. Physiol Rev. 2001;81:299–343. doi: 10.1152/physrev.2001.81.1.299. [DOI] [PubMed] [Google Scholar]
- Willick ML, Kokkinidis L. The effects of ventral tegmental administration of GABAA, GABAB and NMDA receptor agonists on medial forebrain bundle self-stimulation. Behav Brain Res. 1995;70:31–36. doi: 10.1016/0166-4328(94)00181-e. [DOI] [PubMed] [Google Scholar]
- Wu J, Wang Y, Rowan MJ, Anwyl R. Evidence for involvement of the cGMP-protein kinase G signaling system in the induction of long-term depression, but not long-term potentiation, in the dentate gyrus in vitro. J Neurosci. 1998;18:3589–3596. doi: 10.1523/JNEUROSCI.18-10-03589.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi ZX, Stein EA. Baclofen inhibits heroin self-administration behavior and mesolimbic dopamine release. J Pharmacol Exp Ther. 1999;290:1369–1374. [PubMed] [Google Scholar]
- Yang HW, Hu XD, Zhang HM, Xin WJ, Li MT, Zhang T, Zhou LJ, Liu XG. Roles of CaMKII, PKA, and PKC in the induction and maintenance of LTP of C-fiber-evoked field potentials in rat spinal dorsal horn. J Neurophysiol. 2004;91:1122–1133. doi: 10.1152/jn.00735.2003. [DOI] [PubMed] [Google Scholar]
- Yu D, Eldred WD. Nitric oxide stimulates gamma-aminobutyric acid release and inhibits glycine release in retina. The Journal of comparative neurology. 2005;483:278–291. doi: 10.1002/cne.20416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuo M, Hu Y, Schultz C, Kandel ER, Hawkins RD. Role of guanylyl cyclase and cGMP-dependent protein kinase in long-term potentiation. Nature. 1994;368:635–639. doi: 10.1038/368635a0. [DOI] [PubMed] [Google Scholar]