

Abstract
Glycogen synthase kinase 3β (GSK3β) regulates numerous signaling pathways that control a wide range of cellular processes, including cell proliferation, differentiation, apoptosis and metabolism. We report a novel function of GSK3β: It interacts with the inhibitor-of-apoptosis protein (IAP) survivin to modulate its expression, thus regulating apoptosis in human lung cancer cells. A co-immunoprecipitation assay revealed that GSK3β can bind survivin. Activation of GSK3β induced translocation of survivin from the cytoplasm to the nucleus, resulting in G1 cell-cycle arrest and apoptosis, as well as sensitization to the chemotherapeutic drug doxorubicin. In contrast, inactivation of GSK3β, either by transfection of a dominant-negative mutant inhibitor DN-GSK3β or with selective inhibitor LiCl, increased cytoplasmic survivin expression, leading to cell cycle progression and resistance to apoptosis. These results identify a pro-apoptotic role for GSK3β in cancer cells, through its modulation of survivin in subcellular redistribution. This new role suggests that there is a potential for pharmacologic activation of GSK3β to enhance treatment of cancer patients, including those with resistance.
Keywords: GSK3β, Survivin, Cancer, Doxorubicin, Apoptosis
Introduction
Glycogen synthase kinase 3 (GSK3) is a serine/threonine kinase that consists of two highly homologous isoforms, GSK3α and GSK3β [1,2]. GSK3β is critically important in cancer known to have several mechanisms that contribute to the regulation of its activity. GSK3β is activated by phosphorylation of tyrosine 216 and negatively regulated by phosphorylation of serine 9 [3,4]. Akt phosphorylates GSK3β at serine 9; therefore, the PI3K/Akt pathway inhibits GSK3β activation [5]. In addition to phosphorylation, GSK3β's activity is regulated by its subcellular distribution [6]. Most GSK3β is cytoplasmic if cells are unstressed and growing, although a low level remains detectable in the nucleus. Nuclear levels of GSK3β rapidly increase after cellular stress, such as starvation or heat shock. It has also been shown that the GSK3β located in the nucleus is highly active relative to cytoplasmic GSK3β [7]. Lastly, GSK3β is regulated by protein complex formation, such as association with proteins of the Wnt signaling pathway that promotes GSK3β degradation [8].
It is now acknowledged that GSK3β is a critical regulator of numerous signaling pathways that are involved in a wide range of cellular processes, including: metabolism, proliferation, differentiation, survival and apoptosis [9]. So far, the role of GSK3β in the pivotal signaling network that allows for cell survival or apoptosis is not well defined. Previous studies have shown that activation of GSK3β is a key component of some apoptotic signaling cascades. Evidence supporting a pro-apoptotic function for GSK3β was first demonstrated in rat cortical neurons: β-amyloid peptide increased activation of GSK3β, inducing apoptosis that could be inhibited by treatment with GSK3β antisense [10]. Later studies by Pap and Cooper indicated that enforced overexpression of GSK3β in PC12 cells and Rat-1 fibroblasts results in spontaneous apoptosis, and that this GSK3β-induced apoptosis is inhibited by activation of the PI3K/Akt pathway [5]. In addition, recent studies demonstrate that initiating GSK3β-mediated apoptosis can also render human breast cancer and medulloblastoma cells sensitive to death [11-13]. The mechanisms by which GSK3β mediates apoptosis have not been completely understood. A study by Watcharasit et al [14] revealed that nuclear GSK3β becomes associated with p53 after DNA damage and contributes to p53-induced apoptosis. Later studies by the same investigators further demonstrate that GSK3β binds to p53 confined to the nucleus, promoting the p53 tumor suppressor function [15]. This physical interaction between GSK3β and p53 may represent one of the mechanisms used by GSK3β for its pro-apoptotic role.
Survivin, a unique member of the inhibitor-of-apoptosis protein (IAP) family, plays an important role in regulating both apoptosis and cell division [16]. Survivin overexpression is identified as a negative prognostic factor in various cancer types, plus it has been implicated as the reason for resistance to anticancer agents that would otherwise induce cancer cell apoptosis [17]. In cancer patients, survivin is detected as both a cytoplasmic and nuclear protein. Recent studies demonstrate that survivin's anti-apoptotic function is preferentially mediated by cytoplasmic survivin, whereas the presence of nuclear survivin may be indicative of impaired survivin function [18]. In fact, it has been reported that nuclear export is essential for the tumor-promoting activity of survivin to occur [19], and that nuclear localization of survivin can render tumor cells more sensitive to apoptosis due to induction of p53 and Bax [20]. Moreover, clinical research indicates that preferential expression of survivin in the nucleus is of prognostic significance for cancer patients [21-23].
Because it has been found that both GSK3β and survivin's roles in regulating cell survival and apoptosis are dependent on subcellular distribution, we wished to evaluate whether there is a possible correlation between the GSK3β and survivin activities, using the lung cancer cell line A549. In the study described below, we found that GSK3β did physically interact with survivin, and that activation of GSK3β resulted in relocalization of survivin to the nucleus, followed by cell cycle arrest and apoptosis. This pro-apoptotic function of activated GSK3β, achieved through regulation of the nuclear import of survivin, was further confirmed by inhibiting GSK3β with either the selective inhibitor LiCl or transfection of a dominant negative GSK3β mutant that causes cytoplasmic localization of survivin, promoting cell survival.
Materials and methods
Cell culture, reagents and treatments
The human lung adenocarcinoma cell line A549 was obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). A549 cells were cultured in Dulbecco's modified Eagle's medium containing 10% FBS and antibiotics (100 IU/ml penicillin and 50 μg/ml streptomycin), and incubated at 37°C in 5% CO2. The LiCl was purchased from Sigma. Cells were treated with 0-20 mM of LiCl for the time period indicated. The Leptomycin B (LMB) purchased from LC Laboratories was dissolved in ethanol to create a 2 μM stock solution and stored in small aliquots at -20°C. Cells were exposed to 0-10 nM of LMB for the time period indicated, with the final ethanol concentration kept constant in each experiment. Controlled experiments were performed using one set of A549 cells, then repeating at least 3 times.
Plasmids and gene transfection
The GSK3β mutant plasmids used in our experiments, S9A-GSK3β and DN-GSK3β, were generously provided by Professor James R. Woodgett (Ontario Cancer Institute, Canada). These S9A-GSK3β and DN-GSK3β plasmids, the former exhibiting constitutively active GSK3β and the latter a dominant negative GSK3β, have been characterized previously [24,25]. To achieve a transient gene transfection, plates of A549 cells in exponential growth were concurrently transfected with each of the GSK3β mutant plasmids and the empty vector control. Transfection was performed using the Lipofectamine 2000 reagent (Invitrogen, CA, USA) according to the manufacturer's recommendations.
Western blot analysis
Whole cell protein samples were prepared by lysing the desired cells in a buffer composed of 150 mM NaCl, 50 mM Tris (pH 8.0), 5mM EDTA, 1% (v/v) Nonidet p-40, 1 mM phenylmethylsulfonyl fluoride, 20 μg/ml aprotinin, and 25 μg/ml leupeptin for a period of 30 min at 4° C. To detect the cellular localization of survivin and GSK3β, we isolated the nuclear and cytoplasmic fractions using the NE-PER kit (Pierce) according to the manufacturer's instructions. Equal amounts of protein extracted from all samples were resolved in parallel by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) through a gel and transferred to a nitrocellulose filter. After blocking with buffer containing 20 mM Tris-HCl (pH7.5), 500 mM NaCl, and 5% non-fat milk for 1 h at room temperature, the filter was incubated first for 1 h at room temperature with specific antibodies against phospho-GSK3β-ser9 (Cell Signaling Technologies, Beverly, MA, USA), GSK3β, survivin, cyclinD1, caspase-3 and PARP (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Next, the blot was incubated with HRP-labeled secondary antibody. The resulting blots were developed using a chemiluminescent detection system (ECL, Amersham Life Science, Buckinghamshire, England).
Co-immunoprecipitation
Cells were washed with ice-cold PBS containing 1 mM orthovanadate first, and then lysed at 4 °C in a buffer composed of 50 mM Tris at pH7.6, 150 mM NaCl, 1% Nonidet P-40, 10 mM sodium phosphate, 10 mM NaF, 1 mM sodium orthovanadate, 2 mM phenylmethylsulfonyl fluoride, 10 μg/ml aprotinin, 10 μg/ml leupeptin and 10 μg/ml pepstatin. Following centrifugation, 30μg of the clarified cell lysate was incubated with 15μl Protein G plus/Protein A-agarose and specific antibodies. After 24-hour incubation, that agarose blend was centrifuged. The resulting immunoprecipitate pellet was then washed four times with ice-cold lysis buffer, suspended in electrophoresis sample buffer, and boiled for 5 min. This immunoprecipitated protein preparation was further analyzed by western blot, as described above.
Confocal immunofluorescence assay
The two kinds of GSK3β mutant plasmid-transfected cells and the controls were concurrently cultured on coverslips for 24 h, washed with isotonic PBS (pH 7.4) and fixed in absolute methanol for 5-10 min at -20 °C. After the se coverslips were rinsed three times with PBS, the nonspecific binding sites were blocked in PBS containing 5% BSA and 0.2% Triton X-100 for 30 min. After blocking, the cells were placed in a mixture of mouse anti-survivin antibody (1: 50) and rabbit anti-GSK3β antibody in PBS containing 5% BSA/0.2% Triton X-100, and incubated overnight at 4 °C. Next, the cells were washed three times with 0.2% Triton X-100 in PBS and mixed with a PBS solution containing both goat antimouse FITC (1: 100) and goat antirabbit Cy3 (1:200), 5% BSA and 0.2% Triton X-100. The mixture was incubated for 1 h at room temperature, and then washed three times with 0.2% Triton X-100 in PBS. Finally, the nuclei were stained with 1 μg /ml Hoechst 33258 for 15 min. The coverslips were sealed with glycerol and examined under a Leica confocal laser scanning microscope (Mannheim, Germany).
TUNEL assay
After transient transfection of the GSK3β mutant plasmids, the transfected cells were cultured on coverslips for 24 h. Apoptosis was measured using the DeadEnd™ Fluorometric TUNEL System (Promega) and confocal microscopy. All FITC-positive cells were scored as apoptotic cells, within a total of 300 cells counted per slide. The percentage of apoptotic cells is the number of FITC-positive cells per slide/total number of cells counted per slide × 100%.
WST assay
The effect that LiCl had on the cytotoxicity of doxorubicin in A549 cells was determined by water-soluble tetrazolium salt (WST) assay. Briefly, cells were cultured in 96-well microtiter plates with different concentrations of LiCl and doxorubicin for a 44 hr period. Then WST (25 μg/well) was added and the cells were incubated for an additional 4 hr. Following incubation, the optical density (OD) of the wells was read with a microplate reader set at a test wavelength of 450 nm and a reference wavelength of 620 nm. Appropriate controls lacking cells were included to determine background absorbance.
Flow cytometry
Flow cytometry was performed to analyze cell cycle positions and the proportion of annexin-V stained apoptotic cells. Following transfection of GSK3β mutant plasmids or the treatment with LiCl, 5 × 105 cells were collected, rinsed twice with PBS, and fixed in 70% ethanol for 1 h at 4°C. Next, cells were washed twice in PBS, resuspended in 30 μl of phosphate citrate buffer (0.1 M Na citrate/0.2 M Na2HPO4), and incubated for 30 min at room temperature. The samples were re-washed with PBS and then suspended in 0.5 ml PBS containing 20 μg/ml of propidium iodide (PI) and 20 μg/ml of RNase A. After incubating at 4°C for at least 0.5 h, the samples were analyzed using a FACScan (Becton Dickinson) and WinList software (Verity Software House, Inc). For the quantitation of apoptotic cells by annexin-V staining, cells with or without transfections with mutant plasmids or LiCl treatment were washed once with PBS, then stained with FITC-annexin-V and PI according to the manufacturer's instructions. Stained cells were analyzed as outlined above.
Statistical analysis
Data are expressed as the mean ± standard deviation (SD). Statistical significance was examined by one-way analysis of variance (ANOVA) pairwise comparison (SPSS12.0 software). In all analyses, P<0.05 was considered statistically significant.
Results
GSK3β is activated or inactivated by transfection of S9A-GSK3β and DN-GSK3β, respectively, in A549 cells
Skurk et al [26] previously showed that in human umbilical vein endothelial cells, transfection of either the constitutively active GSK3β mutant S9A-GSK3β or the dominant negative GSK3β mutant DN-GSK3β (KM-GSK3β), created decreases or increases, respectively, in the expression of the inactivated form of GSK3β that is phosphorylated on serine 9, p-GSK3β(ser9). In this study, we tested for the expression of p-GSK3β(ser9) in the human lung cancer cell line A549, following transfection with S9A-GSK3β and DN-GSK3β. Consistent with the results obtained in umbilical vein endothelial cells, overexpression due to the transfected S9A-GSK3β mutant in A549 cells led to ser9 dephosphorylation and activation of GSK3β (decreased p-GSK3β(ser9) expression), whereas DN-GSK3β transfection exerted an opposite effect on p-GSK3β(ser9) expression (Fig. 1A). We also evaluated the GSK3β-inhibiting function of LiCl in A549 cells. Similar to transfection of DN-GSK3β, LiCl caused ser9 phosphorylation; therefore, inactivation of GSK3β (Fig. 1B).
Fig. 1.
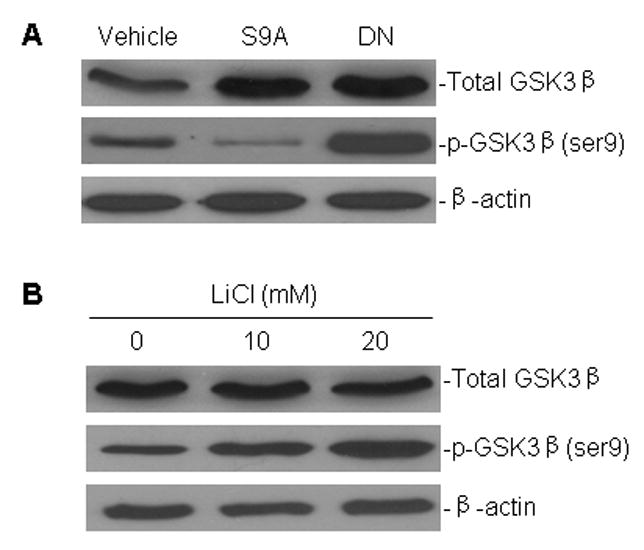
Effect of transfection of GSK3β mutant plasmids and LiCl treatment on the activation of GSK3β in A549 cells, as indicated by GSK3β phosphorylation on serine 9, i.e. p-GSK3β (ser9). A, Cells were transiently transfected with the constitutively active GSK3β mutant S9A-GSK3β (S9A) and dominant negative GSK3β mutant DN-GSK3β (DN) plasmids, as well as an empty vector control (vehicle), for 24 h. Whole cell protein was extracted and subjected to western blot analysis using a specific antibodies against GSK3β and p-GSK3β (ser9). β-actin served as control for equal protein loading. B, Western blot assay for expression of GSK3β and p-GSK3β (ser9) in A549 cells, following treatment with different concentration of LiCl as indicated for 24 h.
GSK3β binds directly to survivin in vitro; activated GSK3β induces nuclear translocation of survivin in A549 cells
Because survivin is another important apoptosis-associated protein, we sought to investigate whether GSK3β could interact directly with survivin. By co-immunoprecipitation analysis, we found that GSK3β was indeed able to physically bind to survivin from cell line A549 (Fig. 2A). This direct interaction between GSK3β and survivin was further confirmed by way of immunoflurescence staining and confocal microscopy in A549 cells. As shown in Fig. 2B, GSK3β and survivin co-localized in both the cytoplasm and the nucleus. Transfection of the constitutively active GSK3β mutant plasmid S9A-GSK3β significantly increased the nuclear accumulation of survivin in A549 cells. To further verify that nuclear translocation of survivin is associated with increased GSK3β activity, the cytoplasmic and nuclear fractions of A549 cells that were either transfected with S9A-GSK3β and the dominant negative GSK3β mutant DN-GSK3β, or treated with the GSK3β inhibitor LiCl, were subjected to western blotting to ascertain the subcellular distribution of survivin in A549 cells under those conditions. Consistent with the results seen by immunofluorescence staining, the western blots confirmed that transfection of S9A-GSK3β increased the level of survivin in the nucleus, while the level of survivin in the cytoplasm was decreased (Fig. 2C). This suggests that increased activity of GSK3β mediates translocation of survivin from the cytoplasm to the nucleus. The GSK3β-mediated nuclear localization of survivin was further demonstrated using assays in which inhibition of GSK3β activity, either by transfection of DN-GSK3β or treatment with LiCl, enhanced the cytoplasmic expression of survivin (Fig. 2C and D). In the western blot assays identifying the cellular distribution of survivin, we found there was a concomitant increase in expression of nuclear GSK3β in S9A-GSK3β-transfected cells. While expression of GSK3β in the nucleus was not reduced, it was significantly increased in the cytoplasm of A549 cells that were either transfected with DN-GSK3β or treated with LiCl. In addition, we found that transfection of S9A-GSK3β downregulated nuclear cyclin D1, whereas transfection of DN-GSK3β or LiCl treatment upregulated nuclear cyclin D1 (Fig. 2C and D).
Fig. 2.
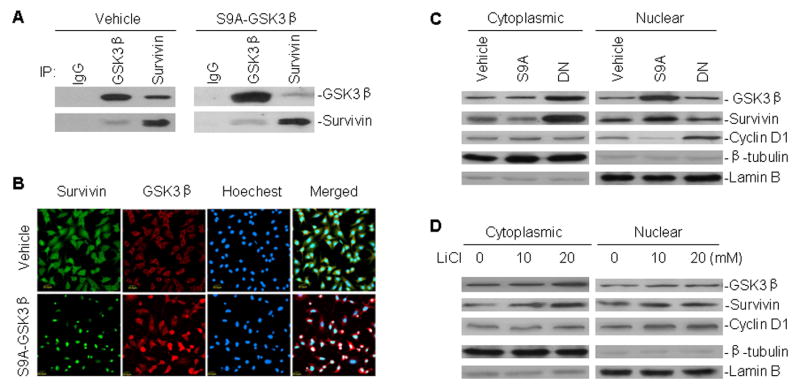
Interaction between GSK3β and survivin, and their subcellular distribution in A549 cells transfected with GSK3β mutant plasmids and treated with LiCl. A, binding of GSK3β to survivin in A549 cells. Cell extracts from A549 transfected with either S9A-GSK3β or control vehicle were immunoprecipitated with antibodies as indicated. Normal mouse or rabbit antibodies served as control (IgG). Proteins in immune complexes were separated on denaturing gels, transferred to filters, and detected by western blotting with anti-GSK3β and anti-survivin antibodies. Western blot and IP antibodies were from different species. B, A549 cells transfected with S9A-GSK3β and vehicle were grown on coverslips for 24 h, fixed in absolute methanol, processed by immunofluorescence staining, and detected by confocal microscopy for the interaction between GSK3β and survivin. C, cytoplasmic and nuclear extracts of A549 cells transfected with S9A-GSK3β (S9A) and DN-GSK3β (DN), subjected to western blot analysis for GSK3β, survivin and cyclin D1 expression. Lamin B and β-tubulin served as controls for equal protein loading of nuclear and cytoplasmic fractions, respectively. D, Western blot assay similar to (C), for subcellular distribution of GSK3β, survivin and cyclin D1 in A549 cells treated with different concentrations of LiCl, as indicated, for 24 h.
GSK3β-mediated nuclear accumulation of survivin induces G1 cell-cycle arrest
Previous studies suggested that nuclear survivin may exert some control over cell division, whereas the cytoplasmic survivin is considered cytoprotective [18]. Therefore, we evaluated the cellular consequences of the GSK3β-mediated nuclear accumulation of survivin in A549 cells. To accomplish this, we examined the cell-cycle progression of A549 cells under conditions in which GSK3β activity was either inhibited or activated, resulting in survivin's localization in the cytoplasm or nucleus, respectively. Upon inhibition of GSK3β activity in A549 cells by treatment with LiCl, the population of cells in G1 phase decreased and the population in S phase increased, as compared to cells not given LiCl treatment (Fig 3A and B). Similarly, inhibition of GSK3β activation by transfection of the dominant negative mutant DN-GSK3β plasmid also decreased the population of cells in G1 phase and increased those in S phase (Fig. 3C and D). In contrast, activation of GSK3β by transfection of S9A-GSK3β induced cell-cycle arrest in G1 phase (Fig. 3C and D). In order to confirm that it truly was the nuclear survivin regulated by GSK3β that rendered arrest in G1, we treated DN-GSK3β-transfected A549 cells with leptomycin B (LMB), a drug that irreversibly blocks the nuclear export of survivin as well as GSK3β, promoting accumulation of these proteins in the nucleus [6,27,28]. Treatment with LMB significantly reversed the previously observed DN-GSK3β-mediated decrease of cells in G1. As seen in Fig. 3C and D, the LMB-treated/DN-GSK3β-transfected A549 cells had G1/S phase populations similar to the A549 cells transfected with S9A-GSK3β.
Fig. 3.
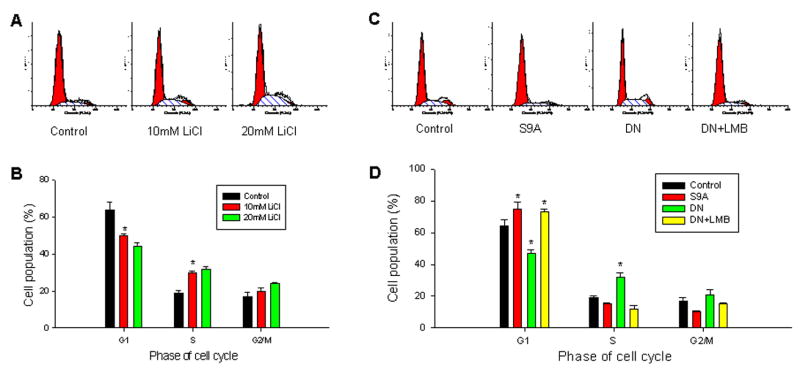
Effect of GSK3β-mediated nuclear localization of survivin on cell cycle progression in A549 cells. A, representative histographs of the cell cycle analysis of A549, following 24 h treatment with various concentrations of LiCl. B, calculation of cell populations (%) in different cell-cycle phases (G1, S and G2/M) for (A). C, representative histographs of cell cycle analysis of A549 transfected with GSK3β mutant plasmids S9A-GSK3β (S9A) and DN-GSK3β (DN). In cells transfected with DN, 10nM Leptomycin B (LMB) was added to the cell culture to block nuclear export of survivin. D, cell populations (%) in different cell-cycle phases (G1, S and G2/M) for (C). Data represent mean ± SD of three independent experiments. Statistical analysis by One-Way ANOVA test for significant differences; *P<0.05.
GSK3β-mediated translocation of survivin from cytoplasm to the nucleus promotes apoptosis
In addition to testing for influences on cell-cycle progression, we also evaluated the effect of the GSK3β-mediated subcellular distribution of survivin on apoptosis. First, we examined the levels of spontaneous apoptosis of A549 cells when transfected with each GSK3β mutant plasmid. As shown in Fig. 4A, the rate of apoptosis (as detected by Annexin V staining) was increased in the A549 cells that were transfected with the constitutively expressing S9A-GSK3β plasmid, whereas it decreased in cells transfected with DN-GSK3β, as compared with cells transfected with a control empty vector. Interestingly, treatment of DN-GSK3β-transfected cells with LMB, the drug that blocks nuclear export of GSK3β and survivin, reversed the inhibition of apoptosis. To further confirm the observed differences in spontaneous apoptosis rates in the A549 cells transfected with S9A-GSK3β and DN-GSK3β in the presence or absence of LMB treatment, we also employed detection by the Fluorometric TUNEL System and confocal microscopy. Results were similar: S9A-GSK3β increased apoptosis, DN-GSK3β decreased apoptosis, and LMB treatment that increased the nuclear expression of GSK3β and survivin reversed the inhibitory effect on apoptosis found in A549 transfected with DN-GSK3β (Fig. 4B and C). Therefore, in the A549 lung cancer cells, increases in nuclear survivin, whether GSK3β-activated or not, resulted in increased spontaneous apoptosis.
Fig. 4.
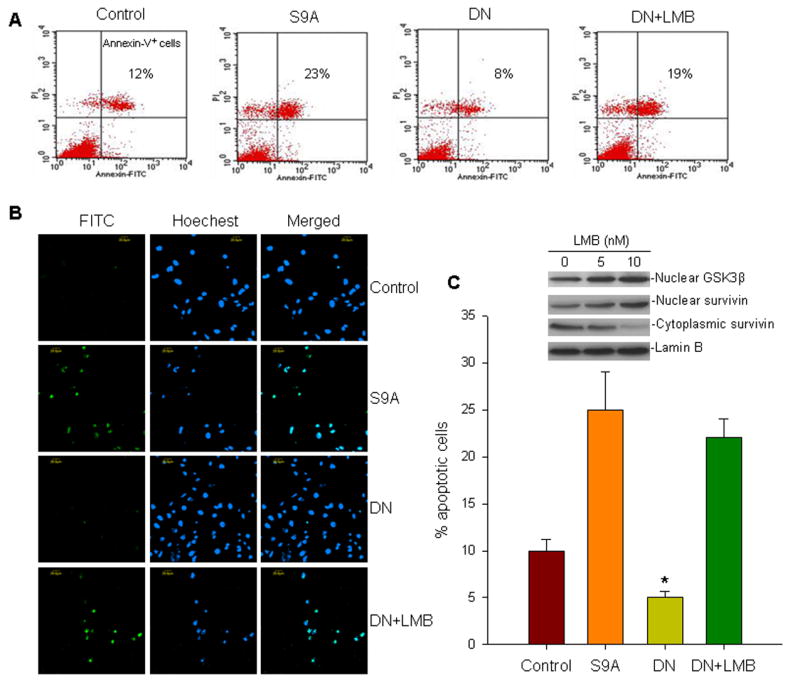
Apoptosis assay of A549 cells transfected with S9A-GSK3β (S9A) and DN-GSK3β (DN) in the presence or absence of LMB. A, representative histographs of Annexin-V analysis examined by flow cytometry. A549 cells were transfected with a control empty vector and the GSK3β mutant plasmids. Cells with DN plasmid were either treated with 10nM LMB or left without treatment. Transfected cells stained with FITC-conjugated Annexin-V and PI were analyzed by flow cytometry. B and C, apoptotic cell analyses of similarly transfected A549 cells treated with LMB as in (A), using the DeadEnd™ Fluorometric TUNEL System and confocal microscopy. A total of 300 cells per slide were scored as either FITC-positive or negative. The percentage of apoptotic cells was determined by the number of FITC-positive cells per slide/total number of cells counted per slide × 100%, as shown in (C). Data represent mean ± SD of three independent experiments; *p<0.01. Expression levels of nuclear GSK3β and survivin as well as cytoplasmic survivin in DN-transfected A549 cells treated with LMB, with Lamin B control, were analyzed by western blot (insert in C).
In addition, we also tested the effect of GSK3β activation on apoptosis induced by the chemotherapeutic drug doxorubicin. Similarly, activation or inactivation of GSK3β was achieved by transfection of S9A-GSK3β or DN-GSK3β. Activation of GSK3β by transfection of S9A-GSK3β sensitized A549 cells to doxorubicin-induced apoptosis, as measured by both activation of caspase-3 and cleavage of the death substrate PARP. As shown in Fig. 5A, there were strong western blot bands indicating caspase-3 activation (17 kD) and PARP cleavage (85 kD) after 24 h of doxorubicin treatment that were observed in S9A-GSK3β-transfected cells, but not in the DN-GSK3β-transfected cells. Likewise, in the quantitative apoptosis assay, there were significantly different percentages of annexin-V positive cells between S9A-GSK3β-transfected and DN-GSK3β-transfected A549 cells after 48 h of doxorubicin treatment. However, when the inhibitor of nuclear export of survivin LMB was added to the DN-GSK3β-transfected cells, promoting nuclear accumulation of survivin, the observed resistance to doxorubicin-induced apoptosis was reversed (Fig. 5B). In the A549 lung cancer cells, increases in nuclear survivin, whether GSK3β-activated or not, resulted in increased sensitivity to apoptosis by doxorubicin.
Fig. 5.
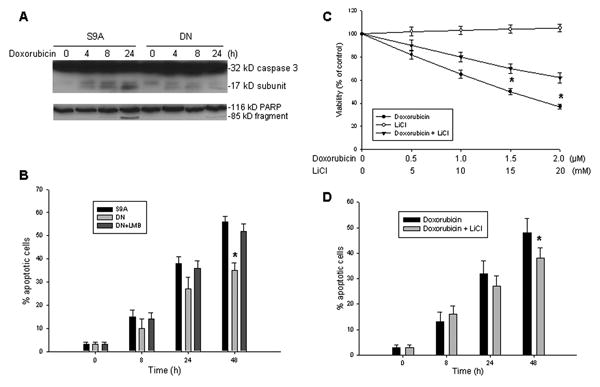
Effects of GSK3β activation on the cytotoxicity and apoptosis induced by doxorubicin, in A549 cells. A, western blot assay for activation of caspase-3 and cleavage of the death substrate PARP in A549 cells transfected either with S9A-GSK3β (S9A) or with DN-GSK3β (DN), treated with 2 μM doxorubicin for the indicated time points. B, quantitative detection of apoptotic A549 cells induced by doxorubicin. A549 cells transfected with S9A and DN in the presence or absence of LMB were treated with 2 μM doxorubicin for the indicated times. Apoptotic cells were detected by flow cytometry. Data represents the mean percentage of annexin-V positive cells from three independent experiments; bars, ± SD. *p<0.01. C, A549 cells treated with different concentrations of doxorubicin and LiCl alone, or with a fixed amount (10 mM) of LiCl and different doses of doxorubicin as indicated, following 48 h incubation. Cell viability determined by WST assay. *p<0.01. D, time-course of apoptosis induced by doxorubicin in combination with LiCl in A549 cells. Cells were treated either with doxorubicin (2 μM) alone or with LiCl (10 mM) added in, for the indicated time. Apoptotic cells were detected by annexin-V staining. *p<0.05.
To further confirm that inhibition of GSK3β influences cells undergoing apoptosis, we tested the effect that downregulation of GSK3β activity by LiCl treatment had on the sensitivity of A549 cells to apoptosis induced by doxorubicin. Treatment with LiCl alone only slightly increased cell survival. However, LiCl treatment greatly increased the resistance of A549 cells to doxorubicin, as is demonstrated in Fig. 5C, where a significant difference was readily notable in mean cell survival after a 48-h treatment, between cells treated with doxorubicin alone and doxorubicin plus LiCl, when doxorubicin concentrations of 1.5 μM and greater were employed (p<0.01). Consistent with these observations, a flow-cytometric apoptosis assay showed that a decreased percentage of A549 cells treated with the combination of doxorubicin and LiCl (from approximately 3% apoptosis in the control to 38% in treated cells) were annexin-V positive at 48-h post-treatment, as compared to doxorubicin alone (from approximately 3% to 50%) (Fig. 5D). In the A549 lung cancer cells, GSK3β activation promoted increased sensitivity to doxorubicin and its inactivation reduced that sensitivity, significantly increasing cellular resistance to doxorubicin.
Discussion
In this study, we have clearly demonstrated that GSK3β has a pro-apoptotic and proliferation-inhibitory effect on the human lung adenocarcinoma cell line A549. Our results are in agreement with other reports stating that GSK3β induces apoptosis in human breast tumor and medulloblastoma cells [11-13]. We employed gene transfection and pharmacologic inhibitor assays to either activate or inactivate GSK3β, to evaluate its roles in regulating cell cycle progression and apoptosis. We found that in A549 cells, an increase in GSK3β activity induced G1 cell-cycle arrest and apoptosis. In contrast, inactivation of GSK3β decreased the proportion of cells experiencing G1 arrest and protected cells from apoptosis, either of spontaneous origin or as induced by the chemotherapeutic drug doxorubicin. An important finding in this study was the discovery that GSK3β can bind to the antiapoptotic factor survivin. A related finding was that activation of GSK3β resulted in the accumulation of survivin in the nucleus. Because nuclear localization of survivin creates loss of its known cytoprotective function, we believe that the accumulation of survivin in the nucleus due to GSK3β regulation contributes, at least in part, to the previously observed pro-apoptotic function of GSK3β in certain tumor cells.
It is well known that GSK3β, an isoform of GSK3 family, is involved in mediating a diverse number of cellular signaling functions that are linked to either positive or negative regulation of cell proliferation, differentiation and survival. Therefore, it is not surprising that GSK3β has also been implicated in the pathological process of cancers. Previous reports demonstrate that GSK3β regulates cell survival in colorectal cancer and melanoma [29-31], but induces apoptosis in breast cancer and medulloblastoma [11-13]. These seemingly contradictory roles of GSK3β as a mediator of both cell survival and apoptosis are likely dependent on the cell types studied, the signaling pathways that are regulated by GSK3β and the activation status of GSK3β in the cells. In addition, a recent study by Meares et al [32] reveals that the subcellular distribution of GSK3β plays a critical role in the molecule's ability to regulate cell survival or apoptosis. Results show that the antiapoptotic effect of GSK3β in TNF-induced apoptosis is mediated only by cytoplasmic, not nuclear, GSK3β. A previous study also notes that nuclear GSK3β levels decrease in response to stimulation by proliferation growth factors, whereas the insults that induce apoptosis can cause accumulation of GSK3β in the nucleus [6]. Additionally, it was found that the active form of GSK3β that is not phosphorylated on serine-9 is in 5- to 8-fold greater quantity in the nucleus than in the cytoplasm of neuroblastoma cells [7]. Consistent with these observations, our results show that nuclear expression of GSK3β increased after transfection with the constitutively active S9A-GSK3β plasmid, resulting in cell-cycle arrest and apoptosis; whereas GSK3β increased in the cytoplasm after either transfection of a dominant negative DN-GSK3β plasmid or treatment with the inhibitor LiCl, leading to cell-cycle progression and protection from apoptosis. These results indicate that both activation and nuclear accumulation of GSK3β are needed for cell cycle arrest and apoptosis to occur. It is already known that the nuclear GSK3β is highly active in phosphorylation and proteolysis of cyclin D1 resulting in cell cycle arrest in G1, and the GSK3β inhibitor LiCl increases the nuclear level of cyclin D1 allowing the cell to progress to S-phase [6, 33]. Consistent with these observations, the present study shows that transfection of the constitutively active GSK3β in A549 cells downregulated nuclear cyclin D1, and treatment of these cells with LiCl increased the nuclear cyclin D1 (Fig. 2C and D). Furthermore, our results demonstrate an additional mechanism by which activated nuclear GSK3β regulates survivin that not only involves in cell cycle arrest but also regulates apoptosis. The discovery of this direct link between GSK3β and survivin should not only shed new light on the basic biology of cancer cells, but should add new targets for the development of therapeutics that can intervene in these important pathways.
A recent advance in the study of survivin is the discovery that survivin is a nuclear-cytoplasmic shuttling protein and that the subcellular distribution of survivin plays a distinct role in the ability of that molecule to regulate cell division and survival. While the expression of survivin in the cytoplasm is cytoprotective, nuclear accumulation of survivin may indeed impair survivin's most well known function [18]. The most recent findings by Connell et al [34] are that nuclear survivin has reduced stability and that sending survivin into the nucleus abolishes resistance to radioactivity. Similarly, our results showed that increasing nuclear survivin attenuated resistance to apoptosis by doxorubicin significantly.
Our laboratory is the first to reveal that GSK3β was able to physically interact with survivin. We also show that activation of GSK3β led to accumulation of survivin in the nucleus. Because GSK3β is a nuclear-cytoplasmic shuttling protein, it contains a bipartite nuclear localization sequence (NLS) that is necessary for its nuclear accumulation, and because the NLS alone is sufficient to drive yellow fluorescent protein into the nucleus [32], we hypothesize that GSK3β is most likely able to drive survivin into the nucleus after binding to it. However, despite having the NLS, most GSK3β is cytoplasmic. Therefore, it remains to be elucidated how and under what conditions the NLS regulates the nuclear translocation of GSK3β that may also carry survivin from the cytoplasm to the nucleus. Our results do indicate that the nuclear accumulation of survivin is closely associated with both activation of GSK3β and the increase in nuclear GSK3β. Therefore, we speculate that NLS-mediated nuclear localization of GSK3β, which can take survivin along with it into the nucleus, is dependent on GSK3β activation. This notion is consistent with previous observations that GSK3β is more highly active in the nucleus than in cytoplasm [7], plus our own experiment results that demonstrate that enforced overexpression of constitutively active GSK3β caused the nuclear accumulation of survivin as well as GSK3β; whereas transfection of dominant negative GSK3β or treatment with the GSK3β inhibitor LiCl did not increase nuclear survivin or GSK3β, but in contrast, increased the cytoplasmic expression of these two proteins.
The interaction between GSK3β and survivin in regulating the nuclear accumulation of survivin should have clinical significance. It is known that the majority of cancer patients whose tumor cells express high levels of cytoplasmic survivin respond poorly to chemotherapeutic treatment, whereas those patients whose tumor cells predominantly express nuclear survivin tend to have a relatively good treatment outcome. Importantly, our study demonstrated that redistribution of survivin into the nucleus can be achieved by increasing GSK3β activity. This novel insight implies that pharmacologic activation of GSK3β could be a promising way to improve the power of today's therapeutics in cancer patients, including the possibility of improving management of cases with apoptosis-resistant cancers.
Acknowledgments
This research was supported by a grant from the National Natural Science Foundation of China (No. 30470757 to RW), grants from the NIH (R01 CA123490 to MZ), the Leukemia and Lymphoma Society (6033-08 to MZ), CURE Childhood Cancer (to MZ), We are most grateful to Professor J.R. Woodgett for providing us with the constitutively active S9A-GSK3β and dominant negative DN-GSK3β mutant plasmids. We thank Kathleen Kite-Powell for manuscript editing.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ali A, Hoeflich KP, Woodgett JR. Glycogen synthase kinase-3: properties, functions, and regulation. Chem Rev. 2001;101:2527–2540. doi: 10.1021/cr000110o. [DOI] [PubMed] [Google Scholar]
- 2.Woodgett JR. Molecular cloning and expression of glycogen synthase kinase-3/factor A. EMBO J. 1990;9:2431–2438. doi: 10.1002/j.1460-2075.1990.tb07419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Frame S, Cohen P. GSK3 takes centre stage more than 20 years after its discovery. Biochem J. 2001;359:1–16. doi: 10.1042/0264-6021:3590001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Doble BW, Woodgett JR. GSK-3: tricks of the trade for a multi-tasking kinase. J Cell Sci. 2003;116:1175–1186. doi: 10.1242/jcs.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pap M, Cooper GM. Role of glycogen synthase kinase-3 in the phosphatidylinositol 3-Kinase/Akt cell survival pathway. J Biol Chem. 1998;273:19929–19932. doi: 10.1074/jbc.273.32.19929. [DOI] [PubMed] [Google Scholar]
- 6.Bijur GN, Jope RS. Proapoptotic stimuli induce nuclear accumulation of glycogen synthase kinase-3 beta. J Biol Chem. 2001;276:37436–37442. doi: 10.1074/jbc.M105725200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bijur GN, Jope RS. Glycogen synthase kinase-3 beta is highly activated in nuclei and mitochondria. Neuroreport. 2003;14:2415–2419. doi: 10.1097/00001756-200312190-00025. [DOI] [PubMed] [Google Scholar]
- 8.Ciani L, Salinas PC. WNTs in the vertebrate nervous system: from patterning to neuronal connectivity. Nat Rev Neurosci. 2005;6:351–362. doi: 10.1038/nrn1665. [DOI] [PubMed] [Google Scholar]
- 9.Grimes CA, Jope RS. The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog Neurobiol. 2001;65:391–426. doi: 10.1016/s0301-0082(01)00011-9. [DOI] [PubMed] [Google Scholar]
- 10.Takashima A, Noguchi K, Sato K, et al. Tau protein kinase I is essential for amyloid beta-protein-induced neurotoxicity. Proc Natl Acad Sci USA. 1993;90:7789–7793. doi: 10.1073/pnas.90.16.7789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alao JP, Stavropoulou AV, Lam EW, et al. Role of glycogen synthase kinase 3 beta (GSK3beta) in mediating the cytotoxic effects of the histone deacetylase inhibitor trichostatin A (TSA) in MCF-7 breast cancer cells. Mol Cancer. 2006;5:40. doi: 10.1186/1476-4598-5-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dong J, Peng J, Zhang H, et al. Meric-Bernstam, Role of glycogen synthase kinase 3beta in rapamycin-mediated cell cycle regulation and chemosensitivity. Cancer Res. 2005;65:1961–1972. doi: 10.1158/0008-5472.CAN-04-2501. [DOI] [PubMed] [Google Scholar]
- 13.Urbanska K, Trojanek J, Del Valle L, et al. Reiss, Inhibition of IGF-I receptor in anchorage-independence attenuates GSK-3beta constitutive phosphorylation and compromises growth and survival of medulloblastoma cell lines. Oncogene. 2007;26:2308–2317. doi: 10.1038/sj.onc.1210018. [DOI] [PubMed] [Google Scholar]
- 14.Watcharasit P, Bijur GN, Song L, et al. Glycogen synthase kinase-3beta (GSK3beta) binds to and promotes the actions of p53. J Biol Chem. 2003;278:48872–48879. doi: 10.1074/jbc.M305870200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Watcharasit P, Bijur GN, Zmijewski JW, et al. Direct, activating interaction between glycogen synthase kinase-3beta and p53 after DNA damage. Proc Natl Acad Sci USA. 2002;99:7951–7955. doi: 10.1073/pnas.122062299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li F, Ambrosini G, Chu EY, et al. Control of apoptosis and mitotic spindle checkpoint by surviving. Nature. 1998;396:580–584. doi: 10.1038/25141. [DOI] [PubMed] [Google Scholar]
- 17.Altieri DC. Survivin, cancer networks and pathway-directed drug discover. Nat Rev Cancer. 2008;8:61–70. doi: 10.1038/nrc2293. [DOI] [PubMed] [Google Scholar]
- 18.Stauber RH, Mann W, Knauer SK. Nuclear and cytoplasmic survivin: molecular mechanism, prognostic and therapeutic potential. Cancer Res. 2007;67:5999–6002. doi: 10.1158/0008-5472.CAN-07-0494. [DOI] [PubMed] [Google Scholar]
- 19.Knauer SK, Krämer OH, Knösel T, et al. Nuclear export is essential for the tumor-promoting activity of surviving. FASEB J. 2007;21:207–216. doi: 10.1096/fj.06-5741com. [DOI] [PubMed] [Google Scholar]
- 20.Temme A, Rodriguez JA, Hendruschk S, et al. localization of survivin renders HeLa tumor cells more sensitive to apoptosis by induction of p53 and Bax. Cancer Lett. 2007;250:177–193. doi: 10.1016/j.canlet.2006.09.020. [DOI] [PubMed] [Google Scholar]
- 21.Grabowski P, Kühnel T, Mühr-Wilkenshoff F, et al. Prognostic value of nuclear survivin expression in oesophageal squamous cell carcinoma. Br J Cancer. 2003;88:115–119. doi: 10.1038/sj.bjc.6600696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grabowski P, Griss S, Arnold CN, et al. Nuclear survivin is a powerful novel prognostic marker in gastroenteropancreatic neuroendocrine tumor disease. Neuroendocrinology. 2005;81:1–9. doi: 10.1159/000084892. [DOI] [PubMed] [Google Scholar]
- 23.Saito T, Arifin MT, Hama S, et al. Survivin subcellular localization in high-grade astrocytomas: simultaneous expression in both nucleus and cytoplasm is negative prognostic marker. J Neurooncol. 2007;82:193–198. doi: 10.1007/s11060-006-9267-1. [DOI] [PubMed] [Google Scholar]
- 24.Stambolic V, Woodgett JR. Mitogen inactivation of glycogen synthase kinase-3 beta in intact cells via serine 9 phosphorylation. Biochem J. 1994;303:701–704. doi: 10.1042/bj3030701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.He X, Saint-Jeannet JP, Woodgett JR, et al. Glycogen synthase kinase-3 and dorsoventral patterning in Xenopus embryos. Nature. 1995;374:617–622. doi: 10.1038/374617a0. [DOI] [PubMed] [Google Scholar]
- 26.Skurk C, Maatz H, Rocnik E, et al. Glycogen-synthase kinase3beta/beta-catenin axis promotes angiogenesis through activation of vascular endothelial growth factor signaling in endothelial cells. Circ Res. 2005;96:308–318. doi: 10.1161/01.RES.0000156273.30274.f7. [DOI] [PubMed] [Google Scholar]
- 27.Knauer SK, Bier C, Habtemichael N, et al. The Survivin-Crm1 interaction is essential for chromosomal passenger complex localization and function. EMBO Rep. 2006;7:1259–1265. doi: 10.1038/sj.embor.7400824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rodriguez JA, Lens SM, Span SW, et al. Subcellular localization and nucleocytoplasmic transport of the chromosomal passenger proteins before nuclear envelope breakdown. Oncogene. 2006;25:4867–4879. doi: 10.1038/sj.onc.1209499. [DOI] [PubMed] [Google Scholar]
- 29.Ghosh JC, Altieri DC. Activation of p53-dependent apoptosis by acute ablation of glycogen synthase kinase-3beta in colorectal cancer cells. Clin Cancer Res. 2005;11:4580–4588. doi: 10.1158/1078-0432.CCR-04-2624. [DOI] [PubMed] [Google Scholar]
- 30.Shakoori A, Ougolkov A, Yu ZW, et al. Deregulated GSK3beta activity in colorectal cancer: its association with tumor cell survival and proliferation. Biochem Biophys Res Commun. 2005;334:1365–1373. doi: 10.1016/j.bbrc.2005.07.041. [DOI] [PubMed] [Google Scholar]
- 31.Smalley KS, Contractor R, Haass NK, et al. An organometallic protein kinase inhibitor pharmacologically activates p53 and induces apoptosis in human melanoma cells. Cancer Res. 2007;67:209–217. doi: 10.1158/0008-5472.CAN-06-1538. [DOI] [PubMed] [Google Scholar]
- 32.Meares GP, Jope RS. Resolution of the nuclear localization mechanism of glycogen synthase kinase-3: functional effects in apoptosis. J Biol Chem. 2007;282:16989–17001. doi: 10.1074/jbc.M700610200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998;12:3499–3511. doi: 10.1101/gad.12.22.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Connell CM, Colnaghi R, Wheatley SP. Nuclear survivin has reduced stability and is not cytoprotective. J Biol Chem. 2008;283:3289–3296. doi: 10.1074/jbc.M704461200. [DOI] [PubMed] [Google Scholar]