

Abstract
Inhalation of the crystalline form of silica is associated with a variety of pathologies from acute lung inflammation to silicosis, in addition to autoimmune disorders and cancer. Basic science researchers looking at the mechanisms involved with the earliest initiators of disease are focused on how the alveolar macrophage (AM) interacts with the inhaled silica particle and the consequences of silica-induced toxicity on the cellular level. Based on experimental results, several rationales have been developed on exactly how crystalline silica particles are toxic to the macrophage cell that is functionally responsible for clearance of the foreign particle. For example, silica is capable of producing reactive oxygen species (ROS) either directly (on the particle surface) or indirectly (produced by the cell as a response to silica) triggering cell-signaling pathways initiating cytokine release and apoptosis. With murine macrophages, reactive nitrogen species (RNS) are produced in the initial respiratory burst in addition to ROS. An alternative explanation for silica toxicity includes lysosomal permeability, where silica disrupts the normal internalization process leading to cytokine release and cell death. Still other research has focused on the cell surface receptors (collectively known as scavenger receptors) involved in silica binding and internalization. The silica-induced cytokine release and apoptosis is described as the function of receptor-mediated signaling rather than free radical damage. Current research ideas on silica toxicity and binding in the alveolar macrophage are reviewed and discussed.
Keywords: Scavenger receptor, Macrophage, Silica, Free radicals, MARCO, SR-A, Immune modulation
Introduction
Silicon (Si) is the second most abundant element on earth next to carbon. A Si atom combined with 2 oxygen (O) atoms creates silicon oxide or silica (SiO2) naturally occurring as quartz or sand. There are multiple crystalline forms and one amorphous form of silica. Inhalation of the crystalline form of silica has been historically associated with the development of a severe respiratory disease, silicosis, which is a lung pneumoconiosis characterized by alveolar proteinosis and diffuse fibrosis resulting in progressively restrictive lung function [1]. Silicosis has been primarily associated with long occupational exposures to crystalline silica that typically occur in sandblasting, silica milling, rock drilling and tunneling [2, 3]. There is evidence that silica exposure can also be linked to the development of autoimmune diseases such as scleroderma (systemic sclerosis), rheumatoid arthritis, chronic renal disease and lupus [4]. Additionally, silica inhalation is believed to be the cause of some rare lung cancers [5], although significant relative risk (RR) of lung cancer is only associated with individuals that already have silicosis from silica exposure [6]. Based on the relative dose-response or exposure-response relationships in experimental animal studies, silica appeared to be a uniquely hazardous particle type [7].
Given the diversity of pathologies associated with silica exposure, it is unlikely that one common mechanism is responsible for all of the possible diseases. Although the exact sequence of events (from silica inhalation to disease) is not known, it is generally accepted that the alveolar macrophage (AM) is a relevant cell type to study [1]. Because the role of the AM is to clear the lung of inhaled debris, it is reasonable to assume that the macrophage is the first cell of the body that will have significant contact with the inhaled silica particle. Upon contact, the AM will bind to the silica and begin to engulf the particle. If the AM survives the silica encounter, it will likely migrate out of the lungs to either the proximal lymph nodes or through the mucosal-ciliary escalator and eventually out of the respiratory tract [8]. If the AM stays in the lung it will migrate to the interstitial space and become an activated interstitial macrophage (IM) that could contribute directly to pathogenesis [9]. Some research indicates that the IM may play important role in the progression of silica-induced lung disease [10, 11].
Researchers studying the AM/silica particle interaction have developed several hypotheses to describe how silica is toxic to AM. Some explanations of toxicity focus on the surface qualities of the silica such as free radicals and surface charge. Others suggest that silica can cause the AM to self-destruct by apoptosis or lysosomal disruption, which could lead to the development of autoantigens. The purpose of this review article is to present all of the current research regarding silica toxicity of the AM because most researchers in this area would agree that the initial toxicity of the AM is an important first step in the development of disease.
SURFACE MODIFCATIONS OF INHALED SILICA BY LUNG SURFACTANT
Before the inhaled silica particles are encountered by AM, lung surfactant composed of phospholipids (PL) and surfactant proteins (SP) could potentially coat the outer surface of the silica particles modifying the surface chemistry and ultimately influence the toxicity. This interaction can be further complicated by free radical modifications of the phospholipids and proteins occurring on surface contact with the silica. Some research has focused on this aspect of silica toxicity by using surfactant components in in vitro and in vivo toxicity models. An illustration of this process can be found in Figure 1.
Figure 1.

Schematic of bronchio-alveolar internal spaceillustrating the source and location of epithelial cells responsible for phospholipid (PL) and surfactant protein (SP) production. Also indicated in this figure: the alveoli as a respiratory endpoint for inhaled silica (SiO2) particles, where phagocytic alveolar macrophages (AM) will internalize the particles for removal from the lung while signaling to the epithelial cells for surfactant overproduction.
Effect of silica exposure on lung surfactant
It has been known for some time that silica inhalation is associated with increased PL and SP in the lung. Early studies in animal models indicated a general increase in surfactant volume, but not composition in response to silica [12, 13], which according to Muller et al, is unique to silica inhalation compared to tobacco smoke and diesel particles [14]. One study found 3–7 fold increases in PL and SP, respectively, occurring in a rat model with acid-washed silica, but not with unwashed silica [15]. Another study found sustained elevations (in weeks) in PL and SP-A in response to silica [16]. Several other studies found silica-induced increases in PL and SP [17, 18] including specific increases in SP-A [19, 20], SP-D [19, 21], Vitamin E [22], and phosphotidyl inositol [23]. In contrast, Seiler et al, found a dose-dependent decrease in phospotidyl glycerol in response to silica [23].
The source of increased surfactant production following silica exposure is the alveolar type II cell and the bronchiolar epithelial cell [1]. There is some evidence that the AM, in response to silica stimulation, signals the type II cell to produce more surfactant [24]. Isolated type II cells following silica-induced lung injury were found to have increased SP-A receptor activity [25], indicating the increase in surfactant production may be receptor regulated. The exact trigger of surfactant overproduction is not well described, but it is believed that there is cell to cell communication between AM, type II cells and epithelial cells resulting in increased PL and SP. Kanj et. al. suggests that surfactant from type II cells can have a direct inhibitory effect on AM beyond simply coating the silica particles [24].
Modification of silica toxicity by lung surfactant
The purpose of the silica-induced surfactant overproduction is a protective mechanism, wherein the raw inhaled silica particles are coated with lung surfactant PL and SP prior to contact with the responsive AM. Several studies have demonstrated the protective effects of surfactant coating on silica toxicity. The synthetic surfactant (Suvanta™) protected AM in vitro and reduced lung injury in vivo while having no effect on neutrophil (PMN) infiltration [26]. The same research group used amiodarone to inhibit phospholipase activity and generate phospholipidosis artificially, which proved to be protective with silica exposure, again having no effect on PMN influx [27]. The surfactant PL component dipalmitoyl phosphatidylcholine (DPPC, also referred to as dipalmitoyl lecithin) has been used as a protective pre-coating agent against silica toxicity [28–30], but was ineffective at inhibiting chysotile asbestos toxicity [29]. In addition, SPA and D have been used to attenuate silica toxicity in vitro [19, 20]. Research clearly indicates that lung surfactant has a prophylactic effect with regard to silica toxicity, and it is not limited to a specific component of the surfactant, as various PL and SP have a similar protective effect.
There is evidence that the protective effect against silica-induced lung damage by lung surfactant coating is only temporary. Research with DPPC as the protective component indicates that the protection lasts for only 3 days in vitro because the particle coating is destroyed by enzymatic digestion (either by phospolipase A2 activity or by AM lysosomal enzymes) [31–33]. One problem with surfactant overproduction is that sustained and uncontrolled over a period of time, it can become a pathological feature of silica exposure. Proteinosis and phospholipidosis of the lungs, secondary to silicosis, is characterized by appearance of tubular myelin-like multilamellated structures in the alveoli and distal airways [34].
THE RECEPTOR-MEDIATED HYPOTHESIS OF SILICA BINDING AND TOXICITY
As inhaled silica particles are encountered by AM in the lung, the physical contact at the cell/particle interface is the first critical step in particle recognition and internalization. The important factors in silica particle recognition and binding are the physical surface characteristics of the particle and the pattern recognition receptors that act as an adhesive [35, 36], binding the silica for further processing by the AM. Some researchers believe that the specific receptors involved in this process are ultimately mediating silica toxicity by internal cell signaling leading to AM apoptosis. Further evidence suggests that these silica-binding receptors are involved in inflammation regulation and normal immune function [37, 38], which can be disrupted by excessive silica exposure.
Macrophage surface receptors involved in silica binding
Macrophage scavenger receptors (SR) are well described in a review article by Murphy et. al. [39]. The SR are cell surface protein receptors that bind a wide variety of ligands. These ligands include, but are not limited to, bacteria, unopsonized particles, and modified (oxidized or acetylated) low-density lipoproteins (ox-LDL or ac-LDL respectively). There are eight known classes of SR designated A through H, which are dissimilar in structure, but related in ligands recognized. Known class A SR consist of three splice variants SR-AI, SR-AII, and SR-AIII, MARCO [40, 41], SRCL [42], and SCARA5 (unique to epithelial cells) [43]. MSR, MSR-1, and CD204 are common synonyms for SR-AI/II. The SR-A’s and MARCO share a similar structure as transmembrane proteins with a N-terminus cytoplasmic domain in addition to an extracellular trimeric collagenous domain, ending with a cysteine-rich C-terminus (SRCR). SR-A also has an α-helical coiled-coil domain not present on MARCO, but MARCO has more collagen repeats. SR-AII lacks the SRCR C-terminus [37, 39, 44].
The SR most associated with AM silica binding are SR-AI, SR-AII and MARCO [35, 36]. Using a CHO cell line transfected with murine SR-AII, our research group determined that the presence of SR-AII was crucial for the cytotoxicity of crystalline silica determined by apoptosis and cell viability [45]. The collagen-like domain of SR-A, regardless of type, has been implicated as the most likely binding site for crystalline silica. This can be deduced by the fact that both SR-AI and SR-AII bind silica, both have the collagen-like domain, but only the SR-AI subclass has the SRCR region making it unlikely that the SRCR region is involved in SR-A binding of silica. The SR-A collagen-like domain, specifically highly conserved lysine clusters, has been identified as the active binding region for other SR ligands [46, 47]. SR ligand recognition depends on charge-dependent conformational interactions throughout the full length of the collagen-like domain [47]. The blocking SR-A antibody, 2F-8 specific for SR-AII, was effective in blocking the cytotoxic effects of silica in vitro [45, 48].
Several studies by Kobzik’s research group have implicated MARCO as a critical binding receptor for non-opsonized inhaled environmental particles and bacteria, using both transfected cell lines and the MARCO−/− null mouse model [49–51]. The MARCO SRCR region is the most likely binding region for crystalline silica (manuscript in preparation). Ojala et. al. propose that multiple MARCO receptors can group together on the surface of the cell allowing the SRCR region to dimerize or oligomerize creating a much larger binding surface area, that is capable of binding physically large ligands such as silica and bacteria [52]. A schematic of the SR and the proposed silica binding sites can be found in Figure 2.
Figure 2. Schematic of A class scavenger receptors involved with silica (SiO2) binding.

The extracellular functional domains and proposed silica binding locations are indicated for SR-AI, SR-AII, and MARCO membrane receptors.
Macrophage surface receptors involved in silica toxicity
Silica-induced toxicity via SR has been described in a number of studies. Iyer et. al. showed that SR-A was necessary for silica initiation of apoptosis using human AM and SR-A inhibitors in an in vitro study [53]. Further evidence from the same group determined that the SR-AII blocking antibody (2F-8) could significantly attenuate caspase activity following silica exposure in murine MH-S cells [48]. In vivo studies using SR-AI/II−/− null mice on the 129SVJ background that were instilled with crystalline silica demonstrated that mice lacking SR-A failed to develop lung fibrosis but did show massive unresolved inflammation [54]. However, we were unable to repeat this experimental result in the C57BL/6 model, indicating a possible strain variation in response to instilled silica (unpublished observation).
Studies using MARCO−/−, SR-AI/II−/−, CD36−/− single and double null combinations on the C57BL/6 mouse model demonstrated that MARCO was exclusively associated with in vitro silica-induced AM apoptosis and cytotoxicity, in addition to silica binding and uptake [55]. The constitutive SR-AI/II expression level could not be determined using the 2F-8 antibody due to a polymorphism unique to the C57BL/6 mouse, and therefore the SR-A lack of association with silica toxicity or binding could not be fully assessed [55]. Clearly, there are variations in the roles of the different SR-A family members on different murine strains as to which SR is constitutively expressed, dominating the initial AM response to SR ligands. Nevertheless, the results are consistent that one or more SR-A family members are involved in triggering AM apoptosis.
Sufficient evidence exists to show the association of SR-A and MARCO with silica binding and toxicity, but the weakness of the receptor-mediated hypothesis is a lack of information about the chain of events following silica binding ultimately resulting in AM cell death. Another unresolved issue with the receptor-mediated hypothesis is that non-toxic particles such as titanium dioxide (TiO2) are known to bind to the same receptors without triggering the death response [36]. There are a few reports addressing the potential signaling pathways/processes involved following SR-A and MARCO binding. One study, using a synthetic bovine SR-A construct, demonstrated that heat-shock proteins HSP90 and HSP70, in addition to glyceraldehyde 3-phosphate dehydrogenase bound to the cytoplasmic N-terminus of the SR [56]. The involvement of HSP could help explain the SR ligand internalization process. Hsu et. al. found SR-A ligands induced tyrosine phosphorylation followed by activation of protein kinase C (PKC) resulted in urokinase-type plasinogen activator expression in THP-1 cells [57]. In a related topic, crocidolite asbestos has also been shown to trigger the PKC (delta isoform specific) pathway leading to apoptosis, but it was not linked with SR activation [58]. Fong and Le identified serine phosphorylation following ac-LDL stimulation and could be disrupted using SR-A receptor mutants on CHO and COS cells at the Ser49 and Asp25 amino acid sites [59].
Although it is not clear how SR can send a differential signal discriminating between various ligands, there is evidence that this could occur. In a study by Hsu et. al., it was demonstrated that two different SR-A ligands (ox-LDL and fucoidan) could trigger differential cell responses using slightly different protein kinase signaling pathways [60]. Most of the research in this area has focused on SR-A receptor signaling. There is obviously a lack of research regarding MARCO signaling, but the receptor protein structure, with the exception of the α-helical coiled-coil domain on SR-A, is very similar especially in the cytoplasmic domain. It is possible that the signaling processes are similar for both receptors, but that remains to be determined.
It would be inaccurate to characterize the SR-A and MARCO receptors as completely redundant surface proteins. There is obviously overlapping functions between the receptors, but some studies suggest differential regulation and function. A study by Jozefowski et. al. suggested that SR-A and MARCO expression were regulated by Th1 and Th2 polarizing factors, as MARCO was increased in the presence of Th1 adjuvants and decreased in the presence of Th2 polarizing factors in J774 macrophage-like cell line in vitro [61]. In addition, MARCO null mice were associated with inhibited IL-12 production, while SR-A null mice were associated with enhanced IL-12 production compared to wild-type mice in response to LPS or IFN-γ [61]. This observation was consistent with their earlier work where the presence of the SR-A receptor suppressed IL-12 release by 25% in AM from C3H/HeJ mice exposed to LPS or IFN-γ [62].
Other pattern recognition receptors may be involved in silica binding and toxicity in the AM. An excellent review article on macrophage pattern recognition receptors is provided by Gordon [63]. Some other possibilities could include the other classes of SR since they share many similar ligands (note: our lab has determined that CD36 is not likely to be involved with silica toxicity [55]). The type 3 compliment receptor may be involved if the silica becomes opsonized prior to cell contact. Toll-like receptors (TLR), mannose receptors, and integrins have not been investigated in silica binding and toxicity but could function in an accessory role. For example, the presence of TLR-4 has been shown to be important for the induction of MARCO expression using TLR-4 mutant mice [64]. Adhesion protein ICAM-1, although not directly involved in silica uptake, is upregulated on AM and other cell types with in vivo and in vitro silica exposure and believed to initiate the inflammatory PMN influx into the lungs [65]. In addition, other receptors with SRCR domains could potentially bind silica particles.
THE FREE RADICAL HYPOTHESIS OF SILICA TOXICITY
When researchers refer to free radical damage initiated by silica inhalation, they are usually describing more than one possible process. There are several possible sources of free radicals resulting from silica internalization including particle-derived reactive oxygen species (ROS), cell-derived ROS and reactive nitrogen species (RNS), and the interaction of particle and cell-derived free radicals producing peroxynitrite (ONOO•−) from nitric oxide (NO•) and superoxide anion (O2•−) [66]. In addition, the AM process of, silica phagocytosis can produce O2•−, hydrogen peroxide (H2O2), and hydroxyl radicals (HO•) [67]. Although not a free radical, silanol groups (SiOH) are also studied because they form hydrogen bonds with oxygen and nitrogen groups in biological membranes that affect hydrophilicity, binding and absorption of silica particles [66, 67]. Free radicals are potentially toxic to AM and surrounding lung tissue because they damage all types of cellular macromolecules including carbohydrates, nucleic acids (mutation), lipids (lipid peroxidation), and proteins via amino acid modifications [68]. Repeated free radical damage could result in disease especially when inflammation is a initial factor [69]. Figure 3 illustrates the free radical toxicity model.
Figure 3. Diagram of the free radical hypothesis of silica toxicity in the alveolar macrophage.
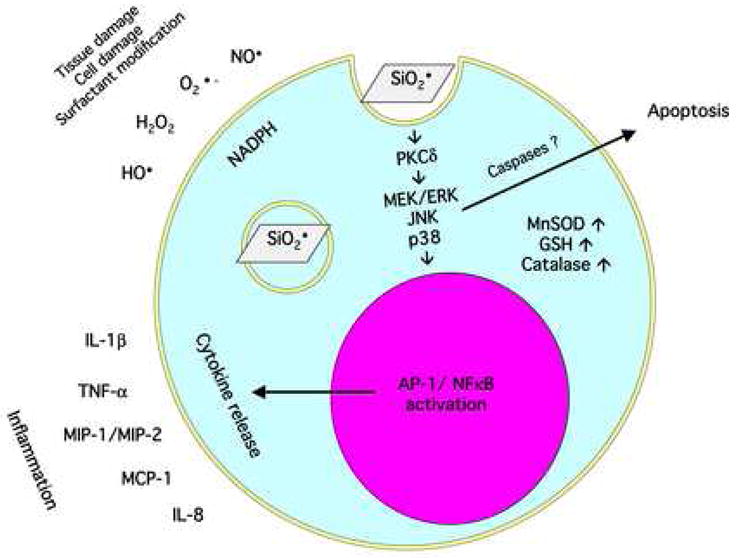
An overview of the effect of silica (SiO2 indicated with surface oxygen radical) endocytosis; the consequential respiratory burst, the signal transduction resulting in inflammatory cytokine release, and the antioxidant compensatory mechanisms are illustrated in this figure.
Particle-derived Free Radicals
The most compelling evidence for oxygen radicals causing silica toxicity is based on the observation that freshly fractured silica particles are more toxic in vitro [70–72], and more inflammatory [73], and fibrogenic in vivo [74] when compared to aged silica. This obviously has relevance to occupational exposure, because in most situations workers will inhale freshly fractured silica dusts. The silicon-oxygen bond is cleaved two ways when it is physically fractured: hemolytic cleavage producing Si•, SiO•, and heterolytic cleavage producing Si+, SiO+ [75]. Oxygen will in turn react with these byproducts producing creating several ROS (SiO2•, SiO3•, and Si+-O•−), which are visible in the Electron Paramagnetic Resonance spectrum of the silica [76]. Other potential free radicals are created on the surface of freshly fractured silica. For instance, Si• can react with gases such as CO2 to produce SiCOO• [77].
Water reacts with freshly fractured silica to produce H2O2, HO•, O•−, and 1O2 [66]. The hydroxyl radical (HO•) can occur by Fenton reaction (when iron traces are available), or by Haber-Weiss cycle (with iron traces and a reductant species present), or directly from surface ROS and water or H2O2 [66]. The Fenton process can be inhibited by the iron chelator desferrioxamineeral, and the ability of fractured silica to produce HO• decreases with time (20 hour half-life) [67]. It is believed that silica contaminated with iron is more cytotoxic than uncontaminated silica [78] because of the significant increase in hydroxyl radicals [79].
Researchers in this area admit that ROS is not completely responsible for silica toxicity [66]. In studies with artificial crystalline silica, the amount of exposed surface and size/shape of the particle also influenced cytotoxic potential [80]. Evidence strongly suggests that silica-derived ROS is directly responsible for in vitro DNA damage [81–83], various morphological changes including apoptosis in in vitro cell cultures [79, 84, 85], and acute lung damage in vivo [71]. The DNA damage is caused primarily by the hydroxyl radical, which is the by-product of iron contaminated silica [86]. Nevertheless, freshly fractured silica, regardless of iron content, can produce some oxidative DNA damage in vitro [86].
Cell-derived Free Radicals
Phagocytic cells (AM), respond to silica with increased oxygen consumption [67], production of superoxide anion O2•−, H2O2 [72, 87], and increased NO• synthesis, in addition to nitric oxide synthase [88]. All of these AM responses are enhanced in the presence of freshly fractured silica [67]. HO• production in rat lung tissue following silica instillation has also been described using titanium dioxide as a negative control particle [89]. Further evidence for silica-induced ROS in rat model lungs comes from observed increases in antioxidant enzymes such as manganese superoxide dismutase (MnSOD) from type II epithelial cells [90], along with increased SOD and glutathione peroxidase mRNA in rat lungs following silica inhalation [91]. Antioxidant depletion has also been described in silica-exposed rat AM in the form of reduced intracellular glutathione (GSH). It was also noted in this study that the GSH precursor (n-acetylcysteine) decreased ROS formation, resulting in reduced membrane permeability changes and DNA damage [92]. Also notable is the possible formation of peroxynitrite from the reaction of NO with O2•−, although to date there is no direct experimental evidence of this reaction occurring with silica inhalation.
Free radical damage is only the initiating event that results in a cascade of cellular responses such as signaling pathway activation, inflammatory cytokine release, and transcription factor activation [66, 86]. For example, silica-induced free radicals activated MEK and ERK phosphorylation in a rat fibroblast cell line [93]. Silica-induced oxidative stress has also been linked to NFκB and AP-1 transcription activation in a number of studies [66, 86]. Two studies by Chen et. al. using the murine macrophage cell line RAW 264.7 demonstrated that silica exposure could induce NFκB activation in a differential manner from lipopolysaccharide (LPS) stimulation with regard to the signaling pathways involved [94, 95]. Shi et. al. proposed that the HO• radical was most likely responsible for the NFκB activation based on using a variety of ROS inhibitors where only SOD failed to inhibit NFκB activation [96]. In vivo evidence of silica-induced NFκB gene expression in luciferase reporter mice was reported by Hubbard et. al. [97]. Luciferase reporter mice have also been used to show AP-1 activation in vivo following silica inhalation through ERK and p38 MAPK signaling pathways [98]. In vitro evidence for silica-initiated oxidative stress-induced AP-1 activation associated with JNK signaling, via c-Jun-NH2-terminal amino kinases, can be found in Shukla et. al. [99]. In a rat AM model, Liu et. al. demonstrated TNF-α and IL-1β release following silica was mediated through phosphotidylcholine-specific phospholipase C regulated in a redox-dependent fashion [100]. The inflammatory cytokines most commonly associated with silica-induced free radicals are TNF-α [101, 102], IL-1β [100], MIP-1, MIP-2, MCP-1 [103], and IL-8 (following TNF-α priming) [104].
Critics of the free radical hypothesis would point out that the lung is normally an oxidative environment where lung cells of many varieties possess inherent antioxidant defenses, such as catalase (eliminating H2O2), GSH (general antioxidant), and SOD (converting O2•− to O2 and H2O2). The hydroxyl radical is the most potentially damaging ROS (due to very short half-life, high reactivity, and lack of effective elimination) to the lung, but it is produced to any significant degree only in the presence of contaminants such as iron (Fenton reaction). Crystalline silica regardless of source, purity, and age is capable of inducing various lung pathologies [6]. Therefore, it is reasonable to assume that free radicals are not the only cause of silica-induced disease.
THE LYSOSOMAL PERMEABILITY HYPOTHESIS OF SILICA TOXICITY
When AM encounter inhaled crystalline silica particles, a process to engulf the particles is initiated immediately. The internalized silica particle is entrapped in a lysosomal compartment where low pH conditions activate strong digestive enzymes that attempt to degrade the particle, as it would if bacteria were being internalized. The silica particle cannot be broken down in the lysosomal compartment, and the resulting failure can lead to a loss of membrane integrity in the process. Some researchers believe that the failed attempt at silica digestion in the AM leads to caspase activation and apoptosis [105, 106]. A model illustrating this hypothesis can be found in Figure 4.
Figure 4. Diagram of the lysosomal permeability hypothesis of silica toxicity in the alveolar macrophage (AM).

The leakage of active cathepsin D from the lysosomal membrane leads to acidic sphingomylinase activity, ceramide generation, mitochondrial depolarization, caspase activation and ultimately cell apoptosis.
One of the earliest observations that internalized silica can disrupt normal endocytosis for the AM was described in a study using bovine AM exposed to silica for 20 hours. The untreated silica caused a significant reduction of lysosomal enzyme (cathepsin B) activity that was inhibited by silica pretreatment with dipalmitoyl lecithin or the presence ammonium chloride [28]. Another study showed that serum-coated silica caused lysosmal rupture in a murine macrophage cell line (J774). It was hypothesized that lysosomal destruction was due to iron-catalyzed peroxidative damage because the iron chelator desferrioxamine partially inhibited the lysosomal destruction [107]. The most comprehensive study on the matter by Thibodeau et. al., using the murine macrophage cell line MH-S, showed lysosomal permeability after only 1 hour of silica exposure using fluorescent markers. A variety of inhibitors of the lysosomal processing resulted in reduced caspase activation and apoptosis [106]. This study concluded that the apoptotic signaling pathway involved lysosomal leaking, cathepsin D and acidic sphingomyelinase, which preceded the mitochondrial depolarization and caspase 3 and 9 activation (reported earlier by the same group) caused by silica exposure [105]. It was further determined that ROS was not the cause of the lysosomal permeability [106].
THE ROLE OF SILICA EXPOSURE IN MACROPHAGE IMMUNE DYSFUNCTION
The development of autoimmune disease is obviously a complex process that involves more than one cell type. Silica exposure has long been associated with a variety of autoimmune problems, including rheumatoid arthritis, lupus, scleroderma, and glomerulonephritis with significant RR of greater than 3.0 [4]. The cause of silica-induced autoimmunity is undetermined at this time, but experimental evidence does exist suggesting silica can alter normal AM immune function. Coupled with the fact that silica induces AM apoptosis along with apoptosis of other cell types (possibly excessive), researchers have established some idea of the early events leading to immune dysfunction. Figure 5 illustrates this concept.
Figure 5. Schematic of the potential sources of autoimmune dysfunction with regard to silica (SiO2) processing and the alveolar macrophage (AM).
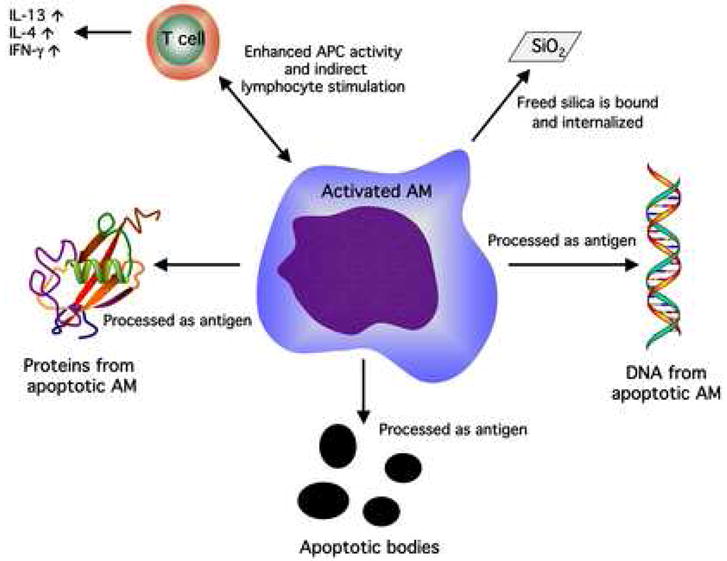
Excessive cell death and apoptosis can lead to a source of autoantigens (apoptotic bodies, self-protein, DNA). The AM/T cell interaction following silica exposure in vitro has been characterized by enhanced T cell activity determined by excessive IL-4 (human only), IFN-γ, and IL-13 release. The free silica particle is indicative of the fact that SiO2 can be internalized more than once by different AM due to its capacity to kill the engulfing cell.
Both ex vivo exposures of AM, and in vitro exposures of bone-marrow-derived macrophages (BMDM), to silica in the murine model have shown increased (relative to basal production) lymphocyte cytokines (IL-13 and IFN-γ) using an antigen-presenting cell (APC) assay in macrophage/lymphocyte co-cultures, [9]. This effect was not necessarily antigen dependent because the lymphocyte-derived cytokine increase could be stimulated in the absence of antigen using CD3 antibody and the presence of silica-treated AM. We have determined that this extra lymphocyte stimulation is probably not the result of secreted AM mediators (using transwell culture chambers isolating AM from lymphocytes), but more likely the product of AM surface expression because direct AM/lymphocyte contact is required (unpublished observation). This result was supported by another study using human AM exposed in vitro to silica resulting in increased lymphocyte cytokines (IL-4, and IFN-γ), using a human APC cell culture model [108].
Related studies have shown that silica exposure (24 hours) to human AM in vitro shifts macrophage phenotypes from a suppressive immune mode to an active immune mode using changes in RFD surface marker expression [109]. An in vivo study using mice reached a similar conclusion as the normal immune suppressive AM population was replaced by immune active dendritic cells (DC) following silica instillation. Further evidence in this study showed that DC preferentially migrated to the lung parenchyma from the alveoli increasing the numbers of activated T lymphocytes in response to silica [110]. Davis et. al. describes a cycle that could possibly ensue once the lung lymphocytes are activated. This would involve the release of IFN-γ and other mediators attracting and activating a secondary population of macrophages creating a cycle or loop of inflammation [111]. An additional study by Borges et. al. demonstrated that Fas ligand mediated lymphocyte apoptosis was a feature of this lung parenchyma inflammation affecting the PMN influx in the murine model in vivo. They also noted enlarged thoracic lymph nodes with immunological abnormalities following silica instillation [112]. The later finding was supported by an earlier study in rats exposed to repeated aerosolized silica, where enlarged lymph nodes were characterized by early T cell infiltration (2 weeks), followed by B cell infiltration (6 weeks) and then finally by macrophages accumulating in granuloma structures surrounded by an even distribution of T and B cells (52 weeks) [113].
The silica-induced apoptosis, regardless of cell type involved, has been suspected to be the source for auto-antigenic material. Brown et. al. established that silica exposure in a lupus-prone New Zealand mixed (NZM) mouse model can accelerate and exacerbate autoimmunity symptoms leading to premature death including the generation of autoantibodies associated with autoimmune disease [114]. This model was further used to show that silica-exposed mice generated autoantibodies to apoptotic cells. By inducing apoptosis in MH-S murine macrophages and staining with serum from NZM mice, they were able to establish that only apoptotic cells or lysates from apoptotic cells reacted to serum containing autoantibodies, unlike necrotic cell or live cells [115]. Finally, the same group successfully reversed the silica-induced exacerbation of autoimmune disease in the NZM mice by using weekly instillations of rottlerin (apoptosis inhibitor disrupting PKCδ pathway) confirming the hypothesis that silica-induced AM apoptosis was probably responsible for auto-antigen production leading to enhanced autoimmune disease [116].
CONCLUSIONS
Several biological rationales describing the binding and toxicity of silica with the AM have been presented in this review article. A summary table describing the relative strengths and weaknesses of each theory of silica toxicity can be found in Table I. It should be noted that none of the hypotheses presented here are mutually exclusive of one another. It is possible that all of the research in this area is the correct interpretation of the toxicological reality silica inhalation presents. Several possibilities exist to explain the diversity of the research results concerning silica toxicity. First, there may simply be more than one mechanism of silica-induced cell death in the AM and other cells. This might help explain why there is a diversity of pathologies linked to silica exposure and inhalation. Second, the different toxicity mechanisms described in this article may work in tandem. For example, SR may be the way silica binds to the AM initially, followed by particle endocytosis complete with a respiratory burst creating ROS and NOS, followed by PKC-mediated MAP kinase signaling cascades resulting in AP-1 and NFκB activation, which ultimately results in cytokine release (IL-1β, MIP-1, MCP-1 and TNF-α). The sequestered silica particle, being bombarded by acidifying digestive enzymes, creates a leaking lysosomal membrane (by unknown mechanism - possibly ROS) and releases cathepsin D into the cytoplasm. This then leads to mitochondrial depolarization, caspase 3 and caspase 9 activation, and apoptosis. Upon cell death the silica particle or particles become cell-dissociated and the silica/AM cycle begins anew. Auto-antigens also result from the excessive AM apoptosis and ultimately lead to chronic inflammation and immune dysfunction. Cell death is obviously not the only pathological consequence to silica exposure. Surviving AM exposed to silica can be altered (activated, for example) in such a way that cellular interactions are contributing to the disease process (e.g., excessive collagen deposition from lung fibroblast triggered by lymphocytes and AM signals).
Table I.
Relative strengths and weaknesses of differing theories involving silica-induced toxicity in the alveolar macrophage.
| Theory of silica AM toxicity | Strengths | Weaknesses | Future Research Directions |
|---|---|---|---|
| Lung surfactant modification of silica toxicity | Fairly well studied. Describes a natural protection mechanism for inhaled particles. | Protective effect is transient. Does not help explain any potential pathogenesis. | Research how silica/surfactant interaction could create secondary toxic species such as modified lipids or modified proteins. |
| Receptor-mediated toxicity | Identifies potential therapeutic target. Helps explain unique toxicity of crystalline silica. | Signaling not well described or understood. | Identify signaling pathways. Determine how differential signaling is possible through the same receptor. Identify other potential silica-binding receptors and any possible interaction between these receptors. Link these observations to pathology. |
| Free radical hypothesis | Well-studied and predominant theory of silica and other inhaled particle toxicity. | Cell damage in general causes secondary ROS production. Free radicals could be an effect rather than a cause. | Link this body of work solidly to specific silica-initiated diseases. Determine whether it is a critical event in vivo. |
| Lysosomal permeability | Describes initiation and process of apoptosis by silica in some detail. | Does not explain how apoptosis is important to the pathogenesis of silica inhalation. | Determine the importance of excessive macrophage apoptosis in the lung. Link this body of work solidly to specific silica-initiated diseases. |
| Immune dysfunction | Potentially identifies an early initiator of fibrotic and autoimmune diseases. Consistent with presence of activated macrophages in humans with disease. | Using a simple model to describe a complex process. Probably involves many different cell types, cell interactions, and genetic predispositions over a period of time. | Requires an ambitious attempt to explain a complex process from silica inhalation to autoimmune disease. Must identify all influences over a relatively long period of time for a variety of disorders. |
The myriad of diseases caused by silica inhalation is obviously a complex process. There must certainly be some interaction with predisposing genetic factors, epigenetic factors, and possibly polymorphisms [117]. In fact, TNF-a and IL-1 gene polymorphisms have been associated with silicosis in resent research findings [118, 119]. Humans exposed to crystalline silica in an occupational setting have variable responses [120]. Not all silica-exposed individuals develop disease. Some diseases, such as silicosis, take years to develop (except for rare extreme exposures that take 2–3 years to develop) [2]. Other diseases occur relatively rapidly following silica exposure as in the case with scleroderma and other silica-induced autoimmune disorders [4]. Understanding how the AM interacts with inhaled silica is a first step in understanding the silica-induced disease process.
Acknowledgments
This publication was made possible by Grant Number P20-RR-017670 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of NCRR or NIH. Additional funding for this research was provided by Grant Number ES 015294 from the NIEHS.
Abbreviations
- Si
silicon
- O
oxygen
- SiO2
silica
- RR
relative risk
- AM
alveolar macrophages
- PL
phospholipids
- SP
surfactant proteins
- PMN
polymorphonuclear leukocyte (neutrophils)
- DPPC
dipalmitoyl phosphatidyl choline
- ROS
reactive oxygen species
- RNS
reactive nitrogen species
- O2•−
superoxide anion
- ONOO•−
peroxynitrite
- H2O2
hydrogen peroxide
- SiOH
silanol group
- HO•
hydroxyl radical
- CO2
carbon dioxide
- DNA
deoxyribonucleic acid
- NO•
nitric oxide
- MnSOD
manganese superoxide dismutase
- GSH
glutathione
- MAPK
mitogen-activated protein kinase
- ERK
extracellular signal-regulated kinase
- MEK
MAP/ERK kinase
- AP-1
activating protein 1
- NFκB
nuclear factor kappa B
- LPS
lipopolysaccaride
- JNK
jun n-terminal kinase
- TNF-α
tumor necrosis factor alpha
- IL-1β
interleukin 1 beta
- MIP
macrophage inflammatory protein
- MCP
monocyte chemotactic protein
- IL-8
interleukin 8
- SR
scavenger receptors
- ox-LDL
oxidized low-density lipoprotein
- ac-LDL
acetylated low-density lipoprotein
- SR-A
scavenger receptor A class
- SRCL
scavenger receptor with c-type lectin
- SCARA5
class A scavenger receptor type 5
- SRCR
scavenger receptor cysteine-rich C-terminal region
- MSR
macrophage scavenger receptor
- MARCO
macrophage receptor with collagenous structure
- CHO
Chinese hamster ovary
- TiO2
titanium dioxide
- HSP
heat-shock protein
- PKC
protein kinase C
- Ser
serine
- Asp
aspartic acid
- Th
T helper
- IL-12
interleukin 12
- IFN-γ
interferon gamma
- TLR
toll-like receptor
- ICAM-1
intercellular adhesion molecule 1
- IL-13
interleukin 13
- APC
antigen-presenting cell
- IL-4
interleukin 4
- DC
dendritic cell
- NZM
New Zealand mice
Biographies
Raymond F. Hamilton Jr. received his graduate and undergraduate degrees from the University of Houston at Clear Lake. Initial biomedical research experience was obtained at the University of Texas Graduate School of Biomedical Sciences as a biostatistician. In 1989 he started working with Andrij Holian at the University of Texas Medical School beginning research in the area of particle-induced lung diseases. He is currently the biostatistician for the Center for Environmental Health Sciences, Department of Biomedical and Pharmaceutical Sciences at the University of Montana. He has authored and coauthored a number of research papers in the area of inhaled particulate toxicity and specifically the effect of silica on macrophage cells.
Sheetal A. Thakur received her Bachelors in Pharmaceutical Sciences from the University of Mumbai (Mumbai, India). She is currently a Ph.D. candidate in Center of Environmental Health Sciences at the University of Montana (Missoula, MT, USA). Her research interests focus on exploring the role of the scavenger receptor, MARCO, in molecular mechanisms underlying silica-induced cell injury.
Andrij Holian received his PhD in Chemistry from Montana State University, and did his postdoctoral training in Biochemistry at The University of Pennsylvania. His currently a professor of Toxicology in the Center for Environmental Health Sciences, Department of Biomedical Sciences at The University of Montana. His research interests focus on immune mechanisms in the development of chronic lung inflammation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Davis GS. The pathogenesis of silicosis. State of the art Chest. 1986;89:166S–169S. doi: 10.1378/chest.89.3_supplement.166s. [DOI] [PubMed] [Google Scholar]
- 2.Castranova V, Vallyathan V, Wallace WE. Silica and silica induced lung diseases. Boca Raton, FL: CRC Press; 1996. [Google Scholar]
- 3.Parks WR. Occupational lung disorders. 2. Boston: Butterworths; 1982. [Google Scholar]
- 4.Cooper GS, Miller FW, Germolec DR. Occupational exposures and autoimmune diseases. Int Immunopharmacol. 2002;2:303–313. doi: 10.1016/s1567-5769(01)00181-3. [DOI] [PubMed] [Google Scholar]
- 5.Peretz A, Checkoway H, Kaufman JD, Trajber I, Lerman Y. Silica, silicosis, and lung cancer. Isr Med Assoc J. 2006;8:114–118. [PubMed] [Google Scholar]
- 6.Pelucchi C, Pira E, Piolatto G, Coggiola M, Carta P, La Vecchia C. Occupational silica exposure and lung cancer risk: a review of epidemiological studies 1996–2005. Ann Oncol. 2006;17:1039–1050. doi: 10.1093/annonc/mdj125. [DOI] [PubMed] [Google Scholar]
- 7.Oberdorster G. Significance of particle parameters in the evaluation of exposure-dose-response relationships of inhaled particles. Inhal Toxicol. 1996;8 (Suppl):73–89. [PubMed] [Google Scholar]
- 8.Lapp NL, Castranova V. How silicosis and coal workers’ pneumoconiosis develop--a cellular assessment. Occup Med. 1993;8:35–56. [PubMed] [Google Scholar]
- 9.Migliaccio CT, Hamilton RF, Jr, Holian A. Increase in a distinct pulmonary macrophage subset possessing an antigen-presenting cell phenotype and in vitro APC activity following silica exposure. Toxicol Appl Pharmacol. 2005;205:168–176. doi: 10.1016/j.taap.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 10.Adamson IY, Letourneau HL, Bowden DH. Comparison of alveolar and interstitial macrophages in fibroblast stimulation after silica and long or short asbestos. Lab Invest. 1991;64:339–344. [PubMed] [Google Scholar]
- 11.Bowden DH, Hedgecock C, Adamson IY. Silica-induced pulmonary fibrosis involves the reaction of particles with interstitial rather than alveolar macrophages. J Pathol. 1989;158:73–80. doi: 10.1002/path.1711580114. [DOI] [PubMed] [Google Scholar]
- 12.Tetley TD, Richards RJ, Harwood JL. Changes in pulmonary surfactant and phosphatidylcholine metabolism in rats exposed to chrysotile asbestos dust. Biochem J. 1977;166:323–329. doi: 10.1042/bj1660323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kornbrust DJ, Hatch GE. Effect of silica and volcanic ash on the content of lung alveolar and tissue phospholipids. Environ Res. 1984;35:140–153. doi: 10.1016/0013-9351(84)90121-x. [DOI] [PubMed] [Google Scholar]
- 14.Muller B, Seifart C, Barth PJ. Effect of air pollutants on the pulmonary surfactant system. Eur J Clin Invest. 1998;28:762–777. doi: 10.1046/j.1365-2362.1998.00342.x. [DOI] [PubMed] [Google Scholar]
- 15.Miles PR, Bowman L, Jones WG, Berry DS, Vallyathan V. Changes in alveolar lavage materials and lung microsomal xenobiotic metabolism following exposures to HCl-washed or unwashed crystalline silica. Toxicol Appl Pharmacol. 1994;129:235–242. doi: 10.1006/taap.1994.1248. [DOI] [PubMed] [Google Scholar]
- 16.Viviano CJ, Bakewell WE, Dixon D, Dethloff LA, Hook GE. Altered regulation of surfactant phospholipid and protein A during acute pulmonary inflammation. Biochim Biophys Acta. 1995;1259:235–244. doi: 10.1016/0005-2760(95)00167-0. [DOI] [PubMed] [Google Scholar]
- 17.Rehn B, Seiler F, Rehn S, Bruch J, Maier M. Investigations on the inflammatory and genotoxic lung effects of two types of titanium dioxide: untreated and surface treated. Toxicol Appl Pharmacol. 2003;189:84–95. doi: 10.1016/s0041-008x(03)00092-9. [DOI] [PubMed] [Google Scholar]
- 18.Seiler F, Rehn B, Rehn S, Bruchs J. Significant differences in the cellular and molecular reactions of rat and hamster lung after quartz exposure. Toxicol Lett. 2001;119:11–19. doi: 10.1016/s0378-4274(00)00289-7. [DOI] [PubMed] [Google Scholar]
- 19.Bridges JP, Davis HW, Damodarasamy M, Kuroki Y, Howles G, Hui DY, McCormack FX. Pulmonary surfactant proteins A and D are potent endogenous inhibitors of lipid peroxidation and oxidative cellular injury. J Biol Chem. 2000;275:38848–38855. doi: 10.1074/jbc.M005322200. [DOI] [PubMed] [Google Scholar]
- 20.Spech RW, Wisniowski P, Kachel DL, Wright JR, Martin WJ., 2nd Surfactant protein A prevents silica-mediated toxicity to rat alveolar macrophages. Am J Physiol Lung Cell Mol Physiol. 2000;278:L713–718. doi: 10.1152/ajplung.2000.278.4.L713. [DOI] [PubMed] [Google Scholar]
- 21.Barbaro M, Cutroneo G, Costa C, Sciorio S, Trimarchi F, Favaloro A, Fenga C, Barbaro Martino L, Spatari G, Abbate C, Bramanti P. Early events of experimental exposure to amorphous and crystalline silica in the rat: time course of surfactant protein D. Ital J Anat Embryol. 2002;107:243–256. [PubMed] [Google Scholar]
- 22.Miles PR, Bowman L, Reasor MJ. Exposure to crystalline silica or treatment with chlorphentermine increases vitamin E levels in rat alveolar lavage materials. J Toxicol Environ Health. 1996;49:511–523. doi: 10.1080/009841096160727. [DOI] [PubMed] [Google Scholar]
- 23.Seiler F, Rehn B, Rehn S, Bruch J. Evidence of a no-effect level in silica-induced rat lung mutagenicity but not in fibrogenicity. Arch Toxicol. 2001;74:716–719. doi: 10.1007/s002040000191. [DOI] [PubMed] [Google Scholar]
- 24.Kanj RS, Kang JL, Castranova V. Interaction between primary alveolar macrophages and primary alveolar type II cells under basal conditions and after lipopolysaccharide or quartz exposure. J Toxicol Environ Health A. 2006;69:1097–1116. doi: 10.1080/14736480500360504. [DOI] [PubMed] [Google Scholar]
- 25.Suwabe A, Panos RJ, Voelker DR. Alveolar type II cells isolated after silica-induced lung injury in rats have increased surfactant protein A (SP-A) receptor activity. Am J Respir Cell Mol Biol. 1991;4:264–272. doi: 10.1165/ajrcmb/4.3.264. [DOI] [PubMed] [Google Scholar]
- 26.Antonini JM, Reasor MJ. Effect of short-term exogenous pulmonary surfactant treatment on acute lung damage associated with the intratracheal instillation of silica. J Toxicol Environ Health. 1994;43:85–101. doi: 10.1080/15287399409531906. [DOI] [PubMed] [Google Scholar]
- 27.Antonini JM, McCloud CM, Reasor MJ. Acute silica toxicity: attenuation by amiodarone-induced pulmonary phospholipidosis. Environ Health Perspect. 1994;102:372–378. doi: 10.1289/ehp.94102372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Patzold S, Schmidt A, Seidel A. Loss of cathepsin B activity in alveolar macrophages after in vitro quartz phagocytosis. J Toxicol Environ Health. 1993;40:547–554. doi: 10.1080/15287399309531818. [DOI] [PubMed] [Google Scholar]
- 29.Schimmelpfeng J, Drosselmeyer E, Hofheinz V, Seidel A. Influence of surfactant components and exposure geometry on the effects of quartz and asbestos on alveolar macrophages. Environ Health Perspect. 1992;97:225–231. doi: 10.1289/ehp.9297225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wallace WE, Keane MJ, Mike PS, Hill CA, Vallyathan V, Regad ED. Contrasting respirable quartz and kaolin retention of lecithin surfactant and expression of membranolytic activity following phospholipase A2 digestion. J Toxicol Environ Health. 1992;37:391–409. doi: 10.1080/15287399209531679. [DOI] [PubMed] [Google Scholar]
- 31.Gao N, Keane MJ, Ong T, Wallace WE. Effects of simulated pulmonary surfactant on the cytotoxicity and DNA-damaging activity of respirable quartz and kaolin. J Toxicol Environ Health A. 2000;60:153–167. doi: 10.1080/009841000156466. [DOI] [PubMed] [Google Scholar]
- 32.Gao N, Keane MJ, Ong T, Ye J, Miller WE, Wallace WE. Effects of phospholipid surfactant on apoptosis induction by respirable quartz and kaolin in NR8383 rat pulmonary macrophages. Toxicol Appl Pharmacol. 2001;175:217–225. doi: 10.1006/taap.2001.9249. [DOI] [PubMed] [Google Scholar]
- 33.Liu X, Keane MJ, Harrison JC, Cilento EV, Ong T, Wallace WE. Phospholipid surfactant adsorption by respirable quartz and in vitro expression of cytotoxicity and DNA damage. Toxicol Lett. 1998;96–97:77–84. doi: 10.1016/s0378-4274(98)00053-8. [DOI] [PubMed] [Google Scholar]
- 34.Hook GE. Alveolar proteinosis and phospholipidoses of the lungs. Toxicol Pathol. 1991;19:482–513. doi: 10.1177/019262339101900416. [DOI] [PubMed] [Google Scholar]
- 35.Palecanda A, Kobzik L. Receptors for unopsonized particles: the role of alveolar macrophage scavenger receptors. Curr Mol Med. 2001;1:589–595. doi: 10.2174/1566524013363384. [DOI] [PubMed] [Google Scholar]
- 36.Kobzik L. Lung macrophage uptake of unopsonized environmental particulates. Role of scavenger-type receptors. J Immunol. 1995;155:367–376. [PubMed] [Google Scholar]
- 37.Gough PJ, Gordon S. The role of scavenger receptors in the innate immune system. Microbes Infect. 2000;2:305–311. doi: 10.1016/s1286-4579(00)00297-5. [DOI] [PubMed] [Google Scholar]
- 38.Peiser L, Gordon S. The function of scavenger receptors expressed by macrophages and their role in the regulation of inflammation. Microbes Infect. 2001;3:149–159. doi: 10.1016/s1286-4579(00)01362-9. [DOI] [PubMed] [Google Scholar]
- 39.Murphy JE, Tedbury PR, Homer-Vanniasinkam S, Walker JH, Ponnambalam S. Biochemistry and cell biology of mammalian scavenger receptors. Atherosclerosis. 2005;182:1–15. doi: 10.1016/j.atherosclerosis.2005.03.036. [DOI] [PubMed] [Google Scholar]
- 40.Arredouani MS, Kobzik L. The structure and function of marco, a macrophage class a scavenger receptor. Cell Mol Biol (Noisy-le-grand) 2004;50 Online Pub:OL657–665. [PubMed] [Google Scholar]
- 41.Elshourbagy NA, Li X, Terrett J, Vanhorn S, Gross MS, Adamou JE, Anderson KM, Webb CL, Lysko PG. Molecular characterization of a human scavenger receptor, human MARCO. Eur J Biochem. 2000;267:919–926. doi: 10.1046/j.1432-1327.2000.01077.x. [DOI] [PubMed] [Google Scholar]
- 42.Nakamura K, Funakoshi H, Tokunaga F, Nakamura T. Molecular cloning of a mouse scavenger receptor with C-type lectin (SRCL)(1), a novel member of the scavenger receptor family. Biochim Biophys Acta. 2001;1522:53–58. doi: 10.1016/s0167-4781(01)00284-6. [DOI] [PubMed] [Google Scholar]
- 43.Jiang Y, Oliver P, Davies KE, Platt N. Identification and characterization of murine SCARA5, a novel class A scavenger receptor that is expressed by populations of epithelial cells. J Biol Chem. 2006;281:11834–11845. doi: 10.1074/jbc.M507599200. [DOI] [PubMed] [Google Scholar]
- 44.Platt N, Gordon S. Is the class A macrophage scavenger receptor (SR-A) multifunctional? - The mouse’s tale. J Clin Invest. 2001;108:649–654. doi: 10.1172/JCI13903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hamilton RF, de Villiers WJ, Holian A. Class A type II scavenger receptor mediates silica-induced apoptosis in Chinese hamster ovary cell line. Toxicol Appl Pharmacol. 2000;162:100–106. doi: 10.1006/taap.1999.8799. [DOI] [PubMed] [Google Scholar]
- 46.Acton S, Resnick D, Freeman M, Ekkel Y, Ashkenas J, Krieger M. The collagenous domains of macrophage scavenger receptors and complement component C1q mediate their similar, but not identical, binding specificities for polyanionic ligands. J Biol Chem. 1993;268:3530–3537. [PubMed] [Google Scholar]
- 47.Andersson L, Freeman MW. Functional changes in scavenger receptor binding conformation are induced by charge mutants spanning the entire collagen domain. J Biol Chem. 1998;273:19592–19601. doi: 10.1074/jbc.273.31.19592. [DOI] [PubMed] [Google Scholar]
- 48.Chao SK, Hamilton RF, Pfau JC, Holian A. Cell surface regulation of silica-induced apoptosis by the SR-A scavenger receptor in a murine lung macrophage cell line (MH-S) Toxicol Appl Pharmacol. 2001;174:10–16. doi: 10.1006/taap.2001.9190. [DOI] [PubMed] [Google Scholar]
- 49.Arredouani M, Yang Z, Ning Y, Qin G, Soininen R, Tryggvason K, Kobzik L. The scavenger receptor MARCO is required for lung defense against pneumococcal pneumonia and inhaled particles. J Exp Med. 2004;200:267–272. doi: 10.1084/jem.20040731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Arredouani MS, Palecanda A, Koziel H, Huang YC, Imrich A, Sulahian TH, Ning YY, Yang Z, Pikkarainen T, Sankala M, et al. MARCO is the major binding receptor for unopsonized particles and bacteria on human alveolar macrophages. J Immunol. 2005;175:6058–6064. doi: 10.4049/jimmunol.175.9.6058. [DOI] [PubMed] [Google Scholar]
- 51.Palecanda A, Paulauskis J, Al-Mutairi E, Imrich A, Qin G, Suzuki H, Kodama T, Tryggvason K, Koziel H, Kobzik L. Role of the scavenger receptor MARCO in alveolar macrophage binding of unopsonized environmental particles. J Exp Med. 1999;189:1497–1506. doi: 10.1084/jem.189.9.1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ojala JR, Pikkarainen T, Tuuttila A, Sandalova T, Tryggvason K. Crystal structure of the cysteine-rich domain of scavenger receptor MARCO reveals the presence of a basic and an acidic cluster that both contribute to ligand recognition. J Biol Chem. 2007;282:16654–16666. doi: 10.1074/jbc.M701750200. [DOI] [PubMed] [Google Scholar]
- 53.Iyer R, Hamilton RF, Li L, Holian A. Silica-induced apoptosis mediated via scavenger receptor in human alveolar macrophages. Toxicol Appl Pharmacol. 1996;141:84–92. doi: 10.1006/taap.1996.0263. [DOI] [PubMed] [Google Scholar]
- 54.Beamer CA, Holian A. Scavenger receptor class A type I/II (CD204) null mice fail to develop fibrosis following silica exposure. Am J Physiol Lung Cell Mol Physiol. 2005;289:L186–195. doi: 10.1152/ajplung.00474.2004. [DOI] [PubMed] [Google Scholar]
- 55.Hamilton RF, Jr, Thakur SA, Mayfair JK, Holian A. MARCO mediates silica uptake and toxicity in alveolar macrophages from C57BL/6 mice. J Biol Chem. 2006;281:34218–34226. doi: 10.1074/jbc.M605229200. [DOI] [PubMed] [Google Scholar]
- 56.Nakamura T, Hinagata J, Tanaka T, Imanishi T, Wada Y, Kodama T, Doi T. HSP90, HSP70, and GAPDH directly interact with the cytoplasmic domain of macrophage scavenger receptors. Biochem Biophys Res Commun. 2002;290:858–864. doi: 10.1006/bbrc.2001.6271. [DOI] [PubMed] [Google Scholar]
- 57.Hsu HY, Hajjar DP, Khan KM, Falcone DJ. Ligand binding to macrophage scavenger receptor-A induces urokinase-type plasminogen activator expression by a protein kinase-dependent signaling pathway. J Biol Chem. 1998;273:1240–1246. doi: 10.1074/jbc.273.2.1240. [DOI] [PubMed] [Google Scholar]
- 58.Shukla A, Stern M, Lounsbury KM, Flanders T, Mossman BT. Asbestos-induced apoptosis is protein kinase C delta-dependent. Am J Respir Cell Mol Biol. 2003;29:198–205. doi: 10.1165/rcmb.2002-0248OC. [DOI] [PubMed] [Google Scholar]
- 59.Fong LG, Le D. The processing of ligands by the class A scavenger receptor is dependent on signal information located in the cytoplasmic domain. J Biol Chem. 1999;274:36808–36816. doi: 10.1074/jbc.274.51.36808. [DOI] [PubMed] [Google Scholar]
- 60.Hsu HY, Chiu SL, Wen MH, Chen KY, Hua KF. Ligands of macrophage scavenger receptor induce cytokine expression via differential modulation of protein kinase signaling pathways. J Biol Chem. 2001;276:28719–28730. doi: 10.1074/jbc.M011117200. [DOI] [PubMed] [Google Scholar]
- 61.Jozefowski S, Arredouani M, Sulahian T, Kobzik L. Disparate regulation and function of the class A scavenger receptors SR-AI/II and MARCO. J Immunol. 2005;175:8032–8041. doi: 10.4049/jimmunol.175.12.8032. [DOI] [PubMed] [Google Scholar]
- 62.Jozefowski S, Kobzik L. Scavenger receptor A mediates H2O2 production and suppression of IL-12 release in murine macrophages. J Leukoc Biol. 2004;76:1066–1074. doi: 10.1189/jlb.0504270. [DOI] [PubMed] [Google Scholar]
- 63.Gordon S. Pattern recognition receptors: doubling up for the innate immune response. Cell. 2002;111:927–930. doi: 10.1016/s0092-8674(02)01201-1. [DOI] [PubMed] [Google Scholar]
- 64.Mukhopadhyay S, Peiser L, Gordon S. Activation of murine macrophages by Neisseria meningitidis and IFN-gamma in vitro: distinct roles of class A scavenger and Toll-like pattern recognition receptors in selective modulation of surface phenotype. J Leukoc Biol. 2004;76:577–584. doi: 10.1189/jlb.0104014. [DOI] [PubMed] [Google Scholar]
- 65.Hubbard AK, Thibodeau M, Giardina C. Cellular and molecular mechanisms regulating silica-induced adhesion molecule expression in mice. J Environ Pathol Toxicol Oncol. 2001;20 (Suppl 1):45–51. [PubMed] [Google Scholar]
- 66.Fubini B, Hubbard A. Reactive oxygen species (ROS) and reactive nitrogen species (RNS) generation by silica in inflammation and fibrosis. Free Radic Biol Med. 2003;34:1507–1516. doi: 10.1016/s0891-5849(03)00149-7. [DOI] [PubMed] [Google Scholar]
- 67.Castranova V. Generation of oxygen radicals and mechanisms of injury prevention. Environ Health Perspect. 1994;102 (Suppl 10):65–68. doi: 10.1289/ehp.94102s1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39:44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 69.Guzik TJ, Korbut R, Adamek-Guzik T. Nitric oxide and superoxide in inflammation and immune regulation. J Physiol Pharmacol. 2003;54:469–487. [PubMed] [Google Scholar]
- 70.Shi XL, Dalal NS, Vallyathan V. ESR evidence for the hydroxyl radical formation in aqueous suspension of quartz particles and its possible significance to lipid peroxidation in silicosis. J Toxicol Environ Health. 1988;25:237–245. doi: 10.1080/15287398809531205. [DOI] [PubMed] [Google Scholar]
- 71.Vallyathan V, Shi XL, Dalal NS, Irr W, Castranova V. Generation of free radicals from freshly fractured silica dust. Potential role in acute silica-induced lung injury. Am Rev Respir Dis. 1988;138:1213–1219. doi: 10.1164/ajrccm/138.5.1213. [DOI] [PubMed] [Google Scholar]
- 72.Vallyathan V, Kang JH, Van Dyke K, Dalal NS, Castranova V. Response of alveolar macrophages to in vitro exposure to freshly fractured versus aged silica dust: the ability of Prosil 28, an organosilane material, to coat silica and reduce its biological reactivity. J Toxicol Environ Health. 1991;33:303–315. doi: 10.1080/15287399109531529. [DOI] [PubMed] [Google Scholar]
- 73.Vallyathan V, Castranova V, Pack D, Leonard S, Shumaker J, Hubbs AF, Shoemaker DA, Ramsey DM, Pretty JR, McLaurin JL, et al. Freshly fractured quartz inhalation leads to enhanced lung injury and inflammation. Potential role of free radicals. Am J Respir Crit Care Med. 1995;152:1003–1009. doi: 10.1164/ajrccm.152.3.7663775. [DOI] [PubMed] [Google Scholar]
- 74.Gothe CJ, Lidstrom L, Swensson A. Influence of mode of disintegration on the fibrogenetic power of quartz particles. Med Lav. 1971;62:375–377. [PubMed] [Google Scholar]
- 75.Fubini B, Giamello E, Volante M, Bolis V. Chemical functionalities at the silica surface determining its reactivity when inhaled. Formation and reactivity of surface radicals. Toxicol Ind Health. 1990;6:571–598. [PubMed] [Google Scholar]
- 76.Fubini B, Bolis V, Giamello E. The surface chemistry of chrushed quartz dusts in relation to its pathogenicity. Inorg Chim Acta Bioinorg Chem. 1987;138:193–197. [Google Scholar]
- 77.Hochstrasser G, Antonini JF. Surface states of pristine silica. 1972;32:644–664. [Google Scholar]
- 78.Castranova V, Vallyathan V, Ramsey DM, McLaurin JL, Pack D, Leonard S, Barger MW, Ma JY, Dalal NS, Teass A. Augmentation of pulmonary reactions to quartz inhalation by trace amounts of iron-containing particles. Environ Health Perspect. 1997;105 (Suppl 5):1319–1324. doi: 10.1289/ehp.97105s51319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fubini B, Fenoglio I, Elias Z, Poirot O. Variability of biological responses to silicas: effect of origin, crystallinity, and state of surface on generation of reactive oxygen species and morphological transformation of mammalian cells. J Environ Pathol Toxicol Oncol. 2001;20 (Suppl 1):95–108. [PubMed] [Google Scholar]
- 80.Fenoglio I, Croce A, Di Renzo F, Tiozzo R, Fubini B. Pure-silica zeolites (Porosils) as model solids for the evaluation of the physicochemical features determining silica toxicity to macrophages. Chem Res Toxicol. 2000;13:489–500. doi: 10.1021/tx990169u. [DOI] [PubMed] [Google Scholar]
- 81.Gilmour PS, Beswick PH, Brown DM, Donaldson K. Detection of surface free radical activity of respirable industrial fibres using supercoiled phi X174 RF1 plasmid DNA. Carcinogenesis. 1995;16:2973–2979. doi: 10.1093/carcin/16.12.2973. [DOI] [PubMed] [Google Scholar]
- 82.Daniel LN, Mao Y, Saffiotti U. Oxidative DNA damage by crystalline silica. Free Radic Biol Med. 1993;14:463–472. doi: 10.1016/0891-5849(93)90103-2. [DOI] [PubMed] [Google Scholar]
- 83.Shi X, Mao Y, Daniel LN, Saffiotti U, Dalal NS, Vallyathan V. Silica radical-induced DNA damage and lipid peroxidation. Environ Health Perspect. 1994;102 (Suppl 10):149–154. doi: 10.1289/ehp.94102s10149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Elias Z, Poirot O, Daniere MC, Terzetti F, Marande AM, Dzwigaj S, Pezerat H, Fenoglio I, Fubini B. Cytotoxic and transforming effects of silica particles with different surface properties in Syrian hamster embryo (SHE) cells. Toxicol In Vitro. 2000;14:409–422. doi: 10.1016/s0887-2333(00)00039-4. [DOI] [PubMed] [Google Scholar]
- 85.Santarelli L, Recchioni R, Moroni F, Marcheselli F, Governa M. Crystalline silica induces apoptosis in human endothelial cells in vitro. Cell Biol Toxicol. 2004;20:97–108. doi: 10.1023/b:cbto.0000027935.45070.75. [DOI] [PubMed] [Google Scholar]
- 86.Vallyathan V, Shi X. The role of oxygen free radicals in occupational and environmental lung diseases. Environ Health Perspect. 1997;105 (Suppl 1):165–177. doi: 10.1289/ehp.97105s1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Vallyathan V, Mega JF, Shi X, Dalal NS. Enhanced generation of free radicals from phagocytes induced by mineral dusts. Am J Respir Cell Mol Biol. 1992;6:404–413. doi: 10.1165/ajrcmb/6.4.404. [DOI] [PubMed] [Google Scholar]
- 88.Blackford JA, Jr, Antonini JM, Castranova V, Dey RD. Intratracheal instillation of silica up-regulates inducible nitric oxide synthase gene expression and increases nitric oxide production in alveolar macrophages and neutrophils. Am J Respir Cell Mol Biol. 1994;11:426–431. doi: 10.1165/ajrcmb.11.4.7522485. [DOI] [PubMed] [Google Scholar]
- 89.Schapira RM, Ghio AJ, Effros RM, Morrisey J, Almagro UA, Dawson CA, Hacker AD. Hydroxyl radical production and lung injury in the rat following silica or titanium dioxide instillation in vivo. Am J Respir Cell Mol Biol. 1995;12:220–226. doi: 10.1165/ajrcmb.12.2.7865220. [DOI] [PubMed] [Google Scholar]
- 90.Holley JA, Janssen YM, Mossman BT, Taatjes DJ. Increased manganese superoxide dismutase protein in type II epithelial cells of rat lungs after inhalation of crocidolite asbestos or cristobalite silica. Am J Pathol. 1992;141:475–485. [PMC free article] [PubMed] [Google Scholar]
- 91.Janssen YM, Marsh JP, Absher MP, Hemenway D, Vacek PM, Leslie KO, Borm PJ, Mossman BT. Expression of antioxidant enzymes in rat lungs after inhalation of asbestos or silica. J Biol Chem. 1992;267:10625–10630. [PubMed] [Google Scholar]
- 92.Zhang Z, Shen HM, Zhang QF, Ong CN. Critical role of GSH in silica-induced oxidative stress, cytotoxicity, and genotoxicity in alveolar macrophages. Am J Physiol. 1999;277:L743–748. doi: 10.1152/ajplung.1999.277.4.L743. [DOI] [PubMed] [Google Scholar]
- 93.Cho YJ, Seo MS, Kim JK, Lim Y, Chae G, Ha KS, Lee KH. Silica-induced generation of reactive oxygen species in Rat2 fibroblast: role in activation of mitogen-activated protein kinase. Biochem Biophys Res Commun. 1999;262:708–712. doi: 10.1006/bbrc.1999.1274. [DOI] [PubMed] [Google Scholar]
- 94.Chen F, Kuhn DC, Sun SC, Gaydos LJ, Demers LM. Dependence and reversal of nitric oxide production on NF-kappa B in silica and lipopolysaccharide-induced macrophages. Biochem Biophys Res Commun. 1995;214:839–846. doi: 10.1006/bbrc.1995.2363. [DOI] [PubMed] [Google Scholar]
- 95.Chen F, Sun SC, Kuh DC, Gaydos LJ, Demers LM. Essential role of NF-kappa B activation in silica-induced inflammatory mediator production in macrophages. Biochem Biophys Res Commun. 1995;214:985–992. doi: 10.1006/bbrc.1995.2383. [DOI] [PubMed] [Google Scholar]
- 96.Shi X, Dong Z, Huang C, Ma W, Liu K, Ye J, Chen F, Leonard SS, Ding M, Castranova V, Vallyathan V. The role of hydroxyl radical as a messenger in the activation of nuclear transcription factor NF-kappaB. Mol Cell Biochem. 1999;194:63–70. doi: 10.1023/a:1006904904514. [DOI] [PubMed] [Google Scholar]
- 97.Hubbard AK, Timblin CR, Shukla A, Rincon M, Mossman BT. Activation of NF-kappaB-dependent gene expression by silica in lungs of luciferase reporter mice. Am J Physiol Lung Cell Mol Physiol. 2002;282:L968–975. doi: 10.1152/ajplung.00327.2001. [DOI] [PubMed] [Google Scholar]
- 98.Ding M, Shi X, Dong Z, Chen F, Lu Y, Castranova V, Vallyathan V. Freshly fractured crystalline silica induces activator protein-1 activation through ERKs and p38 MAPK. J Biol Chem. 1999;274:30611–30616. doi: 10.1074/jbc.274.43.30611. [DOI] [PubMed] [Google Scholar]
- 99.Shukla A, Timblin CR, Hubbard AK, Bravman J, Mossman BT. Silica-induced activation of c-Jun-NH2-terminal amino kinases, protracted expression of the activator protein-1 proto-oncogene, fra-1, and S-phase alterations are mediated via oxidative stress. Cancer Res. 2001;61:1791–1795. [PubMed] [Google Scholar]
- 100.Liu H, Zhang H, Forman HJ. Silica induces macrophage cytokines through phosphatidylcholine-specific phospholipase C with hydrogen peroxide. Am J Respir Cell Mol Biol. 2007;36:594–599. doi: 10.1165/rcmb.2006-0297OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gossart S, Cambon C, Orfila C, Seguelas MH, Lepert JC, Rami J, Carre P, Pipy B. Reactive oxygen intermediates as regulators of TNF-alpha production in rat lung inflammation induced by silica. J Immunol. 1996;156:1540–1548. [PubMed] [Google Scholar]
- 102.Barrett EG, Johnston C, Oberdorster G, Finkelstein JN. Silica-induced chemokine expression in alveolar type II cells is mediated by TNF-alpha-induced oxidant stress. Am J Physiol. 1999;276:L979–988. doi: 10.1152/ajplung.1999.276.6.L979. [DOI] [PubMed] [Google Scholar]
- 103.Barrett EG, Johnston C, Oberdorster G, Finkelstein JN. Antioxidant treatment attenuates cytokine and chemokine levels in murine macrophages following silica exposure. Toxicol Appl Pharmacol. 1999;158:211–220. doi: 10.1006/taap.1999.8716. [DOI] [PubMed] [Google Scholar]
- 104.Stringer B, Kobzik L. Environmental particulate-mediated cytokine production in lung epithelial cells (A549): role of preexisting inflammation and oxidant stress. J Toxicol Environ Health A. 1998;55:31–44. doi: 10.1080/009841098158601. [DOI] [PubMed] [Google Scholar]
- 105.Thibodeau M, Giardina C, Hubbard AK. Silica-induced caspase activation in mouse alveolar macrophages is dependent upon mitochondrial integrity and aspartic proteolysis. Toxicol Sci. 2003;76:91–101. doi: 10.1093/toxsci/kfg178. [DOI] [PubMed] [Google Scholar]
- 106.Thibodeau MS, Giardina C, Knecht DA, Helble J, Hubbard AK. Silica-induced apoptosis in mouse alveolar macrophages is initiated by lysosomal enzyme activity. Toxicol Sci. 2004;80:34–48. doi: 10.1093/toxsci/kfh121. [DOI] [PubMed] [Google Scholar]
- 107.Persson HL. Iron-dependent lysosomal destabilization initiates silica-induced apoptosis in murine macrophages. Toxicol Lett. 2005;159:124–133. doi: 10.1016/j.toxlet.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 108.Hamilton RF, Jr, Pfau JC, Marshall GD, Holian A. Silica and PM1648 modify human alveolar macrophage antigen-presenting cell activity in vitro. J Environ Pathol Toxicol Oncol. 2001;20 (Suppl 1):75–84. [PubMed] [Google Scholar]
- 109.Holian A, Uthman MO, Goltsova T, Brown SD, Hamilton RF., Jr Asbestos and silica-induced changes in human alveolar macrophage phenotype. Environ Health Perspect. 1997;105 (Suppl 5):1139–1142. doi: 10.1289/ehp.97105s51139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Beamer CA, Holian A. Antigen Presenting Cell Population Dynamics During Murine Silicosis. Am J Respir Cell Mol Biol. 2007 doi: 10.1165/rcmb.2007-0099OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Davis GS, Holmes CE, Pfeiffer LM, Hemenway DR. Lymphocytes, lymphokines, and silicosis. J Environ Pathol Toxicol Oncol. 2001;20 (Suppl 1):53–65. [PubMed] [Google Scholar]
- 112.Borges VM, Lopes MF, Falcao H, Leite-Junior JH, Rocco PR, Davidson WF, Linden R, Zin WA, DosReis GA. Apoptosis underlies immunopathogenic mechanisms in acute silicosis. Am J Respir Cell Mol Biol. 2002;27:78–84. doi: 10.1165/ajrcmb.27.1.4717. [DOI] [PubMed] [Google Scholar]
- 113.Friedetzky A, Garn H, Kirchner A, Gemsa D. Histopathological changes in enlarged thoracic lymph nodes during the development of silicosis in rats. Immunobiology. 1998;199:119–132. doi: 10.1016/S0171-2985(98)80068-5. [DOI] [PubMed] [Google Scholar]
- 114.Brown JM, Archer AJ, Pfau JC, Holian A. Silica accelerated systemic autoimmune disease in lupus-prone New Zealand mixed mice. Clin Exp Immunol. 2003;131:415–421. doi: 10.1046/j.1365-2249.2003.02094.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Pfau JC, Brown JM, Holian A. Silica-exposed mice generate autoantibodies to apoptotic cells. Toxicology. 2004;195:167–176. doi: 10.1016/j.tox.2003.09.011. [DOI] [PubMed] [Google Scholar]
- 116.Brown JM, Schwanke CM, Pershouse MA, Pfau JC, Holian A. Effects of rottlerin on silica-exacerbated systemic autoimmune disease in New Zealand mixed mice. Am J Physiol Lung Cell Mol Physiol. 2005;289:L990–998. doi: 10.1152/ajplung.00078.2005. [DOI] [PubMed] [Google Scholar]
- 117.Qu Y, Tang Y, Cao D, Wu F, Liu J, Lu G, Zhang Z, Xia Z. Genetic polymorphisms in alveolar macrophage response-related genes, and risk of silicosis and pulmonary tuberculosis in Chinese iron miners. Int J Hyg Environ Health. 2007 doi: 10.1016/j.ijheh.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 118.Yucesoy B, Vallyathan V, Landsittel DP, Sharp DS, Weston A, Burleson GR, Simeonova P, McKinstry M, Luster MI. Association of tumor necrosis factor-alpha and interleukin-1 gene polymorphisms with silicosis. Toxicol Appl Pharmacol. 2001;172:75–82. doi: 10.1006/taap.2001.9124. [DOI] [PubMed] [Google Scholar]
- 119.Yucesoy B, Vallyathan V, Landsittel DP, Simeonova P, Luster MI. Cytokine polymorphisms in silicosis and other pneumoconioses. Mol Cell Biochem. 2002;234–235:219–224. [PubMed] [Google Scholar]
- 120.Rees D, Murray J. Silica, silicosis and tuberculosis. Int J Tuberc Lung Dis. 2007;11:474–484. [PubMed] [Google Scholar]