

Abstract
We have established strong linkage evidence that supports mapping autosomal-dominant sensory/motor neuropathy with ataxia (SMNA) to chromosome 7q22-q32. SMNA is a rare neurological disorder whose phenotype encompasses both the central and the peripheral nervous system. In order to identify a gene responsible for SMNA, we have undertaken a comprehensive genomic evaluation of the region of linkage, including evaluation for repeat expansion and small deletions or duplications, capillary sequencing of candidate genes, and massively parallel sequencing of all coding exons. We excluded repeat expansion and small deletions or duplications as causative, and through microarray-based hybrid capture and massively parallel short-read sequencing, we identified a nonsynonymous variant in the human interferon-related developmental regulator gene 1 (IFRD1) as a disease-causing candidate. Sequence conservation, animal models, and protein structure evaluation support the involvement of IFRD1 in SMNA. Mutation analysis of IFRD1 in additional patients with similar phenotypes is needed for demonstration of causality and further evaluation of its importance in neurological diseases.
Main Text
We have described a five-generation American family of Northern European ancestry with an autosomal-dominant sensory/motor neuropathy with ataxia (SMNA, MIM 607458). There is no evidence of genetic anticipation, mental retardation, or dementia in this family. The unique and complex phenotype of SMNA includes axonal sensory neuropathy present in all affected family members as its earliest sign. Later in the disease, cerebellar and motor-tract dysfunction develops. In persons with cerebellar dysfunction for whom MRI was performed, mild cerebellar atrophy was seen. Both upper and lower motor neurons are implicated in motor-tract dysfunction, as evidenced by Babinski sign, muscle atrophy, and EMG findings. Full phenotypic description and MRI findings were reported previously.1 The phenotype in this family overlaps with those of hereditary ataxias and neuropathies.
Hereditary ataxias are best categorized by the mode of inheritance and the causative gene or linked locus, if the gene has not been identified. The identification of at least 28 genes or loci for autosomal-dominant spinocerebellar ataxias (SCAs)2 attests to the wide heterogeneity of this group of disorders. Hereditary neuropathies have shown even greater clinical and genetic heterogeneity. Charcot-Marie-Tooth (CMT) neuropathy encompassing at least 40 genes or loci is clinically characterized primarily by progressive motor neuropathy but also includes sensory findings. Hereditary sensory neuropathies have prominent autonomic features, and so far five genes or loci have been identified.
Since our initial publication of SMNA, no other families with neurological phenotypes have been reported to map within the same region. We were confident in our mapping study, because the five-generation pedigree provided a LOD score of 6.36 (p = 1/10Lod = 4.36 × 10−7) and the linked haplotype showed complete segregation with the disease. We were not able to narrow the region by identifying additional family members, and the region of interest remained large, from 109 to 131 Mbp on chromosome 7 (NCBI build 36.2). Consequently, our approach has been to comprehensively evaluate the region of interest for the presence of genetic changes that could be causative. The institutional review board of the University of Washington approved research protocols.
To evaluate for nucleotide repeat expansions, we used the University of California Santa Cruz Genome Browser (UCSC GB) “simple repeats” track to query the critical region for the presence of sequences with potential for expansion. We defined those sequences as CTG/CAG, CGG/CCG, GAA/TTC, GAC/GTC, CCTG/CAGG, and ATTCT/AGAAT repeats with more than ten uninterrupted repeat units present in the reference sequence. For eight sequences in the region with potential for expansion, PCR amplification and visualization on agarose gel of DNA from two affected and two unaffected family members did not reveal evidence for repeat expansion.
In order to identify microdeletions and microduplications, we performed comparative genomic hybridization (CGH) on a custom 385 K high-definition microarray. For our region of 22 Mb, this represents an average tiling density of one probe every 57 base pairs. The hybridization experiments were performed at the NimbleGen facility, and data were analyzed with SigMAP software. Initial CGH analysis of one of the affected individuals indicated two putative microdeletions of 2650 and 924 bp, respectively. However, sequencing of these two locations in the same individual revealed heterozygosity for alleles present in dbSNPs, establishing that putative deletions were false positives and excluding microdeletions as causative.
To evaluate genes in the region, we first initiated capillary sequencing of selected candidate genes. Depending on annotation, the region encompasses more than 150 genes. On the basis of the known or presumed gene function, tissue-expression data, similarities to the model-organism genes, existence of animal models, involvement in human diseases, and other cues from the literature, we ranked candidate genes to prioritize sequencing. Primer 3 software was used in designing primers for the exons of selected genes. Exons were PCR amplified and cycle sequenced with ABI Prism BigDye terminator kits. Samples were electrophoresed on an ABI 310 or 3130XL DNA Analyzer and analyzed with Sequencher software by two independent persons as previously described.3 The candidate-gene analysis encompassed exons of 70 genes.
As massively parallel sequencing became available, we sought to expand the analysis to cover all the genes in the region. Based on NCBI build 36.1, UCSC GB gene-annotation algorithms indicated 297 genes with 1300 exons in the region. This total contains overlaps due to different splice variants. We also included intronic and exon flanking sequences and regions that were identified by sno and miRNA track, because they might be relevant for neurological disorders. In total, we identified 3.7 Mb of target bases. With the identified sequence, we designed an oligonucleotide capture array with 385,000 probes (NimbleGen).4 For isolation and sequencing of targeted DNA, shotgun Illumina sequencing libraries were constructed from genomic DNA of an affected male, his affected father, and his unaffected mother (Figure 1A) and amplified in accordance with the manufacturer's protocol. The libraries were hybridized to the capture array, and after washing, hybridized material was eluted, in accordance with a modified version of the manufacturer's protocol. After elution, the enriched sequencing libraries were sequenced on an Illumina Genome Analyzer I.
Figure 1.
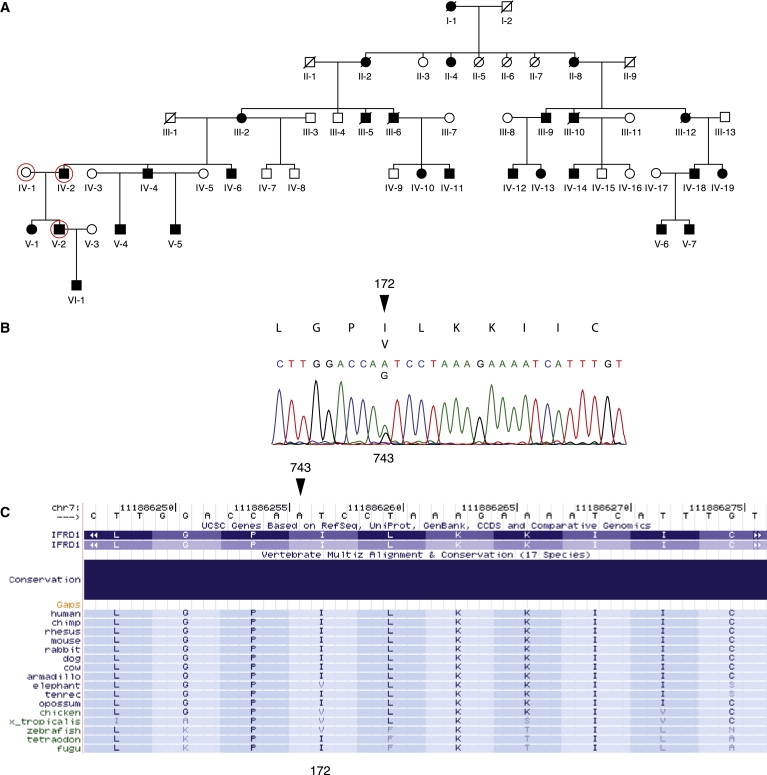
SMNA Family Pedigree and Mutational Analysis
(A) Five-generation SMNA pedigree. Circled individuals were resequenced. Individual VI-1 was recently diagnosed with SMNA.
(B) Sequence analysis of IFRD1 indicating heterozygous nucleotide substitution 743A→G, which causes amino acid Ile172Val change, marked with an arrowhead.
(C) Multiple protein-sequence alignment of IFRD1 with its orthologs from UCSD 17-way conservation panel. Ile172 position is marked with an arrowhead.
The resulting 36 bp single-end sequencing reads were mapped to the human reference genome (build 36.2) with the MAQ software package.5 Consensus calls for variant identification were also carried out with MAQ. A single lane produced 167 Mb, 213 Mb, and 249 Mb of sequence that could be uniquely mapped to the reference genome (MAQ mapping score > 0) for the proband, affected father, and unaffected mother, respectively. Of these, 42% (71 Mb), 37% (79 Mb), and 52% (128 Mb) mapped within the 22 Mb interval of interest. Mean coverage of the 3.7 Mb of targeted bases within the interval was 18-fold, 20-fold, and 32-fold. Applying a MAQ consensus quality threshold of 40 resulted in consensus calling at 82%, 88%, and 92% of the targeted bases. However, the consensus-call rate at coding subsequences was higher. For 141 kb of targeted sequence within the interval defined as coding by the NCBI's CCDS database (Sept 2008 version), 95% and 97% were sufficiently covered for consensus calling with the same thresholds in the proband and affected father, respectively. Across the 3.7 Mb of targeted sequence, 2295, 2429, and 2543 variants were identified. In all three samples, 92% of identified variants were previously documented in dbSNP (version 129). Across the interval, a total of five, four, and three nonsynonymous, heterozygous variants that were not in dbSNP were discovered in the proband, affected father, and unaffected mother, respectively. Three of the variants are present in all affected family members and were confirmed by PCR and capillary sequencing.
A variant, NM_002851.2:g.140329G→A (C7bp121,440,723), in the PTPRZ1 (MIM 176891) gene does not affect a conserved nucleotide (conservation score 0.002). A second variant, NM_152556.2:g.44215T→A (C7bp112,322,954), is in an uncharacterized transcript, C7orf60. This variant has a conservation score of 0.974. Lastly, a NM_001007245.1:g35795A→G (C7bp111,886,256) variant with a conservation score 1.00 in exon 5 of the IFRD1 (MIM 603502) gene was identified. The same IFRD1 variant was also observed during capillary sequencing of candidate genes (Figure 1B). None of the three variants above were observed in 1255 unrelated American healthy control individuals of European descent during testing with TaqMan assays as previously described.6 The 12 exons of the IFRD1 gene were sequenced in 83 case individuals with ataxia without neuropathy, and no new nonsynonymous variants were found.
On the basis of the exclusion of other genomic mechanisms for disease causation and the consideration of IFRD1 biological function, its involvement in pathways where mutations result in phenotypes with neuropathy, and animal knockout models, we propose IFRD1 as a candidate gene for SMNA. The IFRD1 gene has 1791 bp organized in 12 exons and encodes a protein of 451 amino acids. The rat IFRD1 ortholog Pc4 was initially isolated in PC12 cells because it was upregulated during nerve growth factor (NGF)-induced neuronal differentiation.7 In mouse, Tis7/Ifrd1 was identified as an immediate early gene that is induced by tetradecynol phorbol acetate, epidermal growth factor, and fibroblast growth factor stimulation,8,9 and it is 89% and 93% identical to the human ortholog on the nucleotide and protein levels. In human tissues IFRD1 is ubiquitously expressed with high levels of expression in the brain,10 including expression in the cerebellum and spinal cord.11 In the mouse, in early gestation, Ifrd1 is expressed in restricted structures, including the brain, spinal cord and spinal ganglia. At late gestation, the gene is ubiquitously expressed.10 Trophic factor stimulation upregulates Ifrd1 expression and results in translocation of the protein from cytoplasm to the nucleus.12 In the nucleus, Ifrd1 acts as a transcriptional corepressor and interacts with proteins of the SIN3 histone deacetylase complex.13
Genes in the NGF-IFRD1 pathway have roles in other neuropathies. A homozygous mutation in NGF subunit beta NGFB (MIM 162030) has been identified in a Swedish family whose affected members exhibit peripheral neuropathy with a loss of deep pain and temperature perception,14 best fitting into the category of hereditary sensory and autonomic neuropathy type V (HSAN5, MIM 608654). The phenotype of heterozygous carriers does not include loss of pain perception and ranges from asymptomatic cases to disabling polyneuropathy.15 Consistent with this finding, homozygous NGF receptor knockout mice have a phenotype that includes decreased peripheral sensory innervation.16 Mutations in neurotropic tyrosine kinase receptor 1 (NTRK1, MIM 256800) a protein that associates with NGF receptor NGFR (MIM 162010), have been found in multiple families with HSAN4 (MIM 256800), an autosomal-recessive neuropathy that is characterized by mental retardation, anhydrosis, and congenital insensitivity to pain.17 Altogether, these data indicate involvement of NGF signaling in neuronal differentiation and survival and in pathological mechanisms of neuropathies.
Homozygous Ifrd1 knockout mice exhibit muscle atrophy and an increase in the number of central nuclei, a small fiber diameter, and a lower total number of fibers, a phenotype that resembles neurogenic muscle atrophy that was found in one muscle biopsy performed in the SMNA family. The mice also show muscle-regeneration abnormalities, with delays in the reinnervation process and deficits in nerve-evoked stimulations, further implicating neurogenic abnormalities.18 The function of Ifrd1 has been studied in dorsal root ganglion (DRG) neuronal cultures, where it is expressed.19 In Ifrd1−/− DRG cultures, axon initiation, outgrowth, and elongation are diminished. Stimulation of DRGs with NGF results in increased axon initiation, outgrowth, and branching. These experiments further implicate Ifrd1 as a downstream effector of NGF and highlight its role in axonal growth and function and in the DRG sensory system. It is notable that peripheral sensory impairment is a most prominent neurological deficit in our family.
In our family, we observed a missense mutation, NP_001541:p.I172V, which substitutes valine for isoleucine. The identified change, NM_001550:c743A→G, affects a highly conserved nucleotide (conservation score 1.00), as determined by the UCSC browser and the phastCons program that identifies cross-species conserved nucleotides in multiply aligned sequences.20 However, valine is the amino acid at residue 172 in several species, including elephant, chicken, Xenopus tropicalis, and zebrafish (Figure 1C). This change might not severely affect the biochemical properties of the protein, given that both amino acids are aliphatic and hydrophobic. However, isoleucine-to-valine substitutions are pathogenic in other diseases. In Noonan syndrome (MIM 163950), the I282V substitution in PTPN11 (MIM 176876) has been found in multiple cases21 and was shown to perturb the stability of the SH2 tyrosine phosphatase catalytic domain.22 An early onset form of Alzheimer disease (MIM 104300) is caused by mutations in the APP gene (MIM 104760). The most common causal mutation is a reverse V717I substitution, but I716V that affects beta-amyloid plaque formation has also been described.23 Additionally valine-to-isoleucine mutations in the PRNP gene (MIM 176640) have been found in Creutzfeldt-Jakob disease (CJD, MIM 123400). The V180I mutation found in sporadic cases is associated with a distinct phenotype,24 and the V210I mutation was characterized with incomplete penetrance.25,26 Functional studies of V210I have shown that the mechanism of disease is the increase in helical and aggregation propensities of the helix-3 sequence of PRNP27.
To further investigate the I172V protein change and IFRD1 protein structure, we used Phyre.28 Phyre combines different protein homology and analogy detection methods, including known tridimensional structural profiles, in a meta-server in order to identify query-template homologies. Phyre analysis of wild-type (WT) IFRD1 protein identified homologies with structures whose folds are characterized by alpha-alpha superhelix patterns that belong to Armadillo and HEAT repeat families. Homology analysis indicated that amino acids 162–177 form an alpha helix and that amino acid positions P171 and L173 may form a cavity or a cleft (Figure 2). In addition, analysis of WT and I172L mutant proteins predicted differences with respect to their pairing with complex protein structures. Homology of WT IFRD1 to S. ceviseae exportin, complexed with importin and ranGtp, was observed. This homology was not observed for the I172V mutant that, in turn, was found to have homologies to H. sapiens transportin complexed with basic nuclear localization signal proteins. This kind of discrepancy in homologies was not observed when WT protein was compared to an A144V mutant, the only nonsynonymous IFRD1 mutation present in dbSNP. This observation suggests that the mutation might affect the folding properties of IFRD1, as well as its affinity for binding and transport through the nuclear pore complex.
Figure 2.
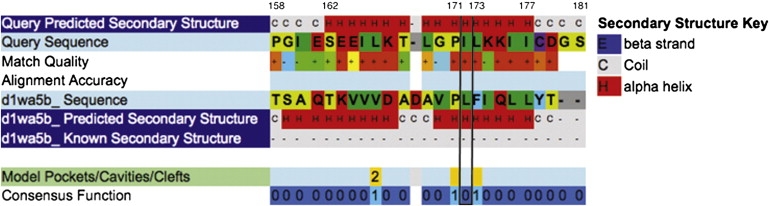
Phyre Analysis of IFRD1 Mutation Region
Phyre analysis of IFRD1 showing that amino acids 162–177 form an alpha helix. Homology with S. cerevisiae Karyopherin alpha protein (d1wa5b_Sequence) indicates that amino acid positions 171 and 173, flanking the mutated residue, may form a cavity or cleft.
In conclusion, by genomic analysis of the region linked to SMNA, we identified IFRD1 as a candidate gene. Evidence that supports IFRD1 as the candidate gene includes the strong statistical evidence for linkage in the region (p = 4.36 × 10−7); absence of a genomic mechanism that might cause the disease, such as repeat expansions, deletions, and/or duplications; and extensive coding-sequence analysis of the region. Additionally, on the basis of its role in the NGF pathway and animal studies, IFRD1 is a strong a priori biological candidate for SMNA. Mutation in genes that are part of NGF pathway, NTRK1 and NGFB, cause HSAN IV and HSAN V. Existing knockout-animal models have neurological phenotypes such as muscle atrophy and axonal-growth dysregulation, consistent with its possible role in a neurodegenerative disorder like SMNA. Our bioinformatic analysis of mutant IFRD1 indicates that the observed mutation might affect binding and transport through the nuclear pore complexes. Altered membrane trafficking of signaling molecules has been proposed as a mechanism that when dysregulated can lead to neuropathies because of extreme polarity and size of peripheral neurons. The weakness of the study is that we have not unequivocally demonstrated that mutation in IFRD1 causes SMNA. It remains possible that IFRD1 I172V is a rare variant and that some other change in the original interval undetected by our approach is causative. Also, we have not excluded changes in PTPRZ1 and C7orf60 as causative. However, PTPRZ1 is unlikely to be causal, given that the gene and the identified variant are poorly conserved. The C7orf60 gene and the variant identified are not as well conserved as IFRD1. Functional studies of the IFRD1 I172V mutant gene could provide support for its pathogenicity and provide additional insights into mechanisms by which the mutant protein causes disease. However, ultimately, proof that mutations in IFRD1 cause SMNA will be provided by identification of other familial and sporadic cases with similar phenotypes. For 83 patients with ataxia available in our laboratory we have screened all the exons of IFRD1 and have not identified any additional mutations. However, neuropathy is not a component of ataxias in our sample. Further screening of IFRD1 for mutation in patients with related phenotypes that include neuropathy is warranted.
Acknowledgments
We are grateful to the members of the family for their participation in these studies and to Elizabeth Thompson for her intellectual contributions during the early stages of the research. Skillful technical assistance was provided by Ruolan Qiu, Choli Lee, Emily Turner, Sarah Summer, Zarshid Arbibi, Ruben Burbank, Catherine Morgan, Jane Ranchalis, and John Wolf. We thank Chang-En Yu for generous provision of control samples. The research was supported in part by funds and resources from the National Organization for Rare Disorders, the Department of Veterans Affairs, the Mary Gates and the Herschel and Caryl Roman Endowments for Students, and NIH/NHGRI grant R21HG004749 to J.S.
Web Resources
The URLs for data presented herein are as follows:
GeneReviews at GeneTests, http://www.genetests.org
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
References
- 1.Brkanac Z., Fernandez M., Matsushita M., Lipe H., Wolff J., Bird T.D., Raskind W.H. Autosomal dominant sensory/motor neuropathy with Ataxia (SMNA): Linkage to chromosome 7q22-q32. Am. J. Med. Genet. 2002;114:450–457. doi: 10.1002/ajmg.10361. [DOI] [PubMed] [Google Scholar]
- 2.Bird TD Hereditary Ataxia Overview.
- 3.Chen D.H., Brkanac Z., Verlinde C.L., Tan X.J., Bylenok L., Nochlin D., Matsushita M., Lipe H., Wolff J., Fernandez M. Missense mutations in the regulatory domain of PKC gamma: A new mechanism for dominant nonepisodic cerebellar ataxia. Am. J. Hum. Genet. 2003;72:839–849. doi: 10.1086/373883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Albert T.J., Molla M.N., Muzny D.M., Nazareth L., Wheeler D., Song X., Richmond T.A., Middle C.M., Rodesch M.J., Packard C.J. Direct selection of human genomic loci by microarray hybridization. Nat. Methods. 2007;4:903–905. doi: 10.1038/nmeth1111. [DOI] [PubMed] [Google Scholar]
- 5.Li H., Ruan J., Durbin R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008;18:1851–1858. doi: 10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brkanac Z., Chapman N.H., Matsushita M.M., Chun L., Nielsen K., Cochrane E., Berninger V.W., Wijsman E.M., Raskind W.H. Evaluation of candidate genes for DYX1 and DYX2 in families with dyslexia. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 2007;144B:556–560. doi: 10.1002/ajmg.b.30471. [DOI] [PubMed] [Google Scholar]
- 7.Tirone F., Shooter E.M. Early gene regulation by nerve growth factor in PC12 cells: Induction of an interferon-related gene. Proc. Natl. Acad. Sci. USA. 1989;86:2088–2092. doi: 10.1073/pnas.86.6.2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Varnum B.C., Lim R.W., Herschman H.R. Characterization of TIS7, a gene induced in Swiss 3T3 cells by the tumor promoter tetradecanoyl phorbol acetate. Oncogene. 1989;4:1263–1265. [PubMed] [Google Scholar]
- 9.Arenander A.T., Lim R.W., Varnum B.C., Cole R., de Vellis J., Herschman H.R. TIS gene expression in cultured rat astrocytes: Multiple pathways of induction by mitogens. J. Neurosci. Res. 1989;23:257–265. doi: 10.1002/jnr.490230303. [DOI] [PubMed] [Google Scholar]
- 10.Buanne P., Incerti B., Guardavaccaro D., Avvantaggiato V., Simeone A., Tirone F. Cloning of the human interferon-related developmental regulator (IFRD1) gene coding for the PC4 protein, a member of a novel family of developmentally regulated genes. Genomics. 1998;51:233–242. doi: 10.1006/geno.1998.5260. [DOI] [PubMed] [Google Scholar]
- 11.Su A.I., Wiltshire T., Batalov S., Lapp H., Ching K.A., Block D., Zhang J., Soden R., Hayakawa M., Kreiman G. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc. Natl. Acad. Sci. USA. 2004;101:6062–6067. doi: 10.1073/pnas.0400782101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guardavaccaro D., Montagnoli A., Ciotti M.T., Gatti A., Lotti L., Di Lazzaro C., Torrisi M.R., Tirone F. Nerve growth factor regulates the subcellular localization of the nerve growth factor-inducible protein PC4 in PC12 cells. J. Neurosci. Res. 1994;37:660–674. doi: 10.1002/jnr.490370514. [DOI] [PubMed] [Google Scholar]
- 13.Vietor I., Vadivelu S.K., Wick N., Hoffman R., Cotten M., Seiser C., Fialka I., Wunderlich W., Haase A., Korinkova G. TIS7 interacts with the mammalian SIN3 histone deacetylase complex in epithelial cells. EMBO J. 2002;21:4621–4631. doi: 10.1093/emboj/cdf461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Einarsdottir E., Carlsson A., Minde J., Toolanen G., Svensson O., Solders G., Holmgren G., Holmberg D., Holmberg M. A mutation in the nerve growth factor beta gene (NGFB) causes loss of pain perception. Hum. Mol. Genet. 2004;13:799–805. doi: 10.1093/hmg/ddh096. [DOI] [PubMed] [Google Scholar]
- 15.Minde J., Andersson T., Fulford M., Aguirre M., Nennesmo I., Remahl I.N., Svensson O., Holmberg M., Toolanen G., Solders G. A novel NGFB point mutation: A phenotype study of heterozygous patients. J. Neurol. Neurosurg. Psychiatry. 2009;80:188–195. doi: 10.1136/jnnp.2007.136051. [DOI] [PubMed] [Google Scholar]
- 16.Lee K.F., Li E., Huber L.J., Landis S.C., Sharpe A.H., Chao M.V., Jaenisch R. Targeted mutation of the gene encoding the low affinity NGF receptor p75 leads to deficits in the peripheral sensory nervous system. Cell. 1992;69:737–749. doi: 10.1016/0092-8674(92)90286-l. [DOI] [PubMed] [Google Scholar]
- 17.Indo Y., Tsuruta M., Hayashida Y., Karim M.A., Ohta K., Kawano T., Mitsubuchi H., Tonoki H., Awaya Y., Matsuda I. Mutations in the TRKA/NGF receptor gene in patients with congenital insensitivity to pain with anhidrosis. Nat. Genet. 1996;13:485–488. doi: 10.1038/ng0896-485. [DOI] [PubMed] [Google Scholar]
- 18.Vadivelu S.K., Kurzbauer R., Dieplinger B., Zweyer M., Schafer R., Wernig A., Vietor I., Huber L.A. Muscle regeneration and myogenic differentiation defects in mice lacking TIS7. Mol. Cell. Biol. 2004;24:3514–3525. doi: 10.1128/MCB.24.8.3514-3525.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dieplinger B., Schiefermeier N., Juchum-Pasquazzo M., Gstir R., Huber L.A., Klimaschewski L., Vietor I. The transcriptional corepressor TPA-inducible sequence 7 regulates adult axon growth through cellular retinoic acid binding protein II expression. Eur. J. Neurosci. 2007;26:3358–3367. doi: 10.1111/j.1460-9568.2007.05951.x. [DOI] [PubMed] [Google Scholar]
- 20.Siepel A., Bejerano G., Pedersen J.S., Hinrichs A.S., Hou M., Rosenbloom K., Clawson H., Spieth J., Hillier L.W., Richards S. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tartaglia M., Martinelli S., Stella L., Bocchinfuso G., Flex E., Cordeddu V., Zampino G., Burgt I., Palleschi A., Petrucci T.C. Diversity and functional consequences of germline and somatic PTPN11 mutations in human disease. Am. J. Hum. Genet. 2006;78:279–290. doi: 10.1086/499925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martinelli S., Torreri P., Tinti M., Stella L., Bocchinfuso G., Flex E., Grottesi A., Ceccarini M., Palleschi A., Cesareni G. Diverse driving forces underlie the invariant occurrence of the T42A, E139D, I282V and T468M SHP2 amino acid substitutions causing Noonan and LEOPARD syndromes. Hum. Mol. Genet. 2008;17:2018–2029. doi: 10.1093/hmg/ddn099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eckman C.B., Mehta N.D., Crook R., Perez-tur J., Prihar G., Pfeiffer E., Graff-Radford N., Hinder P., Yager D., Zenk B. A new pathogenic mutation in the APP gene (I716V) increases the relative proportion of A beta 42(43) Hum. Mol. Genet. 1997;6:2087–2089. doi: 10.1093/hmg/6.12.2087. [DOI] [PubMed] [Google Scholar]
- 24.Jin K., Shiga Y., Shibuya S., Chida K., Sato Y., Konno H., Doh-ura K., Kitamoto T., Itoyama Y. Clinical features of Creutzfeldt-Jakob disease with V180I mutation. Neurology. 2004;62:502–505. doi: 10.1212/01.wnl.0000106954.54011.80. [DOI] [PubMed] [Google Scholar]
- 25.Pocchiari M., Salvatore M., Cutruzzola F., Genuardi M., Allocatelli C.T., Masullo C., Macchi G., Alema G., Galgani S., Xi Y.G. A new point mutation of the prion protein gene in Creutzfeldt-Jakob disease. Ann. Neurol. 1993;34:802–807. doi: 10.1002/ana.410340608. [DOI] [PubMed] [Google Scholar]
- 26.Mouillet-Richard S., Teil C., Lenne M., Hugon S., Taleb O., Laplanche J.L. Mutation at codon 210 (V210I) of the prion protein gene in a North African patient with Creutzfeldt-Jakob disease. J. Neurol. Sci. 1999;168:141–144. doi: 10.1016/s0022-510x(99)00179-3. [DOI] [PubMed] [Google Scholar]
- 27.Thompson A.J., Barnham K.J., Norton R.S., Barrow C.J. The Val-210-Ile pathogenic Creutzfeldt-Jakob disease mutation increases both the helical and aggregation propensities of a sequence corresponding to helix-3 of PrP(C) Biochim. Biophys. Acta. 2001;1544:242–254. doi: 10.1016/s0167-4838(00)00225-9. [DOI] [PubMed] [Google Scholar]
- 28.Bennett-Lovsey R.M., Herbert A.D., Sternberg M.J., Kelley L.A. Exploring the extremes of sequence/structure space with ensemble fold recognition in the program Phyre. Proteins. 2008;70:611–625. doi: 10.1002/prot.21688. [DOI] [PubMed] [Google Scholar]