

Abstract
Primary congenital glaucoma (PCG) is an autosomal-recessive condition characterized by high intraocular pressure (IOP), usually within the first year of life, which potentially could lead to optic nerve damage, globe enlargement, and permanent loss of vision. To date, PCG has been linked to three loci: 2p21 (GLC3A), for which the responsible gene is CYP1B1, and 1p36 (GLC3B) and 14q24 (GLC3C), for which the genes remain to be identified. Here we report that null mutations in LTBP2 cause PCG in four consanguineous families from Pakistan and in patients of Gypsy ethnicity. LTBP2 maps to chromosome 14q24.3 but is around 1.3 Mb proximal to the documented GLC3C locus. Therefore, it remains to be determined whether LTBP2 is the GLC3C gene or whether a second adjacent gene is also implicated in PCG. LTBP2 is the largest member of the latent transforming growth factor (TGF)-beta binding protein family, which are extracellular matrix proteins with multidomain structure. It has homology to fibrillins and may have roles in cell adhesion and as a structural component of microfibrils. We confirmed localization of LTBP2 in the anterior segment of the eye, at the ciliary body, and particularly the ciliary process. These findings reveal that LTBP2 is essential for normal development of the anterior chamber of the eye, where it may have a structural role in maintaining ciliary muscle tone.
Main Text
Primary congenital glaucoma (PCG) is a relatively rare cause of glaucoma that usually manifests within the first year of life. It is characterized by high intraocular pressure (IOP) resulting from an obstruction of aqueous outflow from the anterior segment of the eye, thought to be the result of an anomaly of the trabecular meshwork and anterior chamber angle.1,2 Increased IOP causes irreversible damage to the optic nerve and can lead to blindness if untreated. Affected children typically present with tearing, photophobia, corneal clouding, and enlargement of the globe or cornea.3
In most families, PCG is inherited as an autosomal-recessive condition, and it is more common in communities in which the rate of consanguinity is high. Its prevalence varies from ∼1:10,000 in Western countries4 to ∼1:3,300 in Southern India,5 ∼1:2,500 in Saudi Arabia,6 and ∼1:1,250 in the Gypsy subpopulation of Slovakia.7 Three loci for PCG have been reported: GLC3A (2p21 [MIM #231300]),8 GLC3B (1p36 [MIM %600975]),9 and GLC3C (14q24, I.R. Stoilov and M. Sarfarazi, 2002, A.R.V.O., abstract; A. Booth et al., 2008, A.R.V.O., abstract).10 Mutations in Cytochrome P4501B1 (CYP1B1 [MIM ∗601771]) at the GLC3A locus account for up to 50% of cases11,12 and more than 100 CYP1B1 mutations have been reported in the Human Gene Mutation Database. No responsible gene has yet been identified at the GLC3B and GLC3C loci. The purpose of the current study was to identify the mutated gene in families with PCG that do not map to the CYP1B1 locus.
Previously, we described three unrelated consanguineous Pakistani PCG families, MEP47, PKGL005, and PKGL025, in which homozygosity mapping and linkage analysis implicated a gene within or close to GLC3C on chromosome 14q24 (A. Booth et al., 2008, A.R.V.O., abstract; Figure S1 available online).10 In addition, we ascertained one further family, PKGL010, consistent with GLC3C linkage (S.R., unpublished data). The study was approved by a UK ethics committee and by the institutional review board at the Centre of Excellence in Molecular Biology, Pakistan. Informed consent was obtained from participants and from the elders of each household. Members of consenting families underwent ophthalmic examination, which revealed that affected individuals have typical PCG disease features (Figure 1).10 All the patients had a history of tearing and photophobia from either shortly after birth (for MEP47 and PKGL025) or within the first 3 years of life (for PKGL005 and PKGL010). Most had undergone multiple, failed surgical procedures without antimetabolite therapy and at the time of examination had visual acuities of count fingers or worse. The clinical notes available recorded raised intraocular pressure in both eyes of all cases at first presentation (up to 56 mmHg), with corneal enlargement (horizontal corneal diameter up to 15 mm) and clouding. Peripheral blood was taken from each family member and genomic DNA was extracted according to standard procedures.
Figure 1.
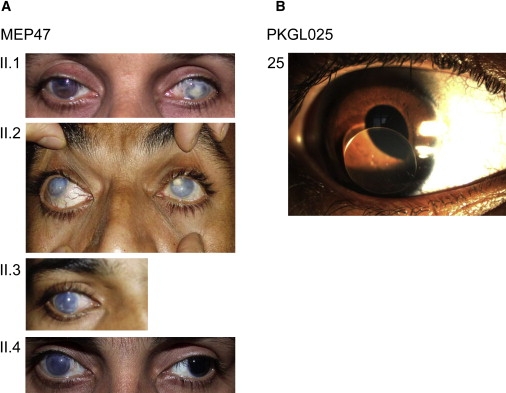
Clinical Features of PCG Patients
(A) For the MEP47 family, photos of the anterior segment of the affected family members are shown. At the time of examination, patients II.1, II.2, II.3, and II.4 were aged 42, 34, 26, and 24 years old, respectively. Case II.1 has Haab's striae in the right eye and a central corneal scar secondary to infection in the left eye. Cases II.2, II.3 (right eye), and II.4 (right eye) have corneal enlargement and clouding. The left eye of case II.4 has an enlarged but clear cornea after surgery.
(B) Slit lamp examination of an affected patient 25 aged 5 years old from family PKGL025 shows dislocation of the lens (ectopia lentis).
Haplotype analysis in these families suggested that the disease gene localizes to a ∼4.2 Mb region, flanked by the markers D14S289 and D14S85, which contains 97 known genes.10 Interestingly, this locus is immediately adjacent to but does not overlap the documented GLC3C locus, first described in a family of Turkish origin, which lies in the interval D14S61 to D14S1000 (I.R. Stoilov and M. Sarfarazi, 2002, A.R.V.O., abstract). Within the interval implicated in the Pakistani families, we systematically viewed the information on each gene with GeneCards and OMIM. The candidate genes were prioritized based on the available literature and their proposed suitability in eye function. The latent transforming growth factor beta binding protein 2 gene (LTBP2 [MIM ∗602091]) was selected as a strong candidate based on its distribution in the anterior segment of the eye.13 The primer pairs used in the PCR amplifications and sequencing are depicted in Table S1. Upon sequencing LTBP2, we identified premature nonsense mutations segregating with PCG in each family (Figures 2 and 3). In MEP47, a homozygous single base pair deletion was identified in exon 1, causing a frameshift and putative stop codon 140 amino acids downstream (c.412 delG; p.A138PfsX278). The sequence variation was confirmed in family members on PCR-amplified DNA with the use of primers 1BF and 1BR followed by SmaI restriction enzyme digestion. In the patients, a SmaI site was destroyed to give a 342 bp product instead of the 218 and 124 bp that would be expected for wild-type sequence. In PKGL005, a homozygous single base change converts an arginine residue to a premature stop codon in exon 4 (c.895C → T; p.R299X). This mutation was confirmed in family members on PCR-amplified DNA with the primer pairs 4IIF and 4R and digestion with the enzyme AlwNI. In the patients, an AlwNI site was formed so that the 257 bp PCR would be digested to yield 228 and 29 bp products. In PKGL025, a homozygous 14 base pair deletion in exon 6 causes a frameshift and putative stop codon 181 amino acids downstream (c.1243-1256 del; p.E415RfsX596). The sequence variation was confirmed in family members by restriction digestions with BsmI on PCR-amplified DNA with the use of primer pairs 6F and 6IIR. In the patients, a BsmI site was destroyed to give a 372 bp product instead of 245 and 141 bp after digestion. Finally, in PKGL010, a homozygous single nucleotide substitution creates a stop codon in exon 1 (c.331C → T; p.Q111X). This mutation was confirmed in the DNA of family members after PCR amplification with 1BF and 1BIIR and digestion with the restriction endonuclease, AlwNI. The mutation destroyed a recognition site for this enzyme to give an intact 172 bp product instead of 143 and 29 bp as expected for the wild-type sequence. These mutations segregated with the PCG phenotype in each family and were excluded from a panel of 110 ethnically matched control DNAs. It is interesting to note that although the four Pakistani families all originated from the Punjab province, they each reside in different cities and also belong to different clans (Table S2). The lack of a single founder mutation common to two or more of these families is consistent with their diverse geographical and ethnic origins.
Figure 2.
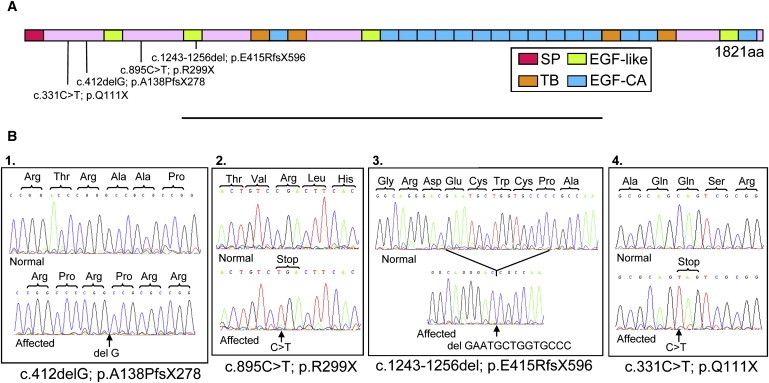
LTBP2 Genetic Analysis in PCG Affecteds
(A) Diagrammatic representation of the LTBP2 protein illustrating the position of the four mutations relative to the full-length 1821 amino acid protein. The domains in the protein are also shown; SP depicts the signal peptide, TB the transforming growth factor beta binding protein-like motif, EGF-like and EGF-CA both represent epidermal growth factor-like domains but the latter also has a calcium binding motif.
(B) Sequence chromatograms of a normal individual and one PCG-affected patient from each of the families. The sequences highlight the mutation in (1) MEP47 (exon 1), (2) PKGL005 (exon 4), (3) PKGL025 (exon 6), and (4) PKGL010 (exon 1).
Figure 3.
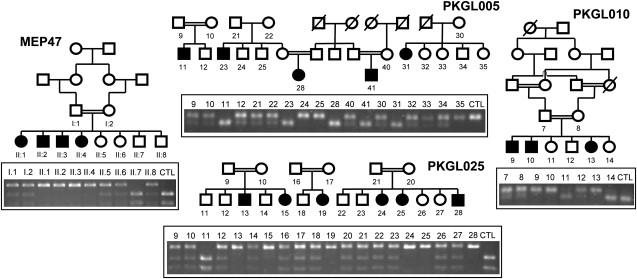
Segregation of the LTBP2 Mutation in Each of the Families
The pedigree structures and individual IDs are shown for MEP47 and PKGL010; however, only the families from which DNA is available are shown for PKGL005 and PKGL025 because the full pedigrees have been published before.10 The agarose gel photographs depicted show segregation of all four mutations with the disease phenotype in each of the respective families as would be expected for a recessive mode of inheritance. Only the PCG-affected members have both LTBP2 alleles mutated as illustrated by the unique banding pattern on the agarose gel. The banding pattern for CTL represents a wild-type control individual.
In addition to investigating the role of LTBP2 in Pakistani PCG families, we also looked in PCG cases from European Gypsies. It has been suggested that the European Gypsies originated from a founder population of Indian subcontinent extraction that migrated from India around 1000 years ago14 and that the high prevalence of PCG in this population7,15 may imply an unidentified ancestral mutation. Recruitment, clinical diagnostic criteria, and CYP1B1 gene investigations in the Gypsy samples were approved by the human research ethics committee, the University of Western Australia, and have been described elsewhere.15 Analysis of LTBP2 in 15 CYP1B1-negative Gypsy patients identified c.895C → T; p.R299X, the mutation segregating in Pakistani family PKGL005, in 8 of the Gypsy cases. The examination of 14 intragenic LTBP2 single-nucleotide polymorphisms revealed an identical haplotype shared between the affected members of the PKGL005 family and the Gypsy PCG patients homozygous for the p.R299X mutation (Table 1), implying that they are distantly related by a common ancestor. The Pakistani PKGL005 family belongs to the Jatt (Jat), a clan/ethnic group of Indo-Aryan descent proposed previously as a possible Gypsy parental population.16 These findings provide further support for this hypothesis and suggest that p.R299X is an ancient founder mutation predating the emergence of European Gypsies from the Jatt. Our data also suggest that p.R299X in LTBP2 is the major PCG founder mutation in the Gypsy population, accounting for more than 50% of CYP1B1-negative and nearly 40% of all PCG cases and is thus of considerable diagnostic significance.
Table 1.
Intragenic LTBP2 SNP Haplotypes
|
PCG Affecteds |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| PCR Amplicon | Chr14 position Ref_Assembly Build 36.3 | SNP ID | Alleles | MEP47 | PKGL025 | PKGL010 | PKGL005 | GYPSY p.R299X homozygotes | GYPSY non-p.R299X | Normal European |
| 1B | 74147995 | ss120037874 | A/C | C | A | A | A | A | A | A |
| 4 | 74092085 | PCG mutation p.R299X | C/T | C | C | C | T | T | C | C |
| 6 | 74088910 | rs11622992 | A/G | G | A | A | G | G | A/G | A/G |
| 9 | 74072528 | rs4899522 | C/T | C | T | C | C | C | C | C |
| 14 | 74061712 | rs862030 | A/G | A | A | G | G | G | G | A/G |
| 14 | 74061608 | rs862031 | C/T | C | C | C | C | C | C | C/T |
| 14 | 74061494 | rs862032 | A/T | T | A | A | A | A | A | A |
| 15 | 74059186 | rs699371 | A/G | G | G | G | G | G | G | A/G |
| 15 | 74059179 | ss120037875 | C/G | G | G | C | C | C | G | G |
| 16 | 74058354 | ss120037876 | C/T | C | C | C | T | T | C | C |
| 18 | 74047763 | ss120037877 | C/G | G | C | C | C | C | C | C |
| 19 | 74046435 | ss120037878 | C/T | T | T | T | C | C | T | T |
| 25 | 74043493 | rs3815329 | C/T | T | T | T | C | C | C/T | T |
| 27 | 74042695 | rs2286411 | C/T | T | C | T | C | C | C/T | C/T |
The PCR amplicon (from Table S1) that was used to locate the SNP, its position in the UCSC genome browser, ID, and allelic variants are shown. Five novel SNPs discovered during this work are shown with the submitter SNP ID (ss) numbers. Haplotypes were constructed based on 14 intragenic SNPs for the PCG affecteds in families MEP47, PKGL025, PKGL010, PKGL005, Gypsies with a homozygous p.R299X mutation, and a Gypsy patient that does not have the p.R299X mutation. The LTBP2 haplotype for a normal European that does not have PCG was also constructed as a control. Note that the PCG affecteds in the family PKGL005 and Gypsies that are p.R299X homozygotes have identical haplotypes across the LTBP2 gene.
The 35 exon LTBP2 gene encodes a 1821 amino acid protein sequence that was analyzed for domains with the SMART program and shown to consist of 20 epidermal growth factor (EGF)-like domains, 4 transforming growth factor beta binding protein (TB)-like modules each containing 8 cysteine residues, and an amino-terminal signal peptide (Figure 2A). Sixteen of the EGF domains have calcium-binding motifs and it has been suggested that these adopt a rod-like molecular arrangement upon calcium binding in order to present a specific surface for protein-protein interactions.17 The cysteine-containing TB domains are unique features of the LTBP-fibrillin superfamily, providing a degree of conformational flexibility.18 LTBP2 has ∼45% amino acid identity to LTBP1 and ∼25% identity to fibrillin1, but unlike other LTBP family members, LTBP2 has been reported not to bind to latent forms of TGF-β.19 The secretory signal targets the protein to the extracellular matrix, where it has cell-adhesive properties,20 a functional role as a docking molecule for elastic fiber assembly,21 and a structural role as a component of fibrillin-rich microfibrils in connective tissues.22 The carboxy-terminal region of LTBP2 competes with LTBP1 for specific binding to the amino-terminal region of fibrillin1, but not fibrillin2, in microfibrils.23
Intriguingly, LTBP2 knockout mice die before embryonic day 6.5.24 In situ hybridization analysis of mouse embryos reveals a pattern of expression primarily restricted to cartilage perichondrium and blood vessels.25 In order to investigate the distribution of the corresponding protein in mouse embryos, we stained serial sections with a rabbit polyclonal antiserum against cow LTBP2 (Figure 4). In brief, whole mouse embryos at E15 were formalin fixed and embedded in paraffin according to standard methods. 4 μm serial sections were incubated either with rabbit polyclonal LTBP2 antibody (LifeSpan Biosciences Inc., Seattle, WA) or the Rabbit Envision Detection system (DAKO, Cambridge, UK). Bound primary antibody was detected with the Rabbit Envision Detection system. The slides were digitized by an Aperio Scanscope (Aperio Technologies Inc., CA) and examined with Imagescope Software (Aperio). LTBP2 was shown to localize to the connective tissues throughout the body and in particular the spinal cord, cardiac, and skeletal muscles as well as the renal and seminiferous tubules. In order to investigate whether the widespread tissue localization may be due to crossreactivity with other LTBP-fibrillin family members, western blot analysis of protein extracts was performed (Figure S2). The antibody recognized a single immunoreactive species of the expected size for LTBP2, confirming the specificity of the antiserum. These observations imply that LTBP2 has a wider structural role in the body, and although this role may be partially fulfilled by other related proteins in the PCG patients, a similar compensatory mechanism does not exist or is not sufficient to sustain life beyond the embryonic stage in mice.
Figure 4.
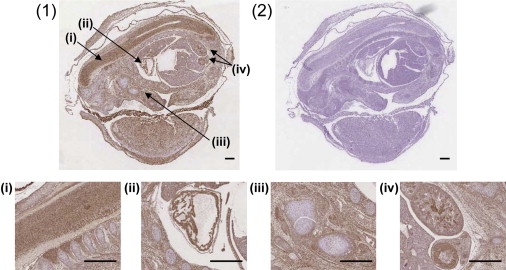
Immunohistological Localization of LTBP2 in Mouse Embryonic Tissue
Section of whole mouse embryo at E15 stained with (1) rabbit polyclonal LTBP2 antibody or (2) the Rabbit Envision Detection system as negative control. LTBP2 immunoreactivity depicted as brown staining was present in the connective tissues throughout the body with an increased localization in the spinal cord (i), the cardiac muscle (ii), the skeletal muscle (iii), and the renal and seminiferous tubules (iv). Scale bars represent 500 μm.
LTBP2 interacts with fibrillin1, mutations in which cause Marfan syndrome (MIM #154700).26 Interestingly, as well as having skeletal and cardiovascular abnormalities, Marfan patients often have ectopia lentis (either partial or complete displacement of the lens) and patients can also develop glaucoma by several different mechanisms.27,28 Furthermore, single-nucleotide polymorphisms in the LTBP2 gene are significantly associated with bone mineral density variation and fracture risk.29 In light of these published findings, we re-examined patients from the PCG families PKGL005, PKGL010, and PKGL025 described herein for signs of skeletal or other abnormalities (Table S3). Bone mineral density was measured in a PCG-affected member from each of these families via dual energy X-ray absorbtiometry (DEXA). These results showed that the PCG-affected individuals as well as the carriers showed mild to moderate osteopenia. Clinical examination of the PCG-affected members showed a high arched palate but other features were not suggestive of classical Marfan syndrome. The echocardiogram investigations were normal in most cases apart from an affected member (patient 10, aged 18 years) from family PKGL010, who had a bicuspid aortic valve and minimal aortic stenosis. Slit lamp examination, where possible, showed that several affected members were aphakic (lens removed by previous surgery), though it was not known whether this was because of cataract formation or ectopia lentis. Two further affected members (patient 25, aged 5 years and patient 28, aged 2 years at the time of examination) from family PKGL025 showed unilateral (Figure 1B) and bilateral ectopia lentis. In MEP47, no history of recurrent fractures or heart problems was reported, but all cases had a Marfanoid habitus with arachnodactyly, joint hypermobility with a positive Steinberg sign, tall stature, and an arm span greater than height. DEXA scanning was not performed in members of this family and corneal clouding made it impossible to identify or exclude ectopia lentis given that the examination was performed in the family home. Whether the skeletal and cardiac features described are related to the lack of functional LTBP2 in these patients requires further investigation; however, ectopia lentis could represent an additional ocular feature for this type of glaucoma.
The precise mechanism by which mutations in LTBP2 lead to PCG is unknown. In order to investigate the distribution of LTBP2 in the anterior segment structures of the eye, sections of mouse and cow eyes were stained with the rabbit polyclonal antiserum (Figure 5). For this, adult mouse and cow eyes were either formalin fixed and paraffin embedded or cryopreserved according to standard procedures. The paraffin-embedded sections (4 μm) were incubated with hematoxylin and eosin, rabbit polyclonal LTBP2 antibody, or the Rabbit Envision Detection system and the slides were processed, digitized, and examined as described before. The cryosections (6 μm) were incubated with hematoxylin and eosin, anti-LTBP2, or a rabbit IgG isotype negative control (Invitrogen Ltd., Paisley, UK). Bound primary antibody was detected with Alexa Fluor 488 conjugated secondary antibody (Invitrogen) and the nuclei were counterstained with DAPI (Invitrogen). Immunofluorescence was analyzed with an Eclipse TE200-E confocal microscope (Nikon Instruments Inc., NY). In both mouse and cow eyes, LTBP2 localized to the ciliary body and particularly the extracellular matrix proteins at the ciliary process. These observations suggest that defects in LTBP2 may increase the elasticity of ciliary body structures and cause changes in the structural support of the surrounding tissues. The defect could be in Schlemm's canal, because its elasticity is thought to influence aqueous outflow.30 Alternatively, a change in the elasticity of the scleral spur might alter the architecture of the trabecular meshwork, because processes from both the ciliary body and the trabecular meshwork insert into the scleral spur.31 Indeed, some glaucoma medications that increase the outflow facility of the eye act by stimulating ciliary muscle contraction against the scleral spur to increase the distension of the trabecular meshwork.32
Figure 5.
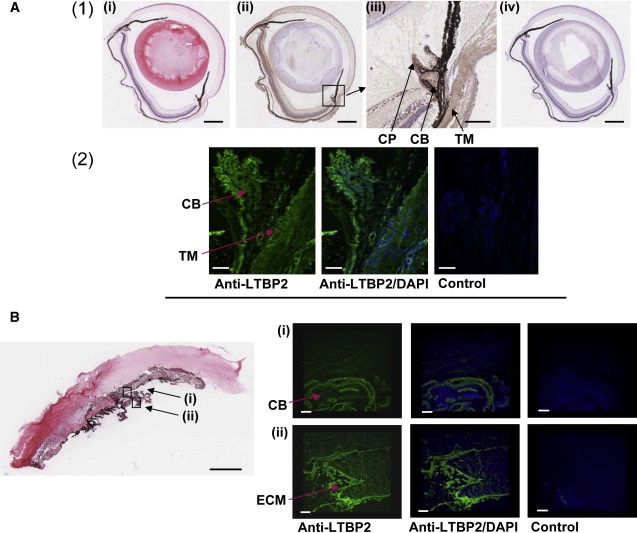
Localization of LTBP2 in the Adult Eye
(A) (1) Formalin-fixed sections of adult mouse eyes stained with hematoxylin and eosin (i), anti-LTBP2 (ii), or the Rabbit Envision Detection system (iv). In (ii), LTBP2 immunoreactivity was observed as brown staining in several structures of the eye including the sclera, retina, cornea, trabecular meshwork, and the ciliary body. The black intense pigment that extends from the retinal pigment epithelium to the iris is melanin (in (i), (ii), and (iv)). Scale bars represent 500 μm. The area surrounding the ciliary body in (ii) has been magnified and is shown in (iii). Note LTBP2 immunoreactivity particularly at the trabecular meshwork (TM), the ciliary body (CB), and the ciliary process (CP). Scale bar represents 100 μm. (2) Cryopreserved sections of adult mouse eyes stained either with anti-LTBP2 or a rabbit IgG isotype negative control. LTBP2 immunoreactivity depicted as green fluorescence was present in the trabecular meshwork (TM), the ciliary body (CB), and particularly the ciliary process. Scale bars represent 50 μm.
(B) Cow eye histological section showing the cornea, iris, and ciliary body stained with hematoxylin and eosin. Scale bar represents 1500 μm. The lens dislocated during the sectioning and the adjacent alignment of the iris with the cornea in the panel is an artifact of tissue preparation. Cryosections stained with anti-LTBP2 or rabbit IgG negative control are shown. Note that the green fluorescence corresponding to LTBP2 immunoreactivity is abundant in the ciliary body (CB) (i) and particularly intense in the extracellular matrix structures (ECM) at the ciliary process (ii). Scale bars represent 150 μm.
LTBP2 is the second gene implicated in PCG to date. Alleles of the other known PCG gene, CYP1B1, have also been associated with early-onset primary open angle glaucoma (POAG [MIM #137760].33,34 POAG, sometimes called chronic glaucoma, is the most common form of glaucoma and can remain undiagnosed for many years while the optic nerve slowly deteriorates.35 It is thought to be a genetically complex trait with both environmental and inherited components.36 Previous studies have suggested possible loci for POAG37,38 and a quantitative trait locus for IOP39 on chromosome 14q, though neither study suggested an association precisely to the LTBP2 region. Furthermore, there are two reports in the literature of cytogenetic studies that suggest a tentative link between chromosome 14q and glaucoma.40,41 It would therefore be interesting to investigate whether LTBP2 gene polymorphisms or mutations in LTBP2 could contribute to raised IOP in POAG.
In summary, we have identified homozygous null mutations in LTBP2 as a cause of PCG in human patients. These findings might have implications for the clinical management of childhood blindness and give new insights into the formation of the anterior structures of the eye, suggesting that LTBP2 may have an important structural role in maintaining the shape of the ciliary body and its surrounding structures. This finding also highlights LTBP2 as a candidate susceptibility gene for the more common and therefore more clinically significant form of glaucoma, POAG.
Acknowledgments
We thank the families for their support and cooperation in this work. This research was supported by the MRC (grant G0501050), the Yorkshire Eye Research (grant 009), the Higher Education Commission (Islamabad, Pakistan), the Ministry of Science and Technology (Islamabad, Pakistan), the Australia India Strategic Research Fund (grant BF020055), and the COMSTECH.EMRO project of the World Health Organization (No. RAB and GH 06-07_24). C.T. is a Royal Society University Research Fellow.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Ensembl, http://www.ensembl.org/index.html
ExPASy, http://us.expasy.org/
GeneCards, http://www.genecards.org/index.shtml
Human Gene Mutation Database, http://www.hgmd.cf.ac.uk/ac/index.php
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Primer3, http://frodo.wi.mit.edu/
UCSC, http://genome.ucsc.edu/
Unigene, http://www.ncbi.nlm.nih.gov/sites/entrez?db = unigene
References
- 1.Kupfer C., Kaiser-Kupfer M.I. Observations on the development of anterior chamber angle with reference to the pathogenesis of congenital glaucomas. Am. J. Ophthalmol. 1979;88:424–426. doi: 10.1016/0002-9394(79)90643-3. [DOI] [PubMed] [Google Scholar]
- 2.Anderson D.R. The development of the trabecular meshwork and its abnormality in primary infantile glaucoma. Trans. Am. Ophthalmol. Soc. 1981;79:458–485. [PMC free article] [PubMed] [Google Scholar]
- 3.Ho C.L., Walton D.S. Primary congenital glaucoma: 2004 update. J. Pediatr. Ophthalmol. Strabismus. 2004;41:271–288. doi: 10.3928/01913913-20040901-11. [DOI] [PubMed] [Google Scholar]
- 4.Francois J. Congenital glaucoma and its inheritance. Ophthalmologica. 1980;181:61–73. doi: 10.1159/000309028. [DOI] [PubMed] [Google Scholar]
- 5.Dandona L., Williams J.D., Williams B.C., Rao G.N. Population-based assessment of childhood blindness in Southern India. Arch. Ophthalmol. 1998;116:545–546. [PubMed] [Google Scholar]
- 6.Jaffar M.S. Care of the infantile glaucoma patient. In: Reineck R.D., editor. Ophthalmology Annual. Raven Press; New York: 1988. p. 15. [Google Scholar]
- 7.Gencik A., Gencikova A., Ferak V. Population genetical aspects of primary congenital glaucoma. I. Incidence, prevalence, gene frequency, and age of onset. Hum. Genet. 1982;61:193–197. doi: 10.1007/BF00296440. [DOI] [PubMed] [Google Scholar]
- 8.Sarfarazi M., Akarsu A.N., Hossain A., Turacli M.E., Aktan S.G., Barsoum-Homsy M., Chevrette L., Sayli B.S. Assignment of a locus (GLC3A) for primary congenital glaucoma (Buphthalmos) to 2p21 and evidence for genetic heterogeneity. Genomics. 1995;30:171–177. doi: 10.1006/geno.1995.9888. [DOI] [PubMed] [Google Scholar]
- 9.Akarsu A.N., Turacli M.E., Aktan S.G., Barsoum-Homsy M., Chevrette L., Sayli B.S., Sarfarazi M. A second locus (GLC3B) for primary congenital glaucoma (Buphthalmos) maps to the 1p36 region. Hum. Mol. Genet. 1996;5:1199–1203. doi: 10.1093/hmg/5.8.1199. [DOI] [PubMed] [Google Scholar]
- 10.Firasat S., Riazuddin S.A., Hejtmancik J.F., Riazuddin S. Primary congenital glaucoma localizes to chromosome 14q24.2–24.3 in two consanguineous Pakistani families. Mol. Vis. 2008;14:1659–1665. [PMC free article] [PubMed] [Google Scholar]
- 11.Stoilov I., Akarsu A.N., Sarfarazi M. Identification of three different truncating mutations in cytochrome P4501B1 (CYP1B1) as the principal cause of primary congenital glaucoma (Bupthalmos) in families linked to the GLC3A locus on chromosome 2p21. Hum. Mol. Genet. 1997;6:641–647. doi: 10.1093/hmg/6.4.641. [DOI] [PubMed] [Google Scholar]
- 12.Stoilov I., Akarsu A.N., Alozie I., Child A., Barsoum-Hmsy M., Turacli M.E., Or M., Lewis R.A., Ozdemir N., Brice G. Sequence analysis and homology modelling suggest that primary congenital glaucoma on 2p21 results from mutations disrupting either the hinge region or the conserved core structures of cytochrome P4501B1. Am. J. Hum. Genet. 1998;62:573–584. doi: 10.1086/301764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schlotzer-Schrehardt U., Zenkel M., Kuchle M., Sakai L.Y., Naumann G.O.H. Role of transforming growth factor-β1 and its latent form binding protein in pseudoexfoliation syndrome. Exp. Eye Res. 2001;73:765–780. doi: 10.1006/exer.2001.1084. [DOI] [PubMed] [Google Scholar]
- 14.Morar B., Gresham D., Angelicheva D., Tournev I., Gooding R., Guergueltcheva V., Schmidt C., Abicht A., Lochmuller H., Tordai A. Mutation history of the Roma/Gypsies. Am. J. Hum. Genet. 2004;75:596–609. doi: 10.1086/424759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sivadorai P., Cherninkova S., Bouwer S., Kamenarova K., Angelicheva D., Seeman P., Hollingsworth K., Mihaylova V., Oscar A., Dimitrova G. Genetic heterogeneity and minor CYP1B1 involvement in the molecular basis of primary congenital glaucoma in Gypsies. Clin. Genet. 2008;74:82–87. doi: 10.1111/j.1399-0004.2008.01024.x. [DOI] [PubMed] [Google Scholar]
- 16.Hancock I. The emergence of Romani as a koine outside of India. In: Acton T., editor. Scholarship and Gypsy Struggle: Commitment in Romani Studies. University of Hertfordshire Press; Hatfield, UK: 2000. pp. 1–13. [Google Scholar]
- 17.Downing A.K., Knott V., Werner J.M., Cardy C.M., Campbell I.D., Handford P.A. Solution structure of a pair of calcium-binding epidermal growth factor-like domains: implications for the Marfan syndrome and other genetic disorders. Cell. 1996;85:597–605. doi: 10.1016/s0092-8674(00)81259-3. [DOI] [PubMed] [Google Scholar]
- 18.Kanzaki T., Olofsson A., Moren A., Wernstedt C., Hellman U., Miyazono K., Claesson-Welsh L., Heldin C.H. TGF-beta 1 binding protein: a component of the large latent complex of TGF-beta 1 with multiple repeat sequences. Cell. 1990;61:1051–1061. doi: 10.1016/0092-8674(90)90069-q. [DOI] [PubMed] [Google Scholar]
- 19.Saharinen J., Keski-Oja J. Specific sequence motif of 8-cys repeats of TGF-beta binding proteins, LTBPs, creates a hydrophobic interaction surface for binding of small latent TGF-beta. Mol. Biol. Cell. 2000;11:2691–2704. doi: 10.1091/mbc.11.8.2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vehvilainen P., Hyytiainen M., Keski-Oja J. Latent transforming growth factor-beta-binding protein 2 is an adhesion protein for melanoma cells. J. Biol. Chem. 2003;278:24705–24713. doi: 10.1074/jbc.M212953200. [DOI] [PubMed] [Google Scholar]
- 21.Hirai M., Horiguchi M., Ohbayashi T., Kita T., Chien K.R., Nakamura T. Latent TGF-β-binding protein 2 binds to DANCE/fibulin-5 and regulates elastic fiber assembly. EMBO J. 2007;26:3283–3295. doi: 10.1038/sj.emboj.7601768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gibson M.A., Hatzinikolas G., Davis E.C., Baker E., Sutherland G.R., Mecham R.P. Bovine latent transforming growth factor β1-binding protein 2: Molecular cloning, identification of tissue isoforms, and immunolocalization to elastin-associated microfibrils. Mol. Cell. Biol. 1995;15:6932–6942. doi: 10.1128/mcb.15.12.6932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hirani R., Hanssen E., Gibson M.A. LTBP-2 specifically interacts with the amino-terminal region of fibrillin-1 and competes with LTBP-1 for binding to this microfibrillar protein. Matrix Biol. 2007;26:213–223. doi: 10.1016/j.matbio.2006.12.006. [DOI] [PubMed] [Google Scholar]
- 24.Shipley J.M., Mecham R.P., Maus E., Bonadio J., Rosenbloom J., McCarthy R.T., Baumann M.L., Frankfater C., Segade F., Shapiro S.D. Developmental expression of latent transforming growth factor β binding protein 2 and its requirement early in mouse development. Mol. Cell. Biol. 2000;20:4879–4887. doi: 10.1128/mcb.20.13.4879-4887.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fang J., Li X., Smiley E., Francke U., Mecham R.P., Bonadio J. Mouse latent TGF-β binding protein-2: Molecular cloning and developmental expression. Biochim. Biophys. Acta. 1997;1354:219–230. doi: 10.1016/s0167-4781(97)00104-8. [DOI] [PubMed] [Google Scholar]
- 26.Dietz H.C., Cutting G.R., Pyeritz R.E., Maslen C.L., Sakai L.Y., Corson G.M., Puffenberger E.G., Hamosh A., Nanthakumar E.J., Curristin S.M. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature. 1991;352:337–339. doi: 10.1038/352337a0. [DOI] [PubMed] [Google Scholar]
- 27.Izquierdo N.J., Traboulsi E.I., Enger C., Maumenee I.H. Glaucoma in the Marfan syndrome. Trans. Am. Ophthalmol. Soc. 1992;90:111–117. [PMC free article] [PubMed] [Google Scholar]
- 28.Challa P., Hauser M.A., Luna C.C., Freedman S.F., Pericak-Vance M., Yang J., McDonald M.T., Allingham R.R. Juvenile bilateral lens dislocation and glaucoma associated with a novel mutation in the fibrillin 1 gene. Mol. Vis. 2006;12:1009–1015. [PubMed] [Google Scholar]
- 29.Cheung C.-L., Sham P.C., Chan V., Paterson A.D., Luk K.D.K., Kung A.W.C. Identification of LTBP2 on chromosome 14q as a novel candidate gene for bone mineral density variation and fracture risk association. J. Clin. Endocrinol. Metab. 2008;93:4448–4455. doi: 10.1210/jc.2007-2836. [DOI] [PubMed] [Google Scholar]
- 30.Tandon P.N., Autar R. Flow of aqueous humor in the canal of Schlemm. Math. Biosci. 1989;93:53–78. doi: 10.1016/0025-5564(89)90013-8. [DOI] [PubMed] [Google Scholar]
- 31.Epstein D.L. Practical aqueos humor dynamics. In: Epstein D.L., Allingham R.R., Schuman J.S., editors. Chandler and Grant's Glaucoma. 4th edn. Williams and Wilkins; Baltimore, MD: 1997. p. 22. [Google Scholar]
- 32.Grierson I., Lee W.R., Abraham S. Effects of pilocarpine on the morphology of the human outflow apparatus. Br. J. Ophthalmol. 1978;62:302–313. doi: 10.1136/bjo.62.5.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vincent A.L., Billingsley G., Buys Y., Levin A.V., Priston M., Trope G., Williams-Lyn D., Heon E. Digenic inheritance of early-onset glaucoma: CYP1B1, a potential modifier gene. Am. J. Hum. Genet. 2002;70:448–460. doi: 10.1086/338709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Melki R., Colomb E., Lefort N., Brezin A.P., Garchon H.-J. CYP1B1 mutations in French patients with early-onset primary open-angle glaucoma. J. Med. Genet. 2004;41:647–651. doi: 10.1136/jmg.2004.020024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Buys Y., Goldberg I., Lambrou G.N., Ritch R. World Glaucoma Day, 6 March 2008: Tackling the glaucoma pandemic. Acta Ophthalmol. (Copenh.) 2008;86:124–125. doi: 10.1111/j.1755-3768.2008.01219.x. [DOI] [PubMed] [Google Scholar]
- 36.Hewitt A.W., Craig J.E., Mackey D.A. Complex genetics of complex traits: The case of primary open-angle glaucoma. Clin. Exp. Ophthalmol. 2006;34:472–484. doi: 10.1111/j.1442-9071.2006.01268.x. [DOI] [PubMed] [Google Scholar]
- 37.Wiggs J.L., Allingham R.R., Hossain A., Kern J., Auguste J., DelBono E.A., Broomer B., Lennon G.F., Hauser M., Pericak-Vance M., Haines J.L. Genome-wide scan for adult onset primary open-angle glaucoma. Hum. Mol. Genet. 2000;9:1109–1117. doi: 10.1093/hmg/9.7.1109. [DOI] [PubMed] [Google Scholar]
- 38.Nemesure B., Jiao X., He Q., Leske M.C., Wu S.Y., Hennis A., Mendell N., Redman J., Garchon H.J., Agarwala R. A genome-wide scan for primary open-angle glaucoma (POAG): The Barbados family study of open-angle glaucoma. Hum. Genet. 2003;112:600–609. doi: 10.1007/s00439-003-0910-z. [DOI] [PubMed] [Google Scholar]
- 39.Rotimi C.N., Chen G., Adeyemo A.A., Jones L.S., Agyenim-Boateng K., Eghan B.A., Jr., Zhou J., Doumatey A., Lashley K., Huang H. Genomewide scan and fine mapping of quantitative trait loci for intraocular pressure on 5q and 14q in West Africans. Invest. Ophthalmol. Vis. Sci. 2006;47:3262–3267. doi: 10.1167/iovs.05-1537. [DOI] [PubMed] [Google Scholar]
- 40.Pfeiffer R.A., Büttinghaus K., Struck H. Partial trisomy 14 following a balanced reciprocal translocation t(14q-;21q+) Humangenetik. 1973;20:187–189. doi: 10.1007/BF00284860. [DOI] [PubMed] [Google Scholar]
- 41.Kousseff B.G. Fahr syndrome and congenital glaucoma: Report of a family and a review. Acta Paediatr. Belg. 1980;33:57–61. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.