

Abstract
Coenzyme Q10 is a mobile lipophilic electron carrier located in the inner mitochondrial membrane. Defects of coenzyme Q10 biosynthesis represent one of the few treatable mitochondrial diseases. We genotyped a patient with primary coenzyme Q10 deficiency who presented with neonatal lactic acidosis and later developed multisytem disease including intractable seizures, global developmental delay, hypertrophic cardiomyopathy, and renal tubular dysfunction. Cultured skin fibroblasts from the patient had a coenzyme Q10 biosynthetic rate of 11% of normal controls and accumulated an abnormal metabolite that we believe to be a biosynthetic intermediate. In view of the rarity of coenzyme Q10 deficiency, we hypothesized that the disease-causing gene might lie in a region of ancestral homozygosity by descent. Data from an Illumina HumanHap550 array were analyzed with BeadStudio software. Sixteen regions of homozygosity >1.5 Mb were identified in the affected infant. Two of these regions included the loci of two of 16 candidate genes implicated in human coenzyme Q10 biosynthesis. Sequence analysis demonstrated a homozygous stop mutation affecting a highly conserved residue of COQ9, leading to the truncation of 75 amino acids. Site-directed mutagenesis targeting the equivalent residue in the yeast Saccharomyces cerevisiae abolished respiratory growth.
Introduction
Coenzyme Q10, or ubiquinone, is a lipophilic molecule located in the inner mitochondrial membrane, where it functions to shuttle electrons from complexes I (NADH:ubiquinone reductase, EC 1.6.5.3) and II (succinate:ubiquinone reductase, EC 1.3.5.1) to complex III (ubiquinol:cytochrome-c reductase, EC 1.10.2.2) in the respiratory chain. Reduced coenzyme Q10 (ubiquinol) also has an important antioxidant role. Other functions of coenzyme Q10 include electron transfer in extramitochondrial electron transport systems (for example, in plasma membranes and lysosomes) and regulation of mitochondrial permeability transition pores.1
Primary coenzyme Q10 deficiency (MIM 607426) is recessively inherited and constitutes a rare but important subgroup of mitochondrial respiratory chain disease, because it is potentially treatable with exogenous coenzyme Q10. Heterogeneous clinical phenotypes have been reported in association with coenzyme Q10 deficiency, including recurrent rhabdomyolysis with seizures,2,3 multisystem disorder of infancy with prominent nephropathy,4,5 ataxia with or without seizures,6 Leigh syndrome,7 and pure myopathy.8 Secondary deficiency of coenzyme Q10 has been implicated in the pathophysiology of statin myopathy, anthracycline toxicity, and Parkinson disease, although the relative contribution of coenzyme Q10 deficiency in these disease processes is debated.9,10
Deficiency of coenzyme Q10 is suspected when activities of respiratory chain complexes I+III and/or II+III are deficient when assayed together (because these assays require the presence of endogenous coenzyme Q10), but activities of individual respiratory chain complexes I, II, and III are all normal when assayed separately (because these assays are independent of coenzyme Q10). The diagnosis is confirmed by measurement of coenzyme Q10 in muscle. Plasma coenzyme Q10 level is an unreliable indicator of total body coenzyme Q10 status, because the former is known to fluctuate with plasma lipid levels.10 However, there is some preliminary evidence that peripheral blood mononuclear cell coenzyme Q10 levels are more closely correlated to muscle coenzyme Q10 levels than are plasma levels, so white blood cell coenzyme Q10 estimation may be a useful primary screening tool for this group of disorders.11
Coenzyme Q10 is a member of a homologous family of ubiquinones that comprise a benzoquinone ring and a polyprenyl “tail” that anchors the molecule to lipid membranes. The numerical subscript used in coenzyme Q nomenclature refers to the number of isoprenyl units in the tail of the molecule. In humans coenzyme Q10 predominates, whereas rodents primarily use coenzyme Q9 and Saccharomyces cerevisiae utilizes coenzyme Q6 as the major isoform. Although the biosynthetic pathway of coenzyme Q10 has not been completely elucidated in humans, it is known that the ring is derived from tyrosine or phenylalanine, whereas the mevalonate pathway generates the decaprenyl tail (Figure 1). The intermediates 4-hydroxybenzoate and decaprenyl diphosphate are then condensed by 4-hydroxybenzoate: polyprenyl transferase, encoded by COQ2 (MIM 609825).1 There are thought to be eight subsequent steps that modify the 6-carbon ring and its side chains to form the functional coenzyme Q10 molecule (Figure 1).
Figure 1.
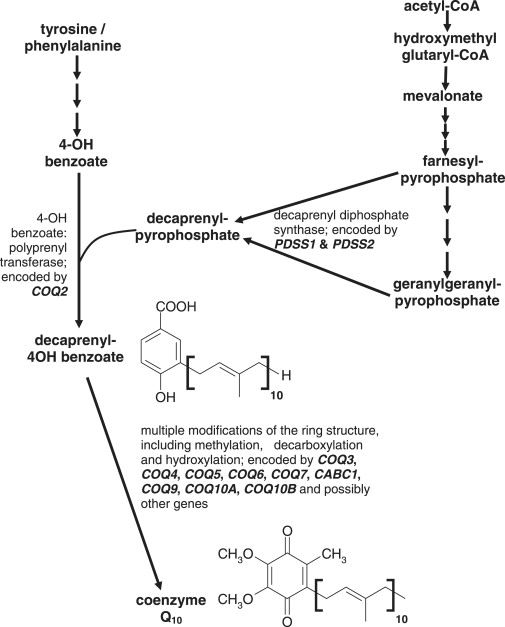
Biosynthetic Pathway of Coenzyme Q10
Genes involved in the biosynthesis of coenzyme Q10 have been characterized in lower organisms (including bacteria, yeast, and nematodes), and the human homologs of 16 of these genes have been identified (Table 1).12 Recently, this has allowed the genetic basis of primary coenzyme Q10 deficiency to be determined in some patients. Mutations in four genes have been reported, in association with a range of clinical phenotypes: mutations in COQ2, PDSS1 (MIM 607429), and PDSS2 (MIM 610564) have been reported in infantile encephalomyopathies,13–16 whereas more recently mutations in CABC1 (MIM 606980) have been associated with juvenile-onset ataxia often accompanied by seizures.17,18 In other patients, the coenzyme Q10 deficiency has been shown to be secondary to other genetic defects: aprataxin mutations in an ataxic form of the disease (MIM 606350)19,20 and mutations in the ETFDH gene (MIM 231657) (which is mutated in multiple acyl CoA dehydrogenase deficiency or glutaric aciduria type II [MIM 231680]) in patients with the myopathic presentation of coenzyme Q10 deficiency.21
Table 1.
Genes Implicated in the Biosynthesis of Human Coenzyme Q10
| Gene | Chromosomal Location | Protein |
|---|---|---|
| PDSS1 | 10p12.1 | decaprenyl-diphosphate synthase subunit 1 |
| PDSS2 | 6q21 | decaprenyl-diphosphate synthase subunit 2 |
| COQ2 | 4q21.23 | 4-hydroxybenzoate polyprenyltransferase |
| COQ3 | 6q16.3 | hexaprenyldihydroxybenzoate methyltransferase |
| COQ4 | 9q34.11 | ubiquinone biosynthesis protein COQ4 homolog |
| COQ5 | 12q24.31 | ubiquinone biosynthesis methyltransferase COQ5 |
| COQ6 | 14q24.3 | ubiquinone biosynthesis monooxygenase COQ6 |
| COQ7 | 16p12.3 | ubiquinone biosynthesis protein COQ7 homolog |
| CABC1 | 1q42.13 | chaperone-activity of bc1 complex-like, mitochondrial precursor (Chaperone-ABC1-like) (aarF domain-containing protein kinase 3) |
| COQ9 | 16q13 | ubiquinone biosynthesis protein COQ9 |
| COQ10A | 12q13.2 | protein COQ10 A |
| COQ10B | 2q33.1 | protein COQ10 B |
| ADCK1 | 14q24.3 | uncharacterized aarF domain-containing protein kinase 1 |
| ADCK2 | 7q34 | uncharacterized aarF domain-containing protein kinase 2 |
| ADCK4 | 19q13.2 | uncharacterized aarF domain-containing protein kinase 4 |
| ADCK5 | 8q24.3 | uncharacterized aarF domain-containing protein kinase 5 |
We now show that a mutation in COQ9, a biosynthetic gene not previously associated with human disease, is a novel cause of coenzyme Q10 deficiency. We report a homozygous nonsense mutation in the COQ9 gene in a patient with neonatal-onset lactic acidosis and multisystem disease. To our knowledge, this is the first report of human coenzyme Q10 deficiency resulting from a mutation in COQ9. It is important to identify the genetic basis of coenzyme Q10 deficiency because more rapid diagnosis allows prompt treatment with exogenous coenzyme Q10 and may improve the outcome of these otherwise devastating and potentially fatal disorders.22
Subjects and Methods
Patient
The clinical details of this patient have previously been reported.5 In brief, the patient was the seventh child of healthy apparently unrelated parents of Pakistani origin who had five other healthy children. An older sister had died on day one of life with a disorder including seizures, acidosis, and aminoaciduria. No diagnosis was made in the older sister. The patient presented at 6 hr of age with poor feeding, hypothermia, increased tone, and lactic acidosis (plasma lactate 19.4 mmol/l, reference range 0.9–1.9). He developed an intractable seizure disorder and was subsequently shown to have severe global developmental delay, with cerebral and cerebellar atrophy on magnetic resonance imaging of the brain. There was also left ventricular hypertrophy and renal tubular dysfunction. Open muscle biopsy performed at 10 months showed type IIB fiber atrophy and lipid accumulation on histological examination and a severe biochemical defect of complexes II+III with normal activities of complexes II and III when assayed separately. HPLC assay confirmed severe deficiency of coenzyme Q10 in skeletal muscle (Table 2).
Table 2.
Coenzyme Q10 Levels in Muscle and Cultured Skin Fibroblasts and Biosynthesis of CoQ10 in Cultured Skin Fibroblasts
| Coenzyme Q10 Levels | Patient | Mean Control ± SD |
|---|---|---|
| Muscle (pmol/mg protein) | 37 | 241 ± 95 (range 140–580, n = 26) |
| Fibroblasts (pmol/mg protein) | 25.3 | 62 ± 14 (range 39–75, n = 5) |
| Fibroblasts (ng/mg protein) | 10.4 | 58 ± 10 (range 41–72, n = 15) |
| Biosynthesis of CoQ10 in fibroblasts (Q10 DPM/mg protein/day) | 304, 480 | 3460 ± 505 (range 3022–4207, n = 6) |
Coenzyme Q10 replacement therapy was started at 11.5 months of age, initially at a dose of 60 mg/day in three divided doses, and increasing after 6 days to 300 mg/day, again in three divided doses. This dose was continued until the patient's death at 2 years of age. Although the plasma lactate appeared to drop after commencing coenzyme Q10 treatment (initially from 7.6 mmol/L to 4.4 mmol/L 11 days after starting treatment, and then to 2.5–2.9 mmol/L after 4 months), there was no associated clinical response. Seizures resistant to quadruple anticonvulsant therapy continued and were associated with severe global developmental delay, microcephaly, and dystonia. Renal tubular function had already improved prior to starting coenzyme Q10, but the cardiomyopathy continued to worsen despite treatment. The patient died just after his second birthday after an intercurrent chest infection.
Muscle and skin biopsies were performed after obtaining informed parental consent, and the study was approved by the Great Ormond Street Hospital for Children NHS Trust and Institute of Child Health Local Research Ethics Committee.
Determination of Coenzyme Q10 Levels and Biosynthetic Activity in Cultured Skin Fibroblasts
After lipid extraction from homogenized cultured skin fibroblasts, coenzyme Q10 levels were determined via reversed-phase HPLC coupled to UV detection at 275 nm11 or electrochemical (EC) detection.14 Rates of coenzyme Q10 biosynthesis were determined by measurement of incorporation of 14C into coenzyme Q10 after culturing skin fibroblasts in 14C-radiolabelled 4-hydroxybenzoate for 24 hr, as previously described.14
SNP Analysis
High-density genome-wide SNP genotyping was performed by hybridizing total genomic DNA extracted from patient fibroblasts on a HumanHap550 array according to the manufacturer's instructions (Illumina Inc.). SNP data was analyzed with BeadStudio v3 (Illumina Inc.) to search for regions of homozygosity.23
Candidate Gene Sequence Analysis
Exons and exon-intron boundaries of the coding sequence of two candidate genes (CABC1 and COQ9) located within regions of homozygosity were PCR amplified from genomic DNA (primers for COQ9 are listed in Supplemental Data available online; primers for CABC1 are available on request). The amplicons were purified and subsequently sequenced bidirectionally, with the ABI BigDye Terminator 3.1 system (Applied Biosystems), on a MegaBace DNA sequencer. Sequence data were analyzed with Sequencher 4.6 software (Gene Codes).
Reverse Transcriptase-PCR Analysis of COQ9 from Fibroblasts
RNA was extracted from fibroblast cultures from the patient and three controls with the RNeasy Mini kit and QIAshredder columns (QIAGEN) according to the manufacturer's instructions. cDNA was generated with the SuperScript III reverse transcriptase kit (Invitrogen) as per the supplied protocol. Two independent RNA extractions were performed. PCR was performed with primers specific for cDNA for COQ9 (in exon 1 and the 3′-UTR) and exon 4 of RPL19 (a housekeeping gene encoding the 60S ribosomal protein L19); primer sequences are available on request. Amplicons were visualized on a 1.5% agarose gel with ethidium bromide. PCR reactions were performed three times with cDNA generated from the two independent extractions.
Modeling of COQ9 Structure
Candidate templates for the COQ9 sequence (Uniprot O75208) were found with the 3DJury metaserver.24 COQ9 was found to be distantly related to a large number of transcriptional regulators from Streptomyces species. Two templates, PDB codes 3c07 and 2qib, were chosen for modeling, the latter in order to fully cover the C terminus, which was uniquely aligned by this template. Residues 1–94 from the sequence were not matched by any template and were not modeled; the Disopred method25 predicts much of this region to be disordered. All of the templates were dimeric according to PQS,26 so we additionally built dimeric models for assessment. Initial alignments from 3DJury were manually refined to avoid indels within elements of secondary structure. Models were generated with MODELER27 and assessed for hydrophobic packing with SolvX28 and other model quality parameters with MODCHECK-HD.29 The best model was selected with these scores and rms deviation from the template structures. Side-chain packing in the final model was optimized with SCWRL.30
Yeast Studies
The yeast COQ9 gene was amplified by PCR from genomic DNA of strain W303-1B. The human COQ9 gene was amplified by PCR of cDNAs that were kindly provided by E. Jacquet (ISCN, CNRS, Gif-sur-Yvette, France) and were obtained by reverse transcription of mRNAs extracted from human cell lines. The COQ9 genes were cloned into a multicopy vector carrying the LEU2 marker and placed under the control of a constitutive promoter (PGK). After checking the sequence of the insert, the plasmids were used for transformation of a Δcoq9 yeast (W303-1A MATa, ade2-1, his3-1,15, leu2-3,112, trp1-1, ura3-1, coq9::URA3), kindly given by A. Tzagoloff (Columbia University, NY). Leu+ transformants were selected on synthetic minimum medium, with a fermentable carbon source (minimum medium: 0.7% yeast nitrogen base, 3% glucose, CSM supplement minus leucine). Respiratory growth was checked on respiratory medium (YPG: 1% yeast extract, 2% peptone, 3% glycerol). Site-directed mutagenesis was performed with the Quickchange Site-Directed Mutagenesis Kit (Stratagene) according to the manufacturer's recommendations.
Results
Biosynthesis of Coenzyme Q10 in Cultured Skin Fibroblasts
In agreement with previous assays of mitochondrial respiratory enzyme activities and coenzyme Q10 levels in muscle,5 HPLC analysis demonstrated low levels of coenzyme Q10 in cultured skin fibroblasts from the patient compared to controls (Table 2) and also revealed a second peak with a slightly shorter retention time than coenzyme Q10. This might represent abnormal accumulation of a biosynthetic intermediate (Figure 2). The rate of biosynthesis of coenzyme Q10, determined with 14C-4-hydroxybenzoate, in cultured skin fibroblasts from the patient was 11% of the mean control activity (Table 2), confirming the presence of a severe primary defect of coenzyme Q10 biosynthesis in this patient.
Figure 2.
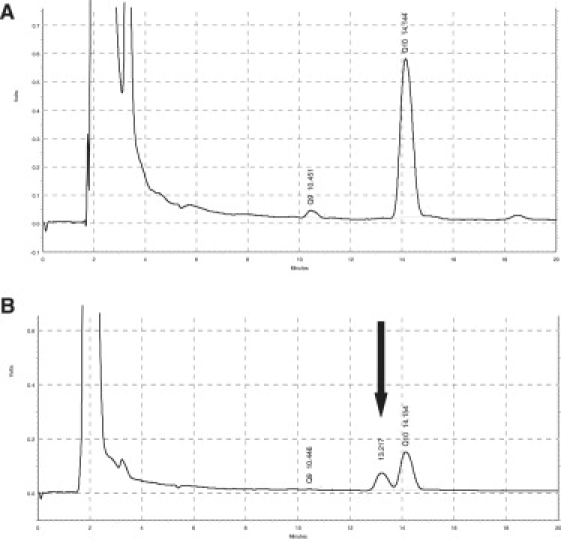
HPLC Analysis of Coenzyme Q10 in Cultured Skin Fibroblasts
Reverse phase EC-HPLC chromatograms obtained from lipid extracts of cultured skin fibroblasts from (A) healthy control fibroblasts and (B) the patient. Retention times are coenzyme Q9 = 10.45 min; coenzyme Q10 = 14.15 min. The patient's trace clearly shows an additional peak at 13.22 min (arrow) not resulting from coenzyme Q9 or coenzyme Q10, likely to be the result of accumulation of a metabolic intermediate of coenzyme Q10 biosynthesis.
SNP Analysis
Analysis of SNP data revealed 16 regions of homozygosity of between 1.5 and 16.8 Mb, of which five were greater than 10 Mb in length (Table 3). The longest region of homozygosity was observed on chromosome 17, but did not contain any candidate genes known to be involved in the biosynthesis of coenzyme Q10. The third and fourth largest regions of homozygosity (13.4 Mb and 10.6 Mb on chromosomes 1 and 16, respectively) each contained a candidate gene implicated in coenzyme Q10 biosynthesis: CABC1 (also known as COQ8) on chromosome 1 and COQ9 on chromosome 16.
Table 3.
Regions of Homozygosity Observed in the Patient
| Chromosome | Flanking SNPs | Length of Homozygous Region Mb | Candidate Genes Located in Region of Homozygosity |
|---|---|---|---|
| 1 | rs12043991, rs1009658 | 13.4 | CABC1 |
| 2 | rs6749901, rs3755021 | 5 | |
| 3 | rs9883258, rs7636942 | 3.2 | |
| 3 | rs1393555, rs9855944 | 10.1 | |
| 5 | rs4498289, rs294580 | 3.3 | |
| 7 | rs41943, rs17595350 | 2.3 | |
| 8 | rs7013926, rs6474040 | 6.6 | |
| 12 | rs4931594, rs10161491 | 1.5 | |
| 13 | rs1314940, rs2025736 | 15.7 | |
| 16 | rs7201926, rs36553 | 10.6 | COQ9 |
| 17 | rs11078078, rs7212579 | 16.8 | |
| 17 | rs2229611, rs6503398 | 1.8 | |
| 19 | rs4807140, rs2240669 | 2.4 | |
| 19 | rs1688021, rs10422365 | 2.9 | |
| 20 | rs6021247, rs6022204 | 1.5 | |
| 22 | rs136026, rs756638 | 4.9 |
DNA Sequence Analysis
The entire coding sequence of CABC1 was wild-type. A homozygous C→T transition was identified in COQ9 at position 730 of the cDNA c.730C→T (Figure 3), predicting a premature stop codon, p.R244X, in exon 7, truncating the terminal 75 amino acids of the protein. Parental DNA was not available for analysis. This mutation was absent in 308 control alleles, including 114 Pakistani control alleles. The arginine at position 244 of the protein is highly conserved through evolution (Figure 4). Sequence analysis in 20 further patients with primary coenzyme Q10 deficiency without a genetic diagnosis did not reveal any mutations in COQ9.
Figure 3.
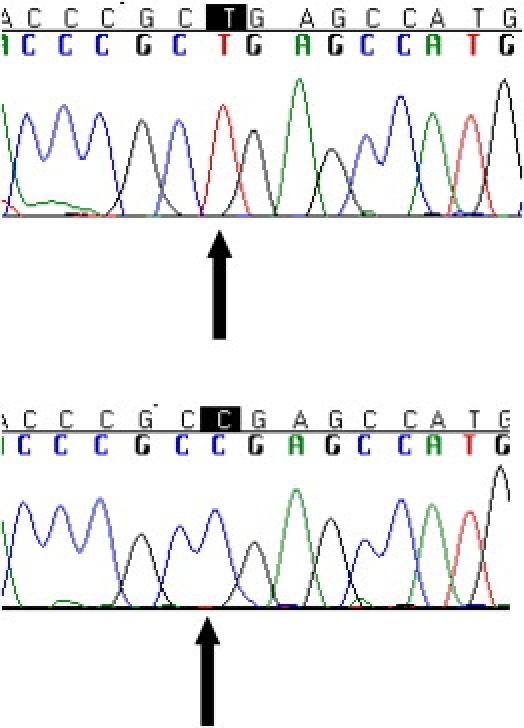
Sequence Analysis of COQ9
Sequencing electropherograms showing homozygous c.730C→T mutation in the patient's genomic DNA (top) and healthy control genomic DNA (bottom). The mutation predicts a change from arginine to a stop codon at amino acid 244 of the protein.
Figure 4.
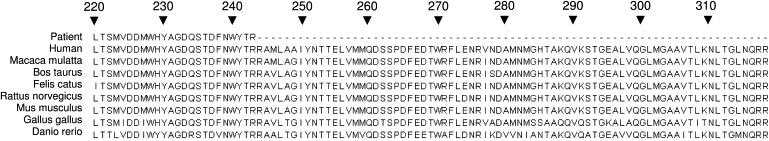
Evolutionary Conservation Data for COQ9
Reverse Transcriptase-PCR Analysis of COQ9 from Fibroblasts
COQ9 cDNA was successfully amplified from fibroblasts in three controls but could not be amplified from the patient's fibroblasts, on three occasions with RNA from two independent extractions (see Supplemental Data). In both patient and controls, amplification of the housekeeping gene encoding the 60S ribosomal protein L19 was observed (Supplemental Data). This result is strongly suggestive of the occurrence of nonsense-mediated decay of COQ9 mRNA in the patient. Consequently it is likely that a negligible amount of COQ9 protein will be formed.
Modeling of COQ9 Structure
Models for 12 alternative template/alignment conformations were generated and evaluated with two quality assessments as described in Subjects and Methods. On the basis that the template structures were homodimeric, we also assessed both monomeric and homodimeric models. The homodimer models were consistently found to score better by the quality assessment methods than did the monomers because the monomer would expose a considerable hydrophobic surface area that is buried in the interface in the dimer model (Figure 5).
Figure 5.
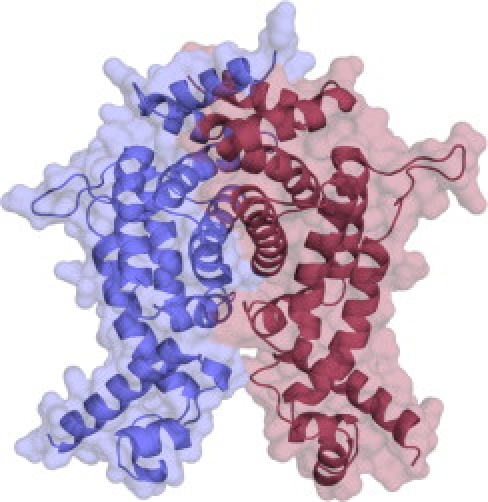
Molecular Model of COQ9 in Homodimeric Form
One monomer is depicted in red, the other in blue. Figure created with Pymol.42
Yeast Studies
The Δcoq9 yeast mutant failed to grow on respiratory medium (Figure 6A) as expected. Transformation of the Δcoq9 yeast mutant with yeast COQ9 inserted into a multicopy yeast expression vector under the control of a constitutive promoter restored respiratory growth to wild-type levels (Figure 6B). Overexpression of human COQ9 with the same vector and promoter did not restore respiratory growth in the yeast Δcoq9 mutant (Figure 6D). However, the introduction of a stop mutation by site-directed mutagenesis at the equivalent position of the human mutation (human p.R244X = yeast p.K191X) into the cloned yeast COQ9 abolished respiratory growth (Figure 6C).
Figure 6.
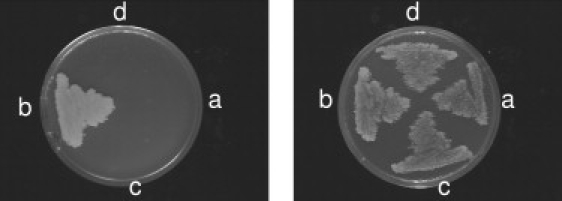
Yeast Studies
Left: Growth on respiratory (glycerol) medium. Right: Growth on fermentable (glucose) medium selective for the plasmids. (a) Δcoq9 mutant+ empty plasmid; (b) Δcoq9 mutant+ pYcoq9 (yeast COQ9); (c) Δcoq9 mutant+ pYcoq9Stop (yeast coq9 K191X); (d) Δcoq9 mutant+ pHcoq9 (human COQ9). COQ9 genes were placed under the control of a constitutive promoter on a multicopy plasmid.
Discussion
We used a strategy involving search for homozygosity by descent at loci of candidate genes known to be involved in coenzyme Q10 biosynthesis to identify the causative genetic defect in a patient with neonatal presentation of primary coenzyme Q10 deficiency. Although the parents of our patient were not known to be related, there is some evidence that extended regions of homozygosity can occur in outbred individuals.31–33 We also reasoned that homozygosity by descent is more likely to be the cause of an extremely rare disorder such as primary coenzyme Q10 deficiency (observed in only 2 of more than 1000 pediatric muscle biopsies from patients with suspected mitochondrial disorders analyzed by our biochemistry laboratory). In fact, the amount of homozygosity in the patient suggests that the parents may have a greater degree of relationship than was elicited in the clinical history.
Biosynthesis of coenzyme Q10 is a complex biological process that is not completely understood in humans. A number of proteins involved in biosynthesis of coenzyme Q in yeast and other lower organisms have been characterized by studies of respiratory-deficient mutants, and the human homologs of 16 of these proteins have been identified (Table 1). The exact function of several of these proteins remains unknown, but four have been implicated in primary deficiency of coenzyme Q10 in humans (Table 4). Clinical features of patients with mutations in these genes are summarized in Table 4. The different clinical phenotype of our patient, and the observation of an extra peak with a slightly shorter retention time than coenzyme Q10 on HPLC analysis, led us to hypothesize that our patient had a different genetic basis to other patients reported in the literature, and that this was likely to be a distal step of coenzyme Q10 biosynthesis, possibly in modification of the 4-hydroxybenzoate ring.
Table 4.
Genotype-Phenotype Correlation in Inherited Deficiency of Coenzyme Q10 Biosynthesis
| Gene | Number of Patients (Families) | Age at Onset | Clinical Features | MRI Features | Lactate mmol/l (Reference Range) | Coenzyme Q10 Level (Reference Range) | Response to Exogenous Q10 Supplementation | References |
|---|---|---|---|---|---|---|---|---|
| PDSS1 | 2 (1) | 1–2 y | deafness, bulimia, obesity, optic atrophy, valvulopathy, macrocephaly, peripheral neuropathy, MR | NR | B 2.1 at 22 y; 3.2 at 14 y (1.0–1.55) | Fb 4 pmol/mg (120 ± 32) | alive at 22 y (mild MR) | 15 |
| PDSS2 | 1 | 3 mo | Leigh syndrome, nephropathy | bilateral symmetrical T2 hyperintensity in BG | B 7.5 (<2.0) | M 4.6 ng/mg (32.1 ± 6.8); Fb 6.7 ng/mg (52.2 ± 9.1) | no clinical response; died at 8 mo (refractory status epilepticus) | 13 |
| COQ2 | 2 (1) | 9–12 mo | encephalomyopathy, nephropathy | cerebellar atrophy and stroke-like lesions | B + CSF N | M 12 ng/mg (32.1 ± 6.7); Fb 18–19 ng/mg (105 ± 14) | dramatic improvement of neurological manifestations; renal transplant at 3 y in proband; renal disease responded to early treatment in younger sibling | 14,22 |
| COQ2 | 2 (2) | 18 mo | steroid-resistant nephropathy | N | B N | M 12 pmol/mg (17.6–49.2); KC 4.5 pmol/mg (26–180) | neurologically normal during 8 mo of treatment | 16 |
| 5 d | acute renal failure, progressive epileptic encephalopathy | cortical and subcortical stroke-like lesions | CSF 11.8 (1.1–2.4) | M 0.8 pmol/mg (17.6–49.2); KC 3.7 pmol/mg (26–180) | no clinical response; died at 6 mo (respiratory failure). | |||
| COQ2 | 1 | birth | neurological distress, liver failure, nephrotic syndrome, anemia, pancytopaenia, IDDM, seizures | NR | B 22.7 (1.0–1.55) | Fb 29 pmol/mg(120 ± 32) | no clinical response; died at 12 d (multiorgan failure) | 15 |
| CABC1 | 2 (2) | <18 mo | proximal muscle weakness, cerebellar ataxia, strabismus, ptosis, seizures | cerebellar atrophy and posterior cerebral cortical and subcortical hyperintensities (both patients) | B 3.0 (1.0–1.55); CSF 4.0 (<2.0) | M 2.6–9.4 pmol/mg (32.2 ± 9.8); Fb 125 pmol/mg (120 ± 32) | severe neurological deterioration and EPC (alive at 16 y and 14 y) | 17 |
| CABC1 | 7 (4) | 3–11 y | cerebellar ataxia, exercise intolerance | cerebellar atrophy | B N – 7.8 (0.5–2.2) | M 12.6 ng/mg (n = 1) (27.6 ± 4.4); Fb 29–69 ng/mg (n = 3) (58.5 ± 4.1) | mild improvement of ataxia (only one patient treated) | 18 |
| COQ9 | 1 | birth | neonatal lactic acidosis, seizures, global developmental delay, hypertrophic cardiomyopathy, renal tubular dysfunction | cerebral + cerebellar atrophy | B 19.4 (0.9–1.9) | M 37 pmol/mg (140–580); Fb 25.3 pmol/mg (39–75) | no clinical response; died at 2 y | 5; this report |
Abbreviations: B, blood; BG, basal ganglia; CSF, cerebrospinal fluid; d, days; EPC, epilepsia partialis continua; Fb, fibroblast; IDDM, insulin-dependent diabetes mellitus; KC, kidney cortex; M, muscle; mo, months; MR, mental retardation; N, normal; NR, not reported; y, years.
Our report confirms that COQ9 is necessary for the biosynthesis of coenzyme Q10 in humans and that its deficiency is associated with an early onset and severe multisystem disorder. Evidence for pathogenicity of the mutation observed in this patient are that it is a nonsense mutation, leading to nonsense-mediated decay of the mRNA, and its absence in >300 control chromosomes. Supportive evidence is provided by the yeast studies, which demonstrated a respiratory defect in yeast in which the equivalent portion of the Coq9 protein had been deleted by site-directed mutagenesis.
The model proposed for the structure depends partly on the quaternary state, because otherwise the conformation of the C-terminal region would not make sense without significant repacking in order to prevent exposure of significant hydrophobic surface area. Although the true quaternary structure is unknown, it has previously been reported that the yeast ortholog of this protein is a homo-oligomer.34 Although the authors state that the mass of the native complex is some three times the mass of the monomeric form, this could be accounted for by complexation with another protein.
The precise function of COQ9 remains obscure. Yeast Coq9p has been localized to the inner mitochondrial membrane, facing the matrix.34 Studies of yeast Coq9p showed that it cosediments with other proteins involved in coenzyme Q biosynthesis, including Coq3p and Coq5p,34 leading some authors to suggest that Coq9p is part of a multienzyme coenzyme Q biosynthetic complex.35 COQ9 mutants in yeast incorporate 4-hydroxybenzoic acid into 3-hexaprenyl-4-hydroxybenzoic acid, indicating that the COQ9 product acts distally to prenylation of 4-hydroxybenzoic acid.34 Our findings support this suggestion and indicate that COQ9 is likely to be involved in an enzymatic modification step of the benzoquinone ring, because of the observation of an extra peak on HPLC analysis of our patient's fibroblasts. We hypothesize that this extra peak represents an accumulating biosynthetic intermediate.
Yeast COQ9 mutants can be suppressed by the overexpression of COQ8, suggesting a genetic or physical interaction of these two genes or their products.34 For example, it has been suggested that the yeast Coq9 polypeptide might be the substrate of the putative kinase ABC1/COQ8.35 The human homolog of ABC1/COQ8 is CABC1 and was recently demonstrated to be mutated in primary coenzyme Q10 deficiency presenting with childhood-onset cerebellar ataxia, with additional features including exercise intolerance and seizures.17,18 The clinical phenotype and disease progression associated with CABC1 mutations appears to be much milder than that observed in our patient with COQ9 mutations, providing evidence that these two proteins are likely to have quite distinct functions. Although it is interesting that cerebellar atrophy is shared by patients with mutations in CABC1 and our patient with COQ9 mutations, ataxia appears to be common to several forms of coenzyme Q10 deficiency, both primary and secondary.2,4,6,7,19,36,37 This may simply reflect low endogenous coenzyme Q10 levels in the cerebellum, leading to increased susceptibility of this tissue to damage in conditions of inherited or acquired coenzyme Q10 deficiency.38
Pathogenic mechanisms underlying manifestations of coenzyme Q10 deficiency remain incompletely understood, but include reduced ATP production, increased production of reactive oxygen species, and impaired de novo pyrimidine biosynthesis.39,40 A recent report has suggested that early treatment is associated with an excellent prognosis in childhood-onset primary coenzyme Q10 deficiency.22 The lack of response to coenzyme Q10 replacement therapy in our patient is most likely explained by the presence of irreversible disease manifestations (such as cerebral and cerebellar atrophy) by the time the diagnosis was established. Other possible explanations include inadequate transfer of exogenous coenzyme Q10 across the blood-brain barrier, variable bioavailability of different coenzyme Q10 formulations,41 and variations between individuals in coenzyme Q10 absorption and/or intramitochondrial uptake of exogenous coenzyme Q10.
In conclusion, characterization of disorders of coenzyme Q10 is of paramount importance because they are potentially treatable. We report a new genetic cause of primary coenzyme Q10 deficiency. Identification of mutations responsible for coenzyme Q10 deficiency allows the possibility of prenatal diagnosis and treatment from birth, which could improve the clinical outcome.
Supplemental Data
Supplemental Data include one figure and one table and can be found with this article online at http://www.ajhg.org/.
Supplemental Data
Web Resources
The URL for data presented herein is as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Acknowledgments
This work was undertaken at GOSH/UCL Institute of Child Health, which received a proportion of funding from the Department of Health's NIHR Biomedical Research Centres funding scheme. We gratefully acknowledge funding from the UK Medical Research Council (grant number G0200335); the Biosapiens Network of Excellence (funded by the European Commission within its FP6 Programme, under the thematic area Life sciences, Genomics and Biotechnology for Health, contract number LSHG-CT-2003-503265); and the Intramural Research Program of the National Institute on Aging, National Institutes of Health, Department of Health and Human Services, project AG000932-01. We also thank Tabitha Owen for her excellent technical assistance.
References
- 1.Turunen M., Olsson J., Dallner G. Metabolism and function of coenzyme Q. Biochim. Biophys. Acta. 2004;1660:171–199. doi: 10.1016/j.bbamem.2003.11.012. [DOI] [PubMed] [Google Scholar]
- 2.Ogasahara S., Engel A.G., Frens D., Mack D. Muscle coenzyme Q deficiency in familial mitochondrial encephalomyopathy. Proc. Natl. Acad. Sci. USA. 1989;86:2379–2382. doi: 10.1073/pnas.86.7.2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sobreira C., Hirano M., Shanske S., Keller R.K., Haller R.G., Davidson E., Santorelli F.M., Miranda A.F., Bonilla E., Mojon D.S. Mitochondrial encephalomyopathy with coenzyme Q10 deficiency. Neurology. 1997;48:1238–1243. doi: 10.1212/wnl.48.5.1238. [DOI] [PubMed] [Google Scholar]
- 4.Rotig A., Appelkvist E.L., Geromel V., Chretien D., Kadhom N., Edery P., Lebideau M., Dallner G., Munnich A., Ernster L. Quinone-responsive multiple respiratory-chain dysfunction due to widespread coenzyme Q10 deficiency. Lancet. 2000;356:391–395. doi: 10.1016/S0140-6736(00)02531-9. [DOI] [PubMed] [Google Scholar]
- 5.Rahman S., Hargreaves I., Clayton P., Heales S. Neonatal presentation of coenzyme Q10 deficiency. J. Pediatr. 2001;139:456–458. doi: 10.1067/mpd.2001.117575. [DOI] [PubMed] [Google Scholar]
- 6.Musumeci O., Naini A., Slonim A.E., Skavin N., Hadjigeorgiou G.L., Krawiecki N., Weissman B.M., Tsao C.Y., Mendell J.R., Shanske S. Familial cerebellar ataxia with muscle coenzyme Q10 deficiency. Neurology. 2001;56:849–855. doi: 10.1212/wnl.56.7.849. [DOI] [PubMed] [Google Scholar]
- 7.van Maldergem L., Trijbels F., DiMauro S., Sindelar P.J., Musumeci O., Janssen A., Delberghe X., Martin J.J., Gillerot Y. Coenzyme Q-responsive Leigh's encephalopathy in two sisters. Ann. Neurol. 2002;52:750–754. doi: 10.1002/ana.10371. [DOI] [PubMed] [Google Scholar]
- 8.Lalani S.R., Vladutiu G.D., Plunkett K., Lotze T.E., Adesina A.M., Scaglia F. Isolated mitochondrial myopathy associated with muscle coenzyme Q10 deficiency. Arch. Neurol. 2005;62:317–320. doi: 10.1001/archneur.62.2.317. [DOI] [PubMed] [Google Scholar]
- 9.Lamperti C., Naini A.B., Lucchini V., Prelle A., Bresolin N., Moggio M., Sciacco M., Kaufmann P., DiMauro S. Muscle coenzyme Q10 level in statin-related myopathy. Arch. Neurol. 2005;62:1709–1712. doi: 10.1001/archneur.62.11.1709. [DOI] [PubMed] [Google Scholar]
- 10.Molyneux S.L., Young J.M., Florkowski C.M., Lever M., George P.M. Coenzyme q10: Is there a clinical role and a case for measurement? Clin. Biochem. Rev. 2008;29:71–82. [PMC free article] [PubMed] [Google Scholar]
- 11.Duncan A.J., Heales S.J., Mills K., Eaton S., Land J.M., Hargreaves I.P. Determination of coenzyme Q10 status in blood mononuclear cells, skeletal muscle, and plasma by HPLC with di-propoxy-coenzyme Q10 as an internal standard. Clin. Chem. 2005;51:2380–2382. doi: 10.1373/clinchem.2005.054643. [DOI] [PubMed] [Google Scholar]
- 12.Rotig A., Mollet J., Rio M., Munnich A. Infantile and pediatric quinone deficiency diseases. Mitochondrion. 2007;7(Suppl 1):S112–S121. doi: 10.1016/j.mito.2007.02.008. [DOI] [PubMed] [Google Scholar]
- 13.Lopez L.C., Schuelke M., Quinzii C.M., Kanki T., Rodenburg R.J., Naini A., DiMauro S., Hirano M. Leigh syndrome with nephropathy and CoQ10 deficiency due to decaprenyl diphosphate synthase subunit 2 (PDSS2) mutations. Am. J. Hum. Genet. 2006;79:1125–1129. doi: 10.1086/510023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Quinzii C., Naini A., Salviati L., Trevisson E., Navas P., DiMauro S., Hirano M. A mutation in para-hydroxybenzoate-polyprenyl transferase (COQ2) causes primary coenzyme Q10 deficiency. Am. J. Hum. Genet. 2006;78:345–349. doi: 10.1086/500092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mollet J., Giurgea I., Schlemmer D., Dallner G., Chretien D., Delahodde A., Bacq D., de Lonlay P., Munnich A., Rotig A. Prenyldiphosphate synthase, subunit 1 (PDSS1) and OH-benzoate polyprenyltransferase (COQ2) mutations in ubiquinone deficiency and oxidative phosphorylation disorders. J. Clin. Invest. 2007;117:765–772. doi: 10.1172/JCI29089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Diomedi-Camassei F., Di Giandomenico S., Santorelli F.M., Caridi G., Piemonte F., Montini G., Ghiggeri G.M., Murer L., Barisoni L., Pastore A. COQ2 nephropathy: a newly described inherited mitochondriopathy with primary renal involvement. J. Am. Soc. Nephrol. 2007;18:2773–2780. doi: 10.1681/ASN.2006080833. [DOI] [PubMed] [Google Scholar]
- 17.Mollet J., Delahodde A., Serre V., Chretien D., Schlemmer D., Lombes A., Boddaert N., Desguerre I., de Lonlay P., de Baulny H.O. CABC1 gene mutations cause ubiquinone deficiency with cerebellar ataxia and seizures. Am. J. Hum. Genet. 2008;82:623–630. doi: 10.1016/j.ajhg.2007.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lagier-Tourenne C., Tazir M., Lopez L.C., Quinzii C.M., Assoum M., Drouot N., Busso C., Makri S., Ali-Pacha L., Benhassine T. ADCK3, an ancestral kinase, is mutated in a form of recessive ataxia associated with coenzyme Q10 deficiency. Am. J. Hum. Genet. 2008;82:661–672. doi: 10.1016/j.ajhg.2007.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Quinzii C.M., Kattah A.G., Naini A., Akman H.O., Mootha V.K., DiMauro S., Hirano M. Coenzyme Q deficiency and cerebellar ataxia associated with an aprataxin mutation. Neurology. 2005;64:539–541. doi: 10.1212/01.WNL.0000150588.75281.58. [DOI] [PubMed] [Google Scholar]
- 20.Le Ber I., Dubourg O., Benoist J.F., Jardel C., Mochel F., Koenig M., Brice A., Lombes A., Durr A. Muscle coenzyme Q10 deficiencies in ataxia with oculomotor apraxia 1. Neurology. 2007;68:295–297. doi: 10.1212/01.wnl.0000252366.10731.43. [DOI] [PubMed] [Google Scholar]
- 21.Gempel K., Topaloglu H., Talim B., Schneiderat P., Schoser B.G., Hans V.H., Palmafy B., Kale G., Tokatli A., Quinzii C. The myopathic form of coenzyme Q10 deficiency is caused by mutations in the electron-transferring-flavoprotein dehydrogenase (ETFDH) gene. Brain. 2007;130:2037–2044. doi: 10.1093/brain/awm054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Montini G., Malaventura C., Salviati L. Early coenzyme Q10 supplementation in primary coenzyme Q10 deficiency. N. Engl. J. Med. 2008;358:2849–2850. doi: 10.1056/NEJMc0800582. [DOI] [PubMed] [Google Scholar]
- 23.Camargos S., Scholz S., Simon-Sanchez J., Paisan-Ruiz C., Lewis P., Hernandez D., Ding J., Gibbs J.R., Cookson M.R., Bras J. DYT16, a novel young-onset dystonia-parkinsonism disorder: Identification of a segregating mutation in the stress-response protein PRKRA. Lancet Neurol. 2008;7:207–215. doi: 10.1016/S1474-4422(08)70022-X. [DOI] [PubMed] [Google Scholar]
- 24.Ginalski K., Elofsson A., Fischer D., Rychlewski L. 3D-Jury: A simple approach to improve protein structure predictions. Bioinformatics. 2003;19:1015–1018. doi: 10.1093/bioinformatics/btg124. [DOI] [PubMed] [Google Scholar]
- 25.Jones D.T., Ward J.J. Prediction of disordered regions in proteins from position specific score matrices. Proteins. 2003;53(Suppl 6):573–578. doi: 10.1002/prot.10528. [DOI] [PubMed] [Google Scholar]
- 26.Brooksbank C., Camon E., Harris M.A., Magrane M., Martin M.J., Mulder N., O'Donovan C., Parkinson H., Tuli M.A., Apweiler R. The European Bioinformatics Institute's data resources. Nucleic Acids Res. 2003;31:43–50. doi: 10.1093/nar/gkg066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sali A., Blundell T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 28.Holm L., Sander C. Evaluation of protein models by atomic solvation preference. J. Mol. Biol. 1992;225:93–105. doi: 10.1016/0022-2836(92)91028-n. [DOI] [PubMed] [Google Scholar]
- 29.Sadowski M.I., Jones D.T. Benchmarking template selection and model quality assessment for high-resolution comparative modeling. Proteins. 2007;69:476–485. doi: 10.1002/prot.21531. [DOI] [PubMed] [Google Scholar]
- 30.Canutescu A.A., Shelenkov A.A., Dunbrack R.L. A graph-theory algorithm for rapid protein side-chain prediction. Protein Sci. 2003;12:2001–2014. doi: 10.1110/ps.03154503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gibbs J.R., Singleton A. Application of genome-wide single nucleotide polymorphism typing: Simple association and beyond. PLoS Genet. 2006;2:e150. doi: 10.1371/journal.pgen.0020150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gibson J., Morton N.E., Collins A. Extended tracts of homozygosity in outbred human populations. Hum. Mol. Genet. 2006;15:789–795. doi: 10.1093/hmg/ddi493. [DOI] [PubMed] [Google Scholar]
- 33.McQuillan R., Leutenegger A.L., Abdel-Rahman R., Franklin C.S., Pericic M., Barac-Lauc L., Smolej-Narancic N., Janicijevic B., Polasek O., Tenesa A. Runs of homozygosity in European populations. Am. J. Hum. Genet. 2008;83:359–372. doi: 10.1016/j.ajhg.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Johnson A., Gin P., Marbois B.N., Hsieh E.J., Wu M., Barros M.H., Clarke C.F., Tzagoloff A. COQ9, a new gene required for the biosynthesis of coenzyme Q in Saccharomyces cerevisiae. J. Biol. Chem. 2005;280:31397–31404. doi: 10.1074/jbc.M503277200. [DOI] [PubMed] [Google Scholar]
- 35.Hsieh E.J., Gin P., Gulmezian M., Tran U.C., Saiki R., Marbois B.N., Clarke C.F. Saccharomyces cerevisiae Coq9 polypeptide is a subunit of the mitochondrial coenzyme Q biosynthetic complex. Arch. Biochem. Biophys. 2007;463:19–26. doi: 10.1016/j.abb.2007.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boitier E., Degoul F., Desguerre I., Charpentier C., Francois D., Ponsot G., Diry M., Rustin P., Marsac C. A case of mitochondrial encephalomyopathy associated with a muscle coenzyme Q10 deficiency. J. Neurol. Sci. 1998;156:41–46. doi: 10.1016/s0022-510x(98)00006-9. [DOI] [PubMed] [Google Scholar]
- 37.Lamperti C., Naini A., Hirano M., De Vivo D.C., Bertini E., Servidei S., Valeriani M., Lynch D., Banwell B., Berg M. Cerebellar ataxia and coenzyme Q10 deficiency. Neurology. 2003;60:1206–1208. doi: 10.1212/01.wnl.0000055089.39373.fc. [DOI] [PubMed] [Google Scholar]
- 38.Quinzii C.M., DiMauro S., Hirano M. Human coenzyme Q10 deficiency. Neurochem. Res. 2007;32:723–727. doi: 10.1007/s11064-006-9190-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lopez-Martin J.M., Salviati L., Trevisson E., Montini G., DiMauro S., Quinzii C., Hirano M., Rodriguez-Hernandez A., Cordero M.D., Sanchez-Alcazar J.A. Missense mutation of the COQ2 gene causes defects of bioenergetics and de novo pyrimidine synthesis. Hum. Mol. Genet. 2007;16:1091–1097. doi: 10.1093/hmg/ddm058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Quinzii C.M., López L.C., Naini A., DiMauro S., Hirano M. Human CoQ10 deficiencies. Biofactors. 2008;32:113–118. doi: 10.1002/biof.5520320113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Molyneux S., Florkowski C., Lever M., George P. The bioavailability of coenzyme Q10 supplements available in New Zealand differs markedly. N. Z. Med. J. 2004;117:U1108. [PubMed] [Google Scholar]
- 42.DeLano, W. The PyMOL Molecular Graphics System. 2002. San Carlos, CA, USA, DeLano Scientific LLC. http://www.pymol.org
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.