

Abstract
We analyzed the current status (as of the end of August 2008) of human mitochondrial genomes deposited in GenBank, amounting to 5140 complete or coding-region sequences, in order to present an overall picture of the diversity present in the mitochondrial DNA of the global human population. To perform this task, we developed mtDNA-GeneSyn, a computer tool that identifies and exhaustedly classifies the diversity present in large genetic data sets. The diversity observed in the 5140 human mitochondrial genomes was compared with all possible transitions and transversions from the standard human mitochondrial reference genome. This comparison showed that tRNA and rRNA secondary structures have a large effect in limiting the diversity of the human mitochondrial sequences, whereas for the protein-coding genes there is a bias toward less variation at the second codon positions. The analysis of the observed amino acid variations showed a tolerance of variations that convert between the amino acids V, I, A, M, and T. This defines a group of amino acids with similar chemical properties that can interconvert by a single transition.
Introduction
The recent increase in high-throughput DNA analysis resulting from new automatic sequencing technologies has had the side effect of condemning the public online mitochondrial DNA (mtDNA) databases to quickly become out of date. The depositing of complete human mtDNA genomes in GenBank,1 to which authors are advised or required to submit sequences prior to publication, is increasing very fast since the first worldwide mtDNA population study appeared in the year 2000.2 One such database is the highly consulted Mitomap database,3 whose lists of polymorphisms remain arguably the most used references for clinical genetic publications. Typically, authors perform an mtDNA screening in a case study versus control group and conduct a search on Mitomap for previous publications of any detected mutation in order to infer novelty and any reported pathological effects of listed mutations. It is difficult (though not impossible) to attribute a pathological effect to a polymorphism that is reported in population surveys, especially those polymorphisms that are frequent in the population or recurrent in recent human evolution, so the population frequency of a polymorphism is essential information for inferring any negative phenotypic influence of that polymorphism. The fundamental role of Mitomap to the mtDNA research community is indisputable, but prior to a recent effort to keep its lists updated,3 (information deposited is based on ∼3000 coding region sequences), many works have been published drawing conclusions about the novelty of mtDNA polymorphisms based on incomplete and outdated information. These biases potentially affect several areas of clinical genetics, because mtDNA variant associations have been tested in many complex traits, such as longevity,4,5 Alzheimer (MIM #104300),6 and male infertility,7 among many other traits.
Previous to the study reported here, the most informative database (in our opinion) for general mtDNA diversity was mtDB—Human Mitochondrial Genome Database8—which reports the mtDNA variation observed in 1865 complete sequences and 839 coding region sequences. This database presents well the information for frequency, location, and type of mutations observed (synonymous or nonsynonymous, if protein coding). The information deposited in this database can be joined to that presented in Mitomap, and together with other online searching queries, should constitute the standard steps conducted by investigators for inferring the novelty of a mutation, as suggested very recently.9
Lately, the same group responsible for Mitomap has launched a new online tool, called Mitomaster.10 These authors argue that simple catalogs, such as lists of pathogenic mutations and haplogroup polymorphisms, are no longer adequate, a position to which we are in agreement. Static catalogs and lists are too quickly outdated in this age, and researchers in this field need the tools to access and use the constantly updated data in centralized databases such as GenBank. Studies based on the set of human mtDNA sequences deposited in GenBank, instead of on the static data reported in databases such as mtDB, will be using a much larger set of sequences and will naturally have better information on the population variation. In a fast-moving field, static databases such as mtDB rapidly become outdated. The statement made above about studies based on incomplete and outdated information, although provocative, is literally true because many just-published studies still refer to mtDB for information on human mtDNA variability. Of course, this does not mean that these studies are in any way invalid, but just that these studies could be improved by using the full amount of the sequence data currently available. In this paper we provide the tools to do that.
It is very important to include information concerning the frequencies observed in global populations (or local populations if relevant) for any polymorphism as a normal part of the evaluation of any potential pathological effect of that polymorphism. Ruiz-Pesini et al.11 developed a formula, which was implemented on Mitomaster, for inferring the potential pathological effect of a polymorphism based on information including the frequency of the polymorphism within the human population and conservation at that site between species. Another obvious parameter to be taken into consideration is the effect of mutations in protein-coding genes—both Mitomaster and the mtDB database present the synonymous or nonsynonymous character of a given polymorphism. This replaces the tedious process of manually consulting the mitochondrial genetic code, the sequence of the revised Cambridge Reference Sequence, to infer whether a position is located in protein-coding genes, and a table with properties for the amino acids. However, the mtDB database supplies this information listed by site and only for the polymorphisms in a fixed set of mtDNA sequences. Mitomaster currently gives this analysis only for a single sequence at a time. It is becoming common for an investigator to need to do this analysis for multiple sequences, and for multiple sites within each sequence. Certainly this is the case when the full set of mtDNA variations within a population study needs to be characterized. The synonymous or nonsynonymous classification of variations within protein-coding genes and the comparison of the site of variations to tRNA and rRNA secondary structure are critical information for inferring potential pathological effects of a variation. Although Mitomaster does supply this information, again it currently does so for only a single sequence at a time. Mitomaster is also not currently downloadable, so data confidentiality cannot be maintained with this particular analysis tool.
The primary aim of this study is to describe the level of diversity observed in the current (as of the end of August 2008) set of human mitochondrial genomes deposited in GenBank, a total of 5140 complete (or at least coding section) sequences. The second aim is to provide a simple downloadable software tool that can be used by others to carry out this analysis themselves in the future as the number of human mtDNA sequences continues to grow, or to carry out the variation analysis on subpopulations of particular interest. With this tool one does not need to be restricted to the existing static online databases and can independently check very large mtDNA sequence databases. The software tool can be downloaded, to protect the confidentiality of any sequences analyzed with it. Finally, the third aim is to compare the observed levels of diversity in the current database of human mtDNA sequences with the extreme limits of all possible transitions and transversions.
Material and Methods
The mtDNA-GeneSyn Computer Tool for Sequence Analysis
The mtDNA-GeneSyn tool for mitochondrial genome analysis was developed for Windows-based platforms and implemented with the C++ language. It is designed to have a simple and intuitive graphical interface and the mtDNA-GeneSyn tool is freely available at the address listed in the Web Resources section. The tool identifies and classifies the mtDNA polymorphisms present in large data sets, with these functions organized in two main menus. The menu “Polymorphisms” allows users to import a file in FASTA format, containing sequences previously aligned versus a reference sequence (which should be the first sequence in the file). This function identifies the positions that are variable relative to the reference, and the list of these variations can be exported as an output file. This output file of the polymorphic positions can also be exported to a binary input file recognized by the Network software.12 Network analyses allow the reconstruction of mtDNA phylogenies and the inference of haplogroup affiliations, and are basically of two kinds: median-joining13 and reduced-median12 networks. When recurrence is expected, as in big data sets, the reduced-median network is the indicated algorithm. But, as far as we are aware, there was no easy and cost-free way of transforming a big sequence data set into the binary input file necessary for this analysis (DnaSP provides only DNAMultistate input format usable in median-joining networks14). So, this function was implemented in mtDNA-GeneSyn in order to facilitate the classification of the phylogeny, in cases where the user is using a personal data set.
The output file with the identified polymorphisms works as the input file for the menu “Classification.” The tool is not limited to human mtDNA analyses, and can be applied to any mammalian mtDNA studies, so the first action when opening the “Classification” menu is to call again the reference sequence, in order to obtain the information about the location of the 37 genes, control region, and the reference sequence itself. The user can choose between the default human revised Cambridge Reference Sequence (rCRS, accession number AC_00002115) or input another mammalian reference sequence in GenBank format (we advise the user to use sequences curated in RefSeq database16). Then the classification of the polymorphisms can be performed, either as a single position or as entire data sets (complete mtDNA sequences in many individuals, such as the total 5140 reported human mtDNA sequences). Analyses of a data set of polymorphisms are partitioned into two main groups, “protein positions” and “nonprotein positions,” and information provided by the tool for each is as exhaustible as possible. Specifically for the “protein positions,” information concerning the protein-coding gene, the type of substitution (synonymous/nonsynonymous), the codon before and after, the amino acid and its properties (polarity and acidity) before and after, the amino acid position inside the protein, and the frequency in the database are provided. For the special cases of positions located in overlapping genes/regions, these are counted twice, because they interfere with the structure of the two genes or regions.
There are two other functions in mtDNA-GeneSyn for analyzing a data set of polymorphisms, which are available only for human sequences. These concern tRNAs and rRNAs and display secondary structures important for the functionality of these gene products. The software allows users to locate polymorphisms inside stem and loop regions in tRNAs and rRNAs. The numbering used for identification of each tRNA region was taken from the database Mamit-tRNA,17 whereas for 12sRNA (MT-RNR1 [MIM ∗561000]) and 16sRNA (MT-RNR2 [MIM ∗561010]) it was inferred from the secondary structures displayed on Mitomaster.
The interface displays a table and a bar graph with the summary information about diversity, but in order to facilitate further analyses intended by users, some versatile output files can be saved: summary accounts of general diversity, information organized by position, and information organized by sample.
Sequence Accession
The complete or coding-region mtDNA sequences published in GenBank amounted by the end of August 2008 to 5140 individuals, after curating the database. In fact, the query “Homo[Organism] AND gene_in_mitochondrion[PROP] AND 14000:19000[SLEN] NOT pseudogene[All Fields]” returns 5147 sequences, but we found that there is some redundancy in the sequences returned and not only Homo sapiens sapiens sequences were returned from this query. We then removed the following sequences from the initial data set: NC_011137 and AM948965, which refer to the recently published Neanderthal complete mtDNA sequence;18 J01415, V00662, and AB055387, which refer to the original Cambridge Reference Sequence,19 the latter replaced by the one published by Andrews et al.15 and identified as AC_000021; X62996, which is an inferred consensus sequence;20 and NC_001807, used as one of the human references in RefSeq database, because it is the same sequence as AF347015.8
For an easy and fast download of sequences in FASTA format we used the program Geneious in subsets of sequences. These subsets of sequences are the most advisable format for their subsequent alignment in BioEdit21 versus the revised CRS,8 via the Clustal W algorithm. The Clustal W algorithm takes considerable time for the alignment of sets of 100 sequences and would be unfeasible for a simultaneous alignment of the total database, which is also unnecessary. The automatic alignment was manually checked, in order to ensure consistency in the inclusion of deletions and insertions at the end of strings of bases (the poly-Cs and poly-CAs stretches). The rCRS contains an artificial value N at position 3107 in order to maintain the standard numbering system for human mitochondrial sequences, which is widely used in medical applications. Because all analyzed sequences are aligned against the rCRS, all of the variations are reported in the standard numbering system based on the rCRS. Note that although the mtDNA-GeneSyn program requires equal length sequences, this requirement is automatically satisfied if the sequences analyzed have been aligned previous to their analysis by mtDNA-GeneSyn, which should always be the case.
Results
The Current Human mtDNA Database
A complete set of the current human mitochondrial genomes available in GenBank was assembled as described in the Material and Methods section. Most of the sequences deposited in GenBank have information concerning the haplogroup and the geographic origin or ethnicity of the subject. When that was not the case, this information was retrieved from the publication or the haplogroup classification was inferred by using Mitomaster.10 Some haplogroup classifications were updated in more recent works, namely most GenBank L sequences were updated22—and in these cases the updated classification was recorded. All this information for each sequence is presented in Table S1 available online.
The compilation of all this information allows the construction of a general informed picture of the present complete or coding-region human mtDNA sequences deposited in GenBank. Most of these sequences were obtained in population genetic surveys performed in worldwide populations. Screenings performed with clinical purposes accounted for 1228 sequences, corresponding to 23.9% of the database, ranging from centenarians, LHON (MIM #535000), diabetes, obesity, male infertile phenotypes, and CADASIL (MIM #125310) to atypical psychosis. Most of the sequences were complete, with only 944 (18.4%) missing the control region information.
The geographical origin of these samples, in a broad sense, covers quite well the world (Figure 1), correcting the initial predominance of Eurasian sequences23,24 in mtDNA studies. Concerning the haplogroup affiliation, some caution is advised in accepting the haplogroup classifications provided by the references and recorded in Table S1. This is mainly a concern if the sequence is from an old publication and if a fine subhaplogroup classification is intended. Nonetheless, the classification presented can give at least a general idea of how many sequences were included for the various haplogroups (Table 1). West Eurasian and East Eurasian haplogroups contributed to 42% and 38%, respectively, of the database, whereas African haplogroups (including the sub-Saharan L, the East African M1, and the North African U6) made up 18% of the sequences. Leading the list of most screened is the West Eurasian haplogroup H, which is by far the most frequent European mtDNA haplogroup, attaining frequencies of ∼40%–50% in most European populations and ∼20% in Near Eastern populations,25 followed by the East Eurasian haplogroups D and M(xM∗,M1).
Figure 1.
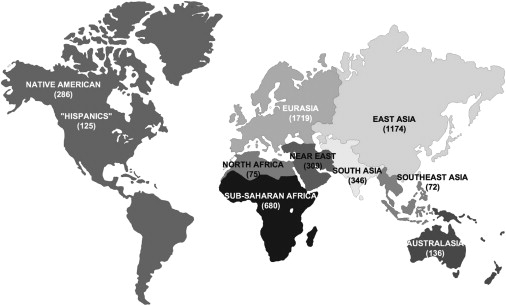
The Geographical Origin of the 5140 mtDNA Sequences Deposited in GenBank, as of the End of August 2008
African-American descendants were included in sub-Saharan Africa and European-American descendants were included in Eurasians given that they were homogeneous groups, bearing haplogroups belonging to these regions, whereas USA “Hispanics” constituted a mixed sample, with most lineages belonging to East Asian haplogroups observed in Native Americans.
Table 1.
Haplogroup Affiliation of the 5140 mtDNA Sequences Deposited in GenBank by the End of August 2008
| Population |
n |
|---|---|
| West Eurasian | |
| pre-HV or R0a | 31 |
| H | 781 |
| HV | 83 |
| I | 50 |
| J | 170 |
| K | 237 |
| N1 | 35 |
| other R | 94 |
| T | 169 |
| U∗ + U1-3 | 73 |
| U4 | 75 |
| U5 | 156 |
| U7-9 | 27 |
| V + pre-V | 87 |
| W | 69 |
| Total | 2137 |
| East Eurasian | |
| A | 257 |
| B | 246 |
| C | 138 |
| D | 507 |
| E | 54 |
| F | 71 |
| G | 100 |
| other M | 391 |
| N9 | 69 |
| other N | 10 |
| O | 1 |
| P | 31 |
| Q | 28 |
| S | 13 |
| Y | 13 |
| Z | 31 |
| Total | 1960 |
| Pan Eurasian | |
| X | 60 |
| Total | 60 |
| Unclear (Eurasian?) | n |
| M∗ | 55 |
| N∗ | 8 |
| Total | 63 |
| African | |
| L0 | 142 |
| L1 | 140 |
| L2 | 210 |
| L3 | 270 |
| other L | 42 |
| M1 | 68 |
| U6 | 48 |
| Total | 920 |
The classification of haplogroups in large population groups followed Macaulay's phylogenetic tree.
Classifying mtDNA Diversity
We estimated the general diversity present in the 5140 human mtDNA sequences for the different genetic regions of the molecule: the protein-coding genes; the control region and a few other noncoding positions scattered throughout the coding region; and the genes with functionally important secondary structures as tRNAs and rRNAs. In order to quantitatively evaluate this broad diversity, we evaluated both the set of all polymorphic sites and the subset of all polymorphisms attaining a threshold of 0.1% frequency in the database, corresponding to at least five hits. Arguably, this threshold avoids most biases resulting from sequencing errors, which potentially are strongest for positions observed in single or very few sequences. As a comparison for the actually observed polymorphisms, another data set was formed including all possible substitutions that can hit each position. This can be easily done by constructing artificial DNA sequences. For instance, to estimate all possible transitions A to G, we can transform all As present in the reference sequence to Gs. The same can be done individually for all remaining transitions (G to A; C to T; and T to C) and for all transversions (A to C; A to T; C to A; C to G; G to C; G to T; T to A; and T to G). These artificial mtDNA sequences can then be analyzed in the mtDNA-GeneSyn tool in the same way as the real sequences. All the raw data are provided in Tables S2–S4.
Variations in Protein-Coding Genes
When considering only substitutions, 4,062 out of 11,395 nucleotide positions (36%) located in protein-coding regions (double counting the small number of substitutions that were found in regions where two genes overlapped) were polymorphic in this sequence database. These polymorphic positions were fairly evenly distributed through the 13 genes (linear squared regression value of 0.892; Figure 2A), although the distribution of observed polymorphic positions was statistically different from the distribution of total protein sizes (χ2 = 100.204; p < 0.001). For each gene, the amount of polymorphic sites was 29% in MT-ND3 (MIM ∗516002) and MT-ND4L (MIM ∗516004); 30% in MT-CO1 (MIM ∗516030) and MT-ND4 (MIM ∗516003); 33% in MT-ND2 (MIM ∗516001); 34% in MT-CO2 (MIM ∗516040) and MT-ND5 (MIM ∗516005); 35% in MT-CO3 (MIM ∗516050); 37% in MT-ND1 (MIM ∗516000); 38% in MT-ND6 (MIM ∗516006); 43% in MT-CYB (MIM ∗516020); 54% in MT-ATP6 (MIM ∗516060); and 57% in MT-ATP8 (MIM ∗516070).
Figure 2.
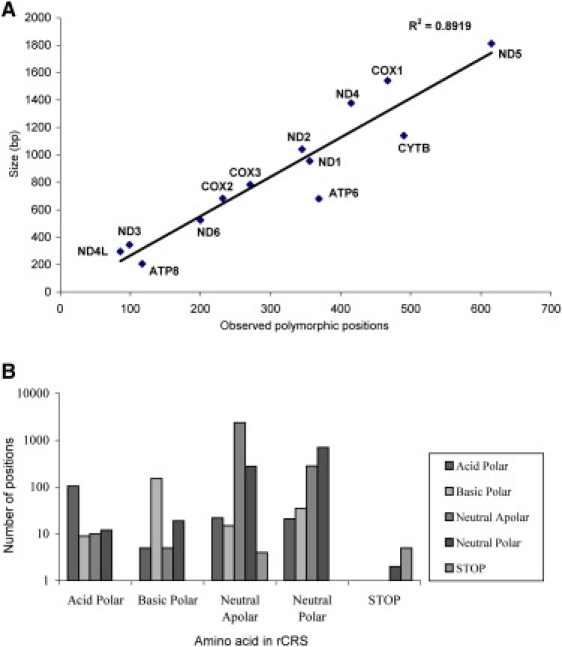
Protein Diversity
Correlation between the observed polymorphic positions and the gene size (bp) in the 13 protein-coding genes (A) and distributions of types of amino acids before and after the substitution (B).
A proportion of 63% of these 4062 polymorphic sites were located on third codon positions, followed by 24% in first codon positions and only 13% in second codon positions. This led to 1366 nonsynonymous versus 2696 synonymous substitutions (ratio of 1:1.97). Most of the polymorphic codons coded for neutral apolar amino acids (66.3%), with 98.5% of those synonymous; followed by neutral polar-coding codons (25.6%); and only 5% and 3% of the observed polymorphic codons were basic polar and acid polar-coding codons, respectively (Figure 2B).
Considering only the subset of polymorphisms with a frequency of at least 0.1%, a total of 1,465 substitutions were observed out of the 11,395 nucleotide positions located in protein-coding regions, corresponding to 13% of all sites. This result confirms the high level of mtDNA diversity that occurs at very low frequencies (below this threshold) in the worldwide human population. These polymorphic positions were also evenly distributed through the 13 protein-coding genes (linear squared regression value of 0.872; χ2 = 49.411; p < 0.001; the set of figures for polymorphisms with a frequency of at least 0.1% is given in Supplemental Data), ranging from 10% to 21% with 10% in MT-CO1, MT-ND3, and MT-ND4L; 11% in MT-CO3; 12% in MT-ND4 and MT-ND5; 13% in MT-CO2 and MT-ND6; 14% in MT-ND2; 15% in MT-ND1; 16% in MT-CYB; 18% in MT-ATP6; and 21% in MT-ATP8. The proportions between the first, second, and third codon positions were, respectively, 23%, 9%, and 68%, yielding 397 nonsynonymous versus 1068 synonymous substitutions (a ratio of 1:2.69). The pattern for polymorphisms in amino acids of different physical properties also followed the general observations in the full set of polymorphisms as described above.
A total of 88% of the complete list of polymorphic positions were transitions, yielding a transversion:transition ratio of 1:7.5, when not accounting for redundancy in the human phylogeny. The dominance of transitions in the evolution of animal mtDNA (not just human mtDNA) has long been recognized.26 This transversion:transition ratio increased to 1:21.2 when considering only the polymorphisms over the 0.1% threshold in the population (95% transitions to 5% transversions), further emphasizing the rarity of transversions. The proportion of transitions was higher in the set of polymorphisms with prevalence >0.1% of the population than in the complete data set, so this may indicate that there is more selection against the transversion mutations.
We compared the set of all observed transitions in the protein-coding genes to all possible transitions from the reference sequence. The observed transitions in the protein-coding region corresponded to 31.5% of the maximum number possible (3,585 out of 11,395), a surprisingly high proportion, with ratios between the four possible transitions highly correlated (Table 2). When comparing observed and maximum values for transitions (Figure 3A) in each of the 13 protein-coding genes, a good correlation (r2 = 0.896) was obtained with MT-CO1 having the lowest proportion of observed transitions compared to all possible ones and MT-ATP6 the largest proportion. When the transitions were partitioned by the type of substitution, the lowest correlating type was G to A (r2 = 0.657; not shown); the other types of transitions showed good correlations between observed and maximum values (r2 = 0.895 for A to G; r2 = 0.946 for C to T; and r2 = 0.819 for T to C). Despite the high correlations, there was a slightly significant difference in the proportions of the four transition types when comparing the observed transitions to the set of all possible transitions (Table 2; p = 0.042 by chi-square test).
Table 2.
Maximum Numbers and Observed Values for Transitions in the 13 Coding-Protein Genes
| Type | Maximum Number (%) | All Observeda (%) | Observedb in >0.1% of Population (%) |
|---|---|---|---|
| G-A | 1339 (11.8) | 412 (11.5) | 250 (17.9) |
| A-G | 3386 (29.7) | 1154 (32.2) | 417 (29.8) |
| C-T | 3785 (33.2) | 1158 (32.3) | 306 (21.9) |
| T-C | 2885 (25.3) | 861 (24.0) | 426 (30.4) |
Compared to the maximum number χ2 = 8.206; p = 0.042.
Compared to the maximum number χ2 = 100.393; p < 0.001.
Figure 3.
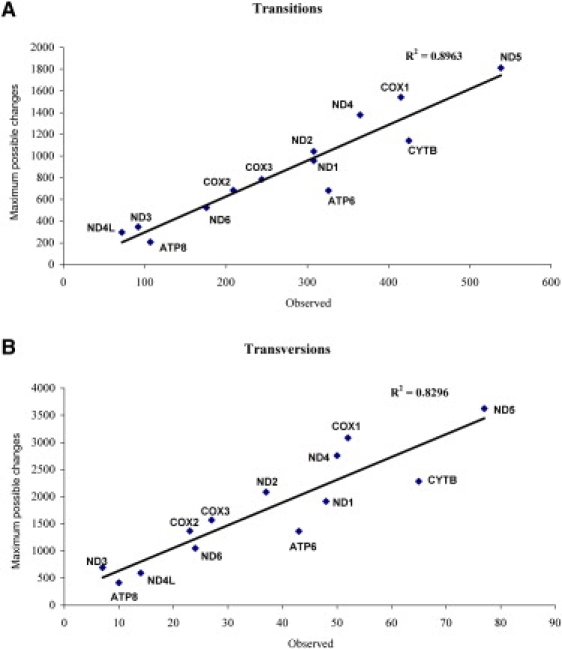
Transitions and Transversions
Correlation between the observed and maximum possible variations for (A) transitions and (B) transversions in the 13 protein-coding genes.
Out of all of the possible sites within protein-coding genes where transversions could have occurred, only 2.1% of these sites were observed to have transversions in this sequence data set (477 in 22790). When the data were broken down into the different types of transversions (Table 3), there was a good correlation between the numbers of observed and the maximum number of transversions. However, there was also a highly significant difference in the proportions of each type of transversion (χ2 = 63.498; p < 0.001), with a considerable excess of C to A and a weaker excess for G to C transversions, leading to the proportional reduction of the other categories. That excess of C to A was shared by all the 13 protein-coding genes (r2 = 0.829), accounting for the good correlation in total transversion among those genes (r2 = 0.830; Figure 3B). Bandelt et al.27 reported a ratio of transversions of 17 to A, 9 to C, 7 to T, and 3 to G (or 5.7:3:2.3:1), in a Eurasian mtDNA tree. Here the values are 183 to A, 95 to C, 80 to T, and 119 to G (or 1.5:0.8:0.7:1). These proportions show some deviations from unity, but not as pronounced as the data of Bandelt et al.27 This difference could be explained by an excess of transversions to G resulting from sequencing errors as reported27 to occur in some published data sets. Those authors report that in cases of a C following a few Gs, such as in GGC, some misclassifications (or what they call phantom mutations) are reported as GGG. But in the total database, only 13 sites were mutations from GGC to GGG (of which only 3 sites are observed to vary at over the threshold 0.1% of the population).
Table 3.
Maximum Numbers and Observed Values for Transversions in the 13 Coding-Protein Genes
| Type | Maximum Number (%) | All Observeda (%) | Observedb in >0.1% of Population (%) |
|---|---|---|---|
| A-C | 3386 (14.9) | 60 (12.6) | 5 (7.6) |
| A-T | 3386 (14.9) | 66 (13.8) | 9 (13.6) |
| C-A | 3785 (16.6) | 140 (29.4) | 27 (40.9) |
| C-G | 3785 (16.6) | 67 (14.0) | 7 (10.6) |
| G-C | 1339 (5.9) | 35 (7.3) | 5 (7.6) |
| G-T | 1339 (5.9) | 14 (2.9) | 3 (4.5) |
| T-A | 2885 (12.7) | 43 (9.0) | 7 (10.6) |
| T-G | 2885 (12.7) | 52 (10.9) | 3 (4.5) |
Compared to the maximum number χ2 = 63.498; p < 0.001.
Compared to the maximum number χ2 = 31.318; p < 0.001.
When the same comparison was made between the maximum possible values for transitions and transversions and the observed polymorphisms with frequency of at least 0.1% in this database (Supplemental Data), the results were quite similar to those for the full set of observed polymorphisms just described. For transversions, there were 34 to A, 10 to C, 12 to T, and 10 to G (or 3.4:1:1.2:1), showing again that the huge difference between values of transversions to A and to G as observed by Bandelt et al.27 in the Eurasian data set is not so strong in this larger worldwide data set.
The observed diversity in terms of synonymous and nonsynonymous substitutions was of 32.5% (2,696 out of 8,291 possible) and 5.3% (1,366 out of 25,894 possible), respectively, compared to all possible substitutions in the protein-coding genes. When the analysis was broken down by each of the 13 protein-coding genes, an extremely high correlation was obtained for synonymous substitutions (r2 = 0.989) but not for nonsynonymous substitutions (r2 = 0.412), with higher observed nonsynonymous substitutions for MT-ATP6 and MT-ATP8 and lower for MT-CO1, MT-ND4, and MT-ND5 (Figure 4). Identical patterns were observed for the smaller subset of polymorphisms with frequencies of 0.1% or more of the population.
Figure 4.
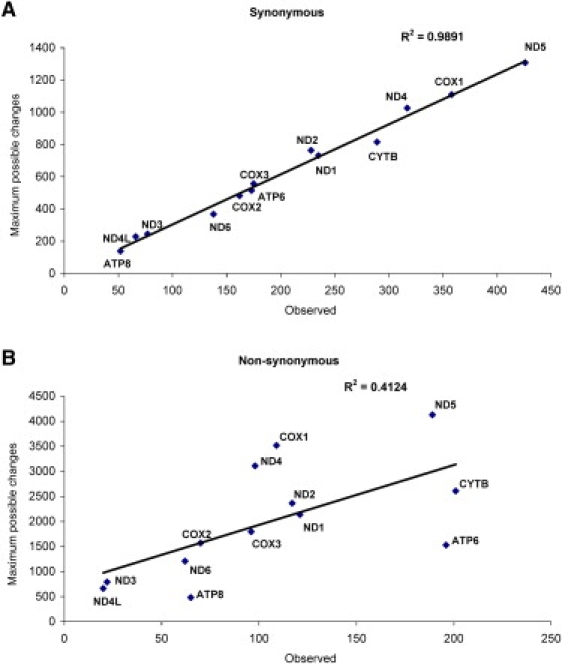
Synonymous and Nonsynonymous Sequence Changes
Correlation between observed and maximum possible variations for (A) synonymous and (B) nonsynonymous substitutions in the 13 protein-coding genes.
To understand the nonsynonymous substitutions, we broke them down into the individual amino acid changes and compared this to the total number of changes to each amino acid that were possible by making all single nucleotide replacements from the reference sequence (Figure 5). This analysis gave us the first result that was not either random or a simple linear relationship between the actual changes and the maximum number of changes. Instead, a group of amino acids (V, A, and T) clearly show a different behavior than the other amino acids. The observed variations to the other amino acids showed a relatively linear relationship to the total possible number of alterations, indicating that these variations are occurring in the human population at roughly an equal rate. In contrast, the V, A, and T group lie farther to the right in the plot, meaning that a higher proportion of all possible changes to these three amino acids is present in the human population. The vast majority of all variations found in the human mtDNA are transitions, so we considered the sets of amino acids with codons that can be interconverted by single transitions via the vertebrate mitochondrial genetic code. This separates the amino acids into only four groups: (V, I, A, M, T), (L, P, F, S(UCN)), (H, Y, C, R, Q, W, stop(UAA/G), and (N, D, G, S(AGU/C), E, K, stop(AGA/G)). Interestingly, in the two larger groups almost all transitions cause a change in the chemical properties (acid-neutral-base or polar-apolar) of the amino acid. In the two smaller groups, all but one of the amino acids in each group has the same properties, neutral apolar. These two groups also are the amino acid variations most often observed in the human population, with the (V, I, A, M, T) group occurring at the highest rate (Figure 5). This is also visible in the analysis of the amino acid chemical properties in Figure 2B, where most of the observed changes are between neutral apolar amino acids.
Figure 5.
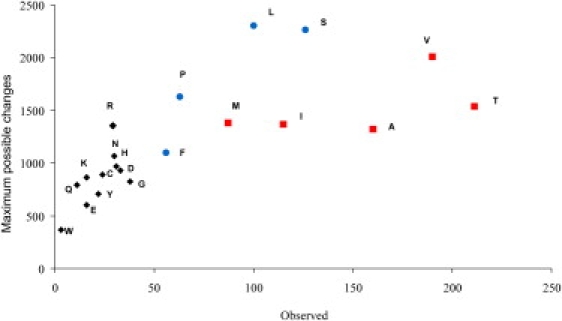
Correlation between Observed and Maximum Possible Variations for Amino Acids Originated from Nonsynonymous Mutations
The amino acids from group (VIAMT) are represented by red squares and the ones from group (FLPS) are represented by blue circles; the remaining amino acids are represented by black diamonds.
Potentially the most severe nonsynonymous point mutations are those occurring in stop codons. In this database of human mtDNA sequences, nine substitutions affected stop codons. Of these, five continue to mean stop, whereas, most interestingly, the other four increase the protein size. Two of these occur in MT-CO1, converting the stop codon to a basic polar amino acid, extending the open-reading frame by three codons into the adjacent tRNA-Ser (UCN) gene (MT-TS1 [MIM ∗590080]), and thus extending the corresponding protein by three amino acids. One of these variations is located at position 7444 and has been associated with LHON disease28 (MIM #535000) but is seen in many population samples from different haplogroup backgrounds and has been reported in diverse publications (the 27 samples bearing it in the database are distributed by haplogroups A2, H, H3, HV, D4, D4e1, L2a, L1b, L1b1a, L3e, L3f1b2, M7a, M38, and V with accession numbers DQ282408, EF657747, EF657594, AM260606, AM260607, AM260608, AM260609, AM260610, AM260611, AM260612, AP009431, AP010714, EF184618, EF184619, DQ112737, EU092893, DQ282507, EU092805, AP010747, AY922286, DQ112936, AF347006, AY339446, AY339447, AY339448, AY339449, and AY339450). The other variable site at position 7445C was seen in patients with hearing loss but was concluded by those authors to be insufficient to cause the phenotype29 (in the database it was observed in one individual belonging to haplogroup D6, accession number EU482325). Another substitution at position 9205 converts the stop codon to a neutral polar amino acid, extending the ATP6 protein by 10 more amino acids coded from the adjacent MT-CO3 gene (Q-W-P-T-N-H-M-P-I-M) and was observed in two individuals with haplogroups X2∗ and A2, from two different data sets (accession numbers EU600328 and EU431081). Notice that although this site is listed in Mitomap as polymorphic, the reference indicated seems to be wrong. The explanation for a termination codon being found in the MT-CO3 gene is that the first position of the MT-CO3 first codon corresponds to the third position of the MT-ATP6 termination codon, generating a reading-frame shift. And finally, the variation at 10765T converts a stop codon to a neutral apolar amino acid, extending the ND4L protein by two more amino acids (L-N) coded from the overlapping MT-ND4 gene, which is naturally in a reading-frame shift relative to ND4L. This substitution was observed in one L0k individual (accession number EF184609).
The opposite effect, amino acid coding codons converting to a stop codon, was observed in five cases: three involving amino acid W at positions 4720 and 5185 in MT-ND2 (the 84th and 239th amino acids, respectively, in a total of 347 amino acids; accession numbers EF661000 and EF660972, respectively), and at position 11403 in MT-ND4 (the 215th amino acid of a 459 amino acid protein; accession number EF661005), all occurring in patients with thyroid cancer. Another involved amino acid M at position 10657G, which has been detected in two patients with thyroid cancer (accession numbers EF660984 and EF660998), occurring at position 63 in MT-ND4L, which has 98 amino acids. The last one involved a K amino acid at position 15606 in MT-CYB (the 287th amino acid of a total of 380), occurring in one individual with thyroid cancer (EF660990). So, whereas extension of a protein by a few amino acids can be seen in the neutral population, the shortening of proteins was observed only in pathological cases.
Variations in the Control Region and Noncoding Sites inside the Coding Region
A total of 692 polymorphisms were observed in the control region, dispersed throughout 560 positions out of the 1123 bp total length. Several positions had three alleles (92 positions) and even four alleles (20 positions). Of these total polymorphisms, 513 were transitions and 179 transversions, leading to a ratio of 1:2.9, not accounting for redundancy. With the threshold of 0.1% frequency, these values were 326 polymorphisms observed throughout 295 positions, being 284 transitions and 42 transversions (ratio of 1:6.8). Again the pattern holds that the transition to transversion ratio increases when only the higher-frequency variations are considered.
For the D loop, the frequency of observed transitions and transversions, relative to possible values, were 45.7% (513 of 1122) and 8% (179 of 2244), respectively, testifying the higher mutation rate of this region relative to the protein-coding region, even not accounting for recurrence. The correlation between the observed and maximum possible numbers of different types of substitutions and different types of transversions were very high (r2 = 0.976 and r2 = 0.768, respectively), indicating no tendency for certain types of substitutions inside each class.
There are some positions along the coding region that are noncoding, summing up to a total of 89 positions. In these positions, 50 polymorphisms were observed, distributed along 43 positions (7 had three alleles, and curiously, 6 were transversions to C and just 1 to A). One important region located in these positions is the assumed L-strand origin of replication (OL), which is located between positions 5721 and 5798, overlapping in the first 9 bases with tRNA-Asp (MT-TD [MIM ∗590015]) and in the last 38 bases with tRNA-Cys (MT-TC [MIM ∗590020]). In the nonoverlapping stretch of the OL, five polymorphisms were observed, all closely located in the middle of this region, which can be important for inferences about the functionality and structure of OL. In fact, compared to a secondary structure calculated by the mfold program,30 all those polymorphic positions are located inside a single predicted loop (Figure 6).
Figure 6.
Distribution of Polymorphic Positions in the Various Structural Regions of the tRNA Genes
tRNAs and rRNAs
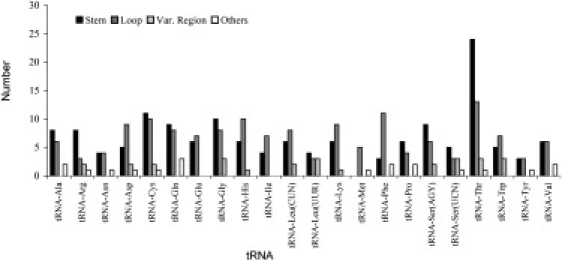
Many of the confirmed mtDNA pathological mutations are located on tRNAs, especially on the stem portions.31 Given that the sizes of the various tRNAs are quite similar, varying only from 65 to 75 bp, there were some interesting differences in variability between the tRNA genes. The genes for the tRNAs Met, Tyr, and Asn (MT-TM [MIM ∗590065], MT-TY [MIM ∗590100], and MT-TN [MIM ∗590010]) showed the fewest polymorphisms (less than 10), whereas all the other tRNAs showed between 11 and 24 polymorphisms, except threonine (MT-TT [MIM ∗590090]), which displayed a double level of polymorphisms, 41 (Figure 7). This renders the relation between observed polymorphisms and size totally uneven (linear squared regression value of 0.08; not shown). The same result occurs when only analyzing those polymorphisms present at a frequency of 0.1% or higher (linear squared regression value of 0.10; data not shown).
Figure 7.
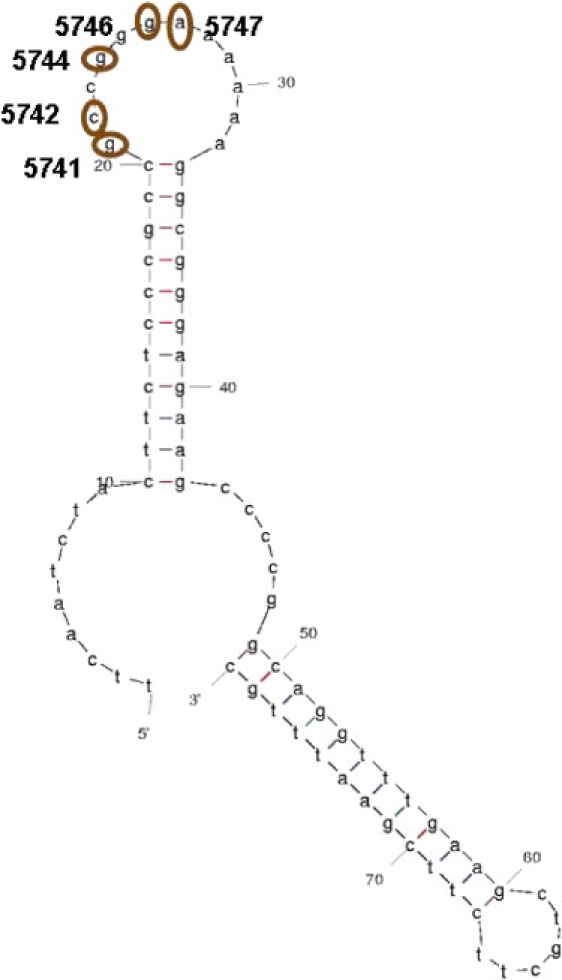
Variability in the Region of the Origin of the Light Strand Related to the Inferred DNA Secondary Structure
Secondary structure of the OL region inferred in mfold28 with the observed polymorphic positions 5741, 5742, 5744, 5746, and 5747 indicated.
This highest amount of polymorphisms observed in MT-TT was previously identified in a worldwide phylogeny based on 277 sequences published.32 The increase in MT-TT diversity occurs mainly in the stem region, which contains almost double the number of polymorphisms than were found in the loops. As can be seen in Figure 8, some polymorphisms in the human population are located in positions identified as 100% and >90% conserved in mammalian tRNAs.32
Figure 8.
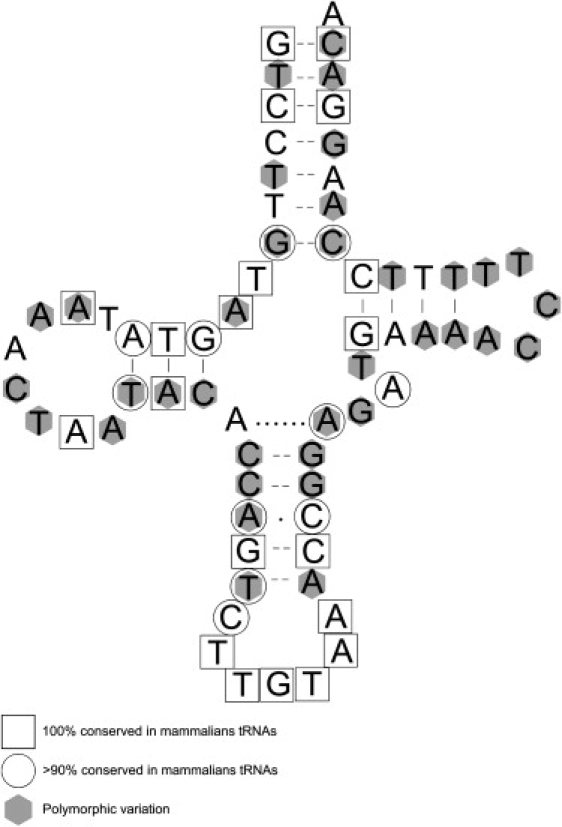
Variability in a tRNA Gene Related to the RNA Secondary Structure
The secondary structure of the threonine tRNA and the observed polymorphic positions in the human database (gray hexagons), and positions that are 100% (square) and >90% (circle) conserved in mammalian species.30
Other tRNA genes also have higher numbers of variations in the stems than in the loops: alanine (MT-TA [MIM ∗590000]), arginine (MT-TR [MIM ∗590005]), cysteine (MT-TC), glutamine (MT-TQ [MIM ∗590030]), glycine (MT-TG [MIM ∗590035]), leucine UUR (MT-TL1 [MIM ∗590050]), proline (MT-TP [MIM ∗590075]), serine AGY (MT-TS2 [MIM ∗590085]), and serine UCN (MT-TS1). The tyrosine and valine tRNA genes (MT-TY and MT-TV [MIM ∗590105]) have the same number of polymorphisms in the stems and loops. Another interesting point is that tRNA-Met (MT-TM) has no polymorphisms in stems (five are located in loops and one is located in other positions), which is consistent with the main function of this tRNA as the initiator of all mtDNA proteins. In total over all tRNA genes, the amount of mutations in stem regions (148) was similar to the amount in the loop regions (150), which seems in contradiction to the functional constraint, but this, again, was estimated without taking into consideration the redundancy of substitutions and the larger proportion of each tRNA gene contained in the stems.
But if these observed values are compared with the maximum possible values, the functional constraint does seem to play an important role in driving tRNA diversity. In fact, when analyzing the locations of all possible substitutions, the total number in stem regions is 2670; for loop regions it is 1332; and for variable regions and other sites 519. In this way, we would expect a ratio of stem:loop of 2 but the observed ratio was 0.99, not accounting for redundancy. The lower variation in the stem regions is consistent with expectations that such regions should be more constrained by selection (in order to maintain the tRNA and rRNA secondary structure) and has been reported recently on an analysis of 2460 human mtDNA sequences.33 The maximum possible values were very similar for all tRNAs, so the high excess of substitutions observed in tRNA-Thr (MT-TT) indicates that the variability in this gene is much higher than might be expected, especially in the stem regions (Figure 6).
There is the possibility that some of those stem polymorphisms could be compensating base changes, where one substitution in one side of the stem that would lead to a less stable stem is corrected by a substitution at the same position in the other side of the stem, stabilizing the tRNA secondary structure. This was observed to occur in the primate lineage in one large secondary structure located in the control region.34 Of course, there is the question whether the time since the emergence of the modern human species (∼200,000 years ago) is enough for these compensating polymorphisms to have occurred. We evaluated this by checking the placement of the detected tRNA polymorphisms against the tRNA secondary structure (Supplemental Data). Of the 34 pairs of polymorphisms that colocate at the same position in both sides of the tRNA stems, only one pair (7528 and 7538G) was observed in the same individual belonging to haplogroup M21 (accession number DQ834257) but causing a mispairing between G-G. So compensating polymorphisms in the tRNA stem structures does not appear as an important feature in the human mtDNA diversity.
Very curiously, there were a few instances of polymorphisms affecting the anticodon of four tRNAs. The variation at site 12170 in the tRNA histidine gene transforms the anticodon GTG in GTA, observed in an individual from haplogroup A2 (accession number EU007838). The polymorphism 12300T transforms TAG to TAT in MT-TL2 (MIM ∗590055) in an L3 individual (accession number EF184640). The variation at site 8324 transforms TTT to TCT in MT-TK (MIM ∗590060) in an R5a2b2 individual (accession number FJ008429). And finally, the polymorphism at position 1633 at MT-TV transforms the anticodon TAC to CAC in an individual from haplogroup N2 (accession number EU787451). All these tRNAs are coded in the H-strand, so the above substitutions at the third position correspond to the first position in the codon, and vice versa, so that the one occurring in MT-TH (MIM ∗590040) would change the amino acid association to Tyr; the substitution in MT-TL2 would change the association from CUN to UUR, remaining a leucine; the substitution in MT-TK would convert to an association with a stop codon; and the substitution in MT-TV would be synonymous. These substitutions leading to different functions for the altered tRNAs give ample reason to suspect that these variations are incorrect.
Of the 22 tRNA substitutions displayed on Mitomap (edition 28 July 2008) as having a confirmed pathological effect, only 4 were observed in our database: the MT-TL1 variation A3243G, only present in patients from diverse diseases (2 MELAS [MIM #540000], 1 OXPHOS deficiency, and 1 thyroid cancer; respective accession numbers DQ862536, DQ826448, DQ489509, and EF661012); the MT-TS1 variation G7497A, present in one patient with OXPHOS deficiency (accession number DQ489514); the MT-TK variation A8344G, observed in one MERRF (MIM #545000) patient (accession number DQ862537) and in one individual from haplogroup V in the population study of Mishmar et al.35 (accession number AY195750); and the MT-TE (MIM ∗590025) variation T14709C, described in the population survey of Herrnstadt et al.,24 belonging to haplogroup T1 (accession number EF657686). Many of the tRNA substitutions listed on Mitomap as “reported” or “unclear” were observed in the GenBank database, some in considerable frequency. This frequency information is important for ascertaining the potential pathological effect of these variations (Table S5).
The rRNA genes presented a higher proportion of polymorphisms on loop regions than on stem regions: 72.6% and 27.4%, respectively, for the 12 s rRNA gene MT-RNR1; and 85.6% and 14.4%, respectively, for the 16 s rRNA gene MT-RNR2 (using the rRNA secondary structure calculated from mfold). The comparison with all possible substitutions also testifies to the prevalence of substitutions in loop regions. For MT-RNR1, the observed and expected ratios between stem:nonstem regions were 0.78 and 0.38, respectively. For MT-RNR2, these values were 0.63 and 0.17, respectively. In both rRNA genes, variations were disproportionally present in the predicted loop structures, as one would expect.
Discussion
The mtDNA-GeneSyn tool allows an easy and fast identification and measurement of the diversity present in large data sets of complete mtDNA genomes. Users have total freedom in analyzing any desired data set. Furthermore, the description and characterization of the current complete human mtDNA genomes deposited in GenBank performed here, and its availability to readers as Supplemental Tables, allows the easy construction of many desired databases, such as a neutral population data set from East Asia, thyroid cancer data, or African L2 haplogroup data. The GeneSyn tool allows the investigator to use up-to-date groups of mtDNA sequences instead of relying on a static database, such as mtDB that has remained at 2704 complete sequences for several years now. The GeneSyn tool is not a web resource and can be downloaded and used locally, so there is no issue with confidentiality of the gene sequences being analyzed, as there is with MitoMaster, which retains all sequences submitted for analysis.
Based on our survey, it is clear that the largest amount of sequences currently deposited in GenBank resulted from population studies. Although this bias can be partially explained by clinical studies still being mainly performed in segments of the mtDNA molecule instead of typing the complete genome, there are clearly many authors from the clinical field who are not submitting sequences to GenBank. The centralized role of GenBank as the main repertoire of human diversity and its utility for researchers must then be reinforced.
The characterization of the diversity present in 5140 complete or coding-region human genomes and its comparison with the maximum possible diversity for human mtDNA can serve as references for case studies and for comparative species evolution. This comparison showed clearly that mutation restrictions related with secondary structure conformation are the main cause for an uneven distribution of mtDNA diversity. These restrictions are strong in tRNAs and rRNAs, genes for which the secondary structure is essential for functionality, but also in regions related with regulation, as the OL. On the other hand, for protein-coding genes, the most obvious feature was the bias against mutation at the second codon position.
Despite the clear and reasonable bias against nonsynonymous variations in the protein coding genes, our analysis indicates that a certain subset of these variations are better tolerated, and therefore are observed at both a higher absolute rate and a higher rate relative to the total number of possible variations to these amino acids (Figure 5). The tolerated variations are among the group of amino acids V, I, A, M, and T, which can be interconverted by transitions and which all but one (T) are neutral apolar amino acids. Curiously, although T is the only polar amino acid in this group, it is also the amino acid change (compared to the reference sequence) that is most often observed in the human sequence data (Figure 5). A detailed discussion of the physical and chemical properties of the amino acids changes observed in a set of 840 human sequences is given by Moilanen and Majamaa.36
This study provides a complete listing of the current knowledge of mtDNA variation in the human population, available as Table S2. This list can be consulted by investigators faced with assessing the importance of a particular sequence variation, which may be found, for example, in a clinical investigation. The analysis also indicates that some nonsynonymous alterations in protein-coding genes (within the V, I, A, M, and T group) have been more easily tolerated in human evolution, and thus may be less likely to be pathogenic alterations in a patient. Perhaps most importantly, this study provides the tools so that this knowledge can be updated by the individual investigator as the number of sequences available grows, as it is certain to do in the near future. These tools and the methods outlined in this paper can also be used by investigators with their own sequence database, either gathered from a particular geographic area or as part of a case-control phenotype study.
The two resources provided in this paper (the current mtDNA variation list and the computational tools for creating such a list from sequence data sets supplied by the user) will be useful for many studies. For example, Stewart et al.37 recently reported exciting new results on selection of mtDNA variants after following heteroplasmic mice through several generations. In that paper they were limited in their comparison of their mouse model to the human mtDNA variation database mtDB, which we discussed in the introduction, instead of utilizing the much larger full data set of human mtDNA variation that was available in GenBank at that time. The information and tools presented here should remove these limitations from future studies. Another example of a large-scale study that could be based on this data would be to model the effects of the observed amino acid changes on the protein structure and function, in order to look for selection effects there (potentially related to the results of Stewart et al.37), and also to explain the distribution of observed human variation in terms of the affected protein domains.
Supplemental Data
Supplemental Data include six figures and five tables and can be found with this article online at http://www.ajhg.org/.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
DNASP, http://www.ub.edu/dnasp/
Geneious, http://www.geneious.com
Macaulay's phylogenetic tree, http://www.stats.gla.ac.uk/∼vincent
Mamit-tRNA, Compilation of mammalian mitochondrial tRNAs, http://mamit-trna.u-strasbg.fr/
MitoMaster, http://mammag.web.uci.edu/twiki/bin/view/Mitomaster
MitoMap, http://www.mitomap.org/
mtDNA GeneSyn, http://www.ipatimup.pt/downloads/mtDNAGeneSyn.zip
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Phylogenetic Network Software, http://www.fluxus-engineering.com/netwinfo.htm
Acknowledgments
This work was financed by a project (PTDC/AFR/71422/2006) funded by FCT (Fundação para a Ciência e a Tecnologia). IPATIMUP is supported by Programa Operacional Ciência, Tecnologia e Inovação (POCTI) e Quadro Comunitário de Apoio III. The authors state that they have no conflicts of interest.
References
- 1.Benson D.A., Karsch-Mizrachi I., Lipman D.J., Ostell J., Wheeler D.L. GenBank. Nucleic Acids Res. 2008;36(Database issue):D25–D30. doi: 10.1093/nar/gkm929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ingman M., Kaessmann H., Pääbo S., Gyllensten U. Mitochondrial genome variation and the origin of modern humans. Nature. 2000;408:708–713. doi: 10.1038/35047064. [DOI] [PubMed] [Google Scholar]
- 3.Ruiz-Pesini E., Lott M.T., Procaccio V., Poole J.C., Brandon M.C., Mishmar D., Yi C., Kreuziger J., Baldi P., Wallace D.C. An enhanced MITOMAP with a global mtDNA mutational phylogeny. Nucleic Acids Res. 2007;35(Database issue):D823–D828. doi: 10.1093/nar/gkl927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.De Benedictis G., Rose G., Carreiri G., De Luca M., Falcone E., Passarino G., Bonafe M., Monti D., Baggio G., Bertolini S. Mitochondrial DNA inherited variants are associated with successful aging and longevity in humans. FASEB J. 1999;13:1532–1536. doi: 10.1096/fasebj.13.12.1532. [DOI] [PubMed] [Google Scholar]
- 5.Bilal E., Rabadan R., Alexe G., Fuku N., Ueno H., Nishigaki Y., Fujita Y., Ito M., Arai Y., Hirose N. Mitochondrial DNA haplogroups D4a is a marker for extreme longevity in Japan. PLoS ONE. 2008;3:e2421. doi: 10.1371/journal.pone.0002421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Elson J.L., Herrnstadt C., Preston G., Thal L., Morris C.M., Edwardson J.A., Beal M.F., Turnbull D.M., Howell N. Does the mitochondrial genome play a role in the etiology of Alzheimer's disease? Hum. Genet. 2006;119:241–254. doi: 10.1007/s00439-005-0123-8. [DOI] [PubMed] [Google Scholar]
- 7.Pereira L., Gonçalves J., Franco-Duarte R., Silva J., Rocha T., Arnold C., Richards M., Macaulay V. No evidence for an mtDNA role in sperm motility: Data from complete sequencing of asthenozoospermic males. Mol. Biol. Evol. 2007;24:868–874. doi: 10.1093/molbev/msm004. [DOI] [PubMed] [Google Scholar]
- 8.Ingman M., Gyllensten U. mtDB: Human Mitochondrial Genome Database, a resource for population genetics and medical sciences. Nucleic Acids Res. 2006;34(Database issue):D749–D751. doi: 10.1093/nar/gkj010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bandelt H.J., Salas A., Taylor R.W., Yao Y.G. Exaggerated status of “novel” and “pathogenic” mtDNA sequence variants due to inadequate database searches. Hum. Mutat. 2008;30:191–196. doi: 10.1002/humu.20846. [DOI] [PubMed] [Google Scholar]
- 10.Brandon M.C., Ruiz-Pesini E., Mishmar D., Procaccio V., Lott M.T., Nguyen K.C., Spolim S., Patil U., Baldi P., Wallace D.C. MITOMASTER: A bioinformatics tool for the analysis of mitochondrial DNA sequences. Hum. Mutat. 2008;30:1–6. doi: 10.1002/humu.20801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ruiz-Pesini E., Mishmar D., Brandon M., Procaccio V., Wallace D.C. Effects of purifying and adaptive selection on regional variation in human mtDNA. Science. 2004;303:223–226. doi: 10.1126/science.1088434. [DOI] [PubMed] [Google Scholar]
- 12.Bandelt H.J., Forster P., Sykes B.C., Richards M.B. Mitochondrial portraits of human populations using median networks. Genetics. 1995;141:743–753. doi: 10.1093/genetics/141.2.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bandelt H.J., Forster P., Röhl A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999;16:37–48. doi: 10.1093/oxfordjournals.molbev.a026036. [DOI] [PubMed] [Google Scholar]
- 14.Rozas J., Rozas R. DnaSP version 3: An integrated program for molecular population genetics and molecular evolution analysis. Bioinformatics. 1999;15:174–175. doi: 10.1093/bioinformatics/15.2.174. [DOI] [PubMed] [Google Scholar]
- 15.Andrews R.M., Kubacka I., Chinnery P.F., Lightowlers R.N., Turnbull D.M., Howell N. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat. Genet. 1999;23:147. doi: 10.1038/13779. [DOI] [PubMed] [Google Scholar]
- 16.Pruitt K.D., Tatusova T., Maglott D.R. NCBI reference sequences (RefSeq): A curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2007;35(Database issue):D61–D65. doi: 10.1093/nar/gkl842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pütz J., Dupuis B., Sissler M., Florentz C. Mamit-tRNA, a database of mammalian mitochondrial tRNA primary and secondary structures. RNA. 2007;13:1184–1190. doi: 10.1261/rna.588407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Green R.E., Malaspinas A.S., Krause J., Briggs A.W., Johnson P.L., Uhler C., Meyer M., Good J.M., Maricic T., Stenzel U. A complete Neandertal mitochondrial genome sequence determined by high-throughput sequencing. Cell. 2008;134:416–426. doi: 10.1016/j.cell.2008.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Anderson S., Bankier A.T., Barrell B.G., de Bruijn M.H., Coulson A.R., Drouin J., Eperon I.C., Nierlich D.P., Roe B.A., Sanger F. Sequence and organization of the human mitochondrial genome. Nature. 1981;290:457–465. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- 20.Marzuki S., Noer A.S., Lertrit P., Thyagarajan D., Kapsa R., Utthanaphol P., Byrne E. Normal variants of human mitochondrial DNA and translation products: The building of a reference data base. Hum. Genet. 1991;88:139–145. doi: 10.1007/BF00206061. [DOI] [PubMed] [Google Scholar]
- 21.Hall T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999;41:95–98. [Google Scholar]
- 22.Behar D.M., Villems R., Soodyall H., Blue-Smith J., Pereira L., Metspalu E., Scozzari R., Makkan H., Tzur S., Comas D. The dawn of human matrilineal diversity. Am. J. Hum. Genet. 2008;82:1130–1140. doi: 10.1016/j.ajhg.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Finnilä S., Lehtonen M.S., Majamaa K. Phylogenetic network for European mtDNA. Am. J. Hum. Genet. 2001;68:1475–1484. doi: 10.1086/320591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Herrnstadt C., Elson J.L., Fahy E., Preston G., Turnbull D.M., Anderson C., Ghosh S.S., Olefsky J.M., Beal M.F., Davis R.E. Reduced-median-network analysis of complete mitochondrial DNA coding-region sequences for the major African, Asian, and European haplogroups. Am. J. Hum. Genet. 2002;70:1152–1171. doi: 10.1086/339933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pereira L., Richards M., Goios A., Alonso A., Albarrán C., Garcia O., Behar D.M., Gölge M., Hatina J., Al-Gazali L. High-resolution mtDNA evidence for the late-glacial resettlement of Europe from an Iberian refugium. Genome Res. 2005;15:19–24. doi: 10.1101/gr.3182305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lanave C., Tommasi S., Preparata G., Saccone C. Transition and transversion rate in the evolution of animal mitochondrial DNA. Biosystems. 1986;19:273–283. doi: 10.1016/0303-2647(86)90004-3. [DOI] [PubMed] [Google Scholar]
- 27.Bandelt H.J., Kong Q.-P., Yao Y.-G., Richards M., Macaulay V. Estimation of mutation rates and coalescence times: some caveats. In: Bandelt H.-J., Macaulay V., Richards M., editors. Mitochondrial DNA and the Evolution of Homo sapiens. Springer-Verlag; Berlin, Germany: 2006. pp. 47–90. [Google Scholar]
- 28.Brown M.D., Yang C.C., Trounce I., Torroni A., Lott M.T., Wallace D.C. A mitochondrial DNA variant, identified in Leber hereditary optic neuropathy patients, which extends the amino acid sequence of cytochrome c oxidase subunit I. Am. J. Hum. Genet. 1992;51:378–385. [PMC free article] [PubMed] [Google Scholar]
- 29.Jin L., Yang A., Zhu Y., Zhao J., Wang X., Yang L., Sun D., Tao Z., Tsushima A., Wu G. Mitochondrial tRNASer(UCN) gene is the hot spot for mutations associated with aminoglycoside-induced and non-syndromic hearing loss. Biochem. Biophys. Res. Commun. 2007;361:133–139. doi: 10.1016/j.bbrc.2007.06.171. [DOI] [PubMed] [Google Scholar]
- 30.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McFarland R., Elson J.L., Taylor R.W., Howell N., Turnbull D.M. Assigning pathogenicity to mitochondrial tRNA mutations: when “definitely maybe” is not good enough. Trends Genet. 2004;20:591–596. doi: 10.1016/j.tig.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 32.Kivisild T., Shen P., Wall D.P., Do B., Sung R., Davis K., Passarino G., Underhill P.A., Scharfe C., Torroni A. The role of selection in the evolution of human mitochondrial genomes. Genetics. 2006;172:373–387. doi: 10.1534/genetics.105.043901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ruiz-Pesini E., Wallace D.C. Evidence for adaptive selection acting on the tRNA and rRNA genes of human mitochondrial DNA. Hum. Mutat. 2006;27:1072–1081. doi: 10.1002/humu.20378. [DOI] [PubMed] [Google Scholar]
- 34.Pereira F., Soares P., Carneiro J., Pereira L., Richards M.B., Samuels D.C., Amorim A. Evidence for variable selective pressures at a large secondary structure of the human mitochondrial DNA control region. Mol. Biol. Evol. 2008;25:2759–2770. doi: 10.1093/molbev/msn225. [DOI] [PubMed] [Google Scholar]
- 35.Mishmar D., Ruiz-Pesini E., Golik P., Macaulay V., Clark A.G., Hosseini S., Brandon M., Easley K., Chen E., Brown M.D. Natural selection shaped regional mtDNA variation in humans. Proc. Natl. Acad. Sci. USA. 2003;100:171–176. doi: 10.1073/pnas.0136972100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moilanen J.S., Majamaa K. Phylogenetic network and physiochemical properties of nonsynonymous mutations in the protein coding genes of human mitochondrial DNA. Mol. Biol. Evol. 2003;20:1195–1210. doi: 10.1093/molbev/msg121. [DOI] [PubMed] [Google Scholar]
- 37.Stewart J.B., Freyer C., Elson J.L., Wredenberg A., Cansu Z., Trifunovic A., Larsson N.-G. Strong purifying selection in transmission of mammalian mitochondrial DNA. PLoS Biol. 2008;6:63–71. doi: 10.1371/journal.pbio.0060010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.