

Abstract
A disulfide relay system (DRS) was recently identified in the yeast mitochondrial intermembrane space (IMS) that consists of two essential components: the sulfhydryl oxidase Erv1 and the redox-regulated import receptor Mia40. The DRS drives the import of cysteine-rich proteins into the IMS via an oxidative folding mechanism. Erv1p is reoxidized within this system, transferring its electrons to molecular oxygen through interactions with cytochrome c and cytochrome c oxidase (COX), thereby linking the DRS to the respiratory chain. The role of the human Erv1 ortholog, GFER, in the DRS has been poorly explored. Using homozygosity mapping, we discovered that a mutation in the GFER gene causes an infantile mitochondrial disorder. Three children born to healthy consanguineous parents presented with progressive myopathy and partial combined respiratory-chain deficiency, congenital cataract, sensorineural hearing loss, and developmental delay. The consequences of the mutation at the level of the patient's muscle tissue and fibroblasts were 1) a reduction in complex I, II, and IV activity; 2) a lower cysteine-rich protein content; 3) abnormal ultrastructural morphology of the mitochondria, with enlargement of the IMS space; and 4) accelerated time-dependent accumulation of multiple mtDNA deletions. Moreover, the Saccharomyces cerevisiae erv1R182H mutant strain reproduced the complex IV activity defect and exhibited genetic instability of the mtDNA and mitochondrial morphological defects. These findings shed light on the mechanisms of mitochondrial biogenesis, establish the role of GFER in the human DRS, and promote an understanding of the pathogenesis of a new mitochondrial disease.
Introduction
Classic mitochondrial disorders result from mutations in the mitochondrial or nuclear DNA that disrupt mitochondrial respiratory function. These diseases typically have brain and skeletal muscle manifestations and are, therefore, often referred to as mitochondrial encephalomyopathies.1
Nuclear DNA mutations leading to mitochondrial diseases have been described in genes encoding respiratory-chain subunits, oxidative phosphorylation assembly factors, proteins involved in mtDNA maintenance, factors related to mitochondrial protein synthesis, biosynthetic enzymes, and proteins promoting mitochondrial biogenesis.2 Many of these proteins are synthesized in the cytosol in the form of precursor proteins and posttranslationally transported to the mitochondria in an unfolded state.
A subgroup of small cysteine-containing proteins, which localize in the intermembrane space (IMS), requires the cooperation of the translocase of the mitochondrial outer membrane (TOM) complex with the Mia40-Erv1 disulfide relay system (DRS) for intramitochondrial import.3 These molecules include (1) proteins with a twin Cx3C motif (two cysteines separated by three other amino acid residues), such as the entire family of small chaperone translocon of the inner membrane (Tim) proteins, namely Tim8, Tim9, Tim10, Tim12, and Tim13 in Saccharomyces cerevisiae; (2) proteins with a twin Cx9C motif, such as the copper chaperone Cox17, which is required for the biogenesis of cytochrome c oxidase (COX), and Cox19 and Cox23, two additional molecules relevant to COX assembly; and (3) other proteins with disulfide bonds, such Cox12, and the copper chaperone for superoxide dismutase 1, Ccs1.4
Proven substrates of the DRS include many proteins relevant to COX biogenesis, as well as many TIM chaperones; therefore, a defect in this pathway is likely to result in pleiotropic effects due to a defective IMS and matrix import of proteins relevant to complex IV biogenesis and a number of yet uncharacterized mitochondrial functions.
We ascertained an inbred Moroccan family with three siblings affected by congenital cataract, progressive muscular hypotonia, sensorineural hearing loss, and developmental delay. Linkage analysis, followed by the sequencing of candidate genes, revealed the presence of a missense mutation in GFER (growth factor, augmenter of liver regeneration ERV1 homolog, S. cerevisiae, MIM 600924) that affects the protein content of the IMS, including some proteins involved in COX biogenesis, and leads to a progressive mitochondrial myopathy.
Subjects and Methods
Subjects
Written informed consent was obtained from the parents of the patients, and investigations were carried out according to the guidelines of the Ethical Committee of “Fondazione I.R.C.C.S. Ospedale Maggiore Policlinico, Mangiagalli e Regina Elena” in Milan, Italy in agreement with Italian and European Union laws.
Subjects II-2, II-4, and II-5 were born of consanguineous Moroccan parents after normal pregnancies. There was no neurological disease or congenital cataract in the family. All patients were born at term, and their birth weights were appropriate for their ages. All patients presented with congenital cataract, muscular hypotonia, sensorineural hearing loss, and developmental delay.
Patient II-2 presented with axial hypotonia and congenital cataract during the first month of life. At the age of one year, II-2 underwent surgery for the cataract with partial benefit. The patient also had a psychomotor delay with the ability to walk at 2 years and to speak at 3 years; he attended basic school and needed tutorial help for difficulties in his studies as a result of a progressive visual deficit. At 11 years old, after several surgeries, the patient suffered from retinal detachment and severe visual impairment. Since the age of 12 years, II-2 has presented with severe progressive hearing loss and bilateral ptosis. At the age of 17 years, neurological examination revealed generalized muscular hypotonia and hypotrophy, especially in the lower limbs, he could walk only with help, he was unable to fix his gaze, and rotatory nystagmus was noted. Proximal and distal muscle strength was normal. The upper- and lower-limb deep-tendon reflexes were weak or absent. No defects of the deep or superficial sensations were noted. Audiological examination showed a severe bilateral hearing deficiency. The ECG and echocardiography were normal. Blood analysis revealed hypoferritinemia (25 ng/ml, range 30–270), hyperamylasemia (351 U/l, range 0–195), and hyperCKemia (195 U/l). Serum lactate was elevated to 24.9 mg/dl (normal range 4.5–19). A muscle biopsy of the right brachial biceps was performed. An MRI of the brain performed at that time showed a thin corpus callosum; the signal from other telencephalic structures and the ventricular size was normal.
Patient II-4 presented with congenital cataract and progressive axial hypotonia. When the patient was 7 years of age, a neurological examination revealed unilateral ptosis, slight sensorineural hearing loss, diffuse muscle hypotrophy, and hypotonia. Blood analysis revealed low serum ferritin levels, hyperamylasemia, and hyperCKemia. The patient's serum lactate was high (34.2 mg/dl). A muscle biopsy of the right brachial biceps was performed.
Patient II-5 had congenital cataract, and a neurological examination performed when the patient was 3 years of age revealed slight axial hypotonia. No muscle biopsy was performed.
Skeletal Muscle Biopsy and Ultrastructural Analysis
After written informed consent was obtained, biopsies of the left biceps muscle were performed in patients II-2 and II-4 according to a protocol approved by the Institutional Review Board of the Fondazione I.R.C.C.S, Ospedale Maggiore Policlinico, Mangiagalli e Regina Elena in Milan, Italy. Ten-micron cryostatic cross sections were processed according to standard histological and histochemical techniques.5 Mitochondrial enzymatic activity was demonstrated by COX and a double reaction for COX and succinate dehydrogenase (SDH).6 A sequential demonstration of COX and SDH activities was performed on 10 μm slices of the cryostat sections from the muscle biopsies. The ultrastructural analysis was performed with a TEM Philips CM-100 microscope as previously described.7 The yeast ultrastructural analysis was performed as previously described.8
mtDNA Analysis
PCR assay for detecting multiple deletions used two primers (forward 7440–7465 and reverse complement 16135–16110) and the following amplification protocol: an initial denaturation at 94°C for 2 min, followed by 25 cycles (94°C for 30 s, 55°C for 30 s, and 68°C for 90 s) and a final extension for 2 min at 72°C (Platinum HiFi Taq Polymerase by Invitrogen, Carlsbad, CA). For the identification of deletion breakpoints, PCR products were subcloned in TOPO-TA cloning vector (Invitrogen) and sequenced on an ABI Prism 3100 Genetic Analyzer via Big Dye Terminator 3.1 protocol (Applied Biosystems, Foster City, CA).
Genetic Studies and Linkage Analysis
Blood-derived DNA samples from all family members were genotyped via the Affymetrix GeneChip Human Mapping 50K Xba240 Array according to protocols provided by the manufacturer (Affymetrix, Santa Clara, CA). Up to 50,000 SNPs were investigated, with an average spacing of approximately 12 kb. Genotypes were called with the GeneChip DNA Analysis Software (GDAS v.3). Mendelian errors were evaluated by Pedcheck and removed from further analysis. A parametric linkage analysis was performed with ALLEGRO software. Haplotypes reconstructed by ALLEGRO were presented graphically by HaploPainter. All data handling was performed with the graphical user interface Easy Linkage.9
A sequence analysis of genes previously reported to be mutated in patients with multiple deletions in mtDNA (POLG, POLG2, ANT1, PEO1) was performed as described above. Results were normal in affected individuals. The coding exons of candidate genes belonging to the identified region were amplified via oligonucleotide primers, which are available upon request.
We amplified the exons and intronic boundaries of GFER, using specific primers (Table S1, available online) and performing an initial denaturation at 95°C for 2 min, followed by 35 cycles of 94°C for 30 s, 59°C for 30 s, and 72°C for 40 s, and a final extension at 72°C for 5 min (Invitrogen Platinum Taq Polymerase plus 10% DMSO and 5 M Betaine). After purification, the PCR products were sequenced as described above.
Healthy control subjects were screened for the identified mutation in the GFER gene via sequence analysis.
Cell Cultures and Transfection Procedures
Cell cultures were established from the patients' skin and muscle biopsies as previously described.10 The control muscle biopsies and cell cultures used in this study were derived from the Telethon Genetic Biobanks Network GTB07001E, Department of Neurological Sciences, University of Milan, Italy). Primary myoblasts were cultured in skeletal muscle cell growth medium (PromoCell GmbH, Heidelberg, Germany) at 37°C in 5% CO2. Primary fibroblasts were cultured in DMEM medium (Invitrogen) with 15% FBS at 37°C in 5% CO2. For determination of the rate of primary cell proliferation, the mean population doubling (MPD) was calculated as previously described.11 A comparison of quantitative parameters between groups was performed with the Mann-Whitney U test. A p value below 0.05 was considered significant.
Wild-type GFER cDNA was retrotranscribed with the use of total RNA extracted from control muscle tissue and cloned into the pAcGFP1-N2 expression vector (Clontech Laboratories, Mountainview, CA). The recombinant vector was propagated in XL-10 Gold Ultracompetent E. coli cells (Stratagene, La Jolla, CA). Plasmid transfection was carried out in patient and control primary fibroblasts via electroporation with the Human Dermal Fibroblast kit (Amaxa, Gaithersburg, MD).
We evaluated GFER by using the HEK293 cell line, which was stably transfected with two vectors overexpressing wild-type and mutated GFER cDNA. Transfections were carried out as described above, and selection was performed through the addition of G418 to the culture medium (500 μg/ml). Rates of transfection and the expression of GFP-tagged GFER were evaluated by fluorescence microscopy and FACS analysis (data not shown).
Biochemical Assays
The specific activities of individual respiratory-chain complexes were measured in cells and muscle homogenates.12 Proteins were extracted from muscle tissue after motor-driven homogenization and from cells after sonication in resuspension buffer. The protein concentration was measured via the Lowry method. The specific activity of each complex was normalized to that of citrate synthase.
GFER mRNA Quantification
Total RNA from the probands' tissues was reverse transcribed (First Strand cDNA Synthesis Kit, GE Healthcare) and quantified by quantitative PCR via the ΔΔCt method on a 7500 Real Time PCR System (Software 2.01, Applied Biosystems, Foster City, CA). The GFER mRNA was evaluated with the Taqman gene expression assay (Hs00193365_m1), and GAPDH was used as the control housekeeping gene (Hs99999905_m1). The expression of GFER was also quantified in several control human tissues contained in Human Total Master Panel II (Clontech Laboratories, Mountain view, CA). The rates of GFER expression were also evaluated in transfected HEK293 cell lines. Overexpression (>50-fold change) was observed in transfected HEK293 cells, and comparable levels were observed in cells containing wild-type or mutated GFER expression vectors (data not shown).
Immunocytochemistry
Myoblasts, fibroblasts, and HEK293 mitochondria were stained with MitoTracker Red (Invitrogen) in fresh culture medium for 30 min at 37°C. For double immunostaining, cells were fixed with 4% formaldehyde for 15 min at 37°C. After rinsing with PBS and preincubation with 0.25% Triton X-100 in PBS, the cultures were incubated with the primary antibodies at 4°C. The following proteins were evaluated: GFER, TIMM13, and COX17 with rabbit polyclonal antibodies (Proteintech Group, Manchester, UK), COX6B1 with mouse monoclonal antibody (Molecular Probes, Invitrogen). After repeated rinses with PBS, the primary unconjugated antibodies were further incubated with R-Phycoerythrin (RPE) or TRITC-conjugated secondary antibodies (1:100; DAKO) in PBS for 2 hr in the dark at room temperature, then rinsed with PBS and coverslipped.
Subcellular Fractionation
Mitochondria were purified through modification of a published protocol.13 HEK293 cells (roughly 10 million cells) were collected and passed 40 times through a 27-gauge needle in a buffer containing 250 mM sucrose, 10 mM Tris, and 0.1 mM EGTA (pH 7.4) and centrifuged at 630 × g for 10 min. The supernatant was then centrifuged at 8000 × g for 15 min for isolation of a crude mitochondrial pellet. The supernatant was subjected to further centrifugation at 48,000 × g for 1 hr 30 min, and the final supernatant was designated the cytosolic fraction. The crude mitochondrial pellet was washed once with the extraction buffer and sedimented at 630 × g for 10 min. The remaining supernatant was further centrifuged at 8000 g for 15 min, and the residual pellet was designated the mitochondrial fraction.
Protein Blot Analyses
Mitochondrial fractions derived from HEK293 cells were analyzed by SDS-PAGE according to Laemmli without the addition of reducing agents in the sample buffer. For reducing conditions, dithiothreitol (DTT) at a final concentration of 15 mM was added during sample preparation, whereas nonreducing analyses were completely DTT free.
The polyacrylamide gels (11%) were run for 2 hr and blotted. The GFER protein was assayed with a commercial polyclonal antibody developed in rabbit (Proteintech Group). After incubation with a peroxidase-conjugated secondary antibody, the GFER signal was detected with ECL-detection reagents (Amersham). The HSP60 (Santa Cruz Biotechnology) and COX4 (Molecular Probes) proteins were used as a normalization control of the mitochondrial fractions.
Yeast Model
Construction of Yeast Strains
Yeast strains were derived from the diploid DWW, obtained from the cross between strains W303-1A (Mata ade2-1 leu2-3, 112 ura3-1 trp1-1 his3-11, 15 can1-100) and W303-1B (Matα ade2-1 leu2-3, 112 ura3-1 trp1-1 his3-11, 15 can1-100). Being that ERV1 is an essential gene, the construction of a null erv1 strain carrying either ERV1 or the erv1R182H allele required a particular procedure. One copy of the ERV1 ORF was deleted in the diploid DWW and substituted with the Kanr marker, resulting in the heterozygous diploid DMT1 (ERV1/Δerv1) strain. The ERV1 gene, amplified by PCR from the W303-1B strain and cloned into the centromeric pFL38 vector containing the auxotrophic marker URA3, was then introduced into DMT1. By sporulation and tetrad analysis, the haploid DMT1-4C/38E strain (Δerv1, carrying a plasmid-borne ERV1 wild-type copy) was then obtained. This strain was transformed with the erv1R182H allele, which was produced by site-directed mutagenesis, via the overlap-extension technique14 and cloned into the centromeric pFL39 vector containing the auxotrophic marker TRP1. The preferred yeast codon encoding histidine (CAT) was used in the sequence of the mutagenic oligonucleotides, maximizing the expression of the mutant allele. It was then possible to isolate the DMT1/39e strain (Δerv1, carrying the plasmid-borne erv1R182H mutant allele) devoid of pFL38ERV1 and completely isogenic with the DMT1-4C/38E strain by plasmid shuffling in the presence of 5-fluoro orotic acid (5FOA, SIGMA), which is toxic to URA3 strains but not to the ura3 mutants.
Miscellaneous Yeast Methods
Cells were cultured in 0.67% yeast nitrogen base (YNB) without amino acids (Difco), supplemented with appropriate amino acids and bases for auxotrophy to a final concentration of 40 mg/ml. Various carbon sources were added at 2% (w/v).
Cytochrome spectra were determined as described previously.15 The spectrum profile was recorded in cells grown at 28°C, so that the required biomass could be obtained, then shifted to 37°C for 15 hr. Spectrophotometric measurements of COX activity were performed for 120 s at 30°C in isolated mitochondria after the decrease of absorbance at 550 nm as a result of cytochrome c oxidation.16 The specific activity was normalized to that of citrate synthase.
For a respiratory-deficient diagnosis (petite colonies), cells from cultures grown for 15 generations at 37°C in YNB supplemented with glucose were plated onto solid YNB medium containing 2% ethanol and 0.3% glucose and maintained for 5 days at 28°C. A mit−-complementation-based analysis of mtDNA was performed on respiratory-deficient colonies as previously described.17
Results
Morphological and Biochemical Studies
The muscle biopsies from subjects II-2 and II-4 exhibited scattered COX-negative fibers (Figures 1A–1D). Biochemical studies revealed a consistent moderate reduction (30%–50% of normal mean values) of complex IV activity in multiple tissues from both probands (Table 1).
Figure 1.
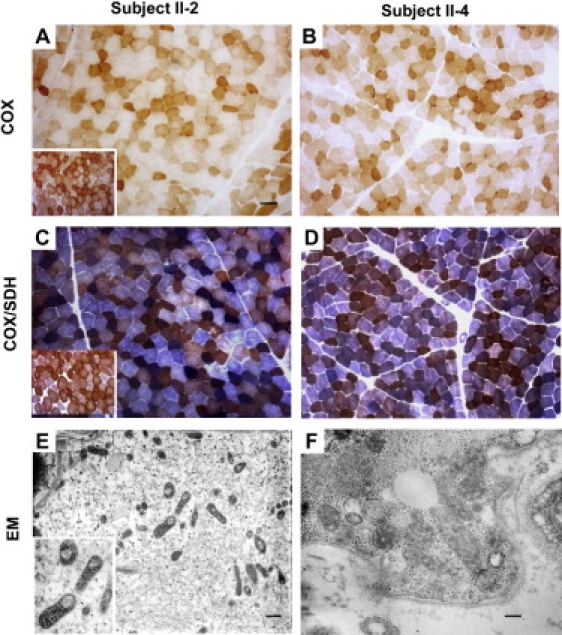
Histochemical and Ultrastructural Analysis of Probands' Skeletal Muscle
(A–D) COX histochemistry reveals the presence of several COX-deficient fibers in probands II-2 and II-4 (A and B). Double staining for COX/SDH. Scattered fibers with increased SDH staining, indicative of mitochondrial proliferation, are present in patient II-2. A COX deficiency is confirmed in both patient II-2 and patient II-4. (C and D) Insets within panels (A) and (C) represent the normal controls.
(E and F) Electron microscopy shows several mitochondria with thickened cristae; many mitochondria are also present with large vacuolization in (E).
Scale bars represent 25 μm (A–D), 700 nm (E), or 250 nm (F).
Table 1.
Activities of the Respiratory-Chain Complex
| CI/CS |
CII/CS |
CI+III/CS |
CII+III/CS |
CIV/CS |
|
|---|---|---|---|---|---|
| Muscle | |||||
| Proband II-2 | 6.6 (46.5%) | 7.2 (62.1%) | 16.4 (47.4%) | 8.0 (65.0%) | 20.1 (44.8%) |
| Normal (n = 38) | 14.2 ± 2.6 | 11.6 ± 1.8 | 34.6 ± 7.8 | 12.3 ± 2.7 | 44.8 ± 5.6 |
| Fibroblasts | |||||
| Proband II-4 | 54.8 | 6.1 | 311.3 | 4.9 | 31.7 (70.0%) |
| Normal (n = 16) | 68.7 ± 22.7 | 6.8 ± 1.2 | 225.78 ± 71 | 4.91 ± 1.2 | 44.8 ± 4.7 |
| Myoblasts | |||||
| Proband II-2 | 33.8 (66.4%) | 4.4 (46.8%) | 255.6 | 5.4 | 27.8 (47.3%) |
| Normal (n = 12) | 50.9 ± 8.5 | 9.4 ± 2.2 | 209.4 ± 27.1 | 6.0 ± 1.2 | 58.8 ± 7.4 |
Mean values (± 1 SD) of the activities of the respiratory-chain complex are normalized to citrate synthase (CS). Activities are expressed as nM/min/mg protein. Activities reduced by at least 30% of the normal mean values are shown in bold. n = number of controls. Biochemical determinations on probands' tissues were performed in triplicate.
Interestingly, patient II-2 displayed a multicomplex alteration, including a complex I and II defect in both muscle and myoblasts, whereas an analysis of patient II-4's fibroblasts revealed only reduced complex IV activity.
The ultrastructural analysis of patient II-2's muscle tissue showed striking mitochondrial structural abnormalities. In addition to being abnormally enlarged, these organelles exhibited increased electron density and thickened cristae. In a subset of mitochondria, large electron-dense vacuolizations were observed, probably representing an abnormal IMS (Figures 1E and 1F). Multiple mtDNA deletions were found by PCR analysis of muscle from patient II-2 (not shown).
Linkage Analysis and Mutation Screening
We performed a genome-wide linkage analysis in all family members, using the Affymetrix GeneChip SNP array. A single region of autozygosity on chromosome 16p13.3 (LOD score 2.657) was identified (Figure 2A). This region (0–12.1 Mb), confirmed by microsatellite analysis (Figure 2B), encompasses 271 genes. Candidate genes were prioritized according to the likelihood score of mitochondrial localization, as implemented in the “Maestro” data set.18
Figure 2.
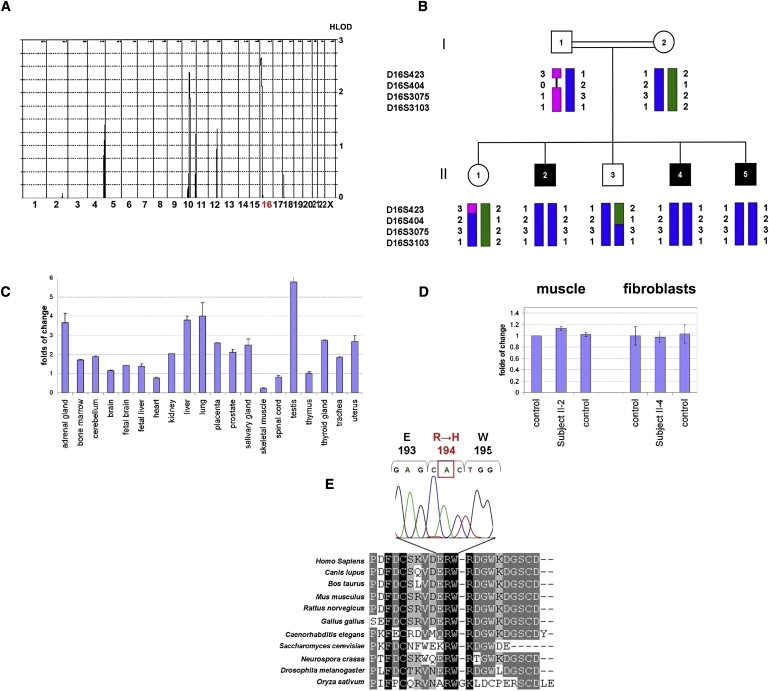
Linkage Analysis, Expression Studies, and Mutation Analysis
(A) Multipoint linkage analysis of the genome-wide scan with the Affymetrix GeneChip Human Mapping 50K Array Xba240.
(B) Family pedigree and haplotype analysis of the GFER locus. Affected individuals are indicated by black symbols.
(C) Quantitative RT-PCR of GFER expression in 20 human control tissues. GAPDH was used as the control housekeeping gene. The thymus mRNA level was used for normalization of the expression data. All determinations have been performed in replicates (n = 6).
(D) Quantitative RT-PCR of GFER expression in muscle and fibroblasts from patients and controls. Results are presented as mean ± SD.
(E) CLUSTALW multiple-alignment sequence of the GFER region containing the mutated residue in our family. The electropherogram of the c.581 G→A mutation in exon 3 of GFER, resulting in a p.R194H substitution, is shown above.
We sequenced the exons and flanking intronic regions of ten genes encoding known or putative mitochondrial proteins: GFER, FAHD1, NDUFB10, NUBP2, DNAJA3, NME4, Magmas, NTHL1, TRAP1, and MPG. Sequencing of GFER revealed a c.581 G→A homozygous mutation in all affected individuals (Figure S1), which results in the p.R194H substitution. This mutation was absent in 380 samples from unrelated European individuals and 183 samples from unrelated Arab individuals (156 Moroccan).
Expression Analysis
The GFER gene is ubiquitously expressed (Figure 2C) in human tissues, the highest expression in the testis and liver and low expression in muscle. Quantitative RT-PCR analysis of the muscle RNA from patient II-2, fibroblast RNA from patient II-4, and relative controls showed no significant difference in the GFER expression level (Figure 2D).
GFER Analysis in Probands and Transfected HEK293 Cells
To test whether the p.R194H mutation (Figure 2E) can affect protein stability and dimer formation, we carried out a immunoblot analysis of HEK293 mitochondrial fractions overexpressing either wild-type or mutant GFER. Immunoblots using the antibody against GFER under reducing and nonreducing conditions showed a reduction of both the GFERR194H monomer and the dimer as compared to the wild-type protein (Figure 3A). Quantitative RT-PCR found no significant differences at the transcriptional level in the total RNA from HEK293 transfected with the wild-type or mutant constructs (data not shown). These results suggest that GFERR194H is less stable in the IMS. Alternatively, being that ERV1 is a substrate of the Mia40-Erv1 import pathway,19 the GFERR194H protein could be either less efficiently imported into or less retained by the IMS. Further study will address this issue.
Figure 3.
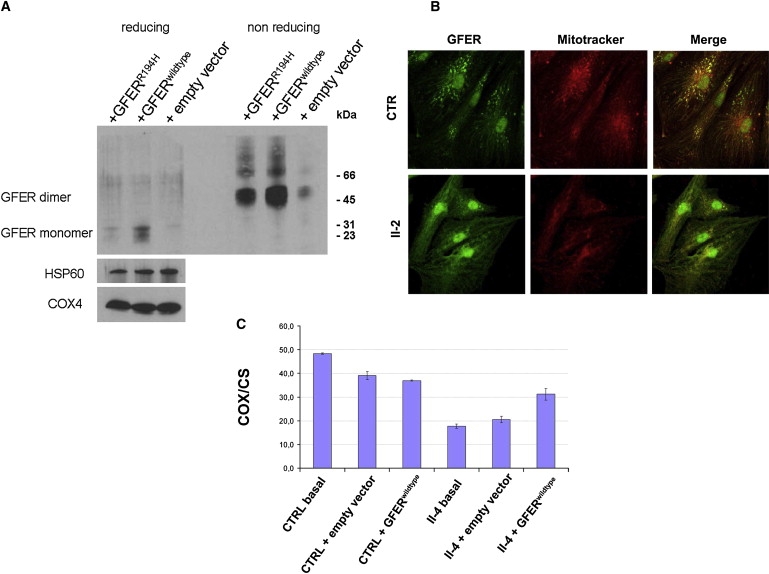
Western Blot Analysis of Overexpressed GFER, Immunocytochemical Analysis of Endogenous GFER in Patient Myoblasts, and Functional Rescue in Patient Fibroblasts
(A) HEK293 cells were stably transfected with vector overexpressing GFERwild-type and GFERR194H cDNA. Immunoblot analysis showed a reduction of GFERR194H in mitochondrial fractions under both reducing (DTT 15 mM) and nonreducing conditions. COX4 and HSP60 were used for normalization.
(B) Antibodies against human GFER showed specific GFER colocalization with MitoTracker and DAPI in control myoblasts, whereas immunofluorescence was almost absent in the cytoplasm. Instead, primary myoblasts from patient II-2 showed a reduced punctuate mitochondrial fluorescence compared to control cells with more diffuse cytoplasmic staining. Scale bar represents 40 μm.
(C) Biochemical functional rescue in proband II-4 fibroblasts that were transfected with GFERwild-type cDNA. The analysis of COX activity (normalized to citrate synthase [CS]) shows a restoration of mitochondrial cytochrome c oxidation (complex IV). Results are presented as mean of triplicate determinations ± SD.
To test whether the p.R194H mutation affects GFER protein localization, we performed an immunocytochemical analysis of myoblasts from patient II-2 and controls. Throughout our experiments, primary cells from patients presented with a lower rate of proliferation compared to control cells (myoblast MPD II-2, 13 ± 2; control MPD, 28 ± 3; p < 0.00001). In control myoblasts, the GFER-specific immunofluorescence pattern colocalized with that of MitoTracker and DAPI and was almost absent in the cytoplasm. Primary myoblasts from patient II-2 showed reduced punctuate mitochondrial fluorescence compared to control cells, with more diffuse cytoplasmic staining (Figure 3B).
Complementation Studies in Human Cells
As previously mentioned, the fibroblasts from patient II-4 exhibited a COX deficiency, whereas other respiratory-chain activities were normal. To evaluate whether the mutated GFER gene was the cause of the complex IV deficiency, we transiently transfected primary fibroblasts from patient II-4 and several controls with GFERwt cDNA. Biochemical analysis revealed a 30% increase in COX activity in GFERwt-transfected patient fibroblasts, normalized to the citrate synthase activity, whereas COX activity remained unchanged in cells transfected with empty vector (Figure 3C). These results demonstrate that the p.R194H mutation is associated with the functional COX defect found in our patients, and they also clearly establish the key role of GFER in complex IV activity in humans.
Analysis of IMS Proteins
To further characterize the role of GFERR194H in mitochondrial biogenesis, we carried out an immunocytochemical analysis of two known DRS substrates, COX17 and TIMM13, as well as COX6B1, in fibroblasts from patient II-4. Confocal analysis revealed reduced colocalization of COX17, TIMM13, and COX6B1 immunostaining with MitoTracker compared to the colocalization signal observed in control cells from healthy donors (Figure 4).
Figure 4.
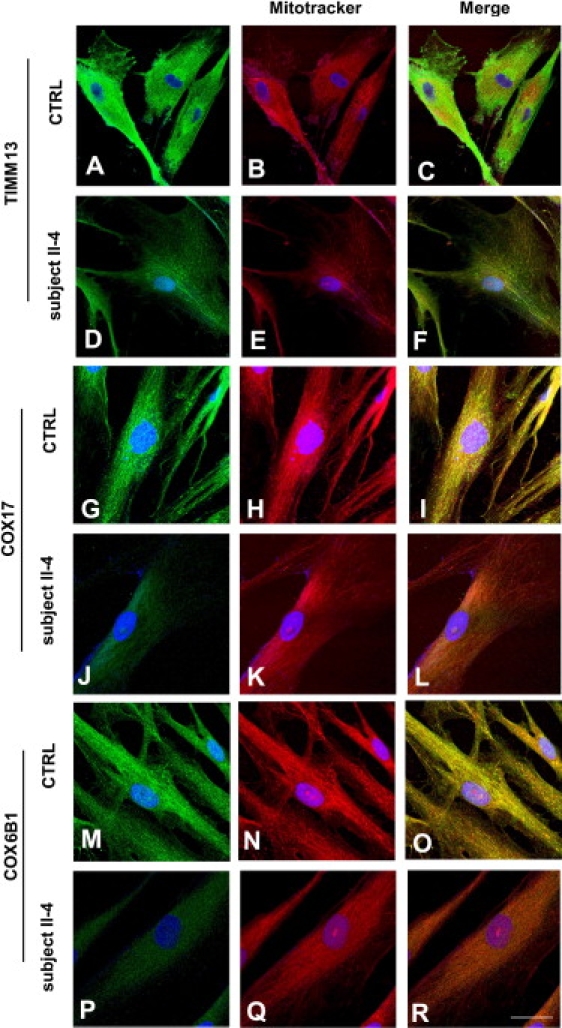
Confocal Immunocytochemical Analysis of IMS Proteins in Patient Fibroblasts
Immunocytochemistry of patient and control fibroblasts, using antibodies against TIMM13, COX17, and COX6B1. Confocal analysis revealed reduced colocalization of COX17, TIMM13, and COX6B1 immunostaining with MitoTracker compared to the colocalization signal observed in control cells from healthy donors. Scale bars represent 40 μm (A–F) or 20 μm (G–R).
Yeast Model
In order to validate the pathogenicity of the human mutation, we set up a recombinant system in S. cerevisiae. The R194H mutation was introduced into the fully conserved R182 position of ScERV1, and this mutant allele was expressed in the null erv1 strain. The resulting erv1R182H mutant strain (Δerv1//erv1R182H) grew on both glucose and oxidative substrates, but it displayed a thermosensitive phenotype. Growth was delayed on solid medium (Figure 5A) and blocked in liquid medium at 37°C (data not shown). Measurement of the mitochondrial cytochrome content is an index of the structural integrity of the respiratory-chain complexes. The strain carrying erv1R182H displayed a significant reduction in cytochrome aa3 but not cytochromes b and c (Figure 5B). Indeed, in mitochondria isolated from the erv1R182H mutant, COX activity was reduced by 45% (Figure 5C).
Figure 5.
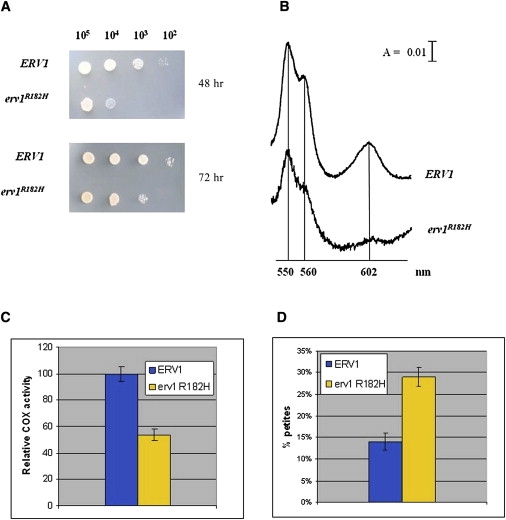
Yeast Model
(A) Phenotypic analysis of the DMT1-4C strain (Δerv1) carrying either ERV1 or the erv1R182H allele. The growth phenotype was that observed at 37°C. Equal amounts of serial dilutions of cells from exponentially grown cultures were spotted onto YNB medium supplemented with 2% glucose.
(B) Cytochrome spectra of cells grown on YNB medium supplemented with 2% glucose at 28°C and then shifted to 37°C overnight (A denotes absorbance).
(C) COX activity measured in the ERV1 and erv1R182H strains.
(D) Petite frequency of Δerv1 strain transformed with wild-type ERV1 and the erv1R182H mutant allele. COX activity and petite frequency determinations were performed in triplicate. The error bars represent SD.
Erv1p is not known to import proteins necessary for the mtDNA replication machinery. Therefore, erv1 mutations should not increase mtDNA mutability. However, Erv1p contributes, either directly or indirectly, to the assembly of components of the inner mitochondrial membrane, where the mtDNA is replicated. To evaluate whether the erv1R182H mutation is associated with mtDNA instability, we measured the frequency of petite mutants in the haploid Δerv1 strain carrying either the wild-type or the erv1R182H allele. The mtDNA instability in S. cerevisiae is associated with the increased segregation of respiratory-deficient petite mutants. In the presence of the erv1R182H allele, the petite frequency was approximately twice that determined in the Δerv1 strain transformed with ERV1 (Figure 5D), suggesting a role for Erv1p in maintaining the integrity of mtDNA. Petite mutants can be either rho− (that is, cells in which the mtDNA is partially deleted) or rhoo (that is, cells in which the mtDNA is completely lost). To evaluate the nature of the petite mutations (rho− versus rhoo), we crossed petite clones with mit− strains, each harboring a different point mutation in three genes encoding respiratory proteins. When rho− mutants are crossed with a mit− mutant, respiratory-competent rho+ cells are produced by recombination, provided that the mit− point mutation maps to a region conserved in the rho− mtDNA. Contrariwise, rhoo mutants, which are completely devoid of mtDNA, are unable to produce rho+ cells. By this analysis, it is possible to establish whether the erv1R182H mutation causes mtDNA deletion or depletion. This complementation test revealed that approximately 70% of the petite clones resulted in rho−, in the presence of both erv1R182H and ERV1 alleles (data not shown), indicating that the mutation determines an increase in mtDNA mutability not associated with increased mtDNA depletion, which would result in an increase in rhoo mutants.
The ultrastructural analysis of the erv1R182H mutant showed clusters of mitochondria with simplified or absent cristae (Figure S2).
Discussion
We describe three patients who presented with progressive myopathy, congenital cataract, sensorineural hearing loss, and developmental delay. In the probands' muscle tissue, the activities of respiratory-chain complexes I, II, and IV were decreased by 50%, 33%, and 50%, respectively, compared to mean control values. Using homozygosity mapping, we identified a missense mutation in the GFER gene affecting a conserved residue, and it was associated with altered protein localization in patient myoblasts and a reduction in the protein level in transfected HEK293 mitochondrial fractions.
Human GFER belongs to the ERV1/ALR protein family, members of which are found in lower and higher eukaryotes.20 The GFER gene codes for two distinct isoforms that are probably synthesized from the same mRNA with the use of different initiation codons. The long isoform (205 aa, 23 kDa) is located mainly in the mitochondrial IMS, and it exists under nonreducing and nondenaturing conditions as a homodimer and a heterodimer. The shorter isoform (125 aa, 15 kDa), which lacks 80 amino acids at its N terminus, is present predominantly in the nucleus.21
The yeast ortholog Erv1p is the key protein of the mitochondrial DRS. Through its flavin-linked sulfhydryl oxidase activity, Erv1p oxidizes the disulfide carrier protein Mia40, which in turn transfers a disulfide to the newly synthesized small cysteine-containing proteins arriving in the IMS22–24 (Figure 6). Erv1p is then reoxidized, transferring its electrons to molecular oxygen via interactions with cytochrome c and COX, linking the DRS to respiratory-chain activity.25 Erv1p depletion prevents the import of those essential proteins and leads to mtDNA aberrations and abnormal mitochondrial morphology.26
Figure 6.
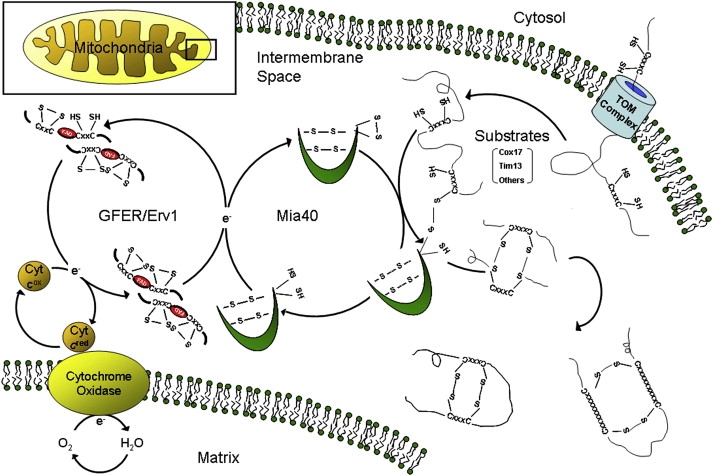
The Mitochondrial Disulfide Relay System
Schematic representation of the mitochondrial DRS, which mediates the import of cysteine-rich substrates into the IMS by an oxidative trapping mechanism. Mia40 performs the oxidation of these proteins and is reoxidized by Erv1/GFER in a disulfide-transfer reaction. For Erv1 to be recycled into its oxidized form, electrons are transferred to cytochrome c, connecting the DRS to the electron-transport chain of the mitochondria.
The role of mammalian GFER in the mitochondrial DRS has been poorly explored. The rat and human GFER proteins act as sulfhydryl oxidases27 and probably play a role similar to that of yeast Erv1p.
Using confocal immunocytochemistry and immunoblot analysis, we showed that GFERR194H is less stable than the wild-type protein within the mitochondria. This relative mitochondrial depletion may disturb the mitochondrial import of several DRS substrates. We showed by confocal analysis that primary fibroblasts from an affected individual probably have a reduced mitochondrial signal for COX17 and TIMM13, which are known DRS substrates in yeast,3 as well as COX6B1, a protein with a twin Cx9C motif. A partial mitochondrial depletion of COX17 and COX6B1 may result in COX-defective biogenesis. Indeed, the copper metallochaperone COX 17 specifically and physically interacts with SCO2 in order to deliver copper to COX.28 Furthermore, the p.R19H change in COX6B1 was recently shown to be associated with the reduced stability of COX holoenzyme and with severe early-onset leukodystrophic encephalopathy, myopathy, and growth retardation.29 In complementation assays, we observed a functional restoration of COX upon the transfection of fibroblasts from patient II-4 with the wild-type cDNA. Moreover, yeast Δerv1/erv1R182H displayed a marked reduction in COX activity. We can also speculate that mutated Erv1/GFER might be less efficient in shuttling electrons to oxidized cytochrome c, from whence they flow through COX to molecular oxygen. In this pathway, the connection of the disulfide relay with the respiratory chain would significantly decrease the efficiency of the oxidase activity.
Taken together, these data establish a role for the GFER mutation in causing a moderate complex IV deficiency in our affected individuals.
The intramitochondrial level of other DRS substrates, as observed for TIMM13, may be equally affected by a defective Mia40-GFER system. The involvement of complexes I and II among the respiratory-chain-deficient activities within the probands' muscle suggests that other components or assembly factors of these two complexes could be less efficiently imported into the inner mitochondrial compartment, perhaps through a TIM-dependent process. The molecular dissection of these faulty mechanisms remains to be elucidated.
Furthermore, both patients' muscle and yeast Δerv1//erv1R182H displayed abnormal ultrastructural mitochondria morphology and mtDNA instability. It is tempting to speculate that still uncharacterized DRS-dependent proteins being imported into the mitochondria with a lower efficiency might cause the observed alteration of mitochondrial morphology. Impairment of the connection between the inner and outer mitochondrial membranes might also affect the link to nucleoids, probably leading to mtDNA disorganization and instability. Furthermore, the mtDNA abnormality could be a consequence of a reduced interaction between Erv1/GFER and cytochrome c. This interaction directly connects the relay system to the respiratory chain and prevents the generation of hydrogen peroxide, which might perturb mtDNA stability. Although we consider the small amount of muscle mtDNA deletions a secondary consequence of a defective GFER-dependent mechanism, it is noteworthy that, in S. cerevisiae, erv1p carrying p.F124S leads to mtDNA depletion and impaired cell survival.26
Human GFER is also known as ALR (augmenter of liver regeneration) or HPO (hepatopoietin), and it is thought to be one of the factors responsible for the extraordinary regenerative capacity of the mammalian liver.30 We did not observe signs of hepatic involvement in patient II-4 on the abdominal ultrasound, although marked hyperamylasemia and hypoferritinemia, without any other apparent cause, were found in all three probands. Except for the congenital cataract, which has already been demonstrated to be associated with some mitochondrial diseases,31 the clinical phenotype and histological and biochemical features described here were more prominent in the oldest sibling, as was the presence of multiple mtDNA deletions. These findings suggest a progressive course for the pathology associated with the identified mutation.
Taken together, these results show a role for GFER in causing an infantile disorder with combined respiratory-chain deficiency, and they unravel the importance of the DRS in the pathogenesis of human mitochondrial diseases.
Supplemental Data
Supplemental Data include two figures and one table and can be found with this article online at http://www.ajhg.org/.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Haplopainter, http://haplopainter.sourceforge.net/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Acknowledgments
We wish to thank the patients and their families for their support and collaboration. This research received support from Associazione Amici del Centro Dino Ferrari. G.P.C. and I F. received funding from Research Grant PRIN 2006 2006069034 from the Italian Ministry of University and Research for the project entitled “An integrated approach to the study of the etiopathogenesis of mitochondrial disorders.” G.P.C. was also funded by the Italian Ministry of Health for the 2009 project entitled “Mitochondrial disorders: From medical genetics to molecular mechanisms, toward the development of therapeutic strategies.” The Telethon Network of Genetics Biobanks (no. GTB 07001E) was the source of muscle, DNA, and cells used in this study. Eurobiobank project QLTR-2001-02769 is also gratefully acknowledged. We thank M. Aguennouz and V. Bonifati for providing DNA from healthy Moroccan subjects. We also thank T. G. Weiss for providing a control GFER antibody.
References
- 1.DiMauro S., Schon E.A. Mitochondrial disorders in the nervous system. Annu. Rev. Neurosci. 2008;31:91–123. doi: 10.1146/annurev.neuro.30.051606.094302. [DOI] [PubMed] [Google Scholar]
- 2.Zeviani M., Carelli V. Mitochondrial disorders. Curr. Opin. Neurol. 2007;20:564–571. doi: 10.1097/WCO.0b013e3282ef58cd. [DOI] [PubMed] [Google Scholar]
- 3.Mesecke N., Terziyska N., Kozany C., Baumann F., Neupert W., Hell K., Herrmann J.M. A disulfide relay system in the intermembrane space of mitochondria that mediates protein import. Cell. 2005;121:1059–1069. doi: 10.1016/j.cell.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 4.Hell K. The Erv1-Mia40 disulfide relay system in the intermembrane space of mitochondria. Biochim. Biophys. Acta. 2008;1783:601–609. doi: 10.1016/j.bbamcr.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 5.Heckmatt J.Z., Dubowitz V. Needle biopsy of skeletal muscle. Muscle Nerve. 1984;7:594. [PubMed] [Google Scholar]
- 6.Sciacco M., Bonilla E. Cytochemistry and Immunocytochemistry of Mitochondria in Tissue Sections. Methods Enzymol. 1996;264:509–521. doi: 10.1016/s0076-6879(96)64045-2. [DOI] [PubMed] [Google Scholar]
- 7.Dubowitz V. Second Edition. Baillière Tindall; Philadelphia: 1985. Muscle Biopsy, a Practical Approach. [Google Scholar]
- 8.Wright R. Transmission electron microscopy of yeast. Microsc. Res. Tech. 2000;51:496–510. doi: 10.1002/1097-0029(20001215)51:6<496::AID-JEMT2>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 9.Hoffmann K., Lindner T.H. EasyLINKAGE-Plus-automated linkage analyses using large-scale SNP data. Bioinformatics. 2005;21:3565–3567. doi: 10.1093/bioinformatics/bti571. [DOI] [PubMed] [Google Scholar]
- 10.Salani S., Lucchiari S., Fortunato F., Crimi M., Corti S., Locatelli F., Bossolasco P., Bresolin N., Comi G.P. Developmental and tissue-specific regulation of a novel dysferlin isoform. Muscle Nerve. 2004;30:366–374. doi: 10.1002/mus.20106. [DOI] [PubMed] [Google Scholar]
- 11.Périé S., Mamchaoui K., Mouly V., Blot S., Bouazza B., Thornell L.E., St Guily J.L., Butler-Browne G. Premature proliferative arrest of cricopharyngeal myoblasts in oculo-pharyngeal muscular dystrophy: Therapeutic perspectives of autologous myoblast transplantation. Neuromuscul. Disord. 2006;16:770–781. doi: 10.1016/j.nmd.2006.07.022. [DOI] [PubMed] [Google Scholar]
- 12.Bresolin N., Zeviani M., Bonilla E., Miller R.H., Leech R.W., Shanske S., Nakagawa M., DiMauro S. Fatal infantile cytochrome c oxidase deficiency: decrease of immunologically detectable enzyme in muscle. Neurology. 1985;35:802–812. doi: 10.1212/wnl.35.6.802. [DOI] [PubMed] [Google Scholar]
- 13.Okado-Matsumoto A., Fridovich I. Subcellular distribution of superoxide dismutases (SOD) in rat liver: Cu,Zn-SOD in mitochondria. J. Biol. Chem. 2001;276:38388–38393. doi: 10.1074/jbc.M105395200. [DOI] [PubMed] [Google Scholar]
- 14.Ho S.N., Hunt H.D., Horton R.M., Pullen J.K., Pease L.R. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 15.Fontanesi F., Palmieri L., Scarcia P., Lodi T., Donnini C., Limongelli A., Tiranti V., Zeviani M., Ferrero I., Viola A.M. Mutations in AAC2, equivalent to human adPEO-associated ANT1 mutations, lead to defective oxidative phosphorylation in Saccharomyces cerevisiae and affect mitochondrial DNA stability. Hum. Mol. Genet. 2004;13:923–934. doi: 10.1093/hmg/ddh108. [DOI] [PubMed] [Google Scholar]
- 16.Wharton D.C., Tzagoloff A. Cytochrome oxidase from beef heart mitochondria. Methods Enzymol. 1967;10:245–250. [Google Scholar]
- 17.Baruffini E., Ferrero I., Foury F. Mitochondrial DNA defects in Saccharomyces cerevisiae caused by functional interactions between DNA polymerase gamma mutations associated with disease in human. Biochim. Biophys. Acta. 2007;1772:1225–1235. doi: 10.1016/j.bbadis.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 18.Calvo S., Jain M., Xie X., Sheth S.A., Chang B., Goldberger O.A., Spinazzola A., Zeviani M., Carr S.A., Mootha V.K. Identification of human mitochondrial disease genes through integrative genomics. Nat. Genet. 2006;38:576–582. doi: 10.1038/ng1776. [DOI] [PubMed] [Google Scholar]
- 19.Terziyska N., Grumbt B., Bien M., Neupert W., Herrmann J.M., Hell K. The sulfhydryl oxidase Erv1 is a substrate of the Mia40-dependent protein translocation pathway. FEBS Lett. 2007;581:1098–1102. doi: 10.1016/j.febslet.2007.02.014. [DOI] [PubMed] [Google Scholar]
- 20.Gatzidou E., Kouraklis G., Theocharis S. Insights on augmenter of liver regeneration cloning and function. World J. Gastroenterol. 2006;12:4951–4958. doi: 10.3748/wjg.v12.i31.4951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li Y., Wei K., Lu C., Li Y., Li M., Xing G., Wei H., Wang Q., Chen J., Wu C. Identification of hepatopoietin dimerization, its interacting regions and alternative splicing of its transcription. Eur. J. Biochem. 2002;269:3888–3893. doi: 10.1046/j.1432-1033.2002.03054.x. [DOI] [PubMed] [Google Scholar]
- 22.Koehler C.M. The small Tim proteins and the twin Cx3C motif. Trends Biochem. Sci. 2004;29:1–4. doi: 10.1016/j.tibs.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 23.Chacinska A., Pfannschmidt S., Wiedemann N., Kozjak V., Sanjuán Szklarz L.K., Schulze-Specking A., Truscott K.N., Guiard B., Meisinger C., Pfanner N. Essential role of Mia40 in import and assembly of mitochondrial intermembrane space proteins. EMBO J. 2004;23:3735–3746. doi: 10.1038/sj.emboj.7600389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tokatlidis K. A disulfide relay system in mitochondria. Cell. 2005;121:965–967. doi: 10.1016/j.cell.2005.06.019. [DOI] [PubMed] [Google Scholar]
- 25.Bihlmaier K., Mesecke N., Terziyska N., Bien M., Hell K., Herrmann J.M. The disulfide relay system of mitochondria is connected to the respiratory chain. J. Cell Biol. 2007;179:389–395. doi: 10.1083/jcb.200707123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Becher D., Kricke J., Stein G., Lisowsky T. A mutant for the yeast scERV1 gene displays a new defect in mitochondrial morphology and distribution. Yeast. 1999;15:1171–1181. doi: 10.1002/(SICI)1097-0061(19990915)15:12<1171::AID-YEA443>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 27.Lisowsky T., Lee J.E., Polimeno L., Francavilla A., Hofhaus G. Mammalian augmenter of liver regeneration protein is a sulfhydryl oxidase. Dig. Liver Dis. 2001;33:173–180. doi: 10.1016/s1590-8658(01)80074-8. [DOI] [PubMed] [Google Scholar]
- 28.Leary S.C., Kaufman B.A., Pellecchia G., Guercin G.H., Mattman A., Jaksch M., Shoubridge E.A. Human SCO1 and SCO2 have independent, cooperative functions in copper delivery to cytochrome c oxidase. Hum Mol Genet. 2004;13:1839–1848. doi: 10.1093/hmg/ddh197. [DOI] [PubMed] [Google Scholar]
- 29.Massa V., Fernandez-Vizarra E., Alshahwan S., Bakhsh E., Goffrini P., Ferrero I., Mereghetti P., D'Adamo P., Gasparini P., Zeviani M. Severe infantile encephalomyopathy caused by a mutation in COX6B1, a nucleus-encoded subunit of cytochrome c oxidase. Am. J. Hum. Genet. 2008;82:1281–1289. doi: 10.1016/j.ajhg.2008.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Francavilla A., Hagiya M., Porter K.A., Polimeno L., Ihara I., Starzl T.E. Augmenter of liver regeneration: its place in the universe of hepatic growth factors. Hepatology. 1994;20:747–757. [PubMed] [Google Scholar]
- 31.van Ekeren G.J., Stadhouders A.M., Smeitink J.A., Sengers R.C. A retrospective study of patients with the hereditary syndrome of congenital cataract, mitochondrial myopathy of heart and skeletal muscle and lactic acidosis. Eur J Pediatr. 1993;152:255–259. doi: 10.1007/BF01956157. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.