

Abstract
Glial cells play an important role in sequestering neuronally released glutamate via Na+-dependent transporters. Surprisingly, these transporters are not operational in glial-derived tumors (gliomas). Instead, gliomas release glutamate, causing excitotoxic death of neurons in the vicinity of the tumor. We now show that glutamate release from glioma cells is an obligatory by-product of cellular cystine uptake via system 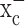 , an electroneutral cystine-glutamate exchanger. Cystine is an essential precursor for the biosynthesis of glutathione, a major redox regulatory molecule that protects cells from endogenously produced reactive oxygen species (ROS). Glioma cells, but not neurons or astrocytes, rely primarily on cystine uptake via system
, an electroneutral cystine-glutamate exchanger. Cystine is an essential precursor for the biosynthesis of glutathione, a major redox regulatory molecule that protects cells from endogenously produced reactive oxygen species (ROS). Glioma cells, but not neurons or astrocytes, rely primarily on cystine uptake via system 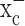 for their glutathione synthesis. Inhibition of system
for their glutathione synthesis. Inhibition of system 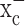 causes a rapid depletion of glutathione, and the resulting loss of ROS defense causes caspase-mediated apoptosis. Glioma cells can be rescued if glutathione status is experimentally restored or if glutathione is substituted by alternate cellular antioxidants, confirming that ROS are indeed mediators of cell death. We describe two potent drugs that permit pharmacological inhibition of system
causes a rapid depletion of glutathione, and the resulting loss of ROS defense causes caspase-mediated apoptosis. Glioma cells can be rescued if glutathione status is experimentally restored or if glutathione is substituted by alternate cellular antioxidants, confirming that ROS are indeed mediators of cell death. We describe two potent drugs that permit pharmacological inhibition of system 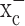 . One of these drugs, sulfasalazine, is clinically used to treat inflammatory bowel disease and rheumatoid arthritis. Sulfasalazine was able to reduce glutathione levels in tumor tissue and slow tumor growth in vivo in a commonly used intracranial xenograft animal model for human gliomas when administered by intraperitoneal injection. These data suggest that inhibition of cystine uptake into glioma cells through the pharmacological inhibition of system
. One of these drugs, sulfasalazine, is clinically used to treat inflammatory bowel disease and rheumatoid arthritis. Sulfasalazine was able to reduce glutathione levels in tumor tissue and slow tumor growth in vivo in a commonly used intracranial xenograft animal model for human gliomas when administered by intraperitoneal injection. These data suggest that inhibition of cystine uptake into glioma cells through the pharmacological inhibition of system 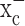 may be a viable therapeutic strategy with a Food and Drug Administration-approved drug already in hand.
may be a viable therapeutic strategy with a Food and Drug Administration-approved drug already in hand.
Keywords: excitotoxicity, glutathione, glutamate transport, glial progenitor cells, sulfasalazine, (S)-4-carboxyphenylglycine
Introduction
Glial-derived tumors (gliomas) are among the most aggressive cancers for which no effective treatment exists. Unlike other cancers, tumor expansion is physically limited by the cavity of the skull. Tumors apparently overcome this problem by vacating space through the active killing of neurons in the vicinity of the tumor. This occurs, at least in part, by the release of excessive amounts of glutamate (Ye and Sontheimer, 1999; Takano et al., 2001; Sontheimer, 2003). Significant increases in extracellular glutamate have been demonstrated in the peritumoral space surrounding experimental tumors in Fisher rats (Behrens et al., 2000; Takano et al., 2001). Importantly, in fully mobilized glioma patients, significantly elevated peritumoral glutamate has been measured by stereotactic microdialysis (Roslin et al., 2003) and by magnetic resonance spectroscopy in patients with oligodendrogliomas (Rijpkema et al., 2003). Moreover, peritumoral seizures present an important sequela associated with malignant glioma (Oberndorfer et al., 2002), and glutamate release from tumor cells may play a major role in seizure initiation (Rothstein, 2002). Glioma cells deficient in glutamate release fail to grow when implanted into host animals, whereas wild-type glioma cells expand rapidly with clear evidence of excitotoxic necrosis surrounding the tumor (Takano et al., 2001). Hence, the evidence that glutamate plays an important role in the growth of these tumors is overwhelming.
The glutamate release from glioma cells has been traced to system 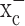 (Ye and Sontheimer, 1999), an abundant but relatively poorly investigated amino acid transporter that transports cystine into the cell in exchange for glutamate being released. System
(Ye and Sontheimer, 1999), an abundant but relatively poorly investigated amino acid transporter that transports cystine into the cell in exchange for glutamate being released. System 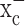 is a heterodimeric protein complex consisting of a catalytic light chain (xCT) and a regulatory heavy chain (4F2hc) (Sato et al., 1999), which is essential for membrane localization of the transporter (Bassi et al., 2001). The light chain, which transports cystine, has recently been cloned (Sato et al., 1999) and shown to belong to the 12-transmembrane domain amino acid transporter proteins. It is highly expressed in glioma cells in which it exists in two splice variants, hxCTa and hxCTb, both of which are upregulated after oxidative stress (Kim et al., 2001).
is a heterodimeric protein complex consisting of a catalytic light chain (xCT) and a regulatory heavy chain (4F2hc) (Sato et al., 1999), which is essential for membrane localization of the transporter (Bassi et al., 2001). The light chain, which transports cystine, has recently been cloned (Sato et al., 1999) and shown to belong to the 12-transmembrane domain amino acid transporter proteins. It is highly expressed in glioma cells in which it exists in two splice variants, hxCTa and hxCTb, both of which are upregulated after oxidative stress (Kim et al., 2001).
System 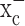 -mediated glutamate release has been implicated in a number of conditions in which excitotoxic cell injury occurs. These include inflammation (Barger and Basile, 2001), virally induced encephalopathy (Espey et al., 1998), and periventricular leukomalacia (Oka et al., 1993). Although it is attractive to speculate that glioma cells may have developed a rather unusual way to co-opt this transporter to inflict excitotoxic injury on the brain (Sontheimer, 2003), we sought here to examine an alternative hypothesis. Specifically, cystine is an essential precursor for the biosynthesis of cellular glutathione (GSH), which is a key regulator of the redox status of the cells (Jefferies et al., 2003). We therefore hypothesize that glutamate release from glioma cells is an obligatory by-product of cystine uptake by glioma cells, which is necessary to maintain the high synthetic rates of glutathione biosynthesis that is typical of glioma cells. Indeed, our data support such an alternative explanation. Importantly, we rather unexpectedly found that system
-mediated glutamate release has been implicated in a number of conditions in which excitotoxic cell injury occurs. These include inflammation (Barger and Basile, 2001), virally induced encephalopathy (Espey et al., 1998), and periventricular leukomalacia (Oka et al., 1993). Although it is attractive to speculate that glioma cells may have developed a rather unusual way to co-opt this transporter to inflict excitotoxic injury on the brain (Sontheimer, 2003), we sought here to examine an alternative hypothesis. Specifically, cystine is an essential precursor for the biosynthesis of cellular glutathione (GSH), which is a key regulator of the redox status of the cells (Jefferies et al., 2003). We therefore hypothesize that glutamate release from glioma cells is an obligatory by-product of cystine uptake by glioma cells, which is necessary to maintain the high synthetic rates of glutathione biosynthesis that is typical of glioma cells. Indeed, our data support such an alternative explanation. Importantly, we rather unexpectedly found that system 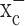 represents the only viable pathway for cystine uptake in glioma cells (but not other brain cells), and hence its pharmacological inhibition leads to selective, apoptotic, caspase-mediated cell death of glioma cells. System
represents the only viable pathway for cystine uptake in glioma cells (but not other brain cells), and hence its pharmacological inhibition leads to selective, apoptotic, caspase-mediated cell death of glioma cells. System 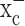 inhibitors hence have tremendous potential to contain glioma growth pharmacologically, and we illustrate this by imaging the in vivo growth of human gliomas xenografted into the brains of severe combined immunodeficient (scid) mice.
inhibitors hence have tremendous potential to contain glioma growth pharmacologically, and we illustrate this by imaging the in vivo growth of human gliomas xenografted into the brains of severe combined immunodeficient (scid) mice.
Materials and Methods
Glioma cells. Glioma cells examined included STTG-1 (CCF-STTG1; American Type Culture Collection, Manassas, VA), D54-MG, U87-MG, U-251MG, and D-65MG (from Dr. D. D. Bigner, Duke University, Durham, NC). We also used primary cultures of glioma cells (GBM62) established from biopsy material from a patient with glioblastoma multiforme, provided by the brain tumor tissue core at the University of Alabama at Birmingham (validated by a pathology report). Glioma cells were cultured in DMEM/F-12 media supplemented with 2 mm glutamine and 7% heat-inactivated fetal bovine serum (FBS). Cortical astrocytes and cortical neurons were prepared from Sprague Dawley rats as described previously (Ye and Sontheimer, 1998).
l-Cystine uptake. l-Cystine uptake was performed using l-[35S]cystine as described previously (Ye et al., 1999).
Cell growth assay. Glioma cells were plated at 10,000 cells per well in 24-well tissue culture plates in DMEM/F-12 supplemented with 2 mm glutamine and 7% FBS. Drug treatments were done in fresh media containing the drug of interest and were initiated 24 h after cell plating. Cells were trypsinized and counted using a hemocytometer. The relative growth rate was calculated as follows: growth (%) = (final test cell number - initial test cell number)/(final control cell number - initial control cell number) × 100.
Cell proliferation. Cell proliferation was measured based on the incorporation of 5-bromo-2′-deoxyuridine (BrdU) into chromosomal DNA as determined using the Cell Proliferation ELISA kit (Roche Applied Science, Indianapolis, IN). Cells were pulsed with BrdU for 2 h. BrdU incorporation was also examined immunohistochemically using anti-BrdU antibodies (10 μg/ml; Roche Applied Science) conjugated with fluorescein and visualized using a Axiovert 200M fluorescence microscope (Zeiss, Oberkochen, Germany).
Cell-cycle flow cytometry. Cell-cycle distribution after drug treatment was determined based on the amount of cellular DNA content using the DNA dye propidium iodide. Glioma cells were incubated for 48 h with DMEM/F-12 supplemented with 2 mm glutamine and 7% FBS containing test reagents. The cells were then harvested and fixed with cold ethanol. Cells were then washed with PBS, treated with 100 μg/ml RNase for 15 min, and incubated with 50 μg/ml propidium iodide for 30 min. The cells were analyzed by exciting at 488 nm and measuring propidium iodide emission at 580 nm using a FACScan flow cytometer (BD Biosciences, San Jose, CA). Cell-cycle distribution was analyzed using the ModFit program (Verity Software, Topsham, ME).
Live/dead assay. Cell death was examined using the Live/Dead Viability/Cytotoxicity kit (Molecular Probes, Eugene, OR) per instructions of the manufacturer. The percentages of live and dead cells were determined using a FACScan flow cytometer (BD Biosciences).
Glutathione assay. Intracellular levels of total glutathione were determined using a glutathione assay kit (Cayman Chemical, Ann Arbor, MI) according to the manufacturer's instructions. Monochlorobimane (ApoAlert glutathione detection kit; BD Biosciences) staining was performed by incubating 20 μm tissue sections from tumor-implanted mice brains with 100 μm monochlorobimane for 10 min on ice (Shih et al., 2003). Slices were washed twice with cold PBS to remove excess monochlorobimane, and fluorescence images were collected immediately at 10× magnification on a Nikon (Melville, NY) Diaphot 300 inverted microscope using OpenLab software. Stained cultures were examined by the same procedure. Cells were viewed directly for fluorescence (Aqua blue; excitation, 436 nm; emission, 480 nm). Quantitative measurements of monochlorobimane/GSH adduct fluorescence were made with NIH ImageJ by taking mean intensities from tumor regions. All values were background subtracted.
Reverse transcription-PCR. Total RNAs were isolated from cell lines and brain biopsies using RNAqueous (Ambion, Austin, TX) according to the instructions of the manufacturer. For the detection of xCT, CD98 (4F2hc), and β-actin RNA transcripts, the OneStep reverse transcription (RT)-PCR kit was used according to the manufacturer's instructions (Qiagen, Valencia, CA). The following pairs of primers were used: xCT, GCTGGCTGGTTTTACCTC (5′ primer) and TGAAAGGACGATGCATATC (3′ primer); CD98, GCTGCTGCTCTTCTGGCTC (5′ primer) and GCCAGTGGCATTCAAATAC (3′ primer); β-actin, CATGCCATCCTGCGTCTG (5′ primer) and CTCCTTCTGCATCCTGTC (3′ primer). Expected amplification product sizes were as follows: 525 bp for xCT, 726 bp for CD98, and 431 bp for β-actin. The reverse transcription reactions were performed at 50°C for 30 min with 250 ng of RNA, followed by 30 cycles of PCR amplification (20 s at 94°C, 20 s at 50°C, and 1 min at 72°C).
Western blot analysis. The expression of CD98 and glutamate transporter 1 (GLT-1) of glioma cell lines and tumor biopsies were examined by Western blot as described previously (Ye et al., 1999). Blots were probed with antibodies to CD98 (0.2 μg/ml; Santa Cruz Biotechnology, Santa Cruz, CA) and to GLT-1 (20 ng/ml; Santa Cruz Biotechnology).
Animal studies. D54-MG and U87-MG glioma cells were established that stably expressed the firefly luciferase gene under the cytomegalovirus (CMV) promoter using a pcDNA3 transfection vector. A total of 2.5 × 105 of these D54-ffLuc human glioma cells was stereotactically implanted through a small burr hole, using a 30 gauge Hamilton syringe, into the intracranium of female nude mice as previously described (Soroceanu et al., 1998). Each animal was also implanted with an electronic biotag (Alltech, Nicholasville, KY) to permit unequivocal identification with a small hand-held electronic reader. After 7 d, animals were randomized into three groups of 12 animals each. One group received 1 ml intraperitoneal saline injections twice daily for 3 weeks. The two test groups received an 8 mg dose of sulfasalazine twice daily for either 1 or 3 weeks in 1 ml of saline. Tumor growth was assessed once weekly by collecting light emission from the tumor regions (in relative photons/second) using a Xenogen (Alameda, CA) IVIS imaging system. Data were captured and quantified using Living Image, version 2.20.1, provided by Xenogen. The data acquisition software ensured that pixels did not become saturated during image collection. Images were collected on mice oriented in the same position (prone) and always at 10 min after intraperitoneal injection of 2.5 mg of endotoxin-free beetle d-luciferin (Promega, Madison, WI). During imaging, mice were maintained under 1.5% enflurane/oxygen gas anesthesia at 37°C. The bioluminescence of each tumor, measured in photons/second, was normalized for each mouse to its level at day 7, and the mean values for all mice in each group were plotted as a function of time. Identical procedures were followed for implanted U87-MG and D54-MG cells.
Results
System 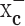 is highly expressed in gliomas
is highly expressed in gliomas
Existing evidence for system 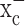 expression and activity in gliomas stems predominantly from tracer studies (Ye and Sontheimer, 1999; Ye et al., 1999). The recent cloning of xCT, the catalytic subunit of
expression and activity in gliomas stems predominantly from tracer studies (Ye and Sontheimer, 1999; Ye et al., 1999). The recent cloning of xCT, the catalytic subunit of 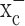 (Sato et al., 1999; Bassi et al., 2001; Kim et al., 2001), made a closer assessment of expression and function feasible. Its distribution in normal brain has been documented by in situ hybridization (Sato et al., 2002), and the obligatory association of xCT with a regulatory subunit 4F2hc has been demonstrated (Bassi et al., 2001). We used a combination of RT-PCR and Western blot analysis to examine expression of RNA and protein in established and frequently used glioma cell lines and compared these data to acute patient-derived tumor biopsies. Representative data from such experiments are summarized in Figure 1. RNA transcripts of both the catalytic (xCT) and the regulatory subunits (4F2hc) of system
(Sato et al., 1999; Bassi et al., 2001; Kim et al., 2001), made a closer assessment of expression and function feasible. Its distribution in normal brain has been documented by in situ hybridization (Sato et al., 2002), and the obligatory association of xCT with a regulatory subunit 4F2hc has been demonstrated (Bassi et al., 2001). We used a combination of RT-PCR and Western blot analysis to examine expression of RNA and protein in established and frequently used glioma cell lines and compared these data to acute patient-derived tumor biopsies. Representative data from such experiments are summarized in Figure 1. RNA transcripts of both the catalytic (xCT) and the regulatory subunits (4F2hc) of system 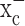 were present in all glioma cell lines examined (D54-MG, STTG1, U-251MG, and U87-MG) and in a primary culture from a human glioma biopsy (GBM62) (Fig. 1a). Importantly, prominent expression of xCT was also found in biopsies from glioma patients and in biopsies obtained from nonmalignant brain tissue (Fig. 1b). These studies used primers that recognize both splice variants of the xCT gene. We also demonstrate the presence of the regulatory subunit 4F2hc at the protein level by Western blot analysis, with prominent expression in all examined glioma lines and in five glioma biopsies (Fig. 1a,c). Antibodies for xCT are not currently available. However, we were able to confirm the previously documented (Ye et al., 1999) absence of the Na+-dependent glutamate transporter GLT-1 from all examined brain tumor biopsies (Fig. 1c) with prominent expression in nonmalignant human brain samples used for comparison. Thus, the proteins composing the system
were present in all glioma cell lines examined (D54-MG, STTG1, U-251MG, and U87-MG) and in a primary culture from a human glioma biopsy (GBM62) (Fig. 1a). Importantly, prominent expression of xCT was also found in biopsies from glioma patients and in biopsies obtained from nonmalignant brain tissue (Fig. 1b). These studies used primers that recognize both splice variants of the xCT gene. We also demonstrate the presence of the regulatory subunit 4F2hc at the protein level by Western blot analysis, with prominent expression in all examined glioma lines and in five glioma biopsies (Fig. 1a,c). Antibodies for xCT are not currently available. However, we were able to confirm the previously documented (Ye et al., 1999) absence of the Na+-dependent glutamate transporter GLT-1 from all examined brain tumor biopsies (Fig. 1c) with prominent expression in nonmalignant human brain samples used for comparison. Thus, the proteins composing the system 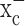 cystine-glutamate exchanger are highly expressed in glioma cell lines and in acute patient-derived glioma tissues.
cystine-glutamate exchanger are highly expressed in glioma cell lines and in acute patient-derived glioma tissues.
Figure 1.
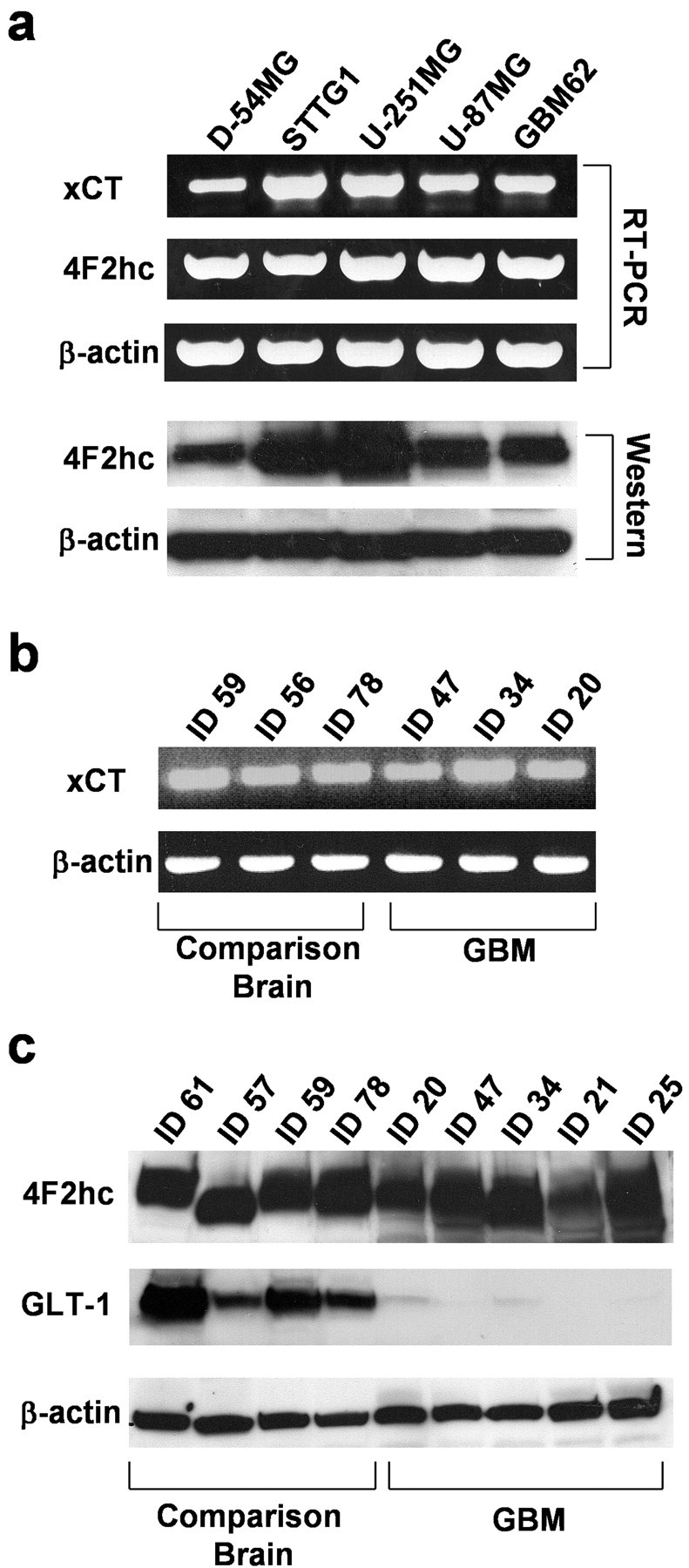
System 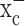 is highly expressed in gliomas. a, Expression of mRNA transcripts of xCT, 4F2hc, and β-actin in human glioma cell lines (D54-MG, STTG-1, U251-MG, and U87-MG) and a glioma primary culture (GBM62) is demonstrated by RT-PCR. The protein expression of 4F2hc determined by Western blot with anti-4F2hc antibodies is illustrated below. b, RT-PCR was performed with RNA extracts from patient biopsies and illustrates expression of mRNA transcripts of xCT in tumors (ID47, ID34, ID20) as well as in comparison brain tissues, which were operated on for other reasons and did not show any evidence of malignancy (ID59, ID56, ID78). c, The expression of 4F2hc, the regulatory subunit of system
is highly expressed in gliomas. a, Expression of mRNA transcripts of xCT, 4F2hc, and β-actin in human glioma cell lines (D54-MG, STTG-1, U251-MG, and U87-MG) and a glioma primary culture (GBM62) is demonstrated by RT-PCR. The protein expression of 4F2hc determined by Western blot with anti-4F2hc antibodies is illustrated below. b, RT-PCR was performed with RNA extracts from patient biopsies and illustrates expression of mRNA transcripts of xCT in tumors (ID47, ID34, ID20) as well as in comparison brain tissues, which were operated on for other reasons and did not show any evidence of malignancy (ID59, ID56, ID78). c, The expression of 4F2hc, the regulatory subunit of system 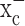 and of the Na+-dependent glutamate transporter GLT-1, was compared between glioma biopsies and comparison brain tissues by Western blot analysis. The expression of GLT-1 was almost completely missing in tumor biopsies, all of which prominently expressed 4F2hc. The origin of human biopsies was as follows: ID61, 22-year-old white male; ID56, unknown; ID57, 1.5-year-old Hispanic male; ID59, 26-year-old white female; ID78, 48-year-old white female; ID20, 45-year-old black female; ID21, 55-year-old white female; ID25, 58-year-old unknown male; ID34, 47-year-old unknown male; ID47, 38-year-old white female. Tissues were obtained in compliance with the Institutional Review Board of the University of Alabama at Birmingham. GBM, Glioblastoma multiforme.
and of the Na+-dependent glutamate transporter GLT-1, was compared between glioma biopsies and comparison brain tissues by Western blot analysis. The expression of GLT-1 was almost completely missing in tumor biopsies, all of which prominently expressed 4F2hc. The origin of human biopsies was as follows: ID61, 22-year-old white male; ID56, unknown; ID57, 1.5-year-old Hispanic male; ID59, 26-year-old white female; ID78, 48-year-old white female; ID20, 45-year-old black female; ID21, 55-year-old white female; ID25, 58-year-old unknown male; ID34, 47-year-old unknown male; ID47, 38-year-old white female. Tissues were obtained in compliance with the Institutional Review Board of the University of Alabama at Birmingham. GBM, Glioblastoma multiforme.
Inhibition of system 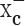 reduces cystine uptake, depleting cells of glutathione
reduces cystine uptake, depleting cells of glutathione
The principal hypothesis in this study is that system 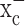 serves as a pipeline for cellular cystine uptake, with glutamate release being an obligatory by-product. Cystine uptake is the rate-limiting step for the synthesis of GSH, which is the main intracellular antioxidant that protects cells from oxidative stress and resulting reactive oxygen species (ROS) (Jefferies et al., 2003). To examine this hypothesis, we first used 35S-labeled cystine and measured intracellular cystine concentrations in the presence and absence of (S)-4-carboxyphenylglycine [(S)-4-CPG] and sulfasalazine, two potent and relatively selective competitive inhibitors of system
serves as a pipeline for cellular cystine uptake, with glutamate release being an obligatory by-product. Cystine uptake is the rate-limiting step for the synthesis of GSH, which is the main intracellular antioxidant that protects cells from oxidative stress and resulting reactive oxygen species (ROS) (Jefferies et al., 2003). To examine this hypothesis, we first used 35S-labeled cystine and measured intracellular cystine concentrations in the presence and absence of (S)-4-carboxyphenylglycine [(S)-4-CPG] and sulfasalazine, two potent and relatively selective competitive inhibitors of system 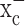 (Ye and Sontheimer, 1999; Patel et al., 2004). Both drugs reduced 35S-labeled cystine uptake by 60-80% in glioma lines and a primary glioma culture (Fig. 2a). This level of inhibition was comparable with the competitive inhibition of this antiporter through impairment of the glutamate gradient by 5 mm
l-glutamate (data not shown). Pharmacological inhibition of cystine uptake was specific for glioma cells, because sulfasalazine exerted only a limited inhibition of cystine uptake in cortical astrocytes (Fig. 2b). Moreover, cystine uptake into cortical neurons (Fig. 2c) was negligible, a finding in agreement with a report showing that neurons preferentially take up cysteine rather than cystine for glutathione biosynthesis (Shanker and Aschner, 2001).
(Ye and Sontheimer, 1999; Patel et al., 2004). Both drugs reduced 35S-labeled cystine uptake by 60-80% in glioma lines and a primary glioma culture (Fig. 2a). This level of inhibition was comparable with the competitive inhibition of this antiporter through impairment of the glutamate gradient by 5 mm
l-glutamate (data not shown). Pharmacological inhibition of cystine uptake was specific for glioma cells, because sulfasalazine exerted only a limited inhibition of cystine uptake in cortical astrocytes (Fig. 2b). Moreover, cystine uptake into cortical neurons (Fig. 2c) was negligible, a finding in agreement with a report showing that neurons preferentially take up cysteine rather than cystine for glutathione biosynthesis (Shanker and Aschner, 2001).
Figure 2.
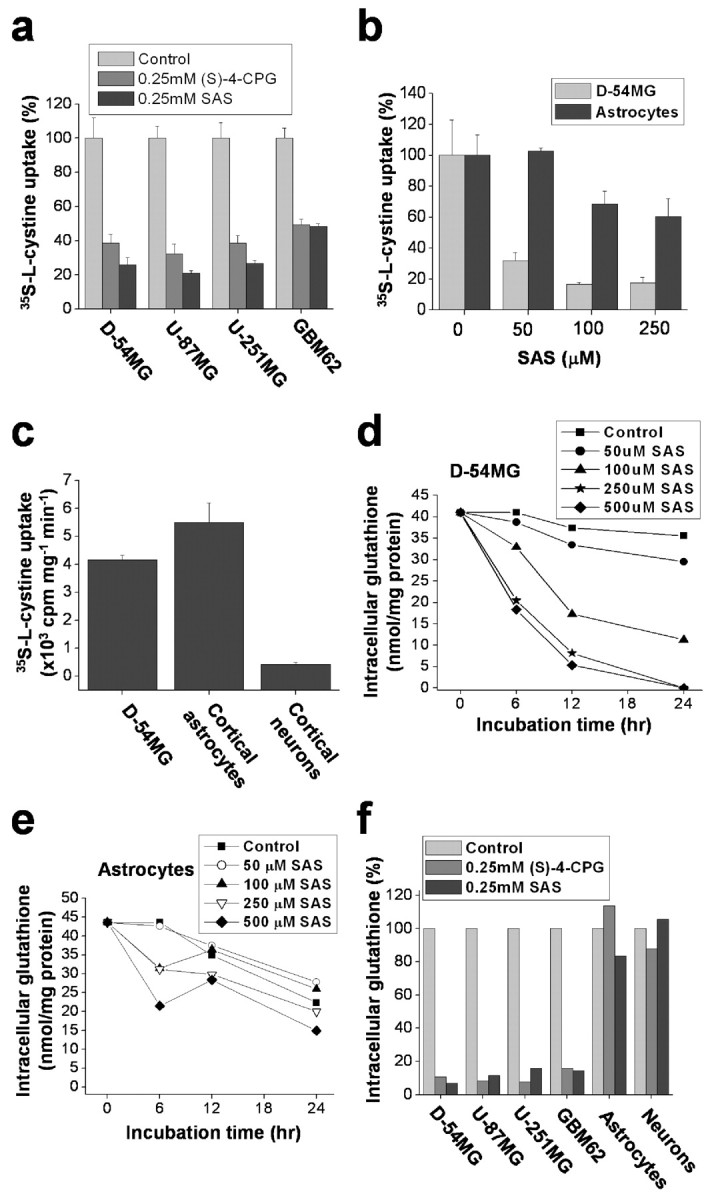
Inhibition of system 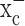 selectively reduces cystine uptake and depletes intracellular glutathione in glioma cells. a, Coincubation with (S)-4-CPG and sulfasalazine (SAS) reduced cystine uptake in glioma cell lines and in a primary glioma cell culture (GBM62). b, Sulfasalazine affects cystine uptake into cortical astrocytes to a much lesser extent than up take into D54-MG glioma cells. c, Cystine uptake in cortical neurons was negligible compared with astrocytes orglioma cells. d, Sulfasalazine caused a time- and dose-dependent decrease in intracellular glutathione in D54-MG cells. e, Intracellular level of glutathione in astrocytes is significantly less affected by sulfasalazine. f, Treatment with (S)-4-CPG sulfasalazine depleted intracellular glutathione in glioma cells but not cortical astrocytes or neurons. Error bars indicate SE.
selectively reduces cystine uptake and depletes intracellular glutathione in glioma cells. a, Coincubation with (S)-4-CPG and sulfasalazine (SAS) reduced cystine uptake in glioma cell lines and in a primary glioma cell culture (GBM62). b, Sulfasalazine affects cystine uptake into cortical astrocytes to a much lesser extent than up take into D54-MG glioma cells. c, Cystine uptake in cortical neurons was negligible compared with astrocytes orglioma cells. d, Sulfasalazine caused a time- and dose-dependent decrease in intracellular glutathione in D54-MG cells. e, Intracellular level of glutathione in astrocytes is significantly less affected by sulfasalazine. f, Treatment with (S)-4-CPG sulfasalazine depleted intracellular glutathione in glioma cells but not cortical astrocytes or neurons. Error bars indicate SE.
We next asked whether limiting cystine uptake via system 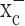 reduces the total intracellular glutathione content (GSH and glutathione disulfide). Sulfasalazine reduced glutathione in a time- and dose-dependent manner with almost complete glutathione depletion after 24 h (Fig. 2d). We obtained similar results with 0.5 mm (S)-4-CPG (data not shown). This depletion of intracellular glutathione by system
reduces the total intracellular glutathione content (GSH and glutathione disulfide). Sulfasalazine reduced glutathione in a time- and dose-dependent manner with almost complete glutathione depletion after 24 h (Fig. 2d). We obtained similar results with 0.5 mm (S)-4-CPG (data not shown). This depletion of intracellular glutathione by system 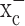 inhibitors was observed in all glioma cells examined; however, system
inhibitors was observed in all glioma cells examined; however, system 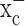 inhibitors had only a small effect on astrocytes (Fig. 2e) and did not significantly reduce intracellular glutathione in neurons (Fig. 2f). These results strongly suggest that system
inhibitors had only a small effect on astrocytes (Fig. 2e) and did not significantly reduce intracellular glutathione in neurons (Fig. 2f). These results strongly suggest that system 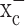 plays an important and essential role in cellular cystine uptake for the synthesis of intracellular glutathione in glioma cells but not astrocytes or neurons.
plays an important and essential role in cellular cystine uptake for the synthesis of intracellular glutathione in glioma cells but not astrocytes or neurons.
Inhibition of cystine uptake via system 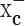 leads to growth inhibition of glioma cells
leads to growth inhibition of glioma cells
A significant body of literature (for review, see Jefferies et al., 2003) suggests a correlation between glutathione redox status and cell growth whereby reduced glutathione levels slow cell growth. To examine whether the depletion of glutathione after system 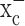 inhibitors affects the growth of glioma cells, we established growth curves for cell lines and primary glioma cultures grown for 5 d in cell culture media in the presence of (S)-4-CPG or sulfasalazine each at their most effective glutathione-depleting concentrations. Both drugs caused dose-dependent growth inhibitions in all glioma cells examined (Fig. 3a,b). Relative growth stagnated or even declined over a 5 d period, indicating that some cell death must have occurred (negative values in Fig. 3a,b). Neither (S)-4-CPG (Fig. 3c) nor sulfasalazine (data not shown) affected the growth of primary astrocyte cultures or the survival of cortical neuronal cultures, consistent with the above finding that neither drug compromised glutathione status in these cells (Fig. 2e,f).
inhibitors affects the growth of glioma cells, we established growth curves for cell lines and primary glioma cultures grown for 5 d in cell culture media in the presence of (S)-4-CPG or sulfasalazine each at their most effective glutathione-depleting concentrations. Both drugs caused dose-dependent growth inhibitions in all glioma cells examined (Fig. 3a,b). Relative growth stagnated or even declined over a 5 d period, indicating that some cell death must have occurred (negative values in Fig. 3a,b). Neither (S)-4-CPG (Fig. 3c) nor sulfasalazine (data not shown) affected the growth of primary astrocyte cultures or the survival of cortical neuronal cultures, consistent with the above finding that neither drug compromised glutathione status in these cells (Fig. 2e,f).
Figure 3.
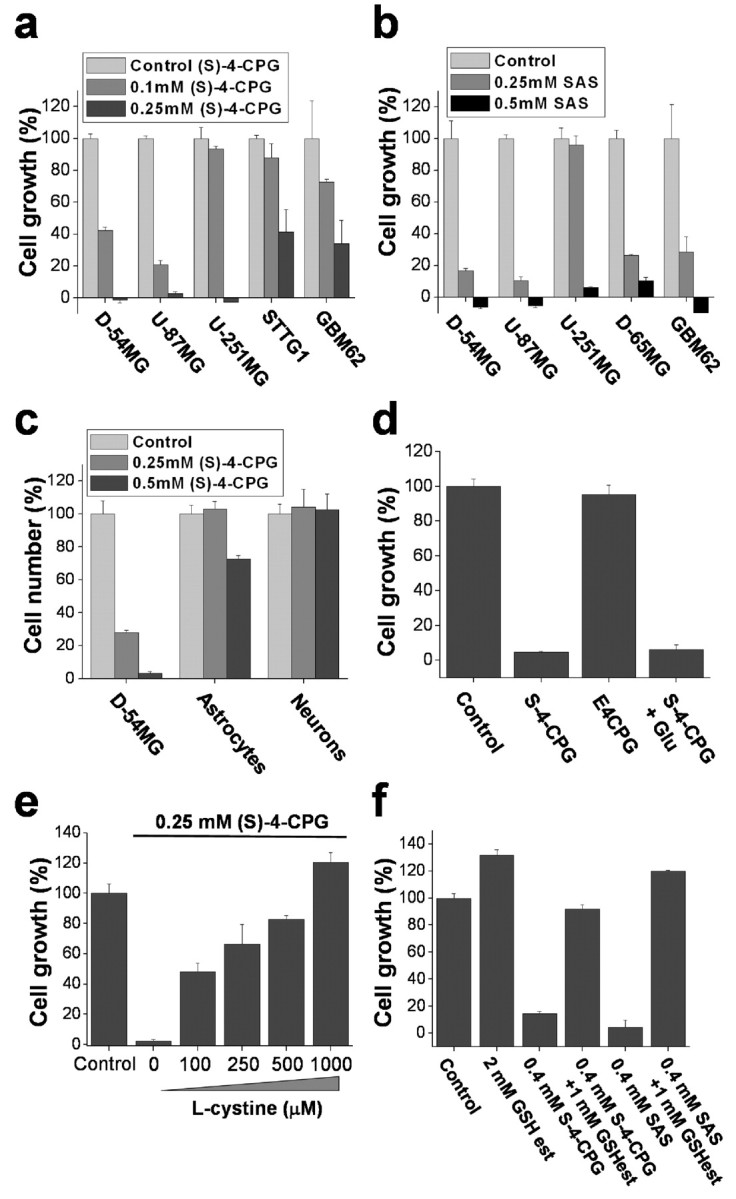
Inhibition of cystine uptake via system 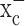 compromises glioma cell growth. a, b, Treatment of glioma cells with (S)-4-CPG (a) or sulfasalazine (SAS) (b) shows dose-dependent growth inhibition in glioma cell lines and primary cultured glioma cells. c, However, (S)-4-CPG does not inhibit the growth of cultured cortical astrocytes or cortical neurons. d, The broad-spectrum mGluR antagonist E4CPG (0.25 mm) does not inhibit cell growth, nor can 1 mm glutamate reverse (S)-4-CPG (0.25 mm)-induced growth inhibition, suggesting that these effects were not mediated via mGluRs. e, Exogenous l-cystine in a dose-dependent manner overcomes the growth inhibition exerted by (S)-4-CPG. f, Similarly, incubation with glutathione ethyl ester, a membrane-permeable form of glutathione, overcomes the growth inhibition by (S)-4-CPG or sulfasalazine in D54-MG cells. Error bars indicate SE.
compromises glioma cell growth. a, b, Treatment of glioma cells with (S)-4-CPG (a) or sulfasalazine (SAS) (b) shows dose-dependent growth inhibition in glioma cell lines and primary cultured glioma cells. c, However, (S)-4-CPG does not inhibit the growth of cultured cortical astrocytes or cortical neurons. d, The broad-spectrum mGluR antagonist E4CPG (0.25 mm) does not inhibit cell growth, nor can 1 mm glutamate reverse (S)-4-CPG (0.25 mm)-induced growth inhibition, suggesting that these effects were not mediated via mGluRs. e, Exogenous l-cystine in a dose-dependent manner overcomes the growth inhibition exerted by (S)-4-CPG. f, Similarly, incubation with glutathione ethyl ester, a membrane-permeable form of glutathione, overcomes the growth inhibition by (S)-4-CPG or sulfasalazine in D54-MG cells. Error bars indicate SE.
(S)-4-CPG is frequently used as antagonist for type I metabotropic glutamate receptors (mGluRs), and hence the growth inhibitory effects of (S)-4-CPG could have been mediated by mGluRs. However, this was ruled out as the broad-spectrum mGluR antagonist ethyl-4-carboxyphenylglycine (E4CPG) (250 μm) did not inhibit glioma cell growth (Fig. 3d), and 1 mm glutamate failed to overcome the inhibitory effect of 0.25 mm (S)-4-CPG (Fig. 3d).
If the inhibitory effects of (S)-4-CPG on cell growth were solely attributable to inhibition of cystine import via system 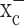 , cell growth should be rescued if cystine levels are experimentally restored. In the presence of 0.25 mm (S)-4-CPG, growth of D54-MG cells indeed recovered by the addition of fourfold excess of cystine (Fig. 3e), but not with glutamate (Fig. 3d), the second substrate for system
, cell growth should be rescued if cystine levels are experimentally restored. In the presence of 0.25 mm (S)-4-CPG, growth of D54-MG cells indeed recovered by the addition of fourfold excess of cystine (Fig. 3e), but not with glutamate (Fig. 3d), the second substrate for system 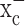 . As would be expected from a competitive inhibitor, cystine addition exhibited a dose-dependent recovery of cell growth in the presence of a fixed (S)-4-CPG concentration (Fig. 3e). Hence, inhibition of cystine uptake via
. As would be expected from a competitive inhibitor, cystine addition exhibited a dose-dependent recovery of cell growth in the presence of a fixed (S)-4-CPG concentration (Fig. 3e). Hence, inhibition of cystine uptake via 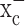 appears to be the primary effect for (S)-4-CPG.
appears to be the primary effect for (S)-4-CPG.
To further delineate the downstream effects of cystine depletion, we examined whether glutathione ethyl ester, a membrane-permeable glutathione analog, could rescue cells from the pharmacological inhibition of cystine uptake. Coincubation with glutathione ethyl ester replenished intracellular glutathione levels in sulfasalazine- and (S)-4-CPG-treated glioma cells (Fig. 3f) and completely restored glioma cell growth. These effects were identical to the rescue afforded by exogenous cystine (Fig. 3e), whereas l-glutamate failed to restore intracellular glutathione levels (Fig. 3d). These data suggest that the glutathione status of the cells limits growth and that both sulfasalazine and (S)-4-CPG affect growth by inhibiting cystine import for glutathione synthesis.
Growth inhibition is attributable to inhibition of DNA synthesis
Growth inhibition may involve reduced cell proliferation, increased cell death, or both. To distinguish between these possibilities, we examined DNA synthesis by measuring incorporation of BrdU into newly synthesized DNA. Treatment of glioma cells for 48 h with 0.25 mm (S)-4-CPG reduced BrdU incorporation into chromosomal DNA by ∼50%, with somewhat larger effects at 0.5 mm (S)-4-CPG (Fig. 4a). Sulfasalazine showed a similar effect on BrdU incorporation (Fig. 4a). The inhibition of DNA synthesis was even more striking when BrdU incorporation was assessed immunocytochemically (Fig. 4b). The presence of BrdU-positive nuclei, indicative of dividing cells, was virtually abolished with treatment of (S)-4-CPG or sulfasalazine (Fig. 4b). These data suggest that cystine uptake inhibition significantly reduces the number of cells in active DNA synthesis.
Figure 4.

System 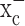 inhibitors block DNA synthesis and increase the percentage of S phase cells. a, b, (S)-4-CPG and sulfasalazine (SAS) inhibit BrdU incorporation into chromosomal DNA in D54-MG cells as assessed by ELISA (a) or immunohistochemistry (b) (representative 20× magnification images). One millimolar cystine (Cys) restored DNA synthesis in the continued presence of either (S)-4-CPG or sulfasalazine. DAPI, 4′,6-Diamidino-2-phenylindole. c, DNA content was determined using a FACS-based assay of propidium iodide. Treatment with either (S)-4-CPG or sulfasalazine significantly increased the cell population in S phase. Error bars indicate SE.
inhibitors block DNA synthesis and increase the percentage of S phase cells. a, b, (S)-4-CPG and sulfasalazine (SAS) inhibit BrdU incorporation into chromosomal DNA in D54-MG cells as assessed by ELISA (a) or immunohistochemistry (b) (representative 20× magnification images). One millimolar cystine (Cys) restored DNA synthesis in the continued presence of either (S)-4-CPG or sulfasalazine. DAPI, 4′,6-Diamidino-2-phenylindole. c, DNA content was determined using a FACS-based assay of propidium iodide. Treatment with either (S)-4-CPG or sulfasalazine significantly increased the cell population in S phase. Error bars indicate SE.
To more closely examine the effect of cystine uptake inhibition on cycling cells, we examined changes in total DNA content after treatment of cells with (S)-4-CPG and sulfasalazine using flow cytometry (Fig. 4c), allowing us to differentiate between the population of cells in S, G1, and G2 phases of the cell cycle. Both drugs yielded a consistent reduction of cells in G2 and a significant increase in S phase cells (Fig. 4c), suggesting S phase arrest, or a slowing down of cell-cycle progression at S phase. Importantly, exogenously added cystine was able to reverse cell-cycle arrest by (S)-4-CPG and sulfasalazine, indicated by normal BrdU incorporation (Fig. 4a), once more demonstrating that these effects were entirely caused by limited cystine availability.
Chronic inhibition of cystine uptake induces caspase-dependent apoptotic cell death
Glutathione is one of several antioxidants that cells use to control reactive oxygen species generated as a by-product of their normal oxidative metabolism. We hypothesize that the above-observed growth arrest was most likely attributable to a lack of antioxidant defense. Consistent with this assumption, we found that glioma cell growth was partially restored by treatment with several well known free radical scavengers including vitamin E, tetramethylpyrroline N-oxide (TMPO) (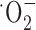 scavenger), and a-phenyl-N-t-butylnitrone (PBN) (
scavenger), and a-phenyl-N-t-butylnitrone (PBN) (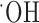 scavenger) (Fig. 5a). Because an accumulation of reactive oxygen species can lead to apoptotic cell death, we further examined whether the system
scavenger) (Fig. 5a). Because an accumulation of reactive oxygen species can lead to apoptotic cell death, we further examined whether the system 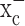 inhibitors likewise induce apoptosis in glioma cells. Exposure of glioma cells to 1 mm (S)-4-CPG for 3 d caused 17% of the cell population to be positive for chromosomal DNA fragmentation, a classic manifestation of apoptosis (Fig. 5b). Apoptosis is often dependent on caspase activities; thus, we examined whether the system
inhibitors likewise induce apoptosis in glioma cells. Exposure of glioma cells to 1 mm (S)-4-CPG for 3 d caused 17% of the cell population to be positive for chromosomal DNA fragmentation, a classic manifestation of apoptosis (Fig. 5b). Apoptosis is often dependent on caspase activities; thus, we examined whether the system 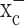 inhibitors induce activated caspase-3, which is the convergent enzyme in the caspase-dependent signaling cascade. Indeed, (S)-4-CPG and sulfasalazine each increased the activated form of caspase-3 (Fig. 5c). As an additional indication that cells underwent the caspase-3-dependent apoptotic cell death, we were able to rescue a significant percentage of cells from death using the panspecific caspase-3 inhibitor Boc-Asp-fluoromethylketone (Boc-D-FMK) (Fig. 5d). These data therefore suggest that cystine depletion via system
inhibitors induce activated caspase-3, which is the convergent enzyme in the caspase-dependent signaling cascade. Indeed, (S)-4-CPG and sulfasalazine each increased the activated form of caspase-3 (Fig. 5c). As an additional indication that cells underwent the caspase-3-dependent apoptotic cell death, we were able to rescue a significant percentage of cells from death using the panspecific caspase-3 inhibitor Boc-Asp-fluoromethylketone (Boc-D-FMK) (Fig. 5d). These data therefore suggest that cystine depletion via system 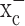 inhibition increases reactive oxygen species in glioma cells, which ultimately leads to caspase-mediated apoptotic cell death. Indeed, prolonged treatment of cells with (S)-4-CPG or sulfasalazine resulted in apoptotic cell death, as examined by flow cytometry (Fig. 5e). In the continued presence of 0.5 mm (S)-4-CPG over a 5 d period, >60% of glioma cells died (Fig. 5e). With 1 mm sulfasalazine, ∼90% of cells died over the 5 d incubation (Fig. 5e). Cell death under all of these conditions could be completely prevented if 1 mm cystine (Fig. 5e) was present, indicating again that limited cystine availability was the key for cell death induced by these system
inhibition increases reactive oxygen species in glioma cells, which ultimately leads to caspase-mediated apoptotic cell death. Indeed, prolonged treatment of cells with (S)-4-CPG or sulfasalazine resulted in apoptotic cell death, as examined by flow cytometry (Fig. 5e). In the continued presence of 0.5 mm (S)-4-CPG over a 5 d period, >60% of glioma cells died (Fig. 5e). With 1 mm sulfasalazine, ∼90% of cells died over the 5 d incubation (Fig. 5e). Cell death under all of these conditions could be completely prevented if 1 mm cystine (Fig. 5e) was present, indicating again that limited cystine availability was the key for cell death induced by these system 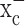 inhibitors.
inhibitors.
Figure 5.
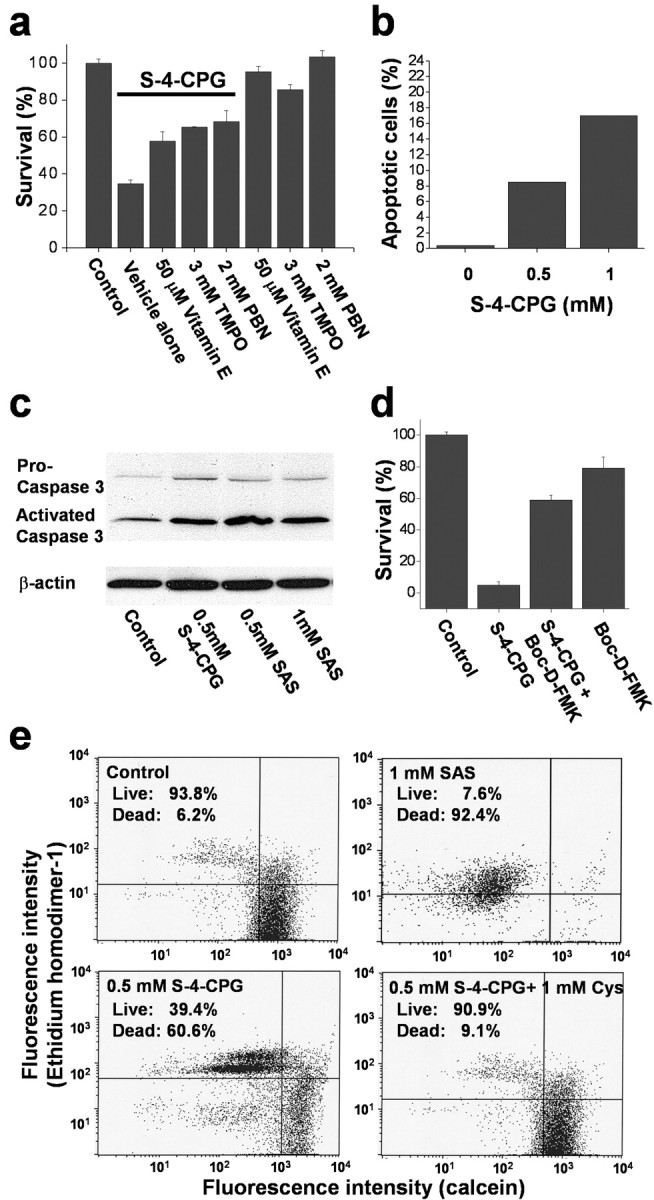
Cystine uptake inhibition induces apoptotic cell death. a, Treatment with free radical scavengers including vitamin E, TMPO (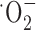 scavenger), and PBN (
scavenger), and PBN (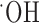 scavenger) partially restores glioma cell growth in the presence of (S)-4-CPG. b, (S)-4-CPG causes DNA fragmentation as indicated by flow cytometric analysis, which is indicative of apoptotic cell death. c, (S)-4-CPG and sulfasalazine (SAS) increase the activated form of caspase-3. Caspase-3 antibodies recognize both the proform (32 kDa) and the activated form (17 kDa). d, The panspecific caspase-3 inhibitor Boc-D-FMK (100 μm) blocked cell death induced by 0.25 mm (S)-4-CPG, further suggesting that cell death invoked caspase-3 activity and hence is presumed to be apoptotic. e, Cell death was also confirmed using the Live/Dead assay kit (Molecular Probes), which, when cells are analyzed by fluorescence-activated cell sorting, show an enhanced number of dead cells that are ethidium homodimer positive, as opposed to live cells that express primarily calcein. Chronic treatment with either (S)-4-CPG or sulfasalazine induced cell death, and exogenously added cystine can overcome cell death in the presence of (S)-4-CPG (c) or sulfasalazine (data not shown). Error bars indicate SE.
scavenger) partially restores glioma cell growth in the presence of (S)-4-CPG. b, (S)-4-CPG causes DNA fragmentation as indicated by flow cytometric analysis, which is indicative of apoptotic cell death. c, (S)-4-CPG and sulfasalazine (SAS) increase the activated form of caspase-3. Caspase-3 antibodies recognize both the proform (32 kDa) and the activated form (17 kDa). d, The panspecific caspase-3 inhibitor Boc-D-FMK (100 μm) blocked cell death induced by 0.25 mm (S)-4-CPG, further suggesting that cell death invoked caspase-3 activity and hence is presumed to be apoptotic. e, Cell death was also confirmed using the Live/Dead assay kit (Molecular Probes), which, when cells are analyzed by fluorescence-activated cell sorting, show an enhanced number of dead cells that are ethidium homodimer positive, as opposed to live cells that express primarily calcein. Chronic treatment with either (S)-4-CPG or sulfasalazine induced cell death, and exogenously added cystine can overcome cell death in the presence of (S)-4-CPG (c) or sulfasalazine (data not shown). Error bars indicate SE.
Sulfasalazine suppresses tumor growth in vivo
The above experiments on isolated cells suggest that growth control of actual tumors in vivo might be achieved using either (S)-4-CPG or sulfasalazine. The latter is a drug that is already clinically used and hence would be a likely candidate for treatment of gliomas in patients. We therefore examined the effect of sulfasalazine on growth control of human gliomas xenografted into the cranium of either female CB-17 scid mice or nude mice, frequently used animal models for malignant gliomas. We assessed the intracranial growth of tumors using the recently developed chemiluminescence methodology for visualizing tumor growth. Specifically, in the first set of data, we stably transfected D54-MG glioma cells with the firefly luciferase gene under the CMV promoter. One-half million cells were stereotactically implanted into the cranium of each mouse. After 7 d, animals were randomized into three groups of 12 animals each. The control group received an intraperitoneal injection of 1 ml/dose saline twice daily. The two test groups received an 8 mg/dose sulfasalazine twice daily for either 1 or 3 weeks. Animals were imaged for tumor size, typically once per week, using a bioluminescence imaging system (IVIS system; Xenogen). Light emission from the tumor regions (in relative photons/second) were quantified using Living Image, version 2.20.1, software provided by Xenogen and plotted as a function of time in Figure 6a. Control animals (squares) showed rapid tumor expansion, as evidenced by an increase in the relative bioluminescence (i.e., normalized for each animal to its bioluminescence value obtained on day 7 when treatment was initiated), which was significantly reduced in both treatment groups. To ensure that these effects were neither cell line specific nor specific to scid mice, we repeated the same experiment after generating a stable luciferase expressing strain of U87-MG, another frequently used glioma cell line for which we established a similar dependence on system 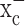 for glutathione synthesis in culture (Figs. 2a,f, 3a,b). These were similarly implanted intracranially but this time into nude mice. Bioluminescence data from one of three representative experiments are displayed in Figure 6b, again comparing 8 mg/ml sulfasalazine treatment to saline injections twice daily. Once again, sulfasalazine was able to reduce tumor growth significantly as judged by bioluminescence imaging, albeit to a somewhat lesser extent. To validate that these images represent, at least qualitatively, changes in tumor size, we killed two animals each from the control and treatment groups at 30 d after tumor induction. Histological sections were obtained and traditional hematoxylin-eosin staining for one example each are shown in Figure 6, c and d. The tumors in both treated animals were significantly smaller than in the two saline control treated animals, which was consistent with imaging data for these four animals. As an additional control experiment, we stained sections obtained from killed animals with antibodies to luciferase to validate the continued presence of strong luciferase expression, in the control and treatment group, and one such example obtained from an animal at 49 d is shown in Figure 6e-g.
for glutathione synthesis in culture (Figs. 2a,f, 3a,b). These were similarly implanted intracranially but this time into nude mice. Bioluminescence data from one of three representative experiments are displayed in Figure 6b, again comparing 8 mg/ml sulfasalazine treatment to saline injections twice daily. Once again, sulfasalazine was able to reduce tumor growth significantly as judged by bioluminescence imaging, albeit to a somewhat lesser extent. To validate that these images represent, at least qualitatively, changes in tumor size, we killed two animals each from the control and treatment groups at 30 d after tumor induction. Histological sections were obtained and traditional hematoxylin-eosin staining for one example each are shown in Figure 6, c and d. The tumors in both treated animals were significantly smaller than in the two saline control treated animals, which was consistent with imaging data for these four animals. As an additional control experiment, we stained sections obtained from killed animals with antibodies to luciferase to validate the continued presence of strong luciferase expression, in the control and treatment group, and one such example obtained from an animal at 49 d is shown in Figure 6e-g.
Figure 6.
Sulfasalazine (SAS) slows tumor growth in vivo. a, Experimental brain tumors were induced in CB-17 scid mice by xenografting D54-ffLuc cells stably expressing the luciferase gene into the brain. Tumor size was determined in vivo every 7 d using a bioluminescence imaging system (IVIS system; Xenogen). Animals were randomized into three groups of 12 animals each. The control group received 1 ml of saline intraperitoneally twice daily, and the two test groups received 8 mg of sulfasalazine (in 1 ml of saline) for either 1 or 3 weeks. Both treatment regimens slowed tumor growth with near-complete inhibition of growth with a 3 week treatment protocol. Animals were retreated for 3 d at 52 d because tumor growth picked up in the animals that received only 1 week of treatment to evaluate continued responsiveness to the drug. b, In a second set of experiments, brain tumors were similarly induced in nude mice by xenografting U87-ffLuc cells that stably expressed the luciferase gene into the brain. The control group received 1 ml of saline intraperitoneally twice daily, the test group received 8 mg of sulfasalazine (in 1 ml of saline) for 3 weeks, followed by one daily dose thereafter. c, d, Representative hematoxylin-eosin staining of mouse brain sections from the same experiment obtained at 30 d. Examples of sections are from saline control (c) and 8 mg/ml SAS treated (d) after 3 weeks of twice-daily injections for both (1.25× magnifications). e-g, Immunocytochemistry using an anti-luciferase conjugated to FITC. Human U87-ffLuc cells transfected with luciferin still showed luciferase expression after 49 d of tumor growth as determined by antibody staining. e is the image of anti-luciferase alone, f is the image of the 4′,6-diamidino-2-phenylindole (DAPI) alone, and g is a merged image of anti-luciferase in green and DAPI-stained nuclei in blue. Error bars indicate SE.
To assess whether sulfasalazine treatment reduced glutathione levels in the tumor of sulfasalazine-treated animals, we used monochlorobimane, a fluorescence-based detection kit to quantitatively determine the reduced form of glutathione (ApoAlert glutathione detection kit; BD Biosciences) (Shih et al., 2003). For these experiments, we killed four mice (two tumor, two controls) at 44 d after tumor induction, and brains were rapidly frozen and cryosectioned to 20 μm, thawed on ice, and then incubated with 100 μm monochlorobimane for 10 min. Fluorescence intensities were examined using a quantitative fluorescence imaging system (see Materials and Methods) comparing side-by-side slices from sulfasalazine-treated and control animals. Based on this assay conducted on 10 slices per animal, glutathione levels were significantly decreased by ∼30% (from 100 ± 5.6 to 71.7 ± 7.5 fluorescence units; p < 0.01; t test) in tumors from sulfasalazine-treated animals compared with controls. In parallel studies, we used the same assay on cultured glioma cells to validate these readouts. A 30% decrease correlates with the glutathione depletion achieved with ∼70 μm sulfasalazine administered for 24 h to cultured cells; the maximal decrease achieved with 250 μm sulfasalazine in cultured U87-MG cells was 68% (data not shown).
To further examine effects of sulfasalazine in vivo at the cellular level, we examined tumor sections with commonly used histological markers for cell death [ApopTag/methyl green (Fig. 7a,b) and terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) (Fig. 7c,d)], and proliferation (Ki-67) (Fig. 7e,f). Multiple fields from several serial sections from two control and two sulfasalazine-treated animals were examined, and representative fields from these experiments are shown in Figure 7. As is typical for World Health Organization (WHO) grade IV gliomas, control tumors present with significant tissue necrosis as visible from the brown ApopTag-DAB reaction product (Fig. 7a). Indeed, necrosis is a hallmark of grade IV gliomas and a necessary feature for their histopathological grading as such (Kleihues et al., 1993). At a higher resolution, control gliomas did present with significant cell death as evident from TUNEL and caspase-3 staining (data not shown). Interestingly, sections from all sulfasalazine-treated tumors showed almost no evidence for necrosis, as shown in a representative example in Figure 7b, and consistent with that, somewhat reduced TUNEL and caspase-3 labeling (data not shown). These sections, however, still showed similar Ki-67 indexes of ∼15, which are considered high-grade glioma (WHO grade IV) (Shibata et al., 1988). Ki-67 values did not differ significantly in a comparison of 14 representative fields from two animals (data not shown). TUNEL values suggested a lower percentage of apoptotic cells in sulfasalazine-treated versus control animals, but in a sampling of 21 sections from two animals each, these values also did not reach statistical significance (data not shown).
Figure 7.
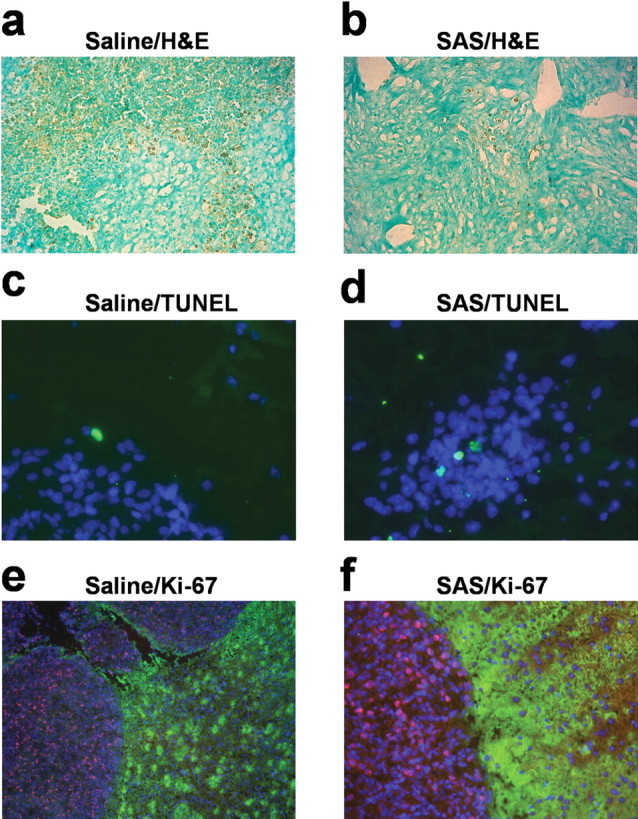
Immunohistochemical analysis of the sulfasalazine effects on in vivo tumors. a, b, DAB-reactive immunohistochemistry showing areas of tissue necrosis using an ApopTag kit based on TUNEL technology (20× magnification). a, A brown reaction product in single cells in the tumor and the background brown signify necrosis in the saline-control mouse (50 d survival). The green color comes from nuclei stained with methyl green. b, The same staining from a sulfasalazine (SAS)-treated mouse brain tumor (56 d survival). H&E, Hematoxylin and eosin. c, d, Representative examples of TUNEL staining (green) of a saline-treated (c) and 8 mg/ml SAS-treated (d) mouse, costained with 4′,6-diamidino-2-phenylindole (DAPI) (40× magnification). e, f, Immunocytochemistry for Ki67 in saline control and sulfasalazine-treated brain tumor sections (20× magnification). Ki-67 staining shows a clear demarcation around the tumor abutted by normal astrocytes stained by GLT-1 antibody shown in green. The Ki67-positive cells are red, and DAPI stains all of the nuclei blue.
Finally, in one pilot experiment with 30 animals injected with U87MGff-luc cells, we have begun to examine animal survival, comparing animals receiving 8 mg/ml sulfasalazine intraperitoneally twice daily throughout the course of the experiment with controls receiving only saline. In this study, half-maximal survival of controls was 44 d and half-maximal survival was not reach by day 77 in sulfasalazine-treated animals with 65% of the treated animals still alive. These results, albeit preliminary, suggest that system  inhibition by sulfasalazine may afford effective suppression of tumor growth in this animal model of intracranial glioma.
inhibition by sulfasalazine may afford effective suppression of tumor growth in this animal model of intracranial glioma.
Discussion
Results presented here suggest that glial-derived tumor cells rely almost exclusively on cystine uptake via system 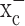 , an electroneutral cystine-glutamate exchanger, to maintain the intracellular antioxidant glutathione, at concentrations required to support cell growth. Pharmacological inhibition of system
, an electroneutral cystine-glutamate exchanger, to maintain the intracellular antioxidant glutathione, at concentrations required to support cell growth. Pharmacological inhibition of system 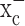 caused glutathione depletion, which in turn led to the progressive development of apoptotic cell death as cells failed to defend themselves against reactive oxygen species (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Importantly, nonmalignant brain cells (i.e., astrocytes and neurons) were unaffected by system
caused glutathione depletion, which in turn led to the progressive development of apoptotic cell death as cells failed to defend themselves against reactive oxygen species (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Importantly, nonmalignant brain cells (i.e., astrocytes and neurons) were unaffected by system 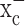 inhibition, suggesting that glutathione synthesis could be selectively inhibited in glioma cells. Most excitingly, sulfasalazine, a Food and Drug Administration (FDA)-approved drug, is available to accomplish this task, and preliminary animal experiments support the feasibility of this approach. The specificity of sulfasalazine for system
inhibition, suggesting that glutathione synthesis could be selectively inhibited in glioma cells. Most excitingly, sulfasalazine, a Food and Drug Administration (FDA)-approved drug, is available to accomplish this task, and preliminary animal experiments support the feasibility of this approach. The specificity of sulfasalazine for system 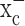 had been suggested by a previous study in lymphoma cells in which its inhibition resulted in similar growth arrest attributable entirely to inhibition of system
had been suggested by a previous study in lymphoma cells in which its inhibition resulted in similar growth arrest attributable entirely to inhibition of system 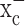 -mediated cystine uptake (Gout et al., 2001).
-mediated cystine uptake (Gout et al., 2001).
System 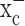 is an abundantly expressed transporter designated to the cellular import of the essential amino acid cystine. The energetically unfavorable uptake of cystine derives its driving force from the obligated coupling to glutamate transport out of the cells for which most cells exhibit a very steep (∼10,000-fold) gradient. Most cells require only a little cystine uptake to sustain cellular protein biosynthesis and the production of the antioxidant glutathione. This is different in cancer cells, which, because of their enhanced growth and metabolism, generate significant concentrations of ROS. If left uncontrolled, these can harm cell membranes, energy metabolism, and DNA. To prevent such ROS damage, many cancer cells, including gliomas (Louw et al., 1997) show increased synthesis of glutathione (Jefferies et al., 2003) requiring enhanced cystine uptake (Wu et al., 2004). This explains the here-observed high levels of expression of both the catalytic and regulatory subunit of system
is an abundantly expressed transporter designated to the cellular import of the essential amino acid cystine. The energetically unfavorable uptake of cystine derives its driving force from the obligated coupling to glutamate transport out of the cells for which most cells exhibit a very steep (∼10,000-fold) gradient. Most cells require only a little cystine uptake to sustain cellular protein biosynthesis and the production of the antioxidant glutathione. This is different in cancer cells, which, because of their enhanced growth and metabolism, generate significant concentrations of ROS. If left uncontrolled, these can harm cell membranes, energy metabolism, and DNA. To prevent such ROS damage, many cancer cells, including gliomas (Louw et al., 1997) show increased synthesis of glutathione (Jefferies et al., 2003) requiring enhanced cystine uptake (Wu et al., 2004). This explains the here-observed high levels of expression of both the catalytic and regulatory subunit of system 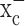 in glioma cells from biopsies and in glioma cell lines. Moreover, cystine uptake experiments suggest that the rate of cystine uptake into glioma cells is ∼500 pmol · min-1 · mg-1 (Ye et al., 1999), a value that is >100-fold greater than that reported for recombinantly expressed system
in glioma cells from biopsies and in glioma cell lines. Moreover, cystine uptake experiments suggest that the rate of cystine uptake into glioma cells is ∼500 pmol · min-1 · mg-1 (Ye et al., 1999), a value that is >100-fold greater than that reported for recombinantly expressed system 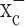 (Bassi et al., 2001; Kim et al., 2001). These data also explain the significant release of glutamate from these tumor cells suggested by several previous studies (Ye and Sontheimer, 1999; Behrens et al., 2000; Takano et al., 2001), because glutamate is coupled in an equimolar ratio to cystine uptake. A compelling case has been made that this glutamate release is required for tumors to expand, because their physical space is limited (Sontheimer, 2003). This glutamate release has also been suggested to contribute to peritumoral seizures and excitotoxicity in the vicinity of the tumor (Sontheimer, 2003). Our data suggest that glutamate release is an obligatory by-product of cystine uptake for the intracellular production of glutathione. The implications are significant, because pharmacological inhibition of cystine supply to glioma cells could have a dual effect. It could stop the growth of the tumor by starving it of GSH as we demonstrate in this study, but at the same time it may reduce glutamate-mediated excitotoxicity and peritumoral seizures. The latter, in particular, would be very exciting, because 69% of low-grade glioma patients present with peritumoral seizures (Oberndorfer et al., 2002).
(Bassi et al., 2001; Kim et al., 2001). These data also explain the significant release of glutamate from these tumor cells suggested by several previous studies (Ye and Sontheimer, 1999; Behrens et al., 2000; Takano et al., 2001), because glutamate is coupled in an equimolar ratio to cystine uptake. A compelling case has been made that this glutamate release is required for tumors to expand, because their physical space is limited (Sontheimer, 2003). This glutamate release has also been suggested to contribute to peritumoral seizures and excitotoxicity in the vicinity of the tumor (Sontheimer, 2003). Our data suggest that glutamate release is an obligatory by-product of cystine uptake for the intracellular production of glutathione. The implications are significant, because pharmacological inhibition of cystine supply to glioma cells could have a dual effect. It could stop the growth of the tumor by starving it of GSH as we demonstrate in this study, but at the same time it may reduce glutamate-mediated excitotoxicity and peritumoral seizures. The latter, in particular, would be very exciting, because 69% of low-grade glioma patients present with peritumoral seizures (Oberndorfer et al., 2002).
The most surprising finding in this study was the exclusive dependence of glioma cells on system 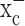 for cystine uptake. Neither astrocytes nor neurons share such an exclusive dependence, and hence, their growth was not impeded when system
for cystine uptake. Neither astrocytes nor neurons share such an exclusive dependence, and hence, their growth was not impeded when system 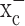 was inhibited. These cells were able to continue their production of glutathione after system
was inhibited. These cells were able to continue their production of glutathione after system 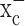 was inhibited, whereas gliomas showed near-complete glutathione depletion after a 48 h block of system
was inhibited, whereas gliomas showed near-complete glutathione depletion after a 48 h block of system 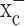 . This is consistent with the finding that neurons derive most of their glutathione from direct uptake of cysteine, the reduced form of cystine, via the Na+-dependent excitatory amino acid transporter (EAAT) family of transporters that are abundantly expressed in neurons and astrocytes (Danbolt, 2001). These transporters have been shown to transport cysteine in addition to glutamate (Flynn and McBean, 2000; McBean, 2002). However, EAAT transporters are lost from the membrane of glioma cells (Fig. 1), thus leaving no back-up system for the supply of cystine/cysteine to glioma cells when system
. This is consistent with the finding that neurons derive most of their glutathione from direct uptake of cysteine, the reduced form of cystine, via the Na+-dependent excitatory amino acid transporter (EAAT) family of transporters that are abundantly expressed in neurons and astrocytes (Danbolt, 2001). These transporters have been shown to transport cysteine in addition to glutamate (Flynn and McBean, 2000; McBean, 2002). However, EAAT transporters are lost from the membrane of glioma cells (Fig. 1), thus leaving no back-up system for the supply of cystine/cysteine to glioma cells when system 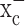 is inhibited. It remains to be seen whether other cell types show a similar reliance on a single amino acid transporter for the biosynthesis of glutathione. Immature embryonic neurons (Ratan et al., 1994) and immortalized neuronal hybridoma cells (Murphy et al., 1989) have been shown to rely on cystine uptake via a cystine glutamate exchanger in their glutathione synthesis. Similarly, glial progenitor cells, which have the potential to give rise to gliomas (Dai and Holland, 2001), have been shown to critically depend on cystine uptake (Yonezawa et al., 1996) in their redox defense, and compromises in oligodendrocyte development have been attributed to glutamate toxicity (Deng et al., 2003) in conjunction with a loss of redox defense (Baud et al., 2004).
is inhibited. It remains to be seen whether other cell types show a similar reliance on a single amino acid transporter for the biosynthesis of glutathione. Immature embryonic neurons (Ratan et al., 1994) and immortalized neuronal hybridoma cells (Murphy et al., 1989) have been shown to rely on cystine uptake via a cystine glutamate exchanger in their glutathione synthesis. Similarly, glial progenitor cells, which have the potential to give rise to gliomas (Dai and Holland, 2001), have been shown to critically depend on cystine uptake (Yonezawa et al., 1996) in their redox defense, and compromises in oligodendrocyte development have been attributed to glutamate toxicity (Deng et al., 2003) in conjunction with a loss of redox defense (Baud et al., 2004).
A significant degree of cellular damage resulting from chemotherapy and radiation therapy is imparted by the increased production of ROS, and enhanced GSH levels have been partially blamed for the enhanced resistance of glioma cells to such treatments. Not surprisingly, therefore, previous studies have examined the ability to control tumor growth in experimental tumor models using chemical inhibitors of glutathione synthesis (for review, see Fojo and Bates, 2003). Unfortunately, these drugs lack specificity and also impair the systemic production of GSH in the liver and other organs and therefore are not good candidates for clinical use. Based on our data, a control of GSH production in glioma cells may, however, be feasible by selectively inhibiting system 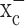 -mediated cystine uptake using (S)-4-CPG or sulfasalazine. Our animal data, albeit preliminary, suggest that sulfasalazine is well tolerated and may be effective in controlling glioma growth in an intracranial model for malignant glioma.
-mediated cystine uptake using (S)-4-CPG or sulfasalazine. Our animal data, albeit preliminary, suggest that sulfasalazine is well tolerated and may be effective in controlling glioma growth in an intracranial model for malignant glioma.
The primary goal of this study was to examine, at the cellular level, the role of system 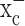 in glioma biology, and the data presented overwhelmingly support the notion that system
in glioma biology, and the data presented overwhelmingly support the notion that system 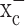 is a principal pathway for glutathione production and redox defense in glioma cells. We embarked on a limited series of translational studies in animals with the primary objective to show “proof of principle,” which we believe to have accomplished. For example, inhibition of system
is a principal pathway for glutathione production and redox defense in glioma cells. We embarked on a limited series of translational studies in animals with the primary objective to show “proof of principle,” which we believe to have accomplished. For example, inhibition of system 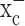 with sulfasalazine in vivo caused tumors to grow more slowly and, based on the histopathological markers, appear to show either an arrest of tumor cells in the cell cycle or a slowing in cell-cycle progression as also observed in our tissue culture studies. Thus, in the treated animals, the tumors have fewer areas of necrosis and somewhat lower rates of apoptosis but appear to still have high levels of the proliferation marker Ki-67. This may be counterintuitive for a more slowly growing tumor. It may, however, signify that sulfasalazine may be slowing the progression of cells through the cell cycle, effectively leading to the same number of cells undergoing a division at any given time (as indicated by similar Ki-67 labeling). However, by spending more time on their doubling, this will still lead to slower overall expansion of the tumor. Such an effect of sulfasalazine has been reported for vascular endothelial cells (Sharon et al., 1992). This would also explain the observed accumulation of DNA synthesis markers in cultured cells (Fig. 4b). Interestingly, the lack of necrosis correlates clinically with much better patient prognosis and much enhanced patient survival (Hammoud et al., 1996; Raza et al., 2002) with a lengthening of mean survival from ∼12 to 49 months.
with sulfasalazine in vivo caused tumors to grow more slowly and, based on the histopathological markers, appear to show either an arrest of tumor cells in the cell cycle or a slowing in cell-cycle progression as also observed in our tissue culture studies. Thus, in the treated animals, the tumors have fewer areas of necrosis and somewhat lower rates of apoptosis but appear to still have high levels of the proliferation marker Ki-67. This may be counterintuitive for a more slowly growing tumor. It may, however, signify that sulfasalazine may be slowing the progression of cells through the cell cycle, effectively leading to the same number of cells undergoing a division at any given time (as indicated by similar Ki-67 labeling). However, by spending more time on their doubling, this will still lead to slower overall expansion of the tumor. Such an effect of sulfasalazine has been reported for vascular endothelial cells (Sharon et al., 1992). This would also explain the observed accumulation of DNA synthesis markers in cultured cells (Fig. 4b). Interestingly, the lack of necrosis correlates clinically with much better patient prognosis and much enhanced patient survival (Hammoud et al., 1996; Raza et al., 2002) with a lengthening of mean survival from ∼12 to 49 months.
As it pertains to the attenuation of glutathione levels, we were able to detect a ∼30% decrease in reduced glutathione in the tumors of sulfasalazine-treated animals compared with controls. This value is less than that observed in isolated cells but comparable with values obtained in other in vivo studies (Freitas et al., 2005). The presence of nonmalignant tissues and blood vessels within the tumor sections, whose glutathione levels are not affected by sulfasalazine, may further contribute to an underestimation of the actual changes that occurred. Finally, animal survival, albeit examined in only 15 treated and 15 control animals was prolonged by sulfasalazine treatment. These findings now warrant a more comprehensive assessment regarding the use of system 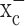 inhibitors in multiple glioma animal models, with a comprehensive pharmacokinetic assessment and thorough animal survival studies. Similarly, the pharmacokinetics and bioavailability of sulfasalazine and (S)-4-CPG in the brain warrant additional assessment, although our data and findings from two previous studies suggest that sulfasalazine does effectively penetrate the blood-brain barrier in mice (Robe et al., 2004) and humans (Noseworthy et al., 1998). The additional pursuit of such studies is compelling in light of the paucity of effective treatments for malignant gliomas on the one hand, and the fact that sulfasalazine is already FDA approved, readily available, and backed by a large database of clinical data for other disease indications.
inhibitors in multiple glioma animal models, with a comprehensive pharmacokinetic assessment and thorough animal survival studies. Similarly, the pharmacokinetics and bioavailability of sulfasalazine and (S)-4-CPG in the brain warrant additional assessment, although our data and findings from two previous studies suggest that sulfasalazine does effectively penetrate the blood-brain barrier in mice (Robe et al., 2004) and humans (Noseworthy et al., 1998). The additional pursuit of such studies is compelling in light of the paucity of effective treatments for malignant gliomas on the one hand, and the fact that sulfasalazine is already FDA approved, readily available, and backed by a large database of clinical data for other disease indications.
Footnotes
This work was supported by National Institutes of Health Grants RO1-NS36692 and P50CA97247 and by a grant from the Goldhirsh Foundation. The authors do not have any competing financial interest.
Correspondence should be addressed to Dr. Harald Sontheimer, Civitan International Research Center 545, 1719 Sixth Avenue South, Birmingham, AL 35294-0021. E-mail: sontheimer@uab.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/257101-10$15.00/0
References
- Barger SW, Basile AS (2001) Activation of microglia by secreted amyloid precursor protein evokes release of glutamate by cystine exchange and attenuates synaptic function. J Neurochem 76: 846-854. [DOI] [PubMed] [Google Scholar]
-
Bassi MT, Gasol E, Manzoni M, Pineda M, Riboni M, Martin R, Zorzano A, Borsani G, Palacin M (2001) Identification and characterisation of human xCT that co-expresses, with 4F2 heavy chain, the amino acid transport activity system
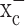 Pflügers Arch
442: 286-296. [DOI] [PubMed] [Google Scholar]
Pflügers Arch
442: 286-296. [DOI] [PubMed] [Google Scholar] - Baud O, Greene AE, Li J, Wang H, Volpe JJ, Rosenberg PA (2004) Glutathione peroxidase-catalase cooperativity is required for resistance to hydrogen peroxide by mature rat oligodendrocytes. J Neurosci 24: 1531-1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens PF, Langemann H, Strohschein R, Draeger J, Hennig J (2000) Extracellular glutamate and other metabolites in and around RG2 rat glioma: an intracerebral microdialysis study. J Neurooncol 47: 11-22. [DOI] [PubMed] [Google Scholar]
- Dai C, Holland EC (2001) Glioma models. Biochim Biophys Acta 1551: M19-M27. [DOI] [PubMed] [Google Scholar]
- Danbolt NC (2001) Glutamate uptake. Prog Neurobiol 65: 1-105. [DOI] [PubMed] [Google Scholar]
- Deng W, Rosenberg PA, Volpe JJ, Jensen FE (2003) Calcium-permeable AMPA/kainate receptors mediate toxicity and preconditioning by oxygen-glucose deprivation in oligodendrocyte precursors. Proc Natl Acad Sci USA 100: 6801-6806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espey MG, Kustova Y, Sei Y, Basile AS (1998) Extracellular glutamate levels are chronically elevated in the brains of LP-BM5-infected mice: a mechanism of retrovirus-induced encephalopathy. J Neurochem 71: 2079-2087. [DOI] [PubMed] [Google Scholar]
- Flynn J, McBean GJ (2000) Kinetic and pharmacological analysis of l-[35S]cystine transport into rat brain synaptosomes. Neurochem Int 36: 513-521. [DOI] [PubMed] [Google Scholar]
- Fojo T, Bates S (2003) Strategies for reversing drug resistance. Oncogene 22: 7512-7523. [DOI] [PubMed] [Google Scholar]
- Freitas RM, Vasconcelos SM, Souza FC, Viana GS, Fonteles MM (2005) Oxidative stress in the hippocampus after pilocarpine-induced status epilepticus in Wistar rats. FEBS J 272: 1307-1312. [DOI] [PubMed] [Google Scholar]
- Gout PW, Buckley AR, Simms CR, Bruchovsky N (2001) Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x(c)-cystine transporter: a new action for an old drug. Leukemia 15: 1633-1640. [DOI] [PubMed] [Google Scholar]
- Hammoud MA, Sawaya R, Shi W, Thall PF, Leeds NE (1996) Prognostic significance of preoperative MRI scans in glioblastoma multiforme. J Neurooncol 27: 65-73. [DOI] [PubMed] [Google Scholar]
- Jefferies H, Coster J, Khalil A, Bot J, McCauley RD, Hall JC (2003) Glutathione. ANZ J Surg 73: 517-522. [DOI] [PubMed] [Google Scholar]
- Kim JY, Kanai Y, Chairoungdua A, Cha SH, Matsuo H, Kim DK, Inatomi J, Sawa H, Ida Y, Endou H (2001) Human cystine/glutamate transporter: cDNA cloning and upregulation by oxidative stress in glioma cells. Biochim Biophys Acta 1512: 335-344. [DOI] [PubMed] [Google Scholar]
- Kleihues P, Burger PC, Scheithauer BW (1993) The new WHO classification of brain tumours. Brain Pathol 3: 255-268. [DOI] [PubMed] [Google Scholar]
- Louw DF, Bose R, Sima AA, Sutherland GR (1997) Evidence for a high free radical state in low-grade astrocytomas. Neurosurgery 41: 1146-1150. [DOI] [PubMed] [Google Scholar]
- McBean GJ (2002) Cerebral cystine uptake: a tale of two transporters. Trends Pharmacol Sci 23: 299-302. [DOI] [PubMed] [Google Scholar]
- Murphy TH, Miyamoto M, Sastre A, Schnaar RL, Coyle JT (1989) Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron 2: 1547-1558. [DOI] [PubMed] [Google Scholar]
- Noseworthy JH, O'Brien P, Erickson BJ, Lee D, Sneve D, Ebers GC, Rice GP, Auty A, Hader WJ, Kirk A, Duquette P, Carter J, Francis G, Metz L, Shuster E (1998) The Mayo Clinic-Canadian Cooperative trial of sulfasalazine in active multiple sclerosis. Neurology 51: 1342-1352. [DOI] [PubMed] [Google Scholar]
- Oberndorfer S, Schmal T, Lahrmann H, Urbanits S, Lindner K, Grisold W (2002) The frequency of seizures in patients with primary brain tumors or cerebral metastases. An evaluation from the Ludwig Boltzmann Institute of Neuro-Oncology and the Department of Neurology, Kaiser Franz Josef Hospital, Vienna. Wien Klin Wochenschr 114: 911-916. [PubMed] [Google Scholar]
- Oka A, Belliveau MJ, Rosenberg PA, Volpe JJ (1993) Vulnerability of oligodendroglia to glutamate: pharamacology, mechanisms, and prevention. J Neurosci 13: 1441-1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
-
Patel SA, Warren BA, Rhoderick JF, Bridges RJ (2004) Differentiation of substrate and non-substrate inhibitors of transport system
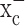 : an obligate exchanger of l-glutamate and l-cystine. Neuropharmacology
46: 273-284. [DOI] [PubMed] [Google Scholar]
: an obligate exchanger of l-glutamate and l-cystine. Neuropharmacology
46: 273-284. [DOI] [PubMed] [Google Scholar] - Ratan RR, Murphy TH, Baraban JM (1994) Oxidative stress induces apoptosis in embryonic cortical neurons. J Neurochem 62: 376-379. [DOI] [PubMed] [Google Scholar]
- Raza SM, Lang FF, Aggarwal BB, Fuller GN, Wildrick DM, Sawaya R (2002) Necrosis and glioblastoma: a friend or a foe? A review and a hypothesis. Neurosurgery 51: 2-12. [DOI] [PubMed] [Google Scholar]
- Rijpkema M, Schuuring J, van der Meulen Y, van der Graaf M, Bernsen H, Boerman R, van der Kogel A, Heerschap A (2003) Characterization of oligodendrogliomas using short echo time 1H MR spectroscopic imaging. NMR Biomed 16: 12-18. [DOI] [PubMed] [Google Scholar]
- Robe PA, Bentires-Alj M, Bonif M, Rogister B, Deprez M, Haddada H, Khac MT, Jolois O, Erkmen K, Merville MP, Black PM, Bours V (2004) In vitro and in vivo activity of the nuclear factor-kappaB inhibitor sulfasalazine in human glioblastomas. Clin Cancer Res 10: 5595-5603. [DOI] [PubMed] [Google Scholar]
- Roslin M, Henriksson R, Bergstrom P, Ungerstedt U, Bergenheim AT (2003) Baseline levels of glucose metabolites, glutamate and glycerol in malignant glioma assessed by stereotactic microdialysis. J Neurooncol 61: 151-160. [DOI] [PubMed] [Google Scholar]
- Rothstein JD (2002) Paving new pathways. Nat Med 8: 938-940. [DOI] [PubMed] [Google Scholar]
- Sato H, Tamba M, Ishii T, Bannai S (1999) Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J Biol Chem 274: 11455-11458. [DOI] [PubMed] [Google Scholar]
-
Sato H, Tamba M, Okuno S, Sato K, Keino-Masu K, Masu M, Bannai S (2002) Distribution of cystine/glutamate exchange transporter, system
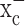 , in the mouse brain. J Neurosci
22: 8028-8033. [DOI] [PMC free article] [PubMed] [Google Scholar]
, in the mouse brain. J Neurosci
22: 8028-8033. [DOI] [PMC free article] [PubMed] [Google Scholar] - Shanker G, Aschner M (2001) Identification and characterization of uptake systems for cystine and cysteine in cultured astrocytes and neurons: evidence for methylmercury-targeted disruption of astrocyte transport. J Neurosci Res 66: 998-1002. [DOI] [PubMed] [Google Scholar]
- Sharon P, Drab EA, Linder JS, Weidman SW, Sabesin SM, Rubin DB (1992) The effect of sulfasalazine on bovine endothelial cell proliferation and cell cycle phase distribution. Comparison with olsalazine, 5-aminosalicylic acid, and sulfapyridine. J Lab Clin Med 119: 99-107. [PubMed] [Google Scholar]
- Shibata T, Burger PC, Kleihues P (1988) Ki-67 immunoperoxidase stain as marker for the histological grading of nervous system tumours. Acta Neurochir Suppl (Wien) 43: 103-106. [DOI] [PubMed] [Google Scholar]
- Shih AY, Johnson DA, Wong G, Kraft AD, Jiang L, Erb H, Johnson JA, Murphy TH (2003) Coordinate regulation of glutathione biosynthesis and release by Nrf2-expressing glia potently protects neurons from oxidative stress. J Neurosci 23: 3394-3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sontheimer H (2003) Malignant gliomas: perverting glutamate and ion homeostasis for selective advantage. Trends Neurosci 26: 543-549. [DOI] [PubMed] [Google Scholar]
- Soroceanu L, Gillespie Y, Khazaeli MB, Sontheimer H (1998) Use of chlorotoxin for targeting of primary brain tumors. Cancer Res 58: 4871-4879. [PubMed] [Google Scholar]
- Takano T, Lin JH, Arcuino G, Gao Q, Yang J, Nedergaard M (2001) Glutamate release promotes growth of malignant gliomas. Nat Med 7: 1010-1015. [DOI] [PubMed] [Google Scholar]
- Wu G, Fang YZ, Yang S, Lupton JR, Turner ND (2004) Glutathione metabolism and its implications for health. J Nutr 134: 489-492. [DOI] [PubMed] [Google Scholar]
- Ye ZC, Sontheimer H (1998) Astrocytes protect neurons from neurotoxic injury by serum glutamate. Glia 22: 237-248. [DOI] [PubMed] [Google Scholar]
- Ye ZC, Sontheimer H (1999) Glioma cells release excitotoxic concentrations of glutamate. Cancer Res 59: 4383-4391. [PubMed] [Google Scholar]
- Ye ZC, Rothstein JD, Sontheimer H (1999) Compromised glutamate transport in human glioma cells: reduction-mislocalization of sodium-dependent glutamate transporters and enhanced activity of cystine-glutamate exchange. J Neurosci 19: 10767-10777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonezawa M, Back SA, Gan X, Rosenberg PA, Volpe JJ (1996) Cystine deprivation induces oligodendroglial death: rescue by free radical scavengers and by a diffusible glial factor. J Neurochem 67: 566-573. [DOI] [PubMed] [Google Scholar]