

Abstract
Cyclooxygenase-2 is often highly expressed in epithelial malignancies and likely has an active role in tumor development. But how it promotes tumorigenesis is not clearly defined. Recent evidence suggests that this may involve transactivation of the epidermal growth factor receptor through E-prostanoid receptors, but reports differ about the mechanism by which this occurs. We found that E-prostanoid receptors 2–4, but not 1, transactivated the epidermal growth factor receptor. This required metalloproteinase activity, leading to release of growth factors from the cell surface. Both transforming growth factor-α and amphiregulin were released in response to overexpression of cyclooxygenase-2, but betacellulin and heparin-binding EGF-like growth factor were not. The metalloproteinase tumor necrosis factor-α converting enzyme was required for proteolytic release of transforming growth factor-α. We also found that addition of epidermal growth factor receptor ligands to HEK293 cells induced cyclooxygenase-2 expression, suggesting that by activating epidermal growth factor receptor signaling, cyclooxygenase-2 potentially creates a self-perpetuating cycle of cell growth. Consistent with this, inhibition of cyclooxygenase-2 reduced growth of epidermal growth factor receptor overexpressing MCF-10A breast epithelial cells in three-dimensional culture.
Keywords: COX-2, EGFR, prostaglandins, E-prostanoid receptor, TACE, growth factors
INTRODUCTION
Substantial evidence supports a role for cyclooxygenase-2 (COX-2) in the development of several types of tumors including colon, head and neck, breast, lung, pancreas, and gastric cancer [1]. COX-2 is usually expressed at high levels in these tumors and its high expression often portends a poor response to treatment and a worse outcome. Clinical evidence demonstrating that COX-2 has an active role in colorectal cancer includes the observation that in some populations, chronic administration of nonsteroidal anti-inflammatory drugs substantially reduces the risk of developing colorectal cancer (reviewed in [2, 3]). Additional studies have demonstrated that cyclooxygenase inhibitors reduce the size and number of intestinal polyps in mice (reviewed in [2]), and deletion of the murine COX-2 gene is protective [4, 5]. While the dysregulated expression of COX-2 appears to be important in multiple stages of the developing cancer, how it contributes to this process is not clear.
Excessive signaling through the epidermal growth factor receptor (EGFR) is thought to be crucial in many types of epithelial cancers (reviewed in [6]). Most often this occurs when either EGFR or the growth factors that bind to it are overexpressed. As with COX-2, high expression of EGFR in tumors correlates with poor survival and resistance to therapy [6]. The growth factors that bind to EGFR are synthesized as large precursors and must be proteolytically released from the cell surface in order to activate the EGFR. This suggests that excessive activity of the proteases that release these growth factors might also be a mechanism by which EGFR signaling is pathological. Indeed, there are numerous examples demonstrating that transgenic expression of transforming growth factor-α (TGFα) in mice causes tumor formation (reviewed in [7]). Several members of the A-Disintegrin and Metalloproteinase (ADAM) family proteolytically release EGFR growth factors. ADAM proteins are often activated through a subset of G protein-coupled receptors (GPCRs). This sequence of events is called transactivation of EGFR because it results in activation of EGFR through a molecule that does not, itself, bind EGFR [8]. Recently, Pai et al. reported that prostaglandin E2 (PGE2), a downstream product of COX-2, transactivated EGFR [9].
There are four receptors for PGE2, called E-prostanoid (EP) 1–4 (reviewed in [10]). The EP receptors all couple to G proteins, and Pai et al. speculated that PGE2 activated a pathway that resulted in proteolytic release of EGFR growth factors. Consistent with this, they found that antibodies that neutralize TGFα abolished transactivation of EGFR by PGE2. Further, inhibitors of metalloproteinases also blocked the PGE2-induced response [9]. Another recent report, however, concluded that PGE2 transactivated EGFR through an intracellular pathway that did not involve metalloproteinases. Instead, Buchanan and coworkers found evidence that Src phosphorylated, and thereby activated, EGFR [11]. The inconsistent requirement for growth factor release noted by these two groups was likely caused by differences in cell types and experimental approaches. Here, we show that PGE2 transactivated EGFR through a subset of EP receptors, which activated metalloproteinases that then released some but not all EGFR ligands. Additionally, we demonstrate that ADAM17, commonly known as tumor necrosis factor-α converting enzyme (TACE), was largely responsible for release of these growth factors. Finally, we show that inhibiting COX-2 reduced growth of mammary epithelial cells overexpressing EGFR.
MATERIALS AND METHODS
Materials
Cell culture medium, antibiotics, serum, epidermal growth factor (EGF), and bovine insulin were from Invitrogen. Cholera toxin was from Biomol and pertussis toxin was from Sigma. Phorbol 12-myristate 13-acetate (PMA), platelet-derived growth factor (PDGF), and hydrocortisone were from Sigma. TGFα, amphiregulin, betacellulin, heparin-binding EGF-like growth factor (HB-EGF), and antibodies against amphiregulin, betacellulin, and HB-EGF were from R&D Systems. Antibodies to detect COX-2 were from Cayman Chemicals. Matrigel (#354230) was from BD PharMingen. PGE2 and AG1478 were from Calbiochem, while GM6001 was from Chemicon.
Cell Culture and Transfection
MCF-10A cells (ATCC) were cultured as described [12]. COS-7 cells (ATCC), HEK 293 cells (ATCC), and either wild-type or TACE-deficient, immortalized mouse embryo fibroblasts (provided by R. Black at Amgen) were propagated in DMEM with 10%FBS. They were transfected using LipofectAmine (Invitrogen) in 6 well plates with COX-2 (in pCDNA1/Amp, 500ng/well for HEK293 cells or 1.5μg/well for fibroblasts) or the empty vector along with TGFα, amphiregulin, betacellulin, or HB-EGF (in pcDNA3.1, 100ng/well for HEK293 cells or 300ng/well for fibroblasts). COS-7 cells were transfected in 6cm plates with a murine EP receptor subtype (EP1, EP2, EP3α, or EP4 in p3X-FLAG, 2.5μg). To measure, EGFR phosphorylation, EGFR (in pcDNA3.1/Myc-His, 0. 5μg, from S. Kuwada, University of Utah) was included in the transfection. The EGFR mutants were generated using a site directed mutagenesis kit (Stratagene) with the following forward primers and reverse complement primers: L858R-5′-CAGATTTTGGGCGGGCCAAACTGCTGGG and delL747-P753insS-5′-CGCTATCAAGGAATCGAAAGCCAACAAGG. To make MCF-10A stable cell lines, cells were transfected with EGFR (1μg/well) and then selected using G418 (Invitrogen, 250μg/mL). Isolated colonies were then propagated for three-dimensional culture experiments.
Assay for Release of Growth Factors
Twenty four hours after transfection, to test the effects of PGE2 (Cayman Chemicals), the cells were starved (DMEM no serum) for three hours with the addition of mAb225 (20μg/ml) during the final 30 minutes. This antibody blocks EGFR to inhibit binding and subsequent internalization of the growth factors. The medium was changed (DMEM, no serum, 20μg/mL mAb225, and PGE2) and then collected two hours later. After collection, the medium was centrifuged (700×g for 5 min.) to remove cellular debris. The adherent cells were washed with cold PBS and then lysed in 200μL of reporter lysis buffer (Promega). To detect TGFα in the medium, we used an ELISA (Oncogene research) and followed the manufacturer’s instructions. To detect amphiregulin, HB-EGF, and betacellulin, we developed sandwich ELISAs using matched antibody sets from R&D Systems. All ELISAs used an unconjugated primary antibody bound to the plate (anti-amphiregulin 1:150, anti-betacellulin 1:400, and anti-HB-EGF 1:800). Cell medium or lysates were then incubated for 2–4 hours, and then following washes (BD OptEIA wash solution, BD Biosciences), a biotin-conjugated secondary antibody (anti-amphiregulin 1:100, anti-betacellulin 1:100, anti-HB-EGF 1:200) was added for 1–2 hours. Following washes, streptavidin-HRP (1:200, R&D Systems) was added for 1 hour. After washes, a colorimetric reaction was initiated with BD OptEIA color substrate (BD Biosciences). All values were normalized to cell lysate protein determined by Pierce BCA protein assay kit and statistical significance was determined using paired, one-tailed t tests.
Assay for COX-2 Expression
HEK 293 cells were starved (DMEM with 0.5%FBS) for 4 hours. The medium was then replaced with DMEM, 0.5%FBS, with or without the agonist (TGFα: 5ng/ml, EGF: 20ng/ml, PMA: 20nM, PDGF: 50ng/ml) and then incubated overnight. The cells were lysed in reporter lysis buffer (Promega) and protein content was determined (Pierce BCA). Lysates (25μg) were separated by 10% SDS-PAGE and COX-2 protein was detected as previously described [13]. To test the effects of wild-type or mutant EGFR expression, the cells were transfected, incubated with 10% serum overnight, and then starved as noted above. To detect COX-2 mRNA, the cells were treated as above and then total RNA was isolated using TRIzol Reagent (Invitrogen) as previously described [13]. RT-PCR to detect COX-2 mRNA was performed as described [14].
Western immunoblotting
Anti-c-Myc #sc-40, anti-pERK1/2 #sc-7383, anti-ERK1 #sc-093, and anti-ERK2 #sc-154 were from Santa Cruz Biotechnology. All other antibodies used for immunoblotting were from Cell Signaling Technologies and were used according to their instructions: anti-EGFR #2232; anti-pEGFR #2234; anti-Akt #9272; anti-pAkt (Ser473) #9271; anti-pAkt (Thr308) #9275, anti-COX-2 #4842.
Three-dimensional cell culture
Stable MCF-10A cell lines expressing either control vector (pcDNA3.1/Myc-His) or EGFR were cultured in Matrigel as described [12]. Digital photos were taken using an Olympus Fluoview confocal microscope. Volumes of the three dimensional structures were calculated using the equation: π/6(largest diameter × [smaller diameter]2).
RESULTS
COX-2 causes release of specific growth factors from the cell surface
Pai and coworkers demonstrated evidence suggesting that PGE2 transactivated EGFR by causing metalloproteinases to release TGFα [9]. At least seven ligands are known to bind and activate EGFR (reviewed in [15]). To examine which EGFR growth factors were released from cells over-expressing COX-2, we expressed COX-2 in HEK293 cells. Release of endogenous growth factors is very difficult to detect because they rapidly bind their receptor and are internalized [16]. To detect release of the growth factor in these experiments we co-transfected the cells with TGFα, amphiregulin, betacellulin, or HB-EGF. Additionally, we added an EGFR neutralizing antibody (mAb225) to the medium to reduce the chance of growth factor internalization. We then measured growth factor released into the medium using ELISAs. We found that expression of COX-2 caused significant release of only TGFα from starved cells (Fig. 1A). These data were consistent with those of Pai et al. who demonstrated that neutralizing antibodies directed against TGFα significantly reduced EGFR transactivation, while antibodies directed against HB-EGF did not [9]. They did not test antibodies directed against amphiregulin or betacellulin.
Figure 1.

COX-2 and PGE2 cause TGFα shedding. A. HEK293 cells were transfected with control vector or COX-2 and either pro-TGFα, pro-amphiregulin, pro-HB-EGF, or pro-betacellulin. The cells were starved overnight and then the medium was replaced and collected 2hrs later and assayed by ELISA to detect the amount of growth factor that was released into the medium over a two hour period. Values shown are mean (+/− standard deviation, n = 3) fold increase in growth factor released into the medium from cells expressing COX-2 compared to vector control. The * indicates that the difference between control and COX-2 growth factor shedding was statistically significant (p<0.05). B. Immortalized embryo fibroblasts from either wild-type or TACEΔZn/ΔZn mice were transfected with pro-TGFα and either a control vector or COX-2. The cells were starved and then the medium was collected after two hours. An ELISA was used to detect TGFα that was released into the medium. Values were normalized to total protein in the cell lysates. The * indicates that the difference between control and COX-2 growth factor shedding was statistically significant (p<0.05). C. HEK293 cells, transfected with pro-TGFα, were starved and then treated with 10μM PGE2 for two hours. The medium was recovered and the amount of TGFα was determined using a specific ELISA. Values were normalized to total protein in the cell lysates (p<0.01, n = 4). Similar results were obtained by normalizing to TGFα in the cell lysates. * indicates that the difference was statistically significant (p<0.01).
TGFα is released predominantly by TACE
Members of the ADAM family of metalloproteinases are thought to be largely responsible for release of EGFR ligands. These are transmembrane proteins that proteolytically release a diverse set of biologically active proteins such as growth factors, cytokines, and their receptors. ADAM17, which is more commonly called TACE, is known to shed most EGFR ligands in addition to several other proteins [17]. Additionally, TACE-deficient mice are very similar to EGFR-deficient mice [18], strongly suggesting that TACE has a prominent role in proteolytic release of most EGFR ligands. To test whether TACE was required for COX-2 to cause release of TGFα, we co-expressed COX-2 with TGFα in murine embryo fibroblasts that were either wild-type or were derived from TACEΔZn/ΔZn mice, in which a portion of the gene encoding TACE had been deleted, causing inactivation of TACE [18]. We found that very little TGFα was released from TACEΔZn/ΔZn fibroblasts, indicating that TACE was required for COX-2 to induce shedding of TGFα. However, there was a slight increase in TGFα release from TACEΔZn/ΔZn fibroblasts in the presence of COX-2 that was likely caused by other ADAM family members, but the majority (>90%) of TGFα release appeared to require TACE. These data are consistent with the report by Pai and coworkers who demonstrated that broad spectrum metalloproteinase inhibitors or neutralizing antibodies directed against TGFα significantly reduced EGFR transactivation caused by PGE2 [9].
Release of growth factors by COX-2 is mimicked by exogenous PGE2
PGE2, a downstream product of the COX-2 reaction, activates the G protein-coupled, EP receptors and can transactivate EGFR. But reports differ on how this occurs. Pai and coworkers, for example, found evidence suggesting that PGE2 activated EGFR through a metalloproteinase, which released TGFα that then activated EGFR [9]. But, Buchanan et al. found that metalloproteinase activity was not required for PGE2 to transactivate EGFR [11]. These differences are not surprising because EGFR can be transactivated through metalloproteinase dependent and independent signaling pathways (reviewed in [8]). To directly examine whether PGE2 could cause TGFα release, we used HEK293 cells, which express EP1-4 (data not shown). We treated the cells with PGE2 and then measured release of TGFα using an ELISA. In these experiments, we found that 10μM PGE2 consistently caused TGFα release into the medium (Fig. 1C). It also caused TGFα shedding at lower concentrations (1.5-fold increase at 1μM PGE2 and 1.6-fold increase at 5μM, n = 2). Since these concentrations of PGE2 were within the range where others have detected transactivation of EGFR [9, 11], our data suggest that PGE2 can transactivate EGFR by causing release of TGFα.
PGE2 transactivates EGFR through a metalloproteinase and a subset of EP receptors
PGE2 binds to four G protein-coupled EP receptors [10]. Each of them has a specific tissue and cell distribution, and each receptor initiates distinct intracellular signaling pathways by activating subsets of G proteins. COS-7 cells have been widely used to characterize EGFR transactivation [15]. To examine which EP receptors could activate EGFR and whether metalloproteinase activity was required, we expressed each of the four EP receptors in COS-7 cells, treated the cells with PGE2, and then measured phosphorylation of Akt at Ser473 in the presence of either an EGFR inhibitor (AG1478) or a broad spectrum metalloproteinase inhibitor (GM6001, Ilomistat). We found that Akt was not phosphorylated in COS-7 cells transfected with the empty vector (Fig 2A). Nor was it phosphorylated in cells expressing EP1. However, Akt was phosphorylated in cells expressing EP2, EP3, or EP4 (Fig. 2A–B). Moreover, the inhibitors had different effects on this phosphorylation. In cells expressing EP2, Akt phosphorylation was completely inhibited by both AG1478 and GM6001, indicating that activation of Akt through EP2 required both EGFR and metalloproteinase activity, respectively. This indicated that EP2 transactivated EGFR through the well-defined pathway involving activation of a metalloproteinase and subsequent release of the growth factor ligands that bind EGFR. EP3 also caused Akt phosphorylation, but this was only partially inhibited by either AG1478 or GM6001, indicating that EP3 caused Akt phosphorylation by metalloproteinase and EGFR-dependent and -independent mechanisms. Finally, Akt was phosphorylated in cells expressing EP4, but this was not inhibited by either AG1478 or GM6001. We also examined phosphorylation of Akt at Thr308 and found similar results (not shown). Additionally, we measured ERK1/2 phosphorylation and found that PGE2 caused ERK1/2 phosphorylation that was not significantly affected by either AG1478 or GM6001, indicating that ERK1/2 activation predominantly occurs directly through the EP receptors rather than through EGFR. We conclude that EP2 and EP3 can activate Akt through a metalloproteinase and EGFR.
Figure 2.

PGE2 transactivates EGFR through specific EP receptors. A. COS-7 cells, transfected with a control empty vector or an EP receptor subtype, were starved and then treated with 10μM PGE2 for 2 minutes in the absence or presence of GM6001 or AG1478. Phospho-Akt (pAkt Ser473) was detected in the cell lysates using a specific antibody. Immunoblots were then reprobed to detect total Akt. Results are representative of three experiments. Each EP receptor has a 3X-FLAG epitope tag, and shown below are typical expression levels of the EP receptors detected with an anti-FLAG antibody. B. Densitometry of pAkt Ser473 immunoblots (n=3) where the pAkt signal was normalized to basal pAkt in cells transfected with control vector. The * indicates that the change was statistically significant (p<0.05) when compared to basal pAkt. TGFα (10ng/mL), the positive control in these experiments, caused a 5.2 (+/−0.4) fold increase in pAkt. C. HEK293 cells with or without pertussis toxin pretreatment (100ng/mL, 16hrs) were treated for 2 minutes with 10μM PGE2 and then pAkt Thr308 and total Akt were detected by immunoblotting. D. COS-7 cells were co-transfected with EGFR and an EP receptor subtype. Starved cells were then treated with PGE2 in the presence or absence of GM6001. Phospho-EGFR (pEGFR) was detected using a specific antibody and then the immunoblots were reprobed to detect total EGFR. Results are representative of at least three experiments. E. Densitometry of pEGFR from four experiments showing the fold increase in pEGFR caused by 10μM PGE2. In all cases, pEGFR was first normalized to total EGFR. In a typical experiment, TGFα (10ng/mL) caused a ~3 fold increase in pEGFR. The * denotes significant change (p<0.03) compared to conditions without PGE2 or GM6001. The ** indicates that GM6001 significantly reduced (p<0.03) pEGFR compared to PGE2 alone.
Some EP receptors couple to Gαi subunits, which are sensitive to pertussis toxin. To test the importance of Gαi subunits, we treated HEK293 cells with pertussis toxin and then examined PGE2-induced ERK1/2 and Akt activation. HEK293 cells express mRNA for all four EP receptors (data not shown). We found that pertussis toxin completely inhibited PGE2-induced Akt phosphorylation (Fig. 2C), indicating that in HEK293 cells, Gαi subunits are important.
The robust, EGFR-independent activation of Akt in cells expressing EP4 was not surprising because G protein-coupled receptors are known to activate phosphatidylinositol 3-kinases, and consequently Akt, by mechanisms that don’t involve transactivation of EGFR [19]. However, we considered the possibility that EP4 might have transactivated EGFR, but that this was masked by EGFR-independent Akt phosphorylation. To more directly assess EGFR activation, we co-expressed EGFR and the EP receptors in COS-7 cells and then assayed the status of EGFR using a phosphorylation-specific antibody. Consistent with the results in Fig. 2A, we found that PGE2 did not cause EGFR phosphorylation in cells expressing EP1, but did cause EGFR phosphorylation in cells expressing EP2 or EP3 (Fig. 2D). Surprisingly, EGFR was also phosphorylated in cells expressing EP4 (Fig. 2D). Using scanning densitometry to quantify the Western blots, we found statistically significant increases in phospho-EGFR in cells expressing EP2, EP3, and EP4 (Fig. 2E). In all cases, the metalloproteinase inhibitor, GM6001 completely abolished EGFR phosphorylation. We conclude that in these conditions, EP receptors 2–4 can transactivate EGFR and that they do so through a metalloproteinase.
EGFR growth factors augment expression of COX-2
Expression of COX-2 can be induced by a number of stimuli including phorbol esters, cytokines, and growth factors (reviewed in [20]). Some reports indicate that growth factors that activate EGFR can increase expression of COX-2. We examined whether TGFα or EGF could enhance expression of COX-2 by treating HEK293 cells with either of these growth factors or PDGF, which does not bind to EGFR. We found that both TGFα and EGF significantly increased expression of COX-2 protein while PDGF did not (Fig 3A). Using RT-PCR, we found that TGFα also increased expression of COX-2 mRNA. Combined with the ability of PGE2 to transactivate EGFR, these data suggested that growth in some tumors may be augmented by, an autocrine loop where COX-2 activates growth factor shedding, which in turn induces the expression of COX-2.
Figure 3.

EGFR growth factors increase the expression of COX-2 protein and mRNA. A. HEK293 cells were starved and then exposed overnight to EGF (20ng/mL), TGFα (5ng/mL), PDGF (50ng/mL), or PMA (20nM). Cell lysates were then analyzed to detect either COX-2 protein or mRNA. Results are representative of at least two experiments. B. HEK293 cells were transfected with the empty vector, wild-type EGFR, L858R EGFR, or delL747-P753insS EGFR. The cells were starved and then exposed overnight to TGFα (5ng/mL). COX-2 or EGFR were then detected by immunoblotting.
Recently, several mutations in the kinase domain of EGFR have been identified in tumors that appear to enhance response to the EGFR inhibitor, Gefitinib [21, 22]. Two of the more common mutations are a point mutation, L858R, and an eighteen base pair in-frame deletion, delL747-P753insS [23]. These mutations appear selectively activate Akt and STAT signaling pathways [23]. To test if these mutations affected expression of COX-2, we transfected HEK293 cells with either a control vector, wild-type EGFR, or one of the two EGFR mutants, treated the cells with TGFα for sixteen hours, and then assessed COX-2 expression by immunoblotting. We found that over-expression of wild-type EGFR increased expression of COX-2, both in basal and stimulated conditions. Over-expressing mutant, active EGFR had an even more profound effect on COX-2 expression (Fig 3B). Together, these results demonstrate that expression of COX-2 can be induced through EGFR and that kinase domain mutations in EGFR further augment COX-2 expression.
Inhibiting COX-2 reduces EGFR-dependent growth in three-dimensional cultures
To test the possibility that inhibiting COX-2 reduces tumor growth caused by EGFR, we created stable MCF10A breast cell lines that over-express EGFR. The cells also expressed COX-2 (Fig. 4A). MCF-10A cells, when grown in three dimensions, form hollow spheres that are structurally similar to normal breast ducts [12]]. We found that over-expression of EGFR in these cells caused them to continue growing beyond spheres to form complicated multi-lobed structures (Fig. 4B). Our previous results suggested a positive feedback loop where EGFR induced COX-2 expression, which in turn caused growth factor shedding that activated EGFR. To examine the effects of interrupting this loop, we treated the cells with 10μg/mL or 50μg/mL celecoxib. These concentrations are above the peak plasma levels (~1μg/mL) after a single dose of celecoxib in fasting adults, but we were unsure of its distribution in Matrigel because celecoxib is highly protein bound and, thus, might have a much lower effective concentration when added to the medium above the Matrigel. We found that celecoxib caused a dose dependent reduction in the size of the three dimensional structures (Fig. 4B–C). Ibuprofen (50μg/mL) had the same effect (data not shown). These results demonstrate an essential role for COX-2 in EGFR tumorigenesis and suggest that COX inhibitors might have an important role for targeted therapy in tumors where EGFR is over-expressed.
Figure 4.
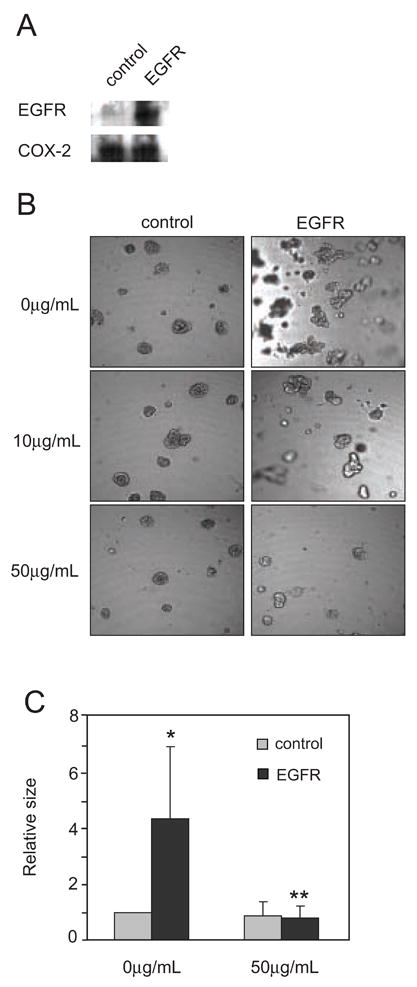
Inhibition of COX-2 reduces EGFR-dependent growth of breast epithelial cells in three-dimensional culture. A. Lysates from stable MCF-10A cell lines expressing either a control empty vector or EGFR were used to detect either EGFR or COX-2 by immunoblotting. B. Stable MCF-10A cells were grown in three-dimensional cultures in the presence or absence of Celecoxib (10μg/ml or 50μg/ml), a selective COX-2 inhibitor. Ibuprofen (50μg/mL) similarly inhibited growth (n = 2). C. Volumes of the three dimensional structures were calculated and normalized to control vector (no EGFR) without 50μg/mL celecoxib (n = 6–9). The * denotes significant change (p<0.02) compared to control cells without Celecoxib, and ** indicates significant change (p<0.01) compared to EGFR cells without Celecoxib.
DISCUSSION
We found that PGE2 transactivated EGFR by inducing release of a subset of its growth factor ligands. This suggests that over-expression of COX-2, which commonly occurs in many types of malignancies, contributes to tumor growth by activating EGFR, which would not only provide a growth signal, but would also enhance survival by activating Akt. Our results are consistent with several recent reports demonstrating activation of EGFR through EP receptors. Pai and coworkers demonstrated that PGE2 transactivated EGFR through release of TGFα [9], while another report [11] showed EGFR transactivation that did not depend on extracellular release of growth factors. Other groups have demonstrated transactivation of ErbB receptors, but they did not address whether or not growth factor release was necessary [24, 25]. While these reports concur that PGE2 can transactivate EGFR, they do not agree on the requirement for metalloproteinase activity. Consistent with a requirement for growth factor shedding, we found that GM6001, a broad spectrum metalloproteinase inhibitor, abolished EGFR transactivation and that PGE2 caused TGFα shedding through TACE. There are a number of possibilities that could explain the different observed requirements for metalloproteinase activity, including the different cell lines that have been used to measure EGFR transactivation. Indeed, given the complexity of GPCR signaling, it would not be surprising that transactivation might occur by different mechanisms in different cell types and in different malignancies.
Surprisingly, we observed that not all of the EGFR ligands were shed from cells expressing COX-2. We found that COX-2 increased basal shedding of TGFα, but did not increase basal shedding of amphiregulin, betacellulin or HB-EGF. We also tested whether COX-2 could augment growth factor shedding stimulated by phorbol esters, which cause shedding of all EGFR ligands. We found that COX-2 expression enhanced phorbol ester-stimulated TGFα shedding 1.7 (+/− 0.8) fold. COX-2 also augmented phorbol ester-stimulated amphiregulin shedding (2.0 +/− 0.4 fold increase), but did not augment betacellulin or HB-EGF shedding. These data indicate that under certain conditions, COX-2 might transactivate EGFR through either TGFα or amphiregulin. TGFα shedding in response to COX-2 expression or PGE2 is consistent with the report by Pai and coworkers [9]. This group, however, did not try to neutralize amphiregulin. There are numerous observations suggesting that amphiregulin contributes to the development of epithelial malignancies[7] and that PGE2 can induce expression of amphiregulin [26–28]. Combined with our data, these observations suggest that COX-2 might also promote tumorigenesis by augmenting amphiregulin shedding.
Several metalloproteinases in the ADAM family are known to shed EGFR ligands from the cell surface. TACE/ADAM17 appears to be largely responsible for basal and stimulated release of most EGFR ligands, while ADAM10 is necessary for basal shedding of betacellulin and EGF [29]. We found that COX-2 did not affect basal shedding of betacellulin, indicating that PGE2 might not activate ADAM10. Conversely, we demonstrated that TACE/ADAM17 was necessary for COX-2 to stimulate release of TGFα. In light of the broad role of TACE/ADAM17 in shedding EGFR ligands, the selective release of TGFα and amphiregulin—but not betacellulin and HB-EGF—in response to PGE2 was surprising. In addition to shedding growth factors, TACE has an important role in releasing a number of biologically active proteins including some cytokines and several different classes of receptors [30]. Little is known about how TACE might selectively shed a subset of its substrates from the cell surface, but it is clear that this must occur, because many of its substrates are concurrently expressed. One possibility is that adaptor proteins couple TACE to specific receptors and growth factor substrates. Suggesting that this might occur, the adaptor protein Eve-1, appears to bind TACE and other ADAMs and was necessary for ectodomain shedding of HB-EGF [31].
We tested the four known EP receptors and found that EP2-4 transactivated EGFR while EP1 did not. There are several reports indicating that EP2 is important for tumorigenesis. For example, ApcΔ716/+ mice had fewer gastrointestinal tumors when crossed with EP2−/− mice [32] and EP2 was necessary for mammary hyperplasia in COX-2 transgenic mice [28]. To our knowledge, there are no reports suggesting that EP3 can transactivate EGFR, but EP4 has been shown to be involved in tumor cell motility [33] and it is over-expressed in tumors from ApcΔ716/+ mice [32]. None of these reports provided a direct link between EP2 or EP4 and EGFR, but combined with our data, they suggest that transactivation of EGFR through these EP receptors might have a role in development of breast and colon cancer and other malignancies. In contrast to EP receptors 2–4, we found that over-expressed EP1 did not transactivate EGFR. However, Han and Wu recently demonstrated that an EP1 receptor agonist induced phosphorylation of EGFR and enhanced proliferation and migration of cholangiocarcinoma cells [24], and Su et al. showed that PGE2 transactivated ErbB2 through EP1 [25]. These differing results likely reflect differences between cell lines, opening the possibility that in the correct context, all four EP receptors can transactivate EGFR.
Once activated by its growth factors, EGFR causes a number of signaling events, many of which coordinate changes in gene transcription. We found increased COX-2 mRNA and protein in cells treated with EGFR agonists. Whether this occurred through a transcriptional event, stabilization of RNA, or both is under investigation. It is interesting to note that the kinase domain mutations in EGFR augmented COX-2 expression, suggesting the possibility that these mutations increase COX-2 expression in vivo. Other groups have demonstrated induction of COX-2 protein and mRNA by growth factors [1]. Combined with the reported induction of amphiregulin by COX-2 [26–28], these results suggest the existence of a self-perpetuating activation loop. COX-2 and EGFR are often concurrently expressed in tumors, indicating that combined inhibition of COX-2 and EGFR might have therapeutic benefits. Indeed, we demonstrated that inhibiting COX-2 significantly reduced in vitro growth of MCF-10A cells overexpressing EGFR, and Torrance et al. demonstrated that combined inhibition of EGFR and cyclooxygenase significantly reduced intestine polyp formation in APCMin/+ mice compared to cyclooxygenase or EGFR inhibition alone [34]. TACE also has a role in tumor formation [35], suggesting that metalloproteinase inhibitors might additionally inhibit tumor growth.
CONCLUSION
In conclusion, we have demonstrated that COX-2 transactivates EGFR through TACE. Of the four growth factors that we tested, only TGFα and amphiregulin were released while betacellulin and HB-EGF were not. Once activated, EGFR can induce expression of COX-2, potentially causing an autocrine loop to develop. We found that inhibiting COX-2 reduced growth of EGFR over-expressing cells in three dimensional cultures, suggesting that interrupting this autocrine loop might have therapeutic benefits.
Acknowledgments
This work was supported by the Huntsman Cancer Foundation, the R. Harold Burton Foundation, the National Institutes of Health Grants R01-CA95463 (to M.K.T.), and P01-CA73992 (to D.M.S.). S.C.U. was supported by a National Institutes of Health, (T32-CA93247). M. A. Al-Salihi was supported by a Pre-doctoral Fulbright Award (2003–05).
Abbreviations
- COX-2
cyclooxygenase-2
- EGFR
epidermal growth factor receptor
- TGFα
transforming growth factor-α
- ADAM
A-Disintegrin and Metalloproteinase
- GPCR
G protein-coupled receptor
- PGE2
prostaglandin E2
- EP
E-prostanoid receptor
- TACE
tumor necrosis factor-α converting enzyme
- EGF
epidermal growth factor
- PMA
phorbol 12-myristate 13-acetate
- PDGF
platelet-derived growth factor
- HB-EGF
heparin-binding EGF-like growth factor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dannenberg AJ, Subbaramaiah K. Cancer Cell. 2003;4:431–436. doi: 10.1016/s1535-6108(03)00310-6. [DOI] [PubMed] [Google Scholar]
- 2.Gupta RA, Dubois RN. Nat Rev Cancer. 2001;1:11–21. doi: 10.1038/35094017. [DOI] [PubMed] [Google Scholar]
- 3.Imperiale TF. N Engl J Med. 2003;348:879–880. doi: 10.1056/NEJMp030005. [DOI] [PubMed] [Google Scholar]
- 4.Oshima M, Dinchuk JE, Kargman SL, Oshima H, Hancock B, Kwong E, Trzaskos JM, Evans JF, Taketo MM. Cell. 1996;87:803–809. doi: 10.1016/s0092-8674(00)81988-1. [DOI] [PubMed] [Google Scholar]
- 5.Chulada PC, Thompson MB, Mahler JF, Doyle CM, Gaul BW, Lee C, Tiano HF, Morham SG, Smithies O, Langenbach R. Cancer Res. 2000;60:4705–4708. [PubMed] [Google Scholar]
- 6.Normanno N, Bianco C, De Luca A, Maiello MR, Salomon DS. Endocrine-Related Cancer. 2003;10:1–21. doi: 10.1677/erc.0.0100001. [DOI] [PubMed] [Google Scholar]
- 7.Normanno N, Bianco C, De Luca A, Salomon DS. Front Bioscience. 2001;6:685–707. doi: 10.2741/normano. [DOI] [PubMed] [Google Scholar]
- 8.Wetzker R, Bohmer FD. Nat Rev Mol Cell Biol. 2003;4:651–657. doi: 10.1038/nrm1173. [DOI] [PubMed] [Google Scholar]
- 9.Pai R, Soreghan B, Szabo IL, Pavelka M, Baatar D, Tarnawski AS. Nat Med. 2002;8:289–293. doi: 10.1038/nm0302-289. [DOI] [PubMed] [Google Scholar]
- 10.Breyer RM, Bagdassarian CK, Myers SA, Breyer MD. Annu Rev Pharmacol Toxicol. 2001;41:661–690. doi: 10.1146/annurev.pharmtox.41.1.661. [DOI] [PubMed] [Google Scholar]
- 11.Buchanan FG, Wang D, Bargiacchi F, Dubois RN. J Biol Chem. 2003;278:35451–35457. doi: 10.1074/jbc.M302474200. [DOI] [PubMed] [Google Scholar]
- 12.Debnath J, Muthuswamy SK, Brugge JS. Methods. 2003;30:256–268. doi: 10.1016/s1046-2023(03)00032-x. [DOI] [PubMed] [Google Scholar]
- 13.Dixon DA, Tolley ND, King PH, Nabors LB, McIntyre TM, Zimmerman GA, Prescott SM. J Clin Invest. 2001;108:1657–1665. doi: 10.1172/JCI12973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kutchera W, Jones DA, Matsunami N, Groden J, McIntyre TM, Zimmerman GA, White RL, Prescott SM. Proc Natl Acad Sci (USA) 1996;93:4816–4820. doi: 10.1073/pnas.93.10.4816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Prenzel N, Fischer OM, Streit S, Hart S, Ullrich A. Endocrine-Related Cancer. 2001;8:11–31. doi: 10.1677/erc.0.0080011. [DOI] [PubMed] [Google Scholar]
- 16.Prenzel N, Zwick E, Daub H, Leserer M, Abraham R, Wallasch C, Ullrich A. Nature. 1999;402:884–888. doi: 10.1038/47260. [DOI] [PubMed] [Google Scholar]
- 17.Seals A, Courtneidge SA. Genes Dev. 2003;17:7–30. doi: 10.1101/gad.1039703. [DOI] [PubMed] [Google Scholar]
- 18.Peschon JJ, Slack JL, Reddy P, Stocking KL, Sunnarborg SW, Black RA. Science. 1998;282:1281–1284. doi: 10.1126/science.282.5392.1281. [DOI] [PubMed] [Google Scholar]
- 19.Gutkind JS. J Biol Chem. 1998;273:1839–1842. doi: 10.1074/jbc.273.4.1839. [DOI] [PubMed] [Google Scholar]
- 20.Dannenberg AJ, Lippman SM, Mann JR, Subbaramaiah K, Dubois RN. J Clin Oncol. 2005;23:254–266. doi: 10.1200/JCO.2005.09.112. [DOI] [PubMed] [Google Scholar]
- 21.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J, Haber DA. N Engl J Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 22.Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y, Eck MJ, Sellers WR, Johnson BE, Meyerson M. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 23.Sordella R, Bell DW, Haber DA, Settleman J. Science. 2004;305:1163–1167. doi: 10.1126/science.1101637. [DOI] [PubMed] [Google Scholar]
- 24.Han C, Wu T. J Biol Chem. 2005;280:24053–24063. doi: 10.1074/jbc.M500562200. [DOI] [PubMed] [Google Scholar]
- 25.Su JL, Shih JY, Yen ML, Jeng YM, Chang CC, Hsieh CY, Wei LH, Yang PC, Kuo ML. Cancer Res. 2004;64:554–564. doi: 10.1158/0008-5472.can-03-1301. [DOI] [PubMed] [Google Scholar]
- 26.Shao J, Evers BM, Sheng H. J Biol Chem. 2004;279:14287–14293. doi: 10.1074/jbc.M313276200. [DOI] [PubMed] [Google Scholar]
- 27.Johansson CC, Yndestad A, Enserink JM, Ree AH, Aukrust P, Tasken K. Endocrinology. 2004;145:5177–5184. doi: 10.1210/en.2004-0232. [DOI] [PubMed] [Google Scholar]
- 28.Chang SH, Ai Y, Breyer RM, Lane TF, Hla T. Cancer Res. 2005;65:4496–4499. doi: 10.1158/0008-5472.CAN-05-0129. [DOI] [PubMed] [Google Scholar]
- 29.Sahin U, Weskamp G, Kelly K, Zhou HM, Higashiyama S, Peschon J, Hartmann D, Saftig P, Blobel CP. J Cell Biol. 2004;164:769–779. doi: 10.1083/jcb.200307137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moss ML, Lambert MH. Essays Biochem. 2002;38:141–153. doi: 10.1042/bse0380141. [DOI] [PubMed] [Google Scholar]
- 31.Tanaka M, Nanba D, Mori S, Shiba F, Ishiguro H, Yoshino K, Matsuura N, Higashiyama S. J Biol Chem. 2004;279:41950–41959. doi: 10.1074/jbc.M400086200. [DOI] [PubMed] [Google Scholar]
- 32.Sonoshita M, Takaku K, Sasaki N, Sugimoto Y, Ushikubi F, Narumiya S, Oshima M, Taketo MM. Nat Med. 2001;7:1048–1051. doi: 10.1038/nm0901-1048. [DOI] [PubMed] [Google Scholar]
- 33.Sheng H, Shao J, Washington MK, Dubois RN. J Biol Chem. 2001;276:18075–18081. doi: 10.1074/jbc.M009689200. [DOI] [PubMed] [Google Scholar]
- 34.Torrance CJ, Jackson PE, Montgomery E, Kinzler KW, Vogelstein B, Wissner A, Nunes M, Frost P, Discafani CM. Nat Med. 2000;6:1024–1028. doi: 10.1038/79534. [DOI] [PubMed] [Google Scholar]
- 35.Borrell-Pages M, Rojo F, Albanell J, Baselga J, Arribas J. EMBO J. 2003;22:1114–1124. doi: 10.1093/emboj/cdg111. [DOI] [PMC free article] [PubMed] [Google Scholar]