

Abstract
Despite the importance of Mcl-1, an anti-apoptotic Bcl-2 family member, in the regulation of apoptosis, little is known regarding its role in nervous system development and injury-induced neuronal cell death. Because germline deletion of Mcl-1 results in peri-implantation lethality, we address the function of Mcl-1 in the nervous system using two different conditional Mcl-1 mouse mutants in the developing nervous system. Here, we show for the first time that Mcl-1 is required for neuronal development. Neural precursors within the ventricular zone and newly committed neurons in the cortical plate express high levels of Mcl-1 throughout cortical neurogenesis. Loss of Mcl-1 in neuronal progenitors results in widespread apoptosis. Double labeling with active caspase 3 and Tuj1 reveals that newly committed Mcl1 deficient neurons undergo apoptosis as they commence migration away from the ventricular zone. Examination of neural progenitor differentiation in vitro demonstrated that cell death in the absence of Mcl1 is cell autonomous. Although conditional deletion of Mcl-1 in cultured neurons does not trigger apoptosis, loss of Mcl-1 sensitizes neurons to an acute DNA damaging insult. Indeed, the rapid reduction of Mcl-1 mRNA and protein levels are early events after DNA damage in neurons, and maintaining high Mcl-1 levels can protect neurons against death. Together, our results are the first to demonstrate the requirement of Mcl-1, an anti-apoptotic Bcl-2 family protein, for cortical neurogenesis and the survival of neurons after DNA damage.
Keywords: neurogenesis, cell death, neuronal progenitors, neuron, apoptosis, development
Introduction
Myeloid cell leukemia 1 (Mcl-1) is an anti-apoptotic member of the Bcl-2 family of proteins. Germline knock-outs (KOs) of Mcl-1 are peri-implantation lethal at embryonic day 3.5 (E3.5) (Rinkenberger et al., 2000), which is the most severe phenotype among the anti-apoptotic Bcl-2 proteins. Furthermore, Mcl-1 is essential for the development and maintenance of B and T lymphocytes and for the survival of hematopoietic stem cells (Opferman et al., 2003, 2005). Mcl-1 is believed to inhibit cell death through interactions with proapoptotic Bcl-2 family members, including multidomained (Youle and Strasser, 2008) and BH3-only (for review, see Letai, 2008). The affinity of interaction between Bcl-2 family members varies significantly, with Mcl-1 showing increased binding affinity for Noxa, PUMA (p53-upregulated modulator of apoptosis), BimEL, and Bcl-2 modifying factor (Chen et al., 2005; Kuwana et al., 2005).
The peri-implantation lethality of Mcl-1 germline mutant mice suggests a unique requirement for Mcl-1 in early embryogenesis (Rinkenberger et al., 2000). Bcl-xl germline deficient mice are also embryonic lethal, at E13, showing increased apoptotic activity in the brain and hematopoietic system (Motoyama et al., 1995). More recent studies have suggested that the requirement for Bcl-xl may vary depending on the cell type examined. For example, conditional deletion of Bcl-xl in catecholaminergic neurons resulted in viable mice with the catecholaminergic neuronal population reduced by one-third (Savitt et al., 2005). Mice with a Bcl-2 targeted deletion show normal neuronal development but exhibit significant loss of sympathetic, motor, and sensory neurons postnatally (Michaelidis et al., 1996). Although these studies demonstrate the importance of Bcl-2 family proteins in the maintenance of certain cell populations within the CNS, none have been shown to have a direct role in neural development.
Despite the importance of Mcl-1 in apoptosis during early development, little is known regarding its role in nervous system development and injury-induced neuronal cell death. Recent studies, however, have shown that Mcl-1 upregulation may play a role in Notch-1-mediated survival of neural precursor cells (Oishi et al., 2004), as well as in maintaining the survival of granule cells during migration and differentiation (Zhang and D'Ercole, 2004). Heterozygous Mcl-1 germline deletion also results in increased susceptibility of neurons to pilocarpine-induced seizure injury (Mori et al., 2004). These studies suggest that Mcl-1 may play an important role in nervous system development, and after acute neuronal injury, however, the impact of Mcl-1 deficiency during nervous system development or after acute injury has never been examined.
In this study, we show for the first time that Mcl-1 is required for neural development. Loss of Mcl-1 in neuronal progenitor cells results in widespread apoptosis of progenitors expressing Nestin and βIII tubulin (Tuj1), demonstrating that Mcl-1 is required for the survival of newborn neurons. We also show that Mcl-1 plays a key role in the regulation of neuronal survival after injury. Loss of Mcl-1 sensitizes neurons to apoptosis induced by DNA damage, whereas maintenance of high Mcl-1 levels protects neurons against death. Together, our results demonstrate that Mcl-1 is required for neural precursor survival and the regulation of injury-induced neuronal cell death.
Materials and Methods
Mice and primary neuronal cultures.
For embryonic time points, the time of plug identification was counted as E0.5. All experiments were approved by the University of Ottawa Animal Care Ethics Committee adhering to the Guidelines of the Canadian Council on Animal Care. Cortical and cerebellar granule neurons (CGNs) were cultured as described previously (Cregan et al., 1999; Fortin et al., 2001).
Generation of transgenic mice.
Floxed Mcl-1 mice were described previously (Opferman et al., 2003). The generation of telencephalon-specific Mcl-1 conditional mutants was accomplished by breeding floxed Mcl-1 mice with Foxg1:Cre mice (Hebert and McConnell, 2000) to generate Foxg1cre/+:Mcl-1flox/flox mice. The generation of nestin-specific conditional mutants, floxed Mcl-1 mice were crossed with Nestin:Cre mice (Berube et al., 2005) to generate Nestin cre/+:Mcl-1 flox/flox mice. The genotyping of these mice was performed as described previously (Hebert and McConnell, 2000; Casanova et al., 2001; Opferman et al., 2003). The floxed Mcl-1 mice were on a C57BL/6 background, the Nestin:Cre lines were on a pure FVBN background, and the Foxg1:Cre mice were a mixed C57BL/6 and FVBN background. Double heterozygous littermates (Cre/+:Mcl-1flox/+) were used as controls for in vivo experiments, because they were indistinguishable from wild-type embryos (results not shown).
Tissue processing, immunohistochemistry, and in situ hybridization.
Tissue fixation and cryoprotection of embryonic tissue was preformed as described previously (Ferguson et al., 2002). Briefly, embryos were dissected and postfixed overnight in 4% PFA and cryoprotected in a sucrose gradient of 12, 16, and 22% sucrose (Sigma-Aldrich) in PBS. Tissue was frozen, and 14 μm coronal cryosections were collected on Superfrost Plus slides (12–550-15; Fisher Scientific). Sections were stained in 0.1% cresyl violet for light microscopy or immunohistochemistry as described previously (Ferguson et al., 2002) for active caspase 3 (AC3) (1:100; Cell Signaling Technology 9664), phospho-Histone H3 (PH3) (1:500; Upstate Biotechnology, 06–570), Nestin (mouse monoclonal, 1:200; Research Diagnostics, 21714), Tuj1 (1:100), and Doublecortin (DCX) (1:200; Santa Cruz Biotechnology). For double immunofluorescence of AC3 and DCX, sections were postfixed in 4.0% paraformaldehyde before staining. PH3-positive cells were counted along a 1000 μm length of the lateral ventricle from the dorsal midline extending laterally at three distinct regions through the rostrocaudal extent of the lateral ventricle. The mean number of PH3-positive cells in Foxg1:Cre Mcl-1 mutant and control brains was compared by Student's t test. In situ hybridization was performed with a riboprobe to Mcl-1 (Rinkenberger et al., 2000) according to previously published protocols (Vanderluit et al., 2004; Ruzhynsky et al., 2007).
Confocal microscopy.
All images were taken using a Zeiss 510 meta confocal microscope. To identify colocalization of active caspase 3 with Tuj1, Nestin, or doublecortin-positive cells, Z stack sections (0.5–1 μm per section) were taken on different fields using an 63× oil immersion objective. The counts were performed individually analyzing six different sections of telencephalon, including ventral (V), dorsal (D), lateral ventral (LV), lateral dorsal (LD), medial ventral (MV), and medial dorsal (MD) areas (see Fig. 5 B). Colocalization for active caspase 3 and doublecortin was counted in the ganglionic eminence. For each animal, two different brain sections were analyzed for three different animals per experiment.
Figure 5.

Mcl-1 deficiency results in increased cell death of Nestin-expressing progenitor cells. A, Confocal photomicrographs of coronal sections from E12.5 Nestin:Cre Mcl-1 mutant and control brains, immunostained for active caspase 3 and Nestin, a marker for neuronal progenitor cells. B, Schematic of the six counting areas around the ventricle: ventral, dorsal, lateral dorsal, lateral ventral, medial dorsal, and medial ventral. C, Quantitative analysis of AC3 and Nestin+ cells in the six different regions. Within the mutant brain, the LV region had significantly greater numbers of Nestin+ apoptotic cells.
Cell culture and recombinant adenovirus infection.
The cDNA for Mcl-1 (mouse) (Rinkenberger et al., 2000) was a gift from Dr. S. Korsmeyer (Dana-Farber Cancer Institute, Harvard Medical School, Boston, MA). Recombinant adenoviral vectors carrying expression cassettes for Mcl-1 or Cre were constructed, purified, and titered as described previously (Cregan et al., 2000). Adenoviral vectors were added to cell suspensions immediately before plating. Multiplicity of infection (MOI) refers to the number of plaque forming units (pfu) per cell, as calculated from viral titers (pfu/μl).
Semiquantitative reverse transcription-PCR analysis.
Total RNA was isolated from cells using Trizol isolation reagent according to the manufacturer instructions (Invitrogen). Pilot experiments were performed to determine the linear range of amplification with respect to quantity of starting template and PCR cycles. The primers used for the detection of genes are shown in Table 1. Total RNA (25–100 ng) was used for cDNA synthesis and targeted gene amplification using the SuperScript One-Step Reverse Transcription (RT)-PCR kit (Invitrogen). cDNA synthesis was performed at 48°C for 45 min followed by a 2 min initial denaturation step at 94°C. This was followed by 24 cycles (Bax), 25 cycles (Mcl-1, S12), 29 cycles (Bcl-2), 32 cycles (Bcl-w), 30 cycles (Bcl-xl), and 30 cycles (Bak) at 94°C for 30 s, 55–64°C for 30 s, and 72°C for 1 min. The resulting products were sequenced to confirm identity.
Table 1.
Primer sequences for RT-PCR gene expression analysis
| Gene | Sequence |
|
|---|---|---|
| 5′-sense-3′ | 5′-anti-sense-3′ | |
| Mcl-1 | TCTGAGATGTTTGGCCTGCGGAGA | TCTAGACTATCTTATTAGATATGCCAGAC |
| Bcl-2 | CTCGTCGCTACCGTCGTGACTTCG | CAGATGCCGGTTCAGGTACTCAGTC |
| Bcl-W | CGAGTTTGAGACCCGTTTCCGCC | GCACTTGTCCCACCAAAGGCTCC |
| Bcl-xL | AGGCAGGCGATGAGTTTGAA | CGGCTCTCGGCTGCTGCATT |
| Bax | AAGCTGAGCGAGTGTCTCCGGCG | GCCACAAAGATGGTCACTGTCTGCC |
| Bak | CACCCCCACCTCATCGTCCATTTC | GCCCACCCCCTCCCCTGTCATA |
| S12 | GGAAGGCATAGCTGCTGG | CCTCGATGACATCCTTGG |
Western blot analysis.
Western blot analysis was performed as described previously (Cregan et al., 1999) with antibodies against Mcl-1 (1:10,000; Rockland Immunochemicals) (Opferman et al., 2003, 2005) and actin (1:2000; SC-1616, Santa Cruz Biotechnology) as a loading control.
Camptothecin treatment and cell viability assays.
For inducing acute DNA damage, neurons were treated with 10 μm camptothecin (CPT) after 2 d in vitro (DIV). Cell survival was measured by the following: LIVE/DEAD viability/cytotoxicity kit (Invitrogen) as described previously (Cregan et al., 2004), and Hoechst nuclear staining with condensed nuclei as an indicator of apoptosis. For Hoechst counts, cells were fixed with 4% PFA in PBS for 30 min and then washed with 1× PBS and stained with Hoechst (33258; Sigma Chemical). For LIVE/DEAD assays and Hoechst counts, data represent the mean and SE from a minimum of three independent experiments.
Cortical progenitor cultures.
Cortical progenitor cells were cultured as described previously (Callaghan et al., 1999). Briefly, cortices were dissected from Foxg1:Cre Mcl1 mutant and littermate control embryos at E12.5. Cortices were mechanically dissociated by trituration, and cell aggregates were plated on polyornithine-coated four-well dishes and cultured in media containing Neurobasal medium (Invitrogen), 0.5 mm glutamine, 0.5% penicillin-streptomycin, 1% N2 supplement (Invitrogen), 2% B27 supplement (Invitrogen), and 10 ng/ml FGF-2 (Sigma). On day one, the media was replaced with fresh media without FGF2. After 3 d, cultures were fixed in 4% PFA, and cell nuclei were stained with Hoechst (Sigma) to identify healthy (diffuse nuclear staining) versus apoptotic (condensed nuclei) cells. The number of apoptotic and healthy cells were quantified per aggregate and represented as the total number of cells per aggregate. Newly committed neurons were identified by immunostaining with antibodies to Tuj1. Cell counts were performed to quantify the total number of Tuj1-positive cells, and the number of apoptotic neurons (Tuj1 positive with condensed nuclei) per aggregate. The number of apoptotic cells/aggregate and the number of apoptotic neurons/total number of neurons in each aggregate were represented as a percentage.
Results
Mcl-1 is required for cortical neurogenesis
During nervous system development, Bcl-2 family proteins play critical roles in shaping the developing brain by regulating the survival of neural precursor cells and neuronal number after neurogenesis (Akhtar et al., 2004). Although Mcl-1 has been shown to play an important role in the development of the immune system, the question as to whether Mcl-1 has any role in nervous system development remains unknown. To address this question, we first asked whether Mcl-1 was expressed in the developing brain and in which population of cells. In situ hybridization with a riboprobe to Mcl-1 was performed on sections from developing mouse brains during the period of cortical neurogenesis from E11 to E15.5 (Fig. 1). Robust expression of Mcl-1 was observed within the neural precursor population in the ventricular zone (VZ) at E11.5. By midneurogenesis at E15.5, Mcl-1 is still highly expressed within the ventricular zone and in postmitotic neurons in the developing cortical plate (CP) (Fig. 1). The high levels of Mcl-1 in both proliferating neural precursors and postmitotic neurons during this critical period of brain development lead us to question whether Mcl-1 regulates the survival of these populations.
Figure 1.

Mcl-1 is expressed in neural precursors and postmitotic neurons in the developing mouse telencephalon. In situ hybridization analysis of Mcl-1 mRNA expression in telencephalic coronal sections of E11.5 (B) and E15.5 (C) mouse embryos. Note the absence of the positive signal in the section of the brain hybridized with sense control riboprobe (A). Scale bar, 250 μm.
To assess the role of Mcl-1 in nervous system development, we used conditional mutants as Mcl-1 germline mutation results in preimplantation lethality (Rinkenberger et al., 2000). Animals in which Cre-recombinase was inserted at the Foxg1 locus (Foxg1:Cre) (Hebert and McConnell, 2000) were interbred with mice carrying a floxed Mcl-1 allele (Opferman et al., 2003). In these animals, Cre is expressed in neural progenitors throughout the developing telencephalon beginning at E8 and reaching maximal expression levels at E17 (Tao and Lai, 1992; Shimamura and Rubenstein, 1997). Previous studies have revealed efficient Cre-mediated recombination of floxed alleles specifically using this Foxg1:Cre mouse model (Hebert and McConnell, 2000; Ferguson et al., 2002). By interbreeding floxed Mcl-1 mice with Foxg1:Cre, we could selectively assess the role of Mcl-1 in neural precursor cells during forebrain development.
Conditional deletion of Mcl-1 specifically within the forebrain resulted in embryonic lethality at E16–E17; however, embryos collected at earlier time points were found at expected Mendelian ratios (Table 2). The expression of Foxg1 in the midbrain-hindbrain junction, a region important for regulating cardiovascular and respiratory function, would result in Cre-mediated Mcl-1 deficiency in this region and may account for the embryonic lethality. Indeed, we and others have shown that Foxg1:Cre-mediated deletion of Rb (Ferguson et al., 2002) or Smo (Fuccillo et al., 2004) results in perinatal lethality.
Table 2.
Recovery of embryos from Mcl-1 conditional mutants
| Mcl-1 conditional mutant | Embryonic time point |
||||||
|---|---|---|---|---|---|---|---|
| E9.5 | E10.5 | E12.5 | E15.5 | E16.5 | E17.5 | E19.5 | |
| Foxg1:Cre Mcl-1 KO | |||||||
| Total number of embryos | 20 | 32 | 11 | 11 | 4 | ||
| Number of knock-outs (%) | 3 (15%) | 8 (25%) | 1 (9%) | 0 (0%) | 0 (0%) | ||
| Expected number Mendelian ratio (%) | 4 (20%) | 7 (21%) | 2 (25%) | 2 (25%) | 1 (25%) | ||
| Nestin:Cre Mcl-1 KO | |||||||
| Total number of embryos | 9 | 30 | 74 | 20 | |||
| Number of knock-outs (%) | 2 (22%) | 8 (27%) | 9 (12%) | 0 (0%) | |||
| Expected number Mendelian ratio (%) | 2 (25%) | 8 (27%) | 17 (24%) | 5 (25%) | |||
To assess the role of Mcl-1 in brain development, embryos were collected at E15.5 corresponding to midneurogenesis. This stage is characterized by a large population of Nestin-expressing progenitors within the VZ and subventricular zone (SVZ), early committed neuroblasts within the SVZ/intermediate zone (IZ), and a growing population of postmitotic neurons in the developing CP. Efficient recombination of the floxed Mcl-1 allele in the telencephalon was confirmed by Western blot analysis (Fig. 2 A). Animals lacking Mcl-1 in the developing telencephalon exhibit a dramatic neural phenotype with the overall size of the mutant telencephalon being significantly smaller relative to littermate controls (Fig. 2 B). Morphological assessment of coronal sections through the telencephalon at E15 revealed a striking reduction in the size of the cortical plate. In contrast to littermate controls in which layering of the VZ/SVZ, IZ, and CP are delineated clearly, Mcl-1 mutant mice lacked a clear distinction between the IZ and CP (Fig. 2 C). These results demonstrate that Mcl-1 is essential for telencephalic development.
Figure 2.
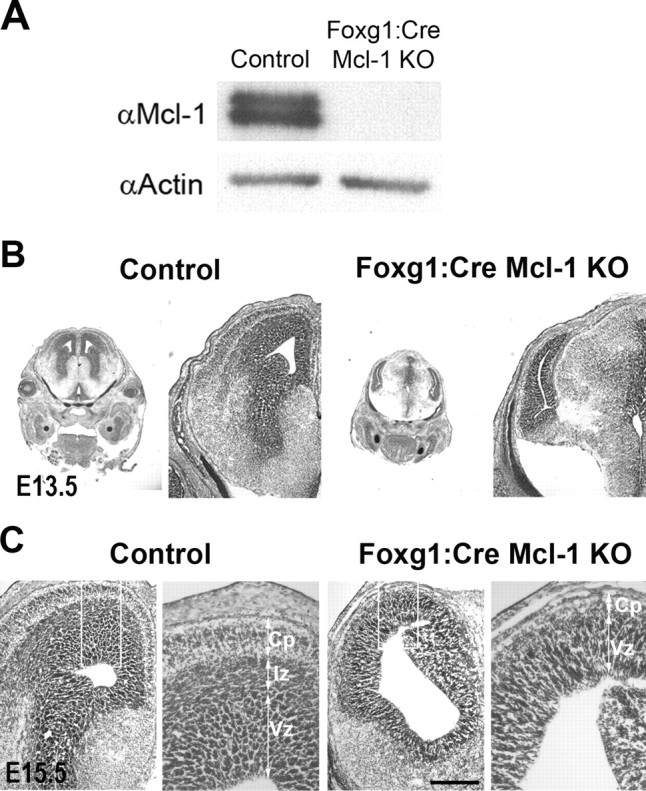
Loss of Mcl-1 in developing neurons results in severe defects in cortical neurogenesis. A, Protein lysates were collected from cortical neurons cultured from Foxg1:Cre mutant or control mice 48 h after plating and were blotted for Mcl-1 expression then for actin as a loading control, showing efficient excision of Mcl-1 in mutant cortical neurons. B, Cresyl violet-stained coronal sections of E13.5 Foxg1:Cre mutant mice and littermate controls showing significant loss of cortical size and morphology. C, Cresyl violet-stained coronal sections of E15.5 Foxg1:Cre mutant mice and littermate controls showing Mcl-1 deficiency has a pronounced effect on CP formation. The right panels are a magnification of the boxed areas. Scale bar, 250 μm.
The dramatic reduction in cortical plate formation led us to question whether this defect occurred before or after precursor commitment to a neuronal fate. A defect before neuronal commitment could be associated with Mcl-1 having a role in neural progenitor proliferation or survival, whereas a defect after commitment would be attributed to Mcl-1 playing a role in the survival of newly born neurons. We first determined whether Mcl-1 has a crucial role in the regulation of uncommitted neural precursor cells. Previous studies have suggested that Mcl-1 may modulate the rate of cell division (Fujise et al., 2000; Jamil et al., 2005). To determine whether Mcl-1 affects neural progenitor proliferation, we quantified the number of cycling cells in Mcl-1 deficient brains using the M-phase cell cycle marker PH3 (Fig 3). Quantitative analysis of the number of cycling cells revealed no significant difference between Mcl-1 deficient (40.2 ± 5.1) and littermate controls (27.5 ± 1.7), demonstrating that Mcl-1 does not perturb neural precursor cell proliferation. We next asked whether Mcl-1 was required for neural precursor cell survival.
Figure 3.

Mcl-1 deficiency does not affect proliferation of cortical progenitor cells. Coronal sections of E15.5 Foxg1:Cre Mcl-1 mutant mice and littermate controls immunostained for PH3. Quantitative analysis of PH3-positive cells in the ventricular zone revealed no significant difference in the number of proliferating cells between mutant and control mice. Scale bar, 100 μm.
We assessed the role of Mcl-1 in neural precursor cell survival in vivo in the Foxg1:Cre Mcl-1 mutant and in a second conditional knock-out in which Cre is expressed in neural precursor cells. Transgenic mice in which Cre-recombinase is expressed under the control of the nestin promoter (Nestin:Cre) (Berube et al., 2005) were interbred with mice carrying a floxed Mcl-1 allele (Opferman et al., 2003). Nestin is expressed in neural progenitors throughout the developing nervous system at E7.5, after preplate formation (Dahlstrand et al., 1995). The use of two conditional mutants allowed us to drive Cre expression very early in development and examine the role of Mcl-1 in neural precursor cells. The absence of Mcl-1 in the developing CNS with the Nestin:Cre resulted in embryonic lethality occurring before E15 (Table 2). Morphological analysis of coronal sections through the brains of E12.5 embryos revealed a similar cortical phenotype as the Foxg1:Cre mutant embryos with a dramatic reduction in cortical plate formation (Fig. 4 A). Deletion of Mcl-1 in neural progenitor cells by either Foxg1:Cre or Nestin:Cre conditional mutants seriously impaired the development of the cortex. The striking reduction in the telencephalon in Mcl-1 mutants suggests that Mcl-1 is required for the survival of precursors in the developing brain.
Figure 4.
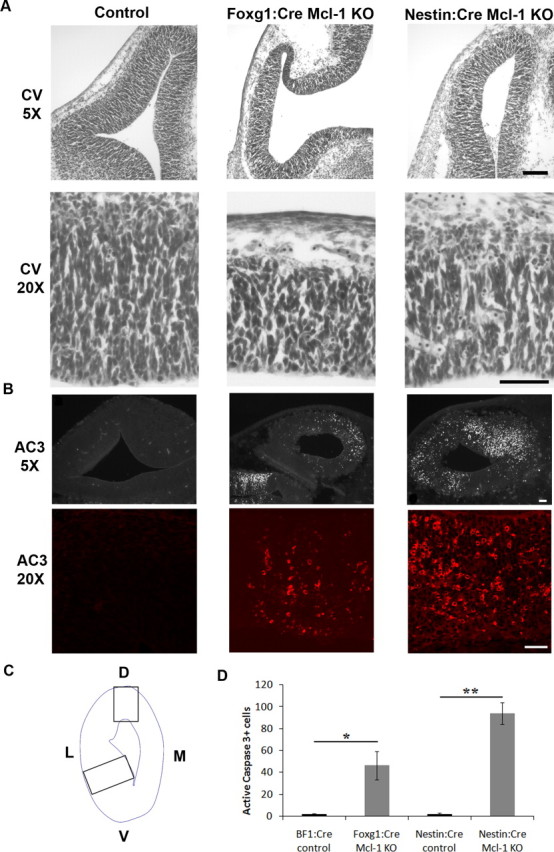
Loss of Mcl-1 in developing neurons results in increased apoptotic activity in neuronal progenitors. A, Cresyl violet-stained coronal sections through the telencephalic hemisphere of E12.5 Foxg1:Cre and Nestin:Cre Mcl-1 mutant embryos and littermate controls showing a significant reduction in the thickness of the developing cortex in the Mcl-1 mutants. The bottom panel represents high-magnification photomicrographs of the developing cortex. B, Coronal sections from E12.5 Foxg1:Cre and Nestin:Cre Mcl-1 mutants and littermate controls were immunostained for AC3, a marker of apoptotic cell death. Numerous active caspase-3-positive cells are dispersed throughout the VZ in Mcl-1 deficient mice. The bottom panels are higher-magnification photomicrographs. C, Schematic representation of areas quantified. D, Quantitative analysis of AC3-positive cells reveals a significant increase in apoptotic cells in mutant animals. L, Lateral; M, medial. Scale bars: A, 100 μm; B, 50 μm. *p < 0.05, **p < 0.01.
To determine whether the observed cell loss in the ventricular zone is caused by apoptosis, sections from Foxg1:Cre and Nestin:Cre mutant embryos along with control littermates at E12.5 were immunostained with antibodies to active caspase 3, a hallmark of classical apoptotic cell death. Numerous active caspase 3-positive cells were present within the VZ/SVZ of Mcl-1 mutant mice, whereas only an occasional apoptotic cell was observed in littermate controls (Fig. 4 B). The total number of active caspase 3-expressing cells was quantified in two distinct regions of the developing cortex (Fig. 4 C). A dramatic increase in the number of apoptotic cells was found in both Foxg1:Cre and Nestin:Cre Mcl-1 deficient brains compared with littermate controls (Fig. 4 D). Our results show that Mcl-1 mutants exhibit extensive apoptotic cell death in the developing cortex relative to littermate controls, which demonstrate an essential role for Mcl-1 in the maintenance of neural precursor survival during cortical development.
Because apoptotic cells are seen throughout the cortex, we next asked which cell types require Mcl-1 for survival: the newly committed neurons or the uncommitted precursors. To distinguish these possibilities, sections were double labeled with antibodies to active caspase 3 and Nestin to identify neural precursor cells, or active caspase 3 and Tuj1 to identify newly committed neurons. Sections were examined for coexpression of cell type-specific markers and active caspase 3, and the percentage of cells exhibiting caspase activation within each population was quantified. We first asked whether uncommitted neural precursor cells required Mcl-1 for survival. Double labeling revealed that a large proportion of Nestin-expressing cells underwent cell death in the absence of Mcl-1 (Fig. 5 A). To determine whether Mcl-1 deficiency resulted in region-specific cell death, we quantified the percentage of Nestin-expressing apoptotic cells in different regions surrounding the lateral ventricles, including the dorsal, lateral dorsal, medial dorsal, lateral ventral, and medial ventral regions (Fig. 5 B). Examination of cell death around the ventricles revealed that a lack of Mcl-1 induces a highly significant increase in cell death of uncommitted neural precursor cells. The highest percentage of precursors double stained for active caspase 3 and Nestin was in the ventral and ventral lateral regions in the Mcl-1 deficient brains with 24.7 ± 7.73 and 32 ± 8.8%, respectively, relative to 0.4 ± 0.41 and 0.5 ± 0.34% of controls (Fig. 5 C). These results demonstrate that neural precursors require Mcl-1 for survival during development and that the ventral populations at E12 are most affected.
Because many caspase 3-expressing cells were found in the intermediate zone where progenitors migrate away from the ventricle, we next asked whether precursor cells undergo apoptosis after commitment to a neuronal fate as they commence migration. To determine whether these newly born neurons required Mcl-1 for survival, coronal sections from E12.5 embryos were double labeled for Tuj1 and active caspase 3, and newly born neurons undergoing cell death were counted and compared with littermate controls (Fig. 6 A). Consistent with a reduction in the cortical plate, the number of newly born neurons undergoing cell death was significantly increased in Mcl-1 mutants, whereas 12.0 ± 0.86% of Tuj1-expressing cells colabeled with the apoptotic marker relative to only 1.2 ± 0.86% in littermate controls. In the lateral ventral region, the percentage of dying cells was 7.03 ± 2.1 versus 1.0 ± 0.33% in the control group (Fig. 6 B). To confirm these findings, we assessed whether committed neurons undergo cell death as they initiate migration to their final destination. Double labeling with doublecortin, a marker for newly migrating populations, and active caspase 3 revealed that these migratory precursor cells were undergoing cell death as they left the ventricular zone and initiated migration (Fig. 7 A). The number of doublecortin-expressing cells undergoing cell death was 64.5 ± 6.4 in Mcl-1 mutant mice relative to 0.3 ± 0.3 in littermate controls. These results explain the reduction in cortical thickness observed in Mcl-1 mutant mice, where Mcl-1 deficient neural precursor cells undergo cell death as they commit to a neuronal fate and migrate away from the ventricular zone.
Figure 6.
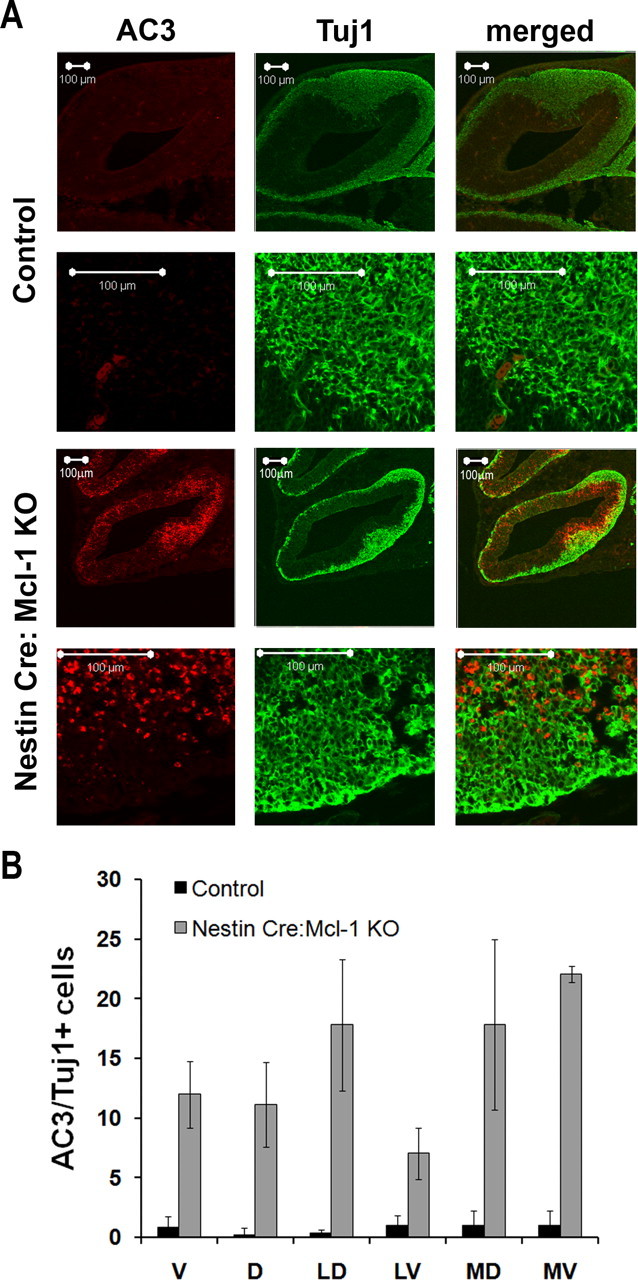
Mcl-1 deficiency results in cell death in newly committed neurons. A, Confocal photomicrographs of coronal sections from E12.5 Nestin:Cre Mcl-1 mutant and control brains immunostained for active caspase 3 and Tuj1, an early neuronal marker. B, Quantitative analysis of AC3/Tuj1-positive cells in each of the regions around the cortex. Nestin:Cre mutant brains have significantly more apoptotic immature neurons relative to control littermates.
Figure 7.
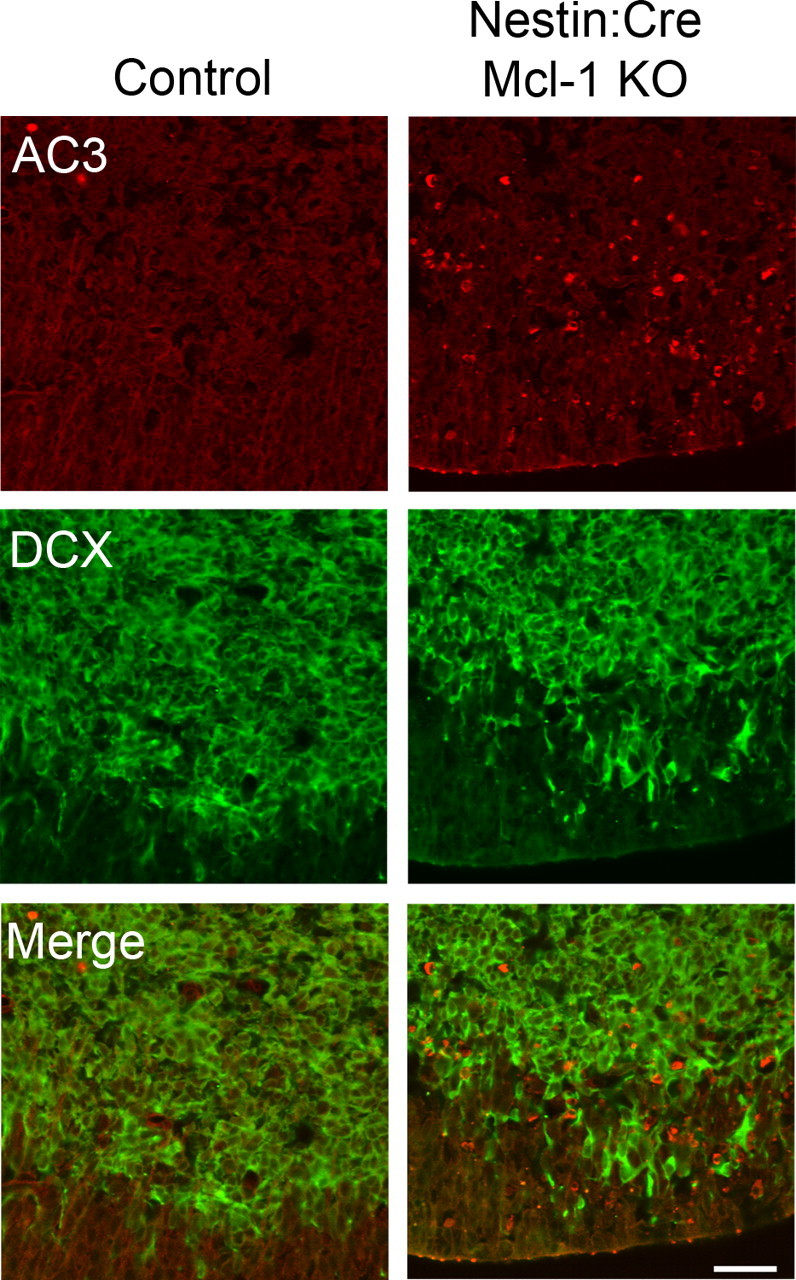
Loss of Mcl-1 results in death of migratory neuroblasts. Confocal photomicrographs of coronal sections from E12.5 Nestin:Cre Mcl-1 mutant and control brains immunostained for active caspase 3 and doublecortin, a migratory neuroblast marker. Quantitative analysis revealed significantly more AC3/DCX-positive cells in Mcl-1 mutant brains relative to control littermates. Scale bar, 40 μm.
To determine whether neural precursor cell death observed in vivo is caused by a cell autonomous or noncell autonomous mechanism, we cultured primary cortical progenitor cells and monitored their survival in vitro as they differentiated. Cortical progenitor cell aggregates from E12.5 Foxg1:Cre Mcl-1 mutant embryos and littermate controls were plated at similar plating densities and allowed to differentiate in vitro. After 3 DIV, significant cell loss was observed in the Mcl-1 deficient cultures, at which point cultures were fixed and stained. Quantification of the total number of cells per aggregate revealed a twofold reduction in Mcl-1 deficient cultures (Fig. 8 A,B). To assess whether the cells in each aggregate were viable, cultures were stained with the Hoechst nuclear stain to identify healthy versus apoptotic cells. Quantification revealed that 52 ± 3% of cells in Mcl-1 deficient cell aggregates were apoptotic versus 15 ± 1% in aggregates from littermate controls. This massive cell death is comparable with the apoptosis observed in vivo and demonstrates that cell death from Mcl1 deficiency is cell autonomous.
Figure 8.
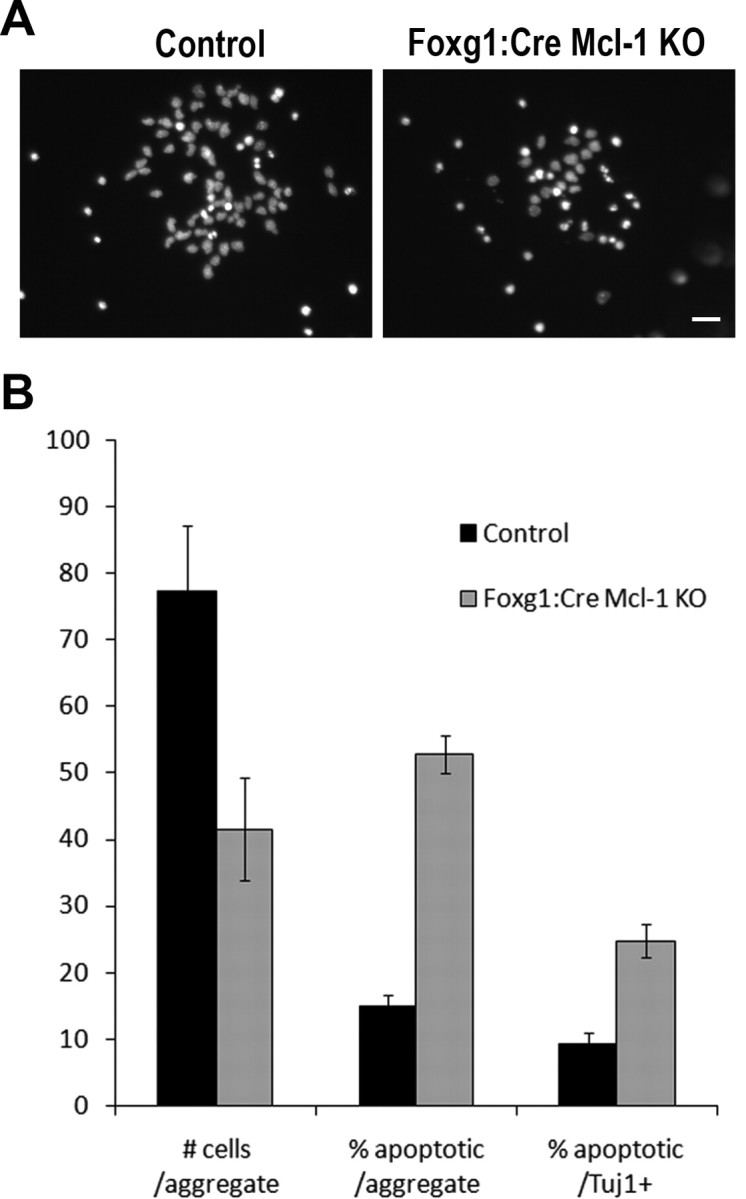
Mcl-1 deficient cortical progenitor cell death is cell autonomous. Cortical progenitor cell aggregates were plated at similar plating densities and allowed to differentiate in culture while survival was monitored. Cell survival was assessed with Hoechst nuclear staining to distinguish both viable (diffuse Hoechst nuclear staining) and apoptotic (condensed nuclei) cells. A, Representative photomicrographs of Hoechst-stained control and Mcl-1 deficient E12.5 cortical precursor cultures after 3 DIV. B, Counts were performed to quantify the total number of cells (viable and apoptotic) per aggregate (represented as mean number of cells/aggregate) and the number of apoptotic cells per aggregate (represented as percentage of apoptotic). Tuj1 immunostaining was performed to determine whether cortical precursors were capable of committing to a neuronal phenotype before dying. The number of apoptotic cells that were also Tuj1 positive (Tuj1+) were counted and compared with the number of Tuj1+ cells per aggregate (represented as percentage of apoptotic/Tuj1+). Results are summarized from six different platings of each genotype derived from two independent animals. Scale bar, 25 μm.
To determine whether Mcl-1 deficient neural precursors in vitro were capable of committing to a neuronal phenotype before apoptosis, cultures were immunostained with antibodies to Tuj1. The percentage of Tuj1-positive cells that are also apoptotic was 2.5-fold higher in Mcl-1 deficient cultures (Fig. 8 B). As apoptosis progresses, cells rapidly lose their phenotypic markers; therefore, our results likely under-represent the true number of apoptotic neurons. Our in vitro findings along with our in vivo observations indicate that Mcl-1 is necessary for the survival of newly committed neurons as well as maintaining the survival of neural progenitor cells. These studies identify a novel cell autonomous requirement for the anti-apoptotic Bcl-2 family protein, Mcl-1, in neurogenesis.
Mcl-1 regulates the onset of cell death after neuronal injury
Because Mcl-1 plays an essential role in the regulation of survival of neural precursor cells and newly committed neurons during telencephalic development, we were unable to evaluate the requirement for Mcl-1 in mature neurons in these mice. To assess whether Mcl-1 is required to maintain survival of postmitotic neurons, cortical neurons were cultured from E14.5 Foxg1:Cre Mcl-1 KO mice and cerebellar granule neurons homozygous for the floxed Mcl-1 allele and then infected with an adenoviral vector expressing Cre-recombinase to induce recombination in vitro (Fig. 9 G). Unlike neural progenitor cells, our results reveal that neither isolated cortical neurons, nor cells lacking Mcl-1 through Cre-mediated recombination of Mcl-1 in vitro undergo apoptosis but remained viable for at least 3–4 d (Fig. 9 A,B). Because Mcl-1 deletion did not induce apoptosis in postmitotic neurons within the timeframe of the experiment, we asked whether Mcl-1 deficiency modulated neuronal sensitivity to acute injury. Although spontaneous apoptosis was not observed in the absence of Mcl-1 in mature postmitotic neurons, Mcl-1 deficient neurons were more sensitive to exposure to a DNA damaging agent, camptothecin (10 μm), than control cells (8.9 ± 1.7% Mcl-1 deficient vs 25.25 ± 3.0% control) (Fig. 9 C,D). Although Mcl-1 deficiency alone did not induce an apoptotic response in more mature neurons, our results suggest that Mcl-1 may play an important role in the regulation of survival in the context of acute neuronal injury.
Figure 9.

Loss of Mcl-1 in neurons results in increased sensitivity to DNA damage-induced cell death. A, Quantitative analysis of cortical neuron survival by LIVE/DEAD assay revealed Mcl-1 mutant cortical neurons survive in culture similarly to cortical neurons cultured from littermate control animals. B, CGNs were cultured from Mcl-1 flox/flox mice and infected with an adenovirus expressing Cre recombinase (AdCre) or green fluorescent protein (GFP) (AdGFP) as a control, and cell survival was assessed at 12 h intervals by LIVE/DEAD assay. Deletion of Mcl-1 in CGNs did not induce cell death, because cell survival was similar in cultures infected with AdGFP. C, Cortical neurons were isolated from Foxg1:Cre Mcl-1 knock-out or control mice, treated with 10 μm CPT, and collected at the times indicated. Neuronal survival was determined by LIVE/DEAD assay (n = 3). Significantly fewer Foxg1:Cre Mcl-1 knock-out neurons were alive at 24 h after camptothecin treatment than neurons from littermate controls (*p < 0.05). D, CGNs were isolated from Mcl-1 flox/flox mice and infected with AdCre or AdGFP (MOI 75). Neuronal survival was determined at the indicated times by LIVE/DEAD assay (n = 3). E, Cortical neurons were cultured from CD1 mice and treated with 10 μm camptothecin, and total RNA was collected at the indicated times and analyzed by semiquantitative RT-PCR for Mcl-1, Bcl-2, Bcl-XL, Bcl-W, Bax, Bak, and S12 transcript expression. CTL, Untreated controls. F, Cortical neurons were treated with 10 μm camptothecin, and protein lysates were collected at indicated times after treatment. Western blot analysis was performed with antibodies for Mcl-1 and actin (loading control). G, Western blot analysis of Mcl-1 protein expression in CGNs infected with AdCre or AdGFP (MOI 75) 48 h after plating, showing efficient excision of Mcl-1 in cultured neurons infected with AdCre.
Because our results reveal that the absence of Mcl-1 accelerates the rate of cell death in response to neuronal injury, we asked whether Mcl-1 or any of the multidomain Bcl-2 family members were transcriptionally regulated after DNA damage induced neuronal cell death. Primary cortical neurons were treated with 10 μm camptothecin after 2 DIV. In contrast to the other Bcl-2 family members where there is no change in mRNA, Mcl-1 was rapidly downregulated after DNA damage (Fig. 9 E). This change in mRNA levels is followed by a steady decline in Mcl-1 protein levels (Fig. 9 F). The rapid downregulation of Mcl-1 before the onset of neuronal cell death suggests that downregulation of Mcl-1 may have a pivotal role regulating the onset of neuronal cell death after injury.
Mcl-1 plays an important role in injury-induced neuronal cell death
If Mcl-1 downregulation is a key event in controlling the onset of apoptosis after acute neuronal injury, then maintaining Mcl-1 levels should protect neurons from cell death. To determine this, we constructed an Mcl-1 adenoviral vector (AdMcl-1). Efficient protein expression of the Mcl-1 vector in neurons was confirmed by Western blot analysis (Fig. 10 A). To assess the protective role of Mcl-1 in neurons, CGNs were infected with AdMcl-1 or a LacZ control adenovirus (AdLacZ) (MOI 75) at the time of plating. After 2 DIV, neurons were treated with 10 μm camptothecin, and cell survival was assessed at 12 h intervals. Cells maintaining high expression of Mcl-1 showed a nearly twofold greater protection against DNA damage-induced cell death relative to cultures expressing LacZ (AdLacZ 24.7 ± 2.3% cell death and AdMcl-1 at 14.8 ± 2.0% cell death); in addition, Mcl-1-mediated resistance to injury is maintained at 48 h (AdLacZ, 53.3 ± 2.3%; AdMcl-1, 25.5 ± 1.9%) after injury (Fig. 10 B). These results show that Mcl-1 downregulation may be involved in controlling the onset of apoptosis after injury, because maintaining Mcl-1 levels in injured neurons can extend neuronal viability. Furthermore, these findings demonstrate a role for Mcl-1 in maintaining neuronal survival against acute injury, and that loss of Mcl-1 increases neuronal sensitivity to DNA damage-induced cell death.
Figure 10.

Sustained expression of Mcl-1 protects neurons against DNA damage-induced cell death. A, Western blot showing efficient transduction of AdMcl-1. CGNs were infected with the indicated MOI of AdMcl-1. Protein lysates were collected 48 h postinfection and immunoblotted for Mcl-1 expression and then for actin as a loading control. B, CGNs were infected with AdMcl-1 or AdLacZ control at 75 MOI at the time of plating and 48 h postinfection were treated with 10 μm CPT. LIVE/DEAD viability/cytotoxicity assay was performed at the indicated times (n = 3).
In these studies, we have shown that Mcl-1 is required to maintain neural precursor survival during forebrain development and after acute neuronal injury. In neuronal progenitors and immature neurons, loss of Mcl-1 results in apoptosis as progenitor cells commence migration away from the ventricular zone. Additionally, we show that Mcl-1 is downregulated in response to DNA damage-induced injury, and maintaining levels of Mcl-1 can reduce the rate of apoptosis, whereas loss of Mcl-1 increases sensitivity to stress. In conclusion, the results of our studies provide the first demonstration of a requirement for the anti-apoptotic Bcl-2 family protein, Mcl-1, for neural development and implicate Mcl-1 as a prime therapeutic target for the protection of neural precursors and neurons from apoptosis.
Discussion
The involvement of Mcl-1 in apoptotic cell death has been well documented (for review, see Craig, 2002; Michels et al., 2005); however, little is known regarding the role of Mcl-1 in the regulation of neuronal apoptosis during development or after acute injury. In this study, the striking cortical phenotype of Mcl-1 conditional mutant mice clearly demonstrates the importance of Mcl-1 in neural development. Specifically, Mcl-1 deficiency results in the apoptotic death of Nestin+ neural progenitors and Tuj1+ newly committed neurons, indicating that Mcl-1 is required for the survival of both cell populations. Furthermore, we also demonstrate a role for Mcl-1 in postmitotic neurons during acute DNA-induced injury. Our results show that Mcl-1 downregulation is an early event in acute neuronal injury and that maintaining Mcl-1 levels can protect neurons against DNA damage-induced cell death. These studies demonstrate a novel mechanism by which neural precursors are regulated in the developing brain and highlight the importance of Mcl-1 in the regulation of neuronal survival.
Neural progenitor cell numbers are tightly regulated by a balance between proliferation and cell death. During embryogenesis, developmental progenitor cell death has been reported to be as high as 50–70% (Blaschke et al., 1996). In the healthy adult brain, with each progenitor cell division, one daughter cell dies by apoptosis (Morshead et al., 1994). Gene knock-out studies have demonstrated the essential role apoptosis plays in regulating the precursor population, where mice deficient for key proapoptotic genes caspase 3, caspase 9, or Apaf-1 exhibit dramatically reduced apoptosis leading to the production of excessive neural precursor cells and brain malformation (Kuida et al., 1996, 1998; Cecconi et al., 1998). Although the apoptotic trigger has yet to be defined, proapoptotic members of the Bcl-2 family have shown some involvement. Single gene deletion of either proapoptotic proteins Bax or Bak does not produce a nervous system phenotype; deletion of both Bax and Bak, however, significantly reduces developmental apoptosis resulting in hypercellularity of specific CNS populations (Lindsten et al., 2000, 2003). The excess cells are located in the germinal zones of the brain (i.e., the SVZ) and have been identified with markers specific for neural precursors (Nestin), astrocytes (GFAP), and neurons (βIII tubulin) (Lindsten et al., 2003). These studies show that apoptosis plays an important role in the regulation of neural precursor cells and their differentiated progeny.
Presently, little is known regarding which anti-apoptotic Bcl-2 family protein is important in maintaining neural precursor cell survival. Previous studies using germline deletions of various Bcl-2 family members have not revealed a crucial role for any single Bcl family member in the development and survival of the CNS. Bcl-2-targeted deficiency studies suggest that Bcl-2 is not essential for development; however, there is loss of peripheral nervous system (PNS) neurons in early postnatal life, including motor, sensory, and sympathetic neuron populations (Michaelidis et al., 1996). This suggests a requirement for Bcl-2 in the maintenance and survival of these postmitotic populations. Bcl-xl germline knock-out mice exhibit extensive neuronal cell death at E13.5 (Motoyama et al., 1995). Unlike the dramatic phenotype of Bcl-xl whole embryo knock-outs, a neuron-specific Bcl-xl conditional mutant mouse revealed a less severe phenotype in which the population of catecholaminergic neurons was reduced by one-third, suggesting that the requirement for Bcl-xL may be cell type specific (Savitt et al., 2005). The results of our studies demonstrate Mcl-1 to be independently essential for neuronal development. In the present study, we identified Mcl-1 as a key regulator of neural precursor cell survival, because conditional mutations of Mcl-1 result in massive apoptosis of neural precursor cells. Mcl-1 is the first anti-apoptotic Bcl-family protein with a critical role in the maintenance of the neural precursor cell population.
In this study, we identify a change in Mcl1 requirement as neural progenitor cells differentiate into postmitotic neurons, and we show that this transitional period requires the anti-apoptotic Bcl-2 family member Mcl-1. In both conditional Mcl-1 mutant mice, we observed widespread apoptosis of Nestin+ neural progenitors and Tuj1+ newly committed neurons. In vitro differentiation of neural progenitors also resulted in extensive apoptosis, indicating a cell autonomous mechanism. In contrast, mature neurons in which Mcl-1 loss was induced by adenovirus delivery of Cre were able to survive in culture for up to 4 d with no evidence of apoptosis. These results indicate that the susceptibility to apoptosis changes during progenitor cell differentiation. Previous studies have shown that the expression profiles of anti-apoptotic and proapoptotic Bcl family proteins change as neural precursors become postmitotic neurons. During nervous system development, proapoptotic Bax and Bid are expressed in neural precursor cells within the ventricular zone, with Bax expression peaking at E12–E15 (Krajewska et al., 2002) corresponding to the early neurogenic interval when we see widespread apoptosis of neural precursors in the Mcl-1 conditional mutants. Although Mcl1 and Bcl-xL have been shown to block Bax- and Bak-mediated apoptosis, Bcl-xL is expressed at very low levels in the neural precursor population (Motoyama et al., 1995; Roth et al., 2000). The extensive neural precursor apoptosis we observe at E12 may be attributed to the relatively high levels of the proapoptotic proteins, such as Bax, and therefore the loss of Mcl-1 has a major impact on their survival. One might predict, therefore, that during neurogenesis, the majority of newly committed (Tuj1+) neurons in Mcl-1 conditional mutants die by apoptosis likely before sufficient Bcl-xL expression, because in the Bcl-xL null mutant, apoptosis is observed in more mature neuronal populations at E12.5 (Motoyama et al., 1995; Roth et al., 1996). The results of this study therefore suggest that Mcl-1 regulates the survival of cells during this transition from progenitor cell to postmitotic neuron.
Although Mcl-1 deficient neurons did not die when Mcl-1 was deleted, these neurons were more sensitive to injury-induced apoptosis. Cells lacking Mcl-1 died at an accelerated rate relative to littermates expressing Mcl-1. After DNA damage-induced injury, cells mount a strong apoptotic response, including a dramatic upregulation of BH3-only proteins, including Puma and Noxa (Cregan et al., 2004). In the context of this changing environment, the absence of Mcl-1 results in a rapid onset of cell death. Indeed, after DNA damage-induced injury, Noxa and Puma are induced, whereas Mcl-1 is rapidly downregulated. Previous studies have shown that Mcl-1 is at the apex of Bcl-2-mediated apoptotic signaling and that the downregulation of Mcl-1 is required for Bax activation (Nijhawan et al., 2003). Consistent with this model, our results show that maintaining Mcl-1 expression can preserve neuronal survival for extended time periods. These studies demonstrate the importance of Mcl-1 in regulating the onset on neuronal apoptosis after acute injury such as DNA damage.
Recent work by Mori et al. (2004) demonstrating increased neuronal sensitivity to seizures in Mcl-1 heterozygous mice has prompted questions regarding the role of Mcl-1 in the regulation of injury-induced neuronal apoptosis. This observed sensitivity suggests that Mcl-1 plays an important role in neuronal survival after insult; however, the mechanism by which this occurs remains unknown. In contrast to the other anti-apoptotic Bcl-2 family members, Mcl-1 is very tightly regulated both transcriptionally and posttranslationally (for review, see Craig, 2002). The rapid reduction in Mcl-1 transcript levels after DNA damage can be attributed to several mechanisms. Previous studies have revealed that camptothecin treatment leads to inactivation of Rb through activation of cyclin-dependent kinases (Park et al., 2000). Rb inactivation results in enhanced activity of E2F transcription factors, which have been shown previously to repress Mcl-1 transcription (Croxton et al., 2002). In addition to regulation by E2F, Mcl-1 is also a regulatory target for HIF1α, (Piret et al., 2005), cAMP response element-binding protein, and sis-inducible element (Wang et al., 1999), which may play important roles in Mcl-1 regulation in acute neuronal injury such as hypoxia. The precise mechanism by which Mcl-1 transcript levels are rapidly downregulated after DNA damage is not known; however, our results suggest that Mcl-1 levels may be sensitive indicators of neuronal injury and impending cell death. This is further supported by the demonstration that sustaining Mcl-1 expression can provide significant protection against neuronal cell death and highlight the importance of Mcl-1 in neuronal cell death regulation.
The results of our studies show a novel requirement for Mcl-1 in the development and maintenance of neurons within the CNS. Our in vivo results demonstrate that Mcl-1 is required for cortical neurogenesis and the survival of newly committed neurons. Additionally, our data show that Mcl-1 regulates apoptotic cell death after a DNA damaging insult. Most importantly, our results implicate Mcl-1 as a key regulatory target with which to expand the neural precursor pool and to maintain neuronal survival in the damaged brain.
Footnotes
This work was supported by a grant from the Heart and Stroke Foundation of Ontario (HSFO) to R.S.S. N.A., A.J.-A., and E.C.C.C. were supported by a Canadian Institutes of Health Research studentship; J.N.L.G. was supported by an HSFO studentship; M.A.K. was supported by an HSFO postdoctoral fellowship; J.L.V. was supported by a career transition award from the Canadian Stem Cell Network; and D.S.P. is an HSFO career investigator. We thank Steve Callaghan for generation of viral vectors and Jason MacLaurin for technical support.
References
- Akhtar RS, Ness JM, Roth KA. Bcl-2 family regulation of neuronal development and neurodegeneration. Biochim Biophys Acta. 2004;1644:189–203. doi: 10.1016/j.bbamcr.2003.10.013. [DOI] [PubMed] [Google Scholar]
- Berube NG, Mangelsdorf M, Jagla M, Vanderluit J, Garrick D, Gibbons RJ, Higgs DR, Slack RS, Picketts DJ. The chromatin-remodeling protein ATRX is critical for neuronal survival during corticogenesis. J Clin Invest. 2005;115:258–267. doi: 10.1172/JCI22329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaschke AJ, Staley K, Chun J. Widespread programmed cell death in proliferative and postmitotic regions of the fetal cerebral cortex. Development. 1996;122:1165–1174. doi: 10.1242/dev.122.4.1165. [DOI] [PubMed] [Google Scholar]
- Callaghan DA, Dong L, Callaghan SM, Hou YX, Dagnino L, Slack RS. Neural precursor cells differentiating in the absence of Rb exhibit delayed terminal mitosis and deregulated E2F 1 and 3 activity. Dev Biol. 1999;207:257–270. doi: 10.1006/dbio.1998.9162. [DOI] [PubMed] [Google Scholar]
- Casanova E, Fehsenfeld S, Mantamadiotis T, Lemberger T, Greiner E, Stewart AF, Schutz G. A CamKIIalpha iCre BAC allows brain-specific gene inactivation. Genesis. 2001;31:37–42. doi: 10.1002/gene.1078. [DOI] [PubMed] [Google Scholar]
- Cecconi F, Alvarez-Bolado G, Meyer BI, Roth KA, Gruss P. Apaf1 (CED-4 homolog) regulates programmed cell death in mammalian development. Cell. 1998;94:727–737. doi: 10.1016/s0092-8674(00)81732-8. [DOI] [PubMed] [Google Scholar]
- Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG, Colman PM, Day CL, Adams JM, Huang DC. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell. 2005;17:393–403. doi: 10.1016/j.molcel.2004.12.030. [DOI] [PubMed] [Google Scholar]
- Craig RW. MCL1 provides a window on the role of the BCL2 family in cell proliferation, differentiation and tumorigenesis. Leukemia. 2002;16:444–454. doi: 10.1038/sj.leu.2402416. [DOI] [PubMed] [Google Scholar]
- Cregan SP, MacLaurin JG, Craig CG, Robertson GS, Nicholson DW, Park DS, Slack RS. Bax-dependent caspase-3 activation is a key determinant in p53-induced apoptosis in neurons. J Neurosci. 1999;19:7860–7869. doi: 10.1523/JNEUROSCI.19-18-07860.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cregan SP, MacLaurin J, Gendron TF, Callaghan SM, Park DS, Parks RJ, Graham FL, Morley P, Slack RS. Helper-dependent adenovirus vectors: their use as a gene delivery system to neurons. Gene Ther. 2000;7:1200–1209. doi: 10.1038/sj.gt.3301208. [DOI] [PubMed] [Google Scholar]
- Cregan SP, Arbour NA, Maclaurin JG, Callaghan SM, Fortin A, Cheung EC, Guberman DS, Park DS, Slack RS. p53 activation domain 1 is essential for PUMA upregulation and p53-mediated neuronal cell death. J Neurosci. 2004;24:10003–10012. doi: 10.1523/JNEUROSCI.2114-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croxton R, Ma Y, Song L, Haura EB, Cress WD. Direct repression of the Mcl-1 promoter by E2F1. Oncogene. 2002;21:1359–1369. doi: 10.1038/sj.onc.1205157. [DOI] [PubMed] [Google Scholar]
- Dahlstrand J, Lardelli M, Lendahl U. Nestin mRNA expression correlates with the central nervous system progenitor cell state in many, but not all, regions of developing central nervous system. Brain Res Dev Brain Res. 1995;84:109–129. doi: 10.1016/0165-3806(94)00162-s. [DOI] [PubMed] [Google Scholar]
- Ferguson KL, Vanderluit JL, Hebert JM, McIntosh WC, Tibbo E, MacLaurin JG, Park DS, Wallace VA, Vooijs M, McConnell SK, Slack RS. Telencephalon-specific Rb knockouts reveal enhanced neurogenesis, survival and abnormal cortical development. EMBO J. 2002;21:3337–3346. doi: 10.1093/emboj/cdf338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortin A, Cregan SP, MacLaurin JG, Kushwaha N, Hickman ES, Thompson CS, Hakim A, Albert PR, Cecconi F, Helin K, Park DS, Slack RS. APAF1 is a key transcriptional target for p53 in the regulation of neuronal cell death. J Cell Biol. 2001;155:207–216. doi: 10.1083/jcb.200105137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuccillo M, Rallu M, McMahon AP, Fishell G. Temporal requirement for hedgehog signaling in ventral telencephalic patterning. Development. 2004;131:5031–5040. doi: 10.1242/dev.01349. [DOI] [PubMed] [Google Scholar]
- Fujise K, Zhang D, Liu J, Yeh ET. Regulation of apoptosis and cell cycle progression by MCL1. Differential role of proliferating cell nuclear antigen. J Biol Chem. 2000;275:39458–39465. doi: 10.1074/jbc.M006626200. [DOI] [PubMed] [Google Scholar]
- Hebert JM, McConnell SK. Targeting of cre to the Foxg1 (BF-1) locus mediates loxP recombination in the telencephalon and other developing head structures. Dev Biol. 2000;222:296–306. doi: 10.1006/dbio.2000.9732. [DOI] [PubMed] [Google Scholar]
- Jamil S, Sobouti R, Hojabrpour P, Raj M, Kast J, Duronio V. A proteolytic fragment of Mcl-1 exhibits nuclear localization and regulates cell growth by interaction with Cdk1. Biochem J. 2005;387:659–667. doi: 10.1042/BJ20041596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krajewska M, Mai JK, Zapata JM, Ashwell KW, Schendel SL, Reed JC, Krajewski S. Dynamics of expression of apoptosis-regulatory proteins Bid, Bcl-2, Bcl-X, Bax and Bak during development of murine nervous system. Cell Death Differ. 2002;9:145–157. doi: 10.1038/sj.cdd.4400934. [DOI] [PubMed] [Google Scholar]
- Kuida K, Zheng TS, Na S, Kuan C, Yang D, Karasuyama H, Rakic P, Flavell RA. Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature. 1996;384:368–372. doi: 10.1038/384368a0. [DOI] [PubMed] [Google Scholar]
- Kuida K, Haydar TF, Kuan CY, Gu Y, Taya C, Karasuyama H, Su MS, Rakic P, Flavell RA. Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell. 1998;94:325–337. doi: 10.1016/s0092-8674(00)81476-2. [DOI] [PubMed] [Google Scholar]
- Kuwana T, Bouchier-Hayes L, Chipuk JE, Bonzon C, Sullivan BA, Green DR, Newmeyer DD. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol Cell. 2005;17:525–535. doi: 10.1016/j.molcel.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Letai AG. Diagnosing and exploiting cancer's addiction to blocks in apoptosis. Nat Rev Cancer. 2008;8:121–132. doi: 10.1038/nrc2297. [DOI] [PubMed] [Google Scholar]
- Lindsten T, Ross AJ, King A, Zong WX, Rathmell JC, Shiels HA, Ulrich E, Waymire KG, Mahar P, Frauwirth K, Chen Y, Wei M, Eng VM, Adelman DM, Simon MC, Ma A, Golden JA, Evan G, Korsmeyer SJ, MacGregor GR, et al. The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Mol Cell. 2000;6:1389–1399. doi: 10.1016/s1097-2765(00)00136-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsten T, Golden JA, Zong WX, Minarcik J, Harris MH, Thompson CB. The proapoptotic activities of Bax and Bak limit the size of the neural stem cell pool. J Neurosci. 2003;23:11112–11119. doi: 10.1523/JNEUROSCI.23-35-11112.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelidis TM, Sendtner M, Cooper JD, Airaksinen MS, Holtmann B, Meyer M, Thoenen H. Inactivation of bcl-2 results in progressive degeneration of motoneurons, sympathetic and sensory neurons during early postnatal development. Neuron. 1996;17:75–89. doi: 10.1016/s0896-6273(00)80282-2. [DOI] [PubMed] [Google Scholar]
- Michels J, Johnson PW, Packham G. Mcl-1. Int J Biochem Cell Biol. 2005;37:267–271. doi: 10.1016/j.biocel.2004.04.007. [DOI] [PubMed] [Google Scholar]
- Mori M, Burgess DL, Gefrides LA, Foreman PJ, Opferman JT, Korsmeyer SJ, Cavalheiro EA, Naffah-Mazzacoratti MG, Noebels JL. Expression of apoptosis inhibitor protein Mcl1 linked to neuroprotection in CNS neurons. Cell Death Differ. 2004;11:1223–1233. doi: 10.1038/sj.cdd.4401483. [DOI] [PubMed] [Google Scholar]
- Morshead CM, Reynolds BA, Craig CG, McBurney MW, Staines WA, Morassutti D, Weiss S, van der Kooy D. Neural stem cells in the adult mammalian forebrain: a relatively quiescent subpopulation of subependymal cells. Neuron. 1994;13:1071–1082. doi: 10.1016/0896-6273(94)90046-9. [DOI] [PubMed] [Google Scholar]
- Motoyama N, Wang F, Roth KA, Sawa H, Nakayama K, Negishi I, Senju S, Zhang Q, Fujii S, Loh DY. Massive cell death of immature hematopoietic cells and neurons in Bcl-x-deficient mice. Science. 1995;267:1506–1510. doi: 10.1126/science.7878471. [DOI] [PubMed] [Google Scholar]
- Nijhawan D, Fang M, Traer E, Zhong Q, Gao W, Du F, Wang X. Elimination of Mcl-1 is required for the initiation of apoptosis following ultraviolet irradiation. Genes Dev. 2003;17:1475–1486. doi: 10.1101/gad.1093903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oishi K, Kamakura S, Isazawa Y, Yoshimatsu T, Kuida K, Nakafuku M, Masuyama N, Gotoh Y. Notch promotes survival of neural precursor cells via mechanisms distinct from those regulating neurogenesis. Dev Biol. 2004;276:172–184. doi: 10.1016/j.ydbio.2004.08.039. [DOI] [PubMed] [Google Scholar]
- Opferman JT, Letai A, Beard C, Sorcinelli MD, Ong CC, Korsmeyer SJ. Development and maintenance of B and T lymphocytes requires antiapoptotic MCL-1. Nature. 2003;426:671–676. doi: 10.1038/nature02067. [DOI] [PubMed] [Google Scholar]
- Opferman JT, Iwasaki H, Ong CC, Suh H, Mizuno S, Akashi K, Korsmeyer SJ. Obligate role of anti-apoptotic MCL-1 in the survival of hematopoietic stem cells. Science. 2005;307:1101–1104. doi: 10.1126/science.1106114. [DOI] [PubMed] [Google Scholar]
- Park DS, Morris EJ, Bremner R, Keramaris E, Padmanabhan J, Rosenbaum M, Shelanski ML, Geller HM, Greene LA. Involvement of retinoblastoma family members and E2F/DP complexes in the death of neurons evoked by DNA damage. J Neurosci. 2000;20:3104–3114. doi: 10.1523/JNEUROSCI.20-09-03104.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piret JP, Minet E, Cosse JP, Ninane N, Debacq C, Raes M, Michiels C. Hypoxia-inducible factor-1-dependent overexpression of myeloid cell factor-1 protects hypoxic cells against tert-butyl hydroperoxide-induced apoptosis. J Biol Chem. 2005;280:9336–9344. doi: 10.1074/jbc.M411858200. [DOI] [PubMed] [Google Scholar]
- Rinkenberger JL, Horning S, Klocke B, Roth K, Korsmeyer SJ. Mcl-1 deficiency results in peri-implantation embryonic lethality. Genes Dev. 2000;14:23–27. [PMC free article] [PubMed] [Google Scholar]
- Roth KA, Motoyama N, Loh DY. Apoptosis of bcl-x-deficient telencephalic cells in vitro . J Neurosci. 1996;16:1753–1758. doi: 10.1523/JNEUROSCI.16-05-01753.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth KA, Kuan C, Haydar TF, D'Sa-Eipper C, Shindler KS, Zheng TS, Kuida K, Flavell RA, Rakic P. Epistatic and independent functions of caspase-3 and Bcl-X(L) in developmental programmed cell death. Proc Natl Acad Sci USA. 2000;97:466–471. doi: 10.1073/pnas.97.1.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzhynsky VA, McClellan KA, Vanderluit JL, Jeong Y, Furimsky M, Park DS, Epstein DJ, Wallace VA, Slack RS. Cell cycle regulator E2F4 is essential for the development of the ventral telencephalon. J Neurosci. 2007;27:5926–5935. doi: 10.1523/JNEUROSCI.1538-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savitt JM, Jang SS, Mu W, Dawson VL, Dawson TM. Bcl-x is required for proper development of the mouse substantia nigra. J Neurosci. 2005;25:6721–6728. doi: 10.1523/JNEUROSCI.0760-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimamura K, Rubenstein JL. Inductive interactions direct early regionalization of the mouse forebrain. Development. 1997;124:2709–2718. doi: 10.1242/dev.124.14.2709. [DOI] [PubMed] [Google Scholar]
- Tao W, Lai E. Telencephalon-restricted expression of BF-1, a new member of the HNF-3/fork head gene family, in the developing rat brain. Neuron. 1992;8:957–966. doi: 10.1016/0896-6273(92)90210-5. [DOI] [PubMed] [Google Scholar]
- Vanderluit JL, Ferguson KL, Nikoletopoulou V, Parker M, Ruzhynsky V, Alexson T, McNamara SM, Park DS, Rudnicki M, Slack RS. p107 regulates neural precursor cells in the mammalian brain. J Cell Biol. 2004;166:853–863. doi: 10.1083/jcb.200403156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JM, Chao JR, Chen W, Kuo ML, Yen JJ, Yang-Yen HF. The antiapoptotic gene mcl-1 is up-regulated by the phosphatidylinositol 3-kinase/Akt signaling pathway through a transcription factor complex containing CREB. Mol Cell Biol. 1999;19:6195–6206. doi: 10.1128/mcb.19.9.6195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- Zhang J, D'Ercole AJ. Expression of Mcl-1 in cerebellar granule neurons is regulated by IGF-I in a developmentally specific fashion. Brain Res Dev Brain Res. 2004;152:255–263. doi: 10.1016/j.devbrainres.2004.07.008. [DOI] [PubMed] [Google Scholar]