

Abstract
Adult height is a model polygenic trait, but there has been limited success in identifying the genes underlying its normal variation. To identify genetic variants influencing adult human height, we used genome-wide association data from 13,665 individuals and genotyped 39 variants in an additional 16,482 samples. We identified 20 variants associated with adult height (P < 5 × 10−7, with 10 reaching P < 1 × 10−10). Combined, the 20 SNPs explain ~3% of height variation, with a ~5 cm difference between the 6.2% of people with 17 or fewer ‘tall’ alleles compared to the 5.5% with 27 or more ‘tall’ alleles. The loci we identified implicate genes in Hedgehog signaling (IHH, HHIP, PTCH1), extracellular matrix (EFEMP1, ADAMTSL3, ACAN) and cancer (CDK6, HMGA2, DLEU7) pathways, and provide new insights into human growth and developmental processes. Finally, our results provide insights into the genetic architecture of a classic quantitative trait.
Adult height is a model polygenic trait. It is the ideal phenotype for genetic studies of quantitative traits in humans, as it is easily and accurately measured and highly heritable, with up to 90% of variation in adult height within a population explained by genetic variation1-5. Final adult height is the result of growth and developmental processes. Identifying genes for human height should therefore provide insights into mechanisms of growth and development, as well as into the genetic architecture of quantitative traits and how best to dissect them.
Despite its strong heritability, there has been little success in identifying the specific genetic variants that influence height in the general population5,6. Some mutations resulting in extreme stature have been identified, but these are rare and cannot explain normal variation of adult height6. Linkage and candidate gene association studies have not identified any robustly associated loci. The advent of genome-wide association (GWA) studies, however, is providing new opportunities for identifying genetic variants influencing adult height.
Recently, using GWA study data from 4,921 individuals, we identified the most convincing example to date of a common variant associated with adult height variation7. The variant was the only one to reach a level of significance suggestive of true association in the GWA study (P = 4 × 10−8), and we confirmed the association in 19,064 adults from four further studies (P = 3 × 10−11). The variant was associated with a 0.4 cm greater height per copy of the allele, explained ~0.3% of the population variation of height, and occurred in the HMGA2 oncogene. In this study, we extend our analyses to a two-staged design comprising 13,665 individuals with GWA study data and 16,482 follow-up individuals.
RESULTS
Height loci identified
We used GWA data from five studies that ranged in size from 1,437 to 3,560 people of European ancestry from the UK and a sixth study of 2,978 Scandinavian individuals for which summary height association statistics have been made publicly available (see URLs section in Methods; Supplementary Table 1 online). All studies were genotyped using the Affymetrix 500K chip. We compared the additive model statistics of 402,951 SNPs that passed quality-control criteria in at least four of the six studies to those expected under the null distribution using quantile-quantile plots, and we found that the sequential addition of each of the six studies resulted in increased deviation of the observed statistics from the null distribution (Fig. 1). As each study was added in, we found (using a cut-off of less than 0.2 for the pairwise linkage disequilibrium (LD) statistic r2) 4 (n = 1,914), 6 (n = 4,892), 12 (n = 6,788), 13 (n = 8,668), 18 (n = 12,228) and 27 (n = 13,665) independent SNPs reaching a P < 1 × 10−5, in contrast to the expected <4 under the null distribution.
Figure 1.

Quantile-quantile plots for the 402,951 SNPs from the genome-wide association meta-analysis as more studies are added in. (a) n = 1,914 (WTCCC-T2D). (b) n = 4,892 (adding DGI). (c) n = 6,788 (adding WTCCC-HT). (d) n = 8,668 (adding WTCCC-CAD). (e) n = 12,228 (adding EPIC-Obesity). (f) n = 13,665 (adding WTCCC-UKBS). Blue line represents the observed P values. The black line is the expected line under the null distribution. The gray bands are 95% concentration bands, which are an approximation to the 95% confidence intervals around the expected line.
In the meta-analysis of 13,665 individuals with GWA data, there were many more significant associations than expected by chance. For example, we observed eight independent signals with a P < 5 × 10−7, where we would expect none under the null distribution, and 27 with a P < 1 × 10−5, where we would expect less than four. Approximately 23 of these loci are therefore likely to represent true positives. The availability of dense genome-wide SNP data allows us to be confident that these results are not due to population stratification. First, individuals of non-European ancestry were excluded. Second, adjusting for residual population structure using EIGENSTRAT8 did not affect the distribution of effect sizes (Supplementary Figure 1 online gives individual study quantile-quantile plots before and after EIGENSTRAT adjustment). Third, the genomic control inflation factor9 for the GWA study meta-analysis was only 1.12, despite the large size of the study (there is a strong relationship between sample size and λ (ref. 10)) and the apparently highly polygenic nature of height. Fourth, 12 of the ancestry informative markers (AIMs) described by the WTCCC, which vary substantially in allele frequency across the UK, did not associate with height (all P > 0.01; the 13th AIM did not pass quality control criteria in this study; Supplementary Table 2 online).
We took 39 SNPs forward into the second stage of our study: the genotyping of an additional 16,482 individuals of European ancestry from four studies (Supplementary Table 1). Of these, 27 represented all the independent (r2 < 0.2) signals with a P < 1 × 10−5, and 11 represented independent regions where there was a SNP with a P < 1 × 10−4 and a gene within flanking recombination hotspots in which mutations have been found to affect length in mouse studies or cause monogenic human phenotypes of extreme stature. Lastly, GWA data from CoLaus (one of our stage 2 cohorts) became available during the course of our analyses, and we took forward a SNP representing a region with the strongest association (P = 4 × 10−8) from that study. Five of the AIMs with the largest differences in allele frequency across the UK11 were also genotyped in stage 2 samples.
In the stage 2 analyses, 20 of the 39 SNPs reached a P < 0.005 (with the same direction of effect as the GWA data), all of which reached a P < 5 × 10−7 in a joint analysis across GWA and stage 2 samples. Although this is an arbitrary statistical cut-off, we chose to focus on these SNPs for reasons previously discussed11, and we note that of the SNPs that reached a P < 5 × 10−7 in ref. 11 and that have been subjected to replication efforts, all have been confirmed. Most of the 20 SNPs had P values substantially lower than 5× 10−7: 17 of the SNPs reached a P < 5 × 10−8, and 10 reached a P < 1 × 10−10 in joint analyses. Of the 19 SNPs that did not reach P < 5 × 10−7, 15 had the same direction of effect in stage 2 as in stage 1 (P = 0.02), suggesting that there are true positives among these. The details of the 20 SNPs are presented in Figure 2 and Table 1; details of the SNPs that did not reach the statistical cut-off are presented in Supplementary Table 3 online. For the 20 SNPs, there was no evidence of heterogeneity across studies when taking into account the number of tests (all P > 0.008). In both joint and stage 2 only analyses, none of the WTCCC AIMs was associated with height, providing further evidence that population stratification is unlikely to have influenced the results (all P > 0.01; see Supplementary Table 2). This means that the associations are likely to reflect true biological effects on height.
Figure 2.

Manhattan plot for the 402,951 SNPs from the stage 1 genomewide association meta-analysis of the WTCCC-T2D, DGI, WTCCC-HT, WTCCC-CAD, EPIC-Obesity and WTCCC-UKBS studies. The red dots represent the SNPs that reached a P < 5 × 10−7 in a joint analysis of stage 1 and stage 2 samples.
Table 1.
Results for the 20 SNPs taken forward into stage 2 that reached P < 5 × 10−7 in joint analyses
| SNP | Candidate gene | Chromosome (position) |
Alleles (1/2) |
MAF | Additive model test P |
Gender test P |
Male s.d. difference (95% CI) |
Female s.d. difference (95% CI) |
R2 (%) |
GWA study P |
Follow-up P | Heterogeneity P |
Overall P excluding DGI |
Overall P |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| rs6440003 | ZBTB38 | 3 (142576907) | A/G | 0.44 | 0.80 | 0.01 | 0.07 (0.04, 0.09) |
0.12 (0.09, 0.14) |
0.32 | 1.3 × 10−14 | 8.7 × 10−12 | 0.52 | 2.7 × 10−23 | 1.8 × 10−24 |
| rs2282978/ rs42046a |
CDK6 | 7 (91898623) | C/T | 0.33 | 0.14 | 0.69 | 0.09 (0.06, 0.12) |
0.08 (0.05, 0.11) |
0.28 | 5.0 × 10−11 | 5.1 × 10−13 | 0.98 | 3.1 × 10−21 | 7.8 × 10−23 |
| rs1042725 | HMGA2 | 12 (64644614) | C/T | 0.49 | 0.70 | 0.34 | 0.05 (0.03, 0.08) |
0.07 (0.05, 0.10) |
0.25 | 5.9 × 10−9 | 8.6 × 10−11 | 0.50 | 1.1 × 10−14 | 2.5 × 10−18 |
| rs6060373 | GDF5 | 20 (33377622) | A/G | 0.38 | 0.17 | 0.70 | -0.08 (-0.11, -0.05) |
-0.07 (-0.10, -0.04) |
0.21 | 2.2 × 10−12 | 1.6 × 10−7 | 0.27 | 2.0 × 10−15 | 1.7 × 10−17 |
| rs16896068 | LCORL | 4 (17621109) | A/G | 0.16 | 0.31 | 0.99 | -0.07 (-0.11, -0.03) |
-0.07 (-0.11, -0.03) |
0.12 | 1.0 × 10−4 | 2.5 × 10−10 | 0.06 | 2.0 × 10−13 | 2.4 × 10−13 |
| rs4549631 | LOC387103 | 6 (127008001) | C/T | 0.50 | 0.62 | 0.85 | 0.06 (0.03, 0.08) |
0.05 (0.03, 0.08) |
0.11 | 1.2 × 10−8 | 4.6 × 10−6 | 0.47 | 2.9 × 10−11 | 4.7 × 10−13 |
| rs3791675 | EFEMP1 | 2 (56022960) | C/T | 0.23 | 0.43 | 0.34 | 0.09 (0.05, 0.12) |
0.06 (0.03, 0.10) |
0.12 | 7.1 × 10−8 | 6.0 × 10−6 | 0.54 | 1.5 × 10−12 | 2.2 × 10−12 |
| rs2814993 | C6orf106 | 6 (34726871) | A/G | 0.15 | 0.18 | 0.87 | 0.09 (0.05, 0.13) |
0.10 (0.06, 0.14) |
0.20 | 8.9 × 10−9 | 5.7 × 10−5 | 0.04 | 4.0 × 10−11 | 4.1 × 10−12 |
| rs10512248 | PTCH1 | 9 (95339258) | G/T | 0.31 | 0.14 | 0.10 | 0.05 (0.02, 0.07) |
0.08 (0.05, 0.11) |
0.19 | 1.5 × 10−6 | 6.0 × 10−6 | 0.82 | 1.0 × 10−9 | 4.2 × 10−11 |
| rs12735613 | SPAG17 | 1 (118596015) | A/G | 0.24 | 0.0090 | 0.02 | -0.08 (-0.11, -0.05) |
-0.03 (-0.06, 0.00) |
0.09 | 3.4 × 10−8 | 8.2 × 10−5 | 0.51 | 2.0 × 10−9 | 4.4 × 10−11 |
| rs11107116 | SOCS2 | 12 (92480972) | G/T | 0.23 | 0.047 | 0.73 | -0.04 (-0.07, -0.01) |
-0.05 (-0.08, -0.02) |
0.06 | 2.5 × 10−5 | 5.6 × 10−6 | 0.41 | 2.3 × 10−8 | 5.6 × 10−10 |
| rs6854783/ rs2055059a |
HHIP | 4 (146000684) | A/G | 0.43 | 0.17 | 0.50 | 0.06 (0.03, 0.08) |
0.04 (0.01, 0.017) |
0.10 | 1.2 × 10−5 | 3.2 × 10−5 | 0.24 | 2.2 × 10−8 | 2.1 × 10−9 |
| rs1390401 | ZNF678 | 1 (224104685) | A/G | 0.18 | 0.0067 | 0.34 | 0.04 (0.01, 0.08) |
0.07 (0.03, 0.10) |
0.09 | 4.3 × 10−6 | 2.0 × 10−4 | 0.58 | 1.4 × 10−6 | 5.4 × 10−9 |
| rs3116602 | DLEU7 | 13 (50009356) | G/T | 0.21 | 0.88 | 0.02 | -0.04 (-0.07, 0.00) |
-0.09 (-0.12, -0.06) |
0.07 | 5.6 × 10−6 | 1.8 × 10−4 | 0.82 | 6.1 × 10−9 | 6.8 × 10−9 |
| rs6686842 | SCMH1 | 1 (41199964) | C/T | 0.44 | 0.30 | 0.97 | -0.05 (-0.08, -0.02) |
-0.05 (-0.08, -0.02) |
0.14 | 8.6 × 10−6 | 3.3 × 10−4 | 0.57 | 4.9 × 10−7 | 1.7 × 10−8 |
| rs10906982 | ADAMTSL3 | 15 (82371586) | A/T | 0.48 | 0.33 | 0.92 | 0.05 (0.02, 0.07) |
0.04 (0.02, 0.07) |
0.07 | 5.4 × 10−7 | 2.1 × 10−3 | 0.57 | 5.3 × 10−7 | 1.7 × 10−8 |
| rs6724465 | IHH | 2 (219769351) | A/G | 0.10 | 0.96 | 0.85 | -0.06 (-0.10, -0.02) |
-0.05 (-0.10, -0.01) |
0.04 | 3.1 × 10−5 | 2.8 × 10−4 | 0.52 | 2.2 × 10−6 | 2.1 × 10−8 |
| rs10935120 |
ANAPC13 or CEP63 |
3 (135715790) | A/G | 0.33 | 0.10 | 0.63 | -0.06 (-0.09, -0.03) |
-0.05 (-0.08, -0.02) |
0.10 | 2.2 × 10−6 | 3.1 × 10−3 | 0.57 | 8.7 × 10−7 | 7.3 × 10−8 |
| rs8041863 | ACAN | 15 (87160693) | A/T | 0.47 | 0.90 | 0.21 | 0.04 (0.01, 0.06) |
0.06 (0.03, 0.09) |
0.03 | 2.2 × 10−5 | 8.6 × 10−4 | 0.02 | 4.9 × 10−9 | 8.1 × 10−8 |
| rs8099594 | DYM | 18 (45245158) | A/G | 0.35 | 0.69 | 0.53 | 0.05 (0.02, 0.08) |
0.04 (0.01, 0.07) |
0.01 | 7.8 × 10−6 | 4.1 × 10−3 | 0.008 | 1.6 × 10−8 | 3.1 × 10−7 |
The results are ordered by the joint-analyses P value. Chromosome positions are based on NCBI build 125. The alleles all refer to the positive strand. Betas are per each additional copy of allele 1. Minor allele frequency (MAF) based on the minor allele (bold and underlined in the alleles column) in the WTCCC-T2D study. R2 (% variation explained) is for follow-up sample only and does not include CoLaus. The additive model test and gender test P values do not include data from DGI or CoLaus.
r2 = 1 proxies used in the stage 2 studies because of assay design issues. Candidate gene is given when monogenic human and/or mouse phenotypes and/or expression results clearly implicate a gene. An overall P value excluding DGI is given because of the small related component of DGI and to provide evidence independent from the accompanying manuscript by Lettre et al.18
Implicated genes and their functions
Because of the correlation between SNPs as a result of LD and the occurrence of many of the 20 SNPs in noncoding regions, we cannot be certain about which genes are involved, but our results implicate genes of many different functions in several different pathways and processes. In ten instances, genes within the region of interest have previously been implicated in the regulation of growth because of known effects from mouse knockouts or human syndromes. LD plots for each region are presented in Supplementary Figure 2 online; Table 2 lists the genes most likely affected by the associated SNPs, the pathways the genes are known to be involved in, and where known, the monogenic syndromes caused by mutations in the associated genes and the phenotypes from knockout mouse models.
Table 2.
Summary of candidate genes in the 20 loci associated with height
| SNP | Candidate or nearest gene(s) |
Monogenic syndrome caused by mutation in gene |
Knockout mouse phenotype | Detailsa |
|---|---|---|---|---|
| rs6440003 |
ZBTB38 (zinc finger and BTB domain-containing protein 38) |
— | — | Transcription factor |
| rs2282978 |
CDK6 (cyclin-dependent kinase-6) |
— | 15% smaller embryos | Involved in the control of the cell cycle. Interacts with D-type G1 cyclins. |
| rs1042725 |
HMGA2 (high-mobility group A2) |
Tall stature, extreme bone and dental overgrowth, and multiple lipomas. |
Pygmy mice | Belongs to the nonhistone chromosomal high mobility group (HMG) protein family. HMG proteins function as chromatin architectural factors. |
| rs6060373 |
GDF5 (growth differentia- tion factor 5) |
Chondrodysplasia (abnormally short and deformed limbs); brachydactyly (short digits) DuPan syndrome; multiple synostoses syndrome. |
Homozygous null mutants show skeleton defects, such as reduced or absent limb bones and joints. |
Involved in bone formation. Also known as cartilage-derived morphogenetic protein 1. |
| rs16896068 |
LCORL (ligand-dependent nuclear receptor corepressor- like protein) |
— | — | May act as transcription activator. |
| rs4549631 | LOC387103 | — | — | Not known. |
| rs3791675 |
EFEMP1 (EGF-containing fibulin-like extracellular matrix protein 1) |
Doyne honeycomb retinal dystrophy; no obvious skeletal defects. |
Normal phenotype | Extracellular matrix. Belongs to the fibulin family. |
| rs2814993 | C6orf106 | — | — | Not known. |
| rs10512248 |
PTCH1 (patched homolog 1 (Drosophila)) |
Gorlin syndrome (basal cell carcinoma); holoprosencephaly. |
Homozygous null mice die during embryogenesis, heterozygotes larger than normal, with hind limb defects. |
Hedgehog signalling. Acts as a receptor for Sonic hedgehog (SHH), Indian hedgehog (IHH) and Desert hedgehog (DHH). |
| rs12735613 |
SPAG1 (sperm associated antigen 17) |
— | — | Not known. |
| rs11107116 |
SOCS2 (suppressor of cyto- kine signaling 2) |
— | Homozygous null mice grow more rapidly. Males are 40% heavier than wild-type littermates; the increase in weight results from general increase in visceral organ weight and long bone length. |
SOCS family proteins form part of a classical negative feedback system that regulates cytokine signal transduction. SOCS2 seems to be a negative regulator in the growth hormone/IGF1 signaling pathway. |
| rs6854783 |
HHIP (Hedgehog interacting protein) |
— | Ectopic expression in transgenic mice results in severe skeletal defects similar to those observed in IHH mutants. |
Hedgehog signaling. Modulates hedgehog signaling through direct interaction with members of the hedgehog family including SHH, IHH and DHH. |
| rs1390401 |
ZNF678 (zinc finger protein 678) |
— | — | Transcription factor. Belongs to the Krüp- pel C2H2-type zinc-finger protein family by similarity. |
| rs3116602 |
DLEU7 (deleted in lympho- cytic leukemia 7) |
— | — | Not known. |
| rs6686842 |
SCMH1 (sex comb on mid- leg homolog 1) |
— | Homozygous null mice present with multiple defects including of skeleton. |
Polycomb protein. A constituent of the mammalian Polycomb repressive complexes 1 involved in chromatin modifications. |
| rs10906982 |
ADAMTSL3 (ADAMTS-like protein 3) |
— | — | Extracellular matrix. Strongly similar to members of the ADAMTS family but lacks metalloprotease and disintegrin-like domains. |
| rs6724465 | IHH (Indian hedgehog) | Brachydactyly; acrocapitofemoral dysplasia (cone-shaped ends of hand and hip bones). |
Homozygous null mice show impaired chondrocyte proliferation and maturation, resulting in dwarfism and numerous skeletal abnormalities. |
Hedgehog signaling. Intercellular signal essential for a variety of patterning events during development. Binds to the patched (PTCH) receptor. |
| rs10935120 |
ANAPC13 (anaphase pro- moting complex subunit 13) |
— | — | Cell cycle. Component of the anaphase promoting complex/cyclosome (APC/C), a cell cycle—regulated E3 ubiquitin ligase that controls progression through mitosis and the G1 phase of the cell cycle. |
| rs8041863 | ACAN (aggrecan) | Autosomal dominant spondylo- epiphyseal dysplasia type Kimberley, characterized by severe, premature osteoarthritis. |
Homozygous mutants are dwarfed at birth. |
Extracellular matrix. A member of the aggrecan/versican proteoglycan family. Part of the extracellular matrix in cartilaginous tissue. |
| rs8099594 | DYM (dymeclin) | Autosomal recessive disorder characterized by abnormal skeletal development and mental retardation. |
— | May have a role in process of intracellular digestion of proteins or in proteoglycan metabolism. |
A candidate gene is listed when monogenic human and/or mouse phenotypes and/or expression results clearly suggest a plausible candidate; otherwise, the nearest gene is given, unless there are no genes within the 500-kb window around the SNP. Information on each gene was obtained from either the OMIM or the Jackson Laboratory websites.
Details are from Uniprot summaries.
In two instances, there is evidence that the SNPs we identified (or those in LD with them) influence gene expression. We used data from the publicly available ‘mRNA by SNP Browser 1.0′ program described recently12 to determine whether any of the SNPs were associated with mRNA expression in lymphocytes. rs2282978, which associates with height at P = 8 × 10−23 and occurs in intron 4 of the CDK6 (cyclin-dependent kinase 6) gene, was associated with CDK6 expression (P = 1 × 10−6). rs1863913, an r2 = 1 proxy for rs10935120 (height P = 7 × 10−8), which occurs in intron 2 of ANAPC13 (anaphase promoting complex 13) and 4.4 kb upstream of CEP63 (centrosomal protein 63), was associated with ANAPC13 (P = 9 × 10−18) and CEP63 (P = 4 × 10−12) expression. There was no evidence for any of the other SNPs affecting transcript expression in these lymphoblastoid cell lines.
The genes implicate a number of biological pathways and processes in the normal determination of human height, including Hedgehog signaling (IHH, HHIP, PTCH1), basic cell cycle regulation (CDK6, one of the cyclin-dependent kinases implicated in cell cycle progression13), extracellular matrix (ADAMTSL3 and EFEMP1) and chromatin rearrangement and polycomb proteins (HMGA2 and SCMH1). Several of the genes are also disrupted in cancers (for example, HMGA2, CDK6, DLEU7), providing further evidence of a link between normal growth and unregulated cell differentiation. For other loci, no gene in the region is an obvious candidate for influencing height, and in one case (rs4549631) only a hypothetical gene, LOC387103, is within a 750-kb window of the SNP.
Of note, rs6060373 (P = 2 × 10−17) is highly correlated (HapMap r2 = 0.89) with a functional SNP in the GDF5 gene that has recently been convincingly shown to alter the risk of osteoarthritis14,15. This allele, which we found to be associated with higher height, is also associated with a decreased risk of hip and knee osteoarthritis. A plausible explanation of these associations is that the variant influences the ‘thickness’ of a person’s cartilage.
Methodological issues
We next carried out a series of analyses to address additional important issues regarding the genetic architecture of human height. Although our results are limited to height, our findings may prove useful in guiding studies of other quantitative traits.
We first tested whether the SNPs representing the 20 loci deviated from an additive model or had different effect sizes in males and females. There was suggestive evidence for deviation from an additive (per allele) mode of inheritance for two of the variants: rs12735613 (P = 0.009) and rs1390401 (P = 0.007). There was also suggestive evidence that rs6440003, the most strongly associated SNP in our study, had a greater effect in females (0.12s.d., 95% CI = 0.09–0.14) than males (0.07s.d., 95% CI = 0.04–0.09), P = 0.01 (Table 1)
Adult height is the result of both growth throughout Adult childhood and loss of height during the aging process. We therefore assessed the influence of age on the 20 robust associations. We did not find any evidence that the effects on height were different in individuals <50 years compared to those aged >50 years (all P > 0.01; similar results were obtained when we used a cut-off of 40 years of age), or when adjusting for age decade (see Supplementary Table 4 online). This suggests that the effects are predominantly on developmental and childhood growth rather than on processes involved in loss of height, although studies of more young adults and children are needed to confirm this.
It has often been stated that gene–gene interactions may have a prominent role in complex traits, but there are few, if any, empirical data to show this. We looked for any evidence of deviation from an additive model of the joint effects between all possible pairs of the 20 loci. When taking into account the number of tests, we did not find any strong evidence for deviation from additivity (all P > 0.017; see Supplementary Table 5 online).
To assess the combined impact of the 20 SNPs on adult height, we analyzed only the UK stage 2 samples. This removes the bias due to the effect of the ‘winner’s curse’16, which we observed in our data: 17 of the 20 SNPs had a larger effect size in the GWA study compared to our follow-up study (P = 0.003 in a test against a 50:50 distribution). Figure 3 shows the linear increase in the average height of individuals with increasing numbers of ‘tall’ alleles, and the normal distribution of the frequency of ‘tall’ alleles. Combined, these 20 SNPs explain ~2.9% of the variance in adult height in the UK stage 2 sample. There is a 0.7s.d. (~5 cm) difference in height between the 6.2% of people with 17 or fewer ‘tall’ alleles compared to the 5.5% of people with 27 or more.
Figure 3.
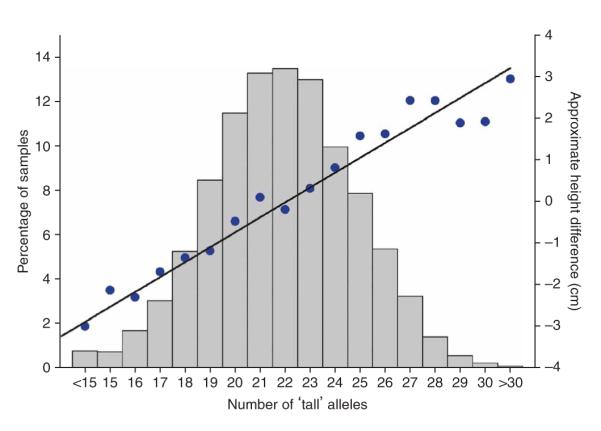
The combined impact of the 20 SNPs with a P < 5 × 10−7. Subjects were classified according to the number of ‘tall’ alleles at each of the 20 SNPs; the mean height for each group is plotted (blue dots). The black line is a linear regression line through these points. The gray bars represent the proportion of the sample with increasing numbers of ‘tall’ alleles. The approximate height difference (cm) was obtained by multiplying the mean Z-score height for each group by 6.82 cm (the approximate average s.d. of height across the samples used in this study).
Power and sample size issues are of primary importance to the field of complex traits genetics. Our results indicate that many tens of thousands of individuals will be needed to reliably detect a large proportion of the variance in some quantitative traits. In this study, real signals emerged only after many individually underpowered GWA studies were combined (Fig. 1 and Supplementary Fig. 3 online). We used the effect sizes observed in the stage 2 samples for each of the 20 SNPs to determine how much power we had to detect the associations in the GWA study (Fig. 4). We had low power to detect some of the SNPs. For example, for four of the SNPs, we had less than 10% power to detect the associations at a P < 1 × 10−5 significance level in the GWA study.
Figure 4.

Power estimates. (a) The power of the genome-wide study to identify the variants that had a P < 5 x 10−7 in the joint analysis at P < 1 × 10−5 using the effect size estimates from the follow-up samples only. (b) The sample size required to identify these variants using the effect size estimates from the follow-up samples only at a P < 5 ~ 10−7 with 80% power. Effect sizes ranged from 0.083s.d. with MAF ~0.44 for rs6440003 to 0.033s.d. with MAF of 0.35 for rs8099594.
Considerable effort and resources have been devoted to identifying regions of the genome that are shared more often than expected by chance between relatives of similar height— the linkage approach to gene identification. We analyzed the overlap between linked regions (lod score >2.0, see URLs section in Methods) and our association results5. We assumed a linked region to be a 10-Mb window around the peak marker for all regions with lod score >2.0. Given the proportion of the genome that these regions cover, we would have expected 3.5 (5.3 × 108 bp covered by linkage regions / 3.0 × 109 bp in the human genome) of the 20 SNPs to have occurred in linked regions by chance alone, and we observed four (P = 0.73); for linked regions with lod scores >3, the corresponding statistics were 0.80 (expected) and 1 (observed), P = 0.81. We did not find any evidence of overrepresentation of significant associations in linked regions: 227 of 79,241 SNPs (0.29%) in linked regions with lod score >2 had P values <0.001, compared to 892 of 323,710 (0.28%) in nonlinked regions, P = 0.60. For linked regions with lod score >3, the corresponding figures are 48 of 22,036 SNPs (0.22%) and 1,071 of 380,915 SNPs (0.28%), respectively, P = 0.08.
DISCUSSION
Our results are consistent with Fisher’s proposal from 1918 that many variants of individually small effect explain the heritability of height17. On the basis of the stage 2 samples, we found that the 20 robustly associated variants alter height by between ~0.2 and 0.6 cm per allele, but they explain only ~3% of the variation in height within the population.
Some of the remaining heritability of height will be explained by additional SNPs with small effect. First, we have shown that some of the SNPs that we took forward into stage 2, but that did not reach a P < 5 × 10−7 on joint analyses, probably represent true associations (for example, an excess of SNPs showed the same direction of effect in stage 2 as in stage 1, P = 0.02). Second, we observed a large effect of the winner’s curse16 and, as such, we had low power to detect some of the SNPs in the GWA part of our study, strongly suggesting that there are many more common variants of a similar effect size yet to be found. Identifying these and variants of even smaller effect will require tens of thousands of individuals (Fig. 4).
To further investigate whether there are more SNPs associated with height to be identified through larger sample size, we compared our results to those presented in the accompanying manuscript from Lettre et al.18 They identify association for several of the loci reported in our study (ZBTB38, HMGA2, GDF5, HHIP, ADAMTSL3 and CDK6), and find suggestive association with a SNP at the FUBP3 locus (P = 8 × 10−7), which we also followed up and found suggestive evidence for (P = 2 × 10−5). FUBP3 therefore likely represents an additional gene associated with height. We produced a quantile-quantile plot for the P values observed in the Lettre et al.18 study for the most-associated 10,000 SNPs from our study, excluding known loci. The deviation of the observed statistics from the null distribution (Fig. 5) clearly indicates that there are many more height-associated SNPs that remain to be identified from GWA studies. Although SNPs will explain some of the residual variation, it is possible that much of the heritability of height will be explained by rare variants or copy number polymorphisms, which are not captured by the GWA approach.
Figure 5.
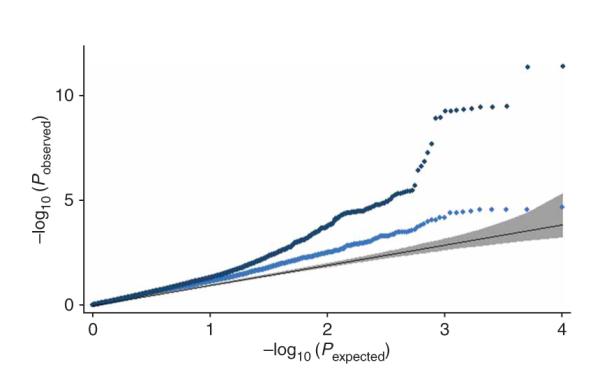
Comparison of results across independent meta-analyses. Quantile-quantile plot for the P values from the accompanying Lettre et al.18 study of the most associated 10,000 SNPs from our study (excluding the DGI component to make the observations independent), including (dark blue dots) and excluding known loci (light blue dots). The black line is the expected line under the null distribution. The gray band represents the 95% concentration bands, which are an approximation to the 95% confidence intervals around the expected line.
As we only tested an additive model and did not carry out sex-specific analyses on a genome-wide level, we were biased away from detecting sex-specific and nonadditive effects in this study. However, we did find some weak evidence that our most-associated SNP had a stronger effect in females (0.12s.d., 95% CI = 0.09–0.14) than males (0.07s.d., 95% CI = 0.04–0.09), P = 0.01, although this finding needs to be replicated. Given that final adult height is highly dichotomized by sex, growth trajectories show clear gender differences, and sex hormones influence height, further studies are needed to investigate more thoroughly the presence of sex-specific effects. It will also be important to test for nonadditive effects within and between loci, and to investigate the role of these and other loci in individuals of non-European ancestry.
We did not find any overlap between previously reported linkage peaks and the results from our GWA study. The variants we have identified have small effects, and as such, it is not surprising that they do not individually explain previously observed linkage peaks. It may be that some of the linkage peaks are explained by low-frequency, relatively high-penetrance alleles, which would not be captured using the GWA approach. However, our findings do not support the idea that genes with common variants associating with height also contain the type of variant that is readily identifiable through the linkage approach.
A limitation of this study is that we have not fine-mapped the identified loci. However, ten of the loci we identified contain genes previously known to be involved in growth from rare human syndromes or animal studies, and we have shown that common variation in or around these genes influences normal human growth. Additionally, two of the variants seem to alter expression of nearby genes (CDK6 and ANAPC13). Further fine-mapping and functional studies of these and the remaining loci will likely provide new insights into growth and development. Mutations in these regions may also explain some monogenic syndromes for which no genes have currently been identified. The observation that half of the identified loci contained candidate genes suggests that combining genome-wide with candidate gene approaches may be a productive way for identifying more loci associated with height.
In conclusion, using 13,665 individuals with genome-wide scan data and 16,482 follow-up subjects, we have identified 20 genomic regions in which common variation influences adult height. The study highlights several important pathways and processes involved in normal growth, and provides insights into the genetic architecture of a classic quantitative trait.
METHODS
Genome-wide association (stage 1) samples
Four of the six genome-wide scan studies were part of the UK Wellcome Trust Case Control Consortium (WTCCC) and have been described in detail previously11. Briefly, these four studies were the type 2 diabetes (WTCCC-T2D), hypertension (WTCCC-HT) and coronary artery (WTCCC-CAD) disease branches and the national blood service (WTCCC-UKBS) controls. A manuscript describing the cohorts used in the Diabetes Genetics Initiative (DGI) 500K genome-wide association study for type 2 diabetes has been published19, and a description of the sample is also available online (see URLs section below). The EPIC Obesity case-cohort study includes 3,847 participants and is nested within the EPIC-Norfolk Study, a population-based cohort study of 25,663 men and women of European ancestry aged 39–79 years recruited in Norfolk, UK between 1993 and 1997. The cases (n = 1,685) were randomly selected from the obese individuals within this cohort and are defined as those with a body mass index >30 kg/m2. The control-cohort consists of 2,566 individuals randomly selected from the EPIC-Norfolk study, and thus, by design, 381 individuals are part of the control-cohort as well as the case group.
Basic anthropometric data for all genome-wide studies are presented in Supplementary Table 1. Extensive quality control steps were taken to exclude poorly performing or samples of non-European descent from analyses. For five of the six GWA studies, these steps are described in detail11,19. For the EPIC-Obesity study, 277 of 3,847 participants were excluded (sample call rate <94%, n = 202; heterozygosity <23% or >30%, n = 36; >5.0% discordance in SNP pairs with r2 = 1 in HapMap, n = 25; ethnic outlier, n = 8; related individuals (concordance with another DNA is >70.0% and <99.0%, 1 selected on the basis of sample call rate), n = 5; duplicate (concordance with another DNA is >99.0%, 1 selected on the basis of sample call rate), n = 1), and 10 individuals did not have genotype data available, such that 3.560 individuals were included in the analyses.
The WTCCC-T2D, WTCCC-HT, DGI and EPIC-Obesity studies measured height using standard anthropometric techniques. For WTCCC-CAD and WTCCC-UKBS, height data was self-reported from questionnaires. The lack of evidence of heterogeneity across all studies for the 20 confirmed loci indicates that the inclusion of self-reported data has not affected the results appreciably.
All subjects gave written informed consent, and the project protocols were approved by the local research ethics committees in the UK.
Stage 2 samples
The UKT2D GCC study has been described previously20. All subjects were of self-reported European descent, living in the Tayside region of Dundee, UK. Height measurements were made as for the WTCCC samples. This study was approved by the Tayside Medical Ethics Committee, and informed consent was obtained from all subjects.
EFSOCH (Exeter Family Study of Childhood Health) is a prospective study of parents and children from a consecutive birth cohort21. Subjects were recruited from a postcode-defined region of Exeter, UK between 2000 and 2004 and were of self-reported European descent. Parental height was measured using a stadiometer by the research midwife at 28 weeks gestation. Ethical approval was given by the North and East Devon Local Research Ethics Committee, and informed consent was obtained from the parents of the newborns.
The MRC British Genetics of Hypertension (BRIGHT) study has been described previously22. Briefly, severely hypertensive individuals were recruited from the Medical Research Council General Practice Framework and other family-physician practices in the UK. All subjects were of self-reported European ancestry up to the level of grandparents. Height was measured by using a Marsden ultrasonic height measure; the standard operating procedure for this is described at the MRC BRIGHT study webpage (see URLs section below).
The CoLaus study has been described in detail previously23. Briefly, it is a single-center, cross-sectional study including a random sample of 6,188 extensively phenotyped subjects of European descent (3,251 women and 2,937 men) aged 35 to 75 years living in Lausanne, Switzerland. Height was measured to the nearest 5 mm using a Seca height gauge.
Statistical methods
All GWA studies were genotyped using the Affymetrix 500K chip. For the WTCCC studies, we used the WTCCC-defined list of 459,446 SNPs that had passed quality control11; additionally, we required a MAF > 0.01, and a Hardy-Weinberg equilibrium P < 1 × 10−4 for each individual GWA study in our analyses. For the EPIC-Obesity study, we included only SNPs that were polymorphic (7,532 excluded), had a call rate ≥90% (31,067 excluded), showed Hardy-Weinberg equilibrium with a P > 10−6 (25,907 excluded) and had MAF ≥5%. We analyzed a total of 338,830 SNPs from the EPIC-Obesity study. The DGI data SNP quality control and exclusion criteria are reported in detail elsewhere19; we used a total of 386,731 SNPs from this study. We note that there is a small familial component to the DGI data, which is not taken into account in the betas and standard errors provided in the publicly available data used in our analyses. The extent of the P-value inflation that is caused by this is small (genomic control λ < 1.1), so it will have marginal effects on the association results, but we have provided results excluding the DGI study in Table 1 to demonstrate the robustness of the associations. We report the 402,951 SNPs which passed quality control in at least four of the six GWA studies.
Individual level genotype data were available from only one GWA study (WTCCC-T2D); only summary height association statistics were available for the other studies. For each GWA study, summary statistics, assuming an additive inheritance model, from linear regression using Z scores (described below) were generated using PLINK24 (WTCCC-T2D, WTCCC-UKBS, WTCCC-CAD, DGI), SAS/Genetics 9.1 (EPIC-Obesity Study) or R (WTCCC-HT).
For each stage 2 study, we examined the associations between genotype and height Z score using linear regression (described below). We carried out stage 2 analyses in Stata/s.e.m. 9.1 for Windows (StataCorp) for all studies, except for CoLaus, for which we used PLINK24.
Height was normally distributed in all cohorts. For the WTCCC GWA studies, UKT2D GCC, BRIGHT and EFSOCH studies, sex-specific height Z scores were generated within each study. Details for the DGI are available on their website. For EPIC-Obesity, height Z scores were created by gender and age decades (<50, 50<60, 60<70, ≥70). For the CoLaus study, height was corrected using a linear model, regressing height simultaneously onto age, sex, ancestry principal components8 and grandparental birthplaces. The residuals were rescaled to have variance 1, and then used as a ‘corrected’ phenotype.
Meta-analysis statistics were generated using the inverse-variance meta-analysis method assuming fixed effects. The Q test was used to test for between-study heterogeneity. We used Stata/s.e.m. 9.1 for Windows (StataCorp) for all meta-analysis calculations.
For the GWA study, EIGENSTRAT8 was run in each individual study on the full set of markers (~400,000 SNPs). Within each study, similar results were obtained when using the first three principal components or the first ten principal components.
All individual level data analyses were done in Stata/s.e.m. 9.1 for Windows (StataCorp). To test for a deviation from an additive mode of inheritance for each of the 39 SNPs that we took forward into stage 2, we carried out a likelihood ratio test of the additive regression model against the full 2 degrees-of-freedom model.
To test for a difference in effect size between genders, we carried out a likelihood ratio test of the additive model against a model that also included a sex-by-genotype interaction term. To test for an influence of age on the effect size, we compared a regression model including dichotomized age (<50 and ≥50) and genotype to a model that also included a dichotomized age-by-genotype interaction term. We also carried out the same analysis using 40 years as a cut-off and age deciles rather than dichotomized age.
For the gene–gene interaction analyses, we assumed additive effects within loci, and compared a joint effects model to a model containing an interaction term using likelihood ratio tests.
For the combined effect analyses, we used only stage 2 UK subjects to reduce the effect of the ‘winner’s curse’16. We only used subjects that had been successfully genotyped at each of the 20 SNPs that reached a P < 5× 10−7, and grouped subjects by the total number of ‘tall’ alleles that they carried. × The mean height (estimated by multiplying the Z-score effect size by 6.82 cm, the average s.d. of adult height across the cohorts used in this study) and frequency were then plotted using SigmaPlot for Windows Version 10.0 (Systat).
Quantile-quantile plots were generated using Stata/s.e.m. 9.1 for Windows. The 95% concentration bands, which are the approximate 95% confidence intervals around the null distribution were generated as described25.
Quanto was used for the power calculation26. To assess the impact of the ‘winner’s curse’, we carried out a binomial distribution test of the number of times the stage 1 result was greater than the stage 2 result, compared to that expected under the null of 50%.
We used linkage data from the website provided by a previous study5, which describes all reports in the literature that achieved lod scores >2 for height. Where a peak marker (or markers) was reported, we called a 10-Mb window around the marker (or markers) a ‘linked region’. Where no peak marker was reported, we used the reported deCODE cM coordinates to determine the linked region. To compare the observed number of occasions that one of the 20 ‘real’ SNPs occurred in a linked region to that expected under the null distribution, we took the total number of base pairs in nonoverlapping linked regions and divided it by the number of base pairs in the human genome (University of California Santa Cruz Genome Browser, NCBI Build 36.1 statistics). The expected number of times that the 20 real SNPs occurred in linked regions is then 20 × (base pairs in linked regions / total number of base pairs in the human genome). We used a Poisson test to determine the significance of the difference in the number of confirmed SNPs observed under linkage peaks compared to the expected number. We carried out this calculation for SNPs with lod scores >3 and those with scores >2.
To determine whether there was any overrepresentation of all associations at P < 0.001 in linked regions, we compared the proportions of these SNPs occurring in linked regions to those not occurring in linked regions. Again, we carried out this calculation for SNPs with lod scores >2 or >3.
Stage 2 genotyping
Genotyping of the UKT2D GCC, BRIGHT and EFOSCH samples was done by KBiosciences using their own novel system of fluorescence-based competitive allele-specific PCR (KASPar). Details of assay design are available from the KBiosciences website. The CoLaus study is a GWA study (for which GWA data were not available in time for this study to be involved in stage 1) and is described in detail elsewhere27.
URLs
Scandinavian study, http://www.broad.mit.edu/diabetes/scandinavs/index.html; Stature Gene Map, http://www.genomeutwin.org/stature_ gene_map.htm; DGI, http://www.broad.mit.edu/diabetes/; MRC BRIGHT study, www.brightstudy.ac.uk.
Supplementary Material
ACKNOWLEDGMENTS
M.N.W. is a Vandervell Foundation Research Fellow. C.L. is a Nuffield Department of Medicine Scientific Leadership Fellow. R.M.F. is funded by a Diabetes UK research studentship. S.B. is supported by the Giorgi-Cavaglieri Foundation and the Swiss National Science Foundation (grant 3100AO-116323/1), which also supports J.S.B. (grant 310000-112552/1). We would like to thank M. Bochud, Z. Kutalik, G. Waeber, K. Song and X. Yuan for their contribution to the Lausanne study. The WTCCC CAD cohort collection was supported by grants from the British Heart Foundation, Medical Research Council and National Health Service Research & Development. N.J.S. holds a chair supported by the British Heart Foundation. We thank the Wellcome Trust for funding. C.W. is funded by the British Heart Foundation (grant number FS/05/061/19501). The BRIGHT study is supported by the Medical Research Council (grant number G9521010D) and the British Heart Foundation (grant number PG02/128).
Footnotes
Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions
Note: Supplementary information is available on the Nature Genetics website.
References
- 1.Macgregor S, Cornes BK, Martin NG, Visscher PM. Bias, precision and heritability of self-reported and clinically measured height in Australian twins. Hum. Genet. 2006;120:571–580. doi: 10.1007/s00439-006-0240-z. [DOI] [PubMed] [Google Scholar]
- 2.Preece MA. The genetic contribution to stature. Horm. Res. 1996;45:56–58. doi: 10.1159/000184849. [DOI] [PubMed] [Google Scholar]
- 3.Silventoinen K, Kaprio J, Lahelma E, Koskenvuo M. Relative effect of genetic and environmental factors on body height: differences across birth cohorts among Finnish men and women. Am. J. Public Health. 2000;90:627–630. doi: 10.2105/ajph.90.4.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Silventoinen K, et al. Heritability of adult body height: a comparative study of twin cohorts in eight countries. Twin Res. 2003;6:399–408. doi: 10.1375/136905203770326402. [DOI] [PubMed] [Google Scholar]
- 5.Perola M, et al. Combined genome scans for body stature in 6,602 European twins: evidence for common Caucasian loci. PLoS Genet. 2007;3:e97. doi: 10.1371/journal.pgen.0030097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Palmert MR, Hirschhorn JN. Genetic approaches to stature, pubertal timing, and other complex traits. Mol. Genet. Metab. 2003;80:1–10. doi: 10.1016/s1096-7192(03)00107-0. [DOI] [PubMed] [Google Scholar]
- 7.Weedon MN, et al. A common variant of HMGA2 is associated with adult and childhood height in the general population. Nat. Genet. 2007;39:1245–1250. doi: 10.1038/ng2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Price AL, et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 9.Devlin B, Roeder K. Genomic control for association studies. Biometrics. 1999;55:997–1004. doi: 10.1111/j.0006-341x.1999.00997.x. [DOI] [PubMed] [Google Scholar]
- 10.Freedman ML, et al. Assessing the impact of population stratification on genetic association studies. Nat. Genet. 2004;36:388–393. doi: 10.1038/ng1333. [DOI] [PubMed] [Google Scholar]
- 11.Wellcome Trust Case Control Consortium Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dixon AL, et al. A genome-wide association study of global gene expression. Nat. Genet. 2007;39:1202–1207. doi: 10.1038/ng2109. [DOI] [PubMed] [Google Scholar]
- 13.Malumbres M, Barbacid M. Mammalian cyclin-dependent kinases. Trends Biochem. Sci. 2005;30:630–641. doi: 10.1016/j.tibs.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 14.Southam L, et al. An SNP in the 5′-UTR of GDF5 is associated with osteoarthritis susceptibility in Europeans and with in vivo differences in allelic expression in articular cartilage. Hum. Mol. Genet. 2007;16:2226–2232. doi: 10.1093/hmg/ddm174. [DOI] [PubMed] [Google Scholar]
- 15.Miyamoto Y, et al. A functional polymorphism in the 5′ UTR of GDF5 is associated with susceptibility to osteoarthritis. Nat. Genet. 2007;39:529–533. doi: 10.1038/2005. [DOI] [PubMed] [Google Scholar]
- 16.Lohmueller KE, Pearce CL, Pike M, Lander ES, Hirschhorn JN. Meta-analysis of genetic association studies supports a contribution of common variants to susceptibility to common disease. Nat. Genet. 2003;33:177–182. doi: 10.1038/ng1071. [DOI] [PubMed] [Google Scholar]
- 17.Fisher RA. The correlation between relatives on the supposition of Mendelian inheritance. Philosoph. Trans. Royal Soc. Edinburgh. 1918;52:399–433. [Google Scholar]
- 18.Lettre G, et al. Identification of ten loci associated with height highlights new biological pathways in human growth. Nat. Genet. 2008 Apr 6; doi: 10.1038/ng.125. advance online publication, doi:10.1038/ng.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Diabetes Genetics Initiative of Broad Institute of Harvard and M.I.T. et al. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science. 2007;316:1331–1336. doi: 10.1126/science.1142358. [DOI] [PubMed] [Google Scholar]
- 20.Zeggini E, et al. Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science. 2007;316:1336–1341. doi: 10.1126/science.1142364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Knight B, Shields BM, Hattersley AT. The Exeter Family Study of Childhood Health (EFSOCH): study protocol and methodology. Paediatr. Perinat. Epidemiol. 2006;20:172–179. doi: 10.1111/j.1365-3016.2006.00701.x. [DOI] [PubMed] [Google Scholar]
- 22.Caulfield M, et al. Genome-wide mapping of human loci for essential hypertension. Lancet. 2003;361:2118–2123. doi: 10.1016/S0140-6736(03)13722-1. [DOI] [PubMed] [Google Scholar]
- 23.Marques-Vidal P, et al. Prevalence and characteristics of vitamin or dietary supplement users in Lausanne, Switzerland: the CoLaus study. Eur. J. Clin. Nutr. 2007 Oct 17; doi: 10.1038/sj.ejcn.1602932. advance online publication, doi: 10.1038/sj.ejcn.1602932. [DOI] [PubMed] [Google Scholar]
- 24.Purcell S, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stirling WD. Enhancements to aid interpretation of probability plots. Statistician. 1982;31:211–220. [Google Scholar]
- 26.Gauderman WJ. Sample size requirements for association studies of gene-gene interaction. Am. J. Epidemiol. 2002;155:478–484. doi: 10.1093/aje/155.5.478. [DOI] [PubMed] [Google Scholar]
- 27.Sandhu MS, et al. LDL-cholesterol concentrations: a genome-wide association study. Lancet. 2008;371:483–491. doi: 10.1016/S0140-6736(08)60208-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.