

Abstract
Pasteurella multocida toxin (PMT) stimulates and subsequently uncouples phospholipase C β1 (PLCβ1) signal transduction through its selective action on the alpha subunit of the Gq protein. Here, we describe the application of an NFAT-β-lactamase reporter assay as a functional readout for PMT-induced activation of the Gq-protein-coupled PLCβ1-IP3-Ca2+ signaling pathway. Use of the NFAT-β-lactamase reporter assay with a cell-permeable fluorogenic substrate provides high sensitivity due to the absence of endogenous β-lactamase activity in mammalian cells. This assay system was optimized for cell density, dose and time exposure of PMT stimulation. It is suited for quantitative characterization of PMT activity in mammalian cells and for use as a high-throughput screening method for PMT deletion and point mutants suitable for vaccine development. This method has application for diagnostic screening of clinical isolates of toxinogenic P. multocida.
Keywords: atrophic rhinitis, dermonecrotic toxin, β-Lactamase, FRET, Gq-protein, NFAT, PLCβ1, PMT, reporter
Introduction
The protein toxin from Pasteurella multocida (PMT) experimentally induces all of the major symptoms of a number of economically important diseases in a wide range of wild and domestic livestock and pets (rabbits, cats and dogs) (Foged, 1992; Harper et al., 2006; Wilson and Ho, 2006), including moderate to severe progressive atrophic rhinitis, dermonecrosis and decreased overall stature and weight gain. P. multocida causes opportunistic infections in humans through bite or scratch wounds or respiratory infections from chronic zoonotic exposure to infected animals (Arashima and Kumasaka, 2005). Although PMT activates dendritic cells, it is a poor adjuvant and appears to suppress the antibody response (Bagley et al., 2005). However, PMT is nonetheless an effective immunogen and mutant derivatives have shown potential for vaccine development (Liao et al., 2006; Petersen et al., 1991; Register et al., 2007; Riising et al., 2002).
PMT is a 1285-amino acid protein that binds to and enters mammalian cells via receptor-mediated endocytosis (Pettit et al., 1993; Rozengurt et al., 1990). PMT has dramatic and differential effects on differentiation and proliferation of fibroblasts, bone cells, epithelial cells and cardiomyocytes (reviewed in (Wilson and Ho, 2004; Wilson and Ho, 2006)). The primary intracellular target of PMT responsible for activation of the PLCβ1 pathway is the free monomeric α subunit of the Gq-protein (Wilson and Ho, 2004; Wilson et al., 1997; Zywietz et al., 2001). PMT-induced stimulation of PLCβ1 by Gαq leads to the hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP2) to inositol 1,4,5-trisphosphate (IP3) and diacylglycerol. Accordingly, the release of these second messengers stimulates Ca2+ mobilization and activates protein kinase phosphorylation. One of the downstream signaling pathways activated by PMT-mediated Ca2+ release is the nuclear factor of activated T cells (NFAT) (Aminova and Wilson, 2007).
A number of cellular and biochemical assays for PMT activity have been reported, including: assays for cytotoxicity based on cell morphology using Vero cells (Pettit et al., 1993; Wilson et al., 2000) and embryonic bovine lung cell (Busch et al., 2001; Pennings and Storm, 1984), assays for stress fiber formation based on fluorescence labeling of actin (Dudet et al., 1996; Lacerda et al., 1996), assays for stimulation of DNA synthesis based on tritiated thymidine incorporation into DNA or increased cell number (Higgins et al., 1992; Rozengurt et al., 1990; Wilson et al., 2000), assays for PLCβ1 activity based on stimulation of Ca2+-dependent Cl− currents in voltage-clamped oocytes (Wilson et al., 1997) or on tritiated inositol incorporation into total cellular inositol phosphates (Murphy and Rozengurt, 1992; Orth et al., 2004; Staddon et al., 1991), and assays for activation of mitogenic signaling or protein kinase cascades based on luciferase reporter gene activation in transfected cells (Aminova and Wilson, 2007; Orth et al., 2007a; Sabri et al., 2000) or on Western blotting (Obreztchikova et al., 2006; Orth et al., 2007a; Orth et al., 2007b; Orth et al., 2005; Sabri et al., 2002; Seo et al., 2000; Wilson et al., 2000). All of these methods are time consuming and laborious, with many also requiring transient transfection of mammalian cells or use of radioactivity. Importantly, they are all difficult to adapt for high-throughput screening.
β-lactamase is a bacterial enzyme that has been developed as a reporter system for detection and quantification of gene expression in mammalian cells resulting from activation of receptor-mediated signal transduction cascades. One such reporter system utilizes Ca2+-dependent NFAT signaling to activate NFAT-specific enhancer-promoter regions upstream of the reporter gene (Zlokarnik, 2000; Zlokarnik et al., 1998). β-lactamase activity within intact cells can be detected by using the membrane-permeable fluorogenic substrate ester, CCF4/AM, which diffuses into the cell, but the ester then is hydrolyzed and the resulting hydrophilic substrate accumulates to provide a high concentration of substrate in the cytosol (Kunapuli et al., 2003). In the CCF4 substrate, fluorescence excitation of the coumarin moiety at 409 nm results in transfer of light energy to the fluorescein acceptor moiety, generating fluorescence resonance energy transfer (FRET) and emission of green light from the cells with a wavelength of 520 nm. Expression of β-lactamase in the cell causes hydrolysis of the substrate and loss of FRET due to physical separation of the coumarin and fluorescein moieties. Consequently, the released coumarin component absorbs the light energy and emits at 447 nm, resulting in the cells appearing blue. Thus, upon excitation at 409 nm, resting cells without β-lactamase activity appear green, while activated cells expressing β-lactamase appear blue under fluorescence microscopy. To calculate the fraction of activated cells, the ratio of blue/green emission (460 nm to 530 nm) is determined (Kunapuli et al., 2003).
Here, we describe the use of the NFAT-coupled β-lactamase blue/green reporter assay as a functional readout for PMT-induced activation of Ca2+ signaling. We tested the blue/green assay for detection of PMT biological activity using bacterially expressed full-length PMT and N-terminal and C-terminal deletion fragments of PMT. In addition, we exogenously expressed full-length PMT as well as deletion and site-specific point mutants of PMT as GFP fusion proteins in NFAT-bla CHO-K1 cells by using mammalian expression vectors. Expression of the expected proteins was confirmed by Western blotting, and the intracellular activity was assessed using the NFAT-β-lactamase blue/green assay.
Materials and Methods
Materials
NFAT-bla CHO-K1 cells, cell-permeable CCF4/AM substrate, zeocin antibiotic, DMEM, non-essential amino acids, dialyzed fetal bovine serum, 1M HEPES buffer, LipofectAMINE reagent and other cell culture reagents were purchased from Invitrogen. Thapsigargin, PEG 400, and probenecid were purchased from Sigma. Restriction enzymes and other molecular biology reagents were obtained from Roche Biochemicals or New England Biolabs. The 96-well, black, clear-bottom, tissue culture-treated assay plates were purchased from Corning Inc.
Purification of recombinant PMT
Bacterially expressed recombinant PMT (rPMT) was prepared, purified and quantified essentially as previously described (Aminova and Wilson, 2007). In brief, rPMT was expressed in E. coli TOP10 cells (Invitrogen) containing the pTHC-toxA vector under control of IPTG induction. Cell extracts were partially purified by Ni2+-NTA-agarose chromatography (Qiagen). Fractions containing toxin were further purified by HiTrapQ anion exchange chromatography (Amersham) using an FPLC (Akta), followed by desalting using a PD-10 column (Amersham). The His6-tag was removed by thrombin treatment using Thrombin Cleavage Capture Kit (Novagen), according to manufacturer’s protocol. The cleaved His6-tag was removed by Ni2+-NTA-agarose chromatography, and the filtrate containing rPMT was further purified by FPLC using first a HiTrapQ anion exchange column, then a Superdex 200 gel filtration column. The pooled rPMT-containing fractions were concentrated by using Centricon filter units (Millipore) and then desalted by PD-10 column with PBS containing 10% glycerol. Concentrations of rPMT protein were determined by quantitative digital image analysis of Pierce GelCode Blue-stained SDS-PAGE gels with BSA as the standard using NIH ImageJ software. The toxin samples were stored at −80°C until use. The biological activity of rPMT was confirmed as previously described using Vero cell assay (Wilson et al., 2000), with an EC50 value of ~2 ng/mL. Controls of heat-inactivated rPMT (65°C for 10 min) showed no detectable toxic activity or effects on Vero, CHO-K1, or HEK-293T cells.
Purification of recombinant N-terminal and C-terminal PMT fragments
An N-terminal fragment of PMT (residues 1–568) with GFP at the C terminus (1–568g) and a C-terminal fragment of PMT (residues 456–1285) with GFP at the N terminus (g456–1285) were constructed by standard cloning methods into the bacterial expression vector pTrcHis (Invitrogen) and then expressed from E. coli BL21 (DE3) cells (Novagen) and purified and quantified as described above.
Plasmid constructs
Deletion mutants were constructed by fragment exchange or PCR cloning using previously described PMT constructs in pET21 vectors (Novagen) (Wilson et al., 1999). Holotoxin and PMT fragments were cloned into pEGFP-C1 vector (Clontech). The primers and cloning strategy used for each of the deletion mutant constructs is summarized in Table 1. PCR-based site-directed mutagenesis (Fisher and Pei, 1997) with published primers (Orth et al., 2003; Ward et al., 1998), followed by digestion with the restriction enzyme DpnI, was used to introduce the single mutations C1165S, H1205L or H1223L into wild type PMT and fragments (g506–1285 and g1005–1285). After sequence verification, the corresponding sequences containing the desired mutations were introduced into constructs encoding full-length PMT and PMT deletion fragments (g506–1285) and (g1005–1285) by fragment exchange.
Table 1.
Constructs of PMT and PMT fragments used in this study
| Cloning vector pEGFP with GFP affinity and fluorescence tag at N-terminus | ||
|---|---|---|
| Construct | Protein (residues) | Cloning strategy |
| g1–505 | GFP-PMT (1–505) | XhoI/KpnI restriction sites of g1–568 construct filled using Klenow fragment and re-ligated |
| g1–568 | GFP-PMT (1–568) | BglII/BamHI |
| g1–1130 | GFP-PMT (1–1130) | EcoRI/ApaI restriction sites of g1–1285 construct filled with Klenow fragment and re-ligated |
| g506–1285 | GFP-PMT (506–1285) | Fragment cloned using XhoI/KpnI sites |
| g576–1285 | GFP-PMT (576–1285) | Fragments amplified by PCR with BglII/ApaI restriction sites |
| g771–1285 | GFP-PMT (771–1285) | |
| g917–1285 | GFP-PMT (917–1285) | |
| g1005–1285 | GFP-PMT (1005–1285) | |
| g1130–1285 | GFP-PMT (1130–1285) | XhoI/EcoRI restriction site of g506–1285 construct filled using Klenow fragment and re-ligated |
| g1–1285 | GFP-PMT (1–1285) | Fragment 1–505 inserted into g506–1285 construct by using BamHI/XhoI sites |
Transient transfection of NFAT-bla CHO-K1 cells
NFAT-bla CHO-K1 cells were maintained in growth medium (DMEM supplemented with 0.292 g/liter L-glutamine, 10% (v/v) dialyzed fetal bovine serum, 25 mM HEPES, 100 U/ml penicillin, and 100 μg/ml streptomycin) at 37 °C in a 5% CO2 humidified atmosphere. Cells were grown to 60–80% confluence before transient transfection in 6-well plate. Transfection was performed using LipofectAMINE reagent, according to the manufacturer’s instructions. Cells were replated 24 h after transfection at 7,000–10,000 cells/well in 100 μl growth medium without antibiotics in a 96-well black-wall, clear-bottom tissue culture-treated assay plate. Expression of all constructs as full-length proteins in mammalian cells was confirmed by Western blotting (data not shown), except for the constructs g771–1285 and g917–1285, which did not appear to be stable and were largely degraded.
β-Lactamase blue/green assay
NFAT-bla CHO-K1 cells were maintained in growth medium supplemented with 100 μg/ml zeocin, according to manufacturer’s instruction. Cells were replated at 7,000–10,000 cells/well in 100 μl growth medium in a 96-well black clear bottom tissue culture-treated assay plate, 16–20 h before PMT treatment. Cells were treated for 16 h with different concentrations of PMT or N-terminal and C-terminal PMT mutants or with 10 nM thapsigargin as a positive control for maximal cellular response. After PMT treatment, 20 μl of the 6 × CCF4/AM loading buffer, prepared according to the manufacturer’s instructions, was added to the cells in 96-well assay plates, and the cells were incubated at room temperature for 2 h. The microplate was then read in a fluorescence bottom-read microplate reader (Bio-Tek Synergy HT Multi Detection Microplate Reader) using an excitation of 405 nm and emissions at 460 and 530 nm (excitation filter = 409/20 nm, blue emission filter = 460/40 nm, green emission filter = 530/30 nm). Fluorescence micrograph images of the cellular response were also obtained by fluorescence microscopy using an Olympus IX70 fluorescence microscope using blue/aqua filter (#41031, Chroma Technology).
Data analysis and statistics
Data analysis for the β-lactamase assays was performed using Gen5 (Bio-Tek) and Excel (Microsoft). The data were plotted as the ratio of the blue/green emissions (460/530 nm) after subtraction of the background values (buffer only without cells). Experimental data points are shown as the mean ± the standard deviation of triplicate values. Data shown are representative from at least three independent experiments.
Results
Use of NFAT-β-lactamase to assay for PMT activation of calcium signaling
PMT stimulates Ca2+ signaling pathways in cells (Wilson and Ho, 2004). In this study, we exploited a Ca2+-dependent NFAT-β-lactamase reporter system as an assay for PMT stimulation of Ca2+-dependent NFAT signaling in mammalian cells. As shown in Fig. 1, untreated control cells fluoresce green due to the presence of intact FRET-substrate. In cells treated with PMT or thapsigargin, where Ca2+-dependent NFAT signaling is activated, the reporter gene for β-lactamase is expressed and hydrolyzes the FRET-substrate, converting it into the product, which fluoresces blue. External treatment with PMT caused a dose and time-dependent increase in NFAT-β-lactamase activity (Fig. 2, A and B). Treatment of cells externally with bacterially expressed and purified N-terminal (1–568g) or C-terminal (g456–1285) fragments did not cause a similar increase in activity (Fig. 3), confirming that full-length toxin protein is required for entry into cells to exhibit full biological activity.
Fig. 1. Fluorescence micrographs of NFAT-bla CHO-K1 cells with PMT-induced β-lactamase expression.
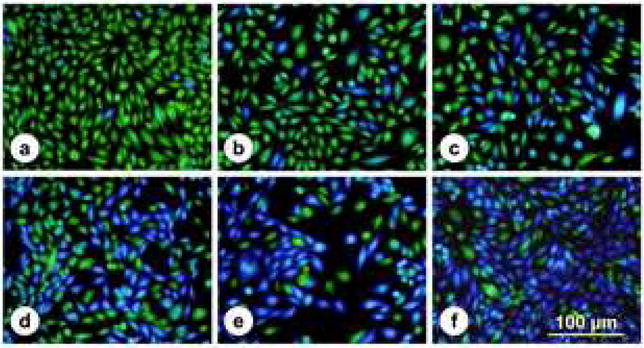
NFAT-bla CHO-K1 cells were grown in 96-well plates and incubated for 16 h with medium or stimulated with increasing doses of PMT: (a) 0, (b) 0.065 nM (10 ng/ml), (c) 0.65 nM (100 ng/ml), (d) 3.25 nM (500 ng/ml), (e) 6.5 nM (1,000 ng/ml), or with (f) 10 nM thapsigargin as a positive control for maximal cellular response. Cells were loaded with CCF4/AM for 2 h at room temperature and visualized by fluorescence microscopy. Non-stimulated cells appeared green and stimulated cells appeared blue. Bar, 100 βm.
Fig. 2. Dose and time course responses of NFAT-bla CHO-K1 Cells to PMT treatment.
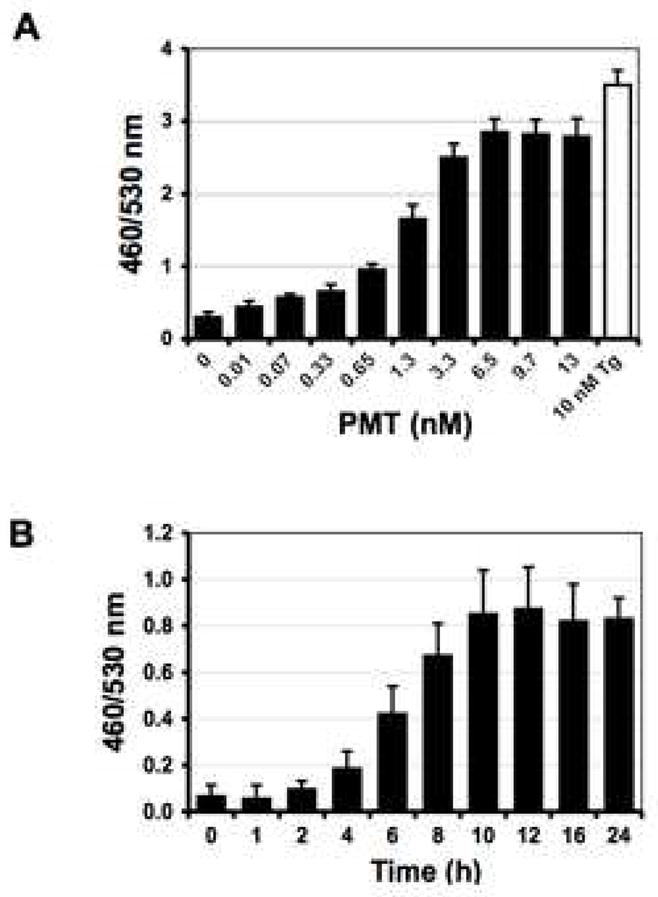
A) Shown is the dose response after 16 h of incubation with the indicated concentrations of PMT at 37°C. Cells were loaded with CCF4/AM for 2 h at room temperature and analyzed by fluorescence microplate reader with excitation at 405 nm and emission at 460 and 530 nm using bottom-read mode. Data were plotted as 460/530 nm ratios versus the concentration of PMT. The EC50 value of PMT determined using this assay was 1.3 nM. B) Shown is a time course of the response to PMT treatment in NFAT-bla CHO-K1 cells. Cells were treated with 0.65 nM PMT at 37°C. At the indicated times, cells were analyzed as described in (A).
Fig. 3. Screening bacterially produced PMT mutants for cellular activity using the β-lactamase blue/green assay.
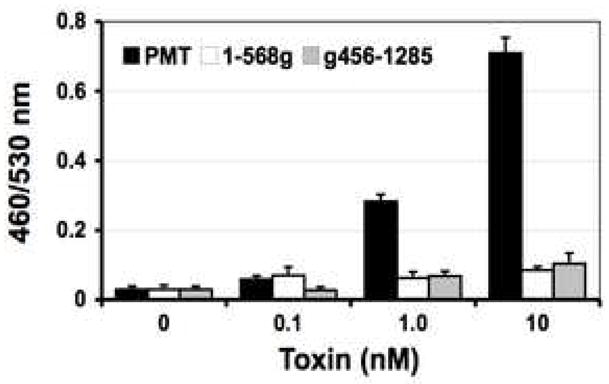
Shown is the dose response to external treatment for 16 h with bacterially expressed and purified PMT or PMT fragments (1–568g, g456–1285). Cells were analyzed as described in Fig. 2A.
Expression of PMT deletion mutants in mammalian cells assessed using NFAT-β-lactamase reporter activity
Exogenous expression of full-length GFP-PMT (g1–1285) in NFAT-bla CHO-K1 cells caused up to a 10-fold increase in reporter activity using the NFAT-β-lactamase blue/green assay (Fig. 4). Whereas none of the N-terminal fragments (g1–505, g1–568, g1–1130) showed activity, exogenous expression of the C-terminal PMT fragments (g506–1285, g576–1285 and g1005–1285) had intracellular activity (Fig. 4B), with the g506–1285 and g576–1285 proteins having activity comparable to that found for full-length GFP-PMT (g1-1285). The presence of GFP had no apparent effect on the blue/green readings (Fig. 4A).
Fig. 4. Screening exogenously expressed PMT mutants for intracellular activity using the β-lactamase blue/green assay.

NFAT-bla CHO-K1 cells were transiently transfected with a mammalian expression vector (pEGFP-C1) encoding GFP-fusions with full-length PMT or N-terminal or C-terminal fragments of PMT. A) Fluorescence micrographs showing the expression of GFP-toxin proteins in NFAT-bla CHO-K1 cells after transfection with pEGFP (control GFP alone), g1–568, g1–1285 or g506–1285 for 24-hours (upper panel). The cells were then replated at higher density for blue/green assay (lower panel). The presence of GFP did not appear to interfere with the blue/green readings. Bar, 50βm. B) Comparison of exogenously expressed GFP-toxin proteins for activation of NFAT signaling in NFAT-bla CHO-K1 cells using the blue/green assay. Transfected cells were analyzed as described in Fig. 2A. C) Comparison of exogenously expressed GFP-toxin point mutants for intracellular activity using the blue/green assay. Full-length PMT or PMT fragments, expressed in NFAT-bla CHO-K1 cells as GFP-fusion proteins with or without point mutations (C1165S or H1205L and H1223L) known to ablate biological activity of PMT, were assayed for intracellular activity as described in Fig. 2A.
Point mutations of PMT or PMT fragments assessed using NFAT-β-lactamase activity
To verify that the β-lactamase reporter activity observed for PMT and C-terminal fragments was not a result of overexpression of foreign protein, but was specific to PMT activity, we also generated single amino acid substitution mutations (C1165S, H1205L, and H1223L), which were previously reported to abolish PMT intracellular activity (Busch et al., 2001; Orth et al., 2003; Pullinger et al., 2001). Exogenous expression of these point mutants as GFP fusions in NFAT-bla CHO-K1 cells completely abolished PMT-mediated NFAT-β-lactamase reporter activity (Fig. 4C), conforming the specific response to PMT activity.
Discussion
Our previous studies showed that PMT induced a Ca2+-dependent chloride current in Xenopus oocytes, which involved Gq-coupled PLCβ1 activation (Wilson et al., 1997). Stimulation of this Ca2+-dependent chloride current by PMT occurred upon microinjection of PMT into the voltage-clamped oocytes, which resulted in a rapid response within seconds after injection. However, the action of PMT on intact mammalian cells requires first binding to cell surface receptors, followed by internalization and delivery of the toxin into the cytosol, where its cellular targets are located. This process requires full-length toxin protein; deletion fragments are inactive. In addition to the oocyte microinjection assay for PMT-stimulated calcium signaling described above, there are other cellular and biochemical assays for PMT activity, including assays based on: cytotoxicity (Busch et al., 2001; Pennings and Storm, 1984; Pettit et al., 1993; Wilson et al., 2000), visualization of cellular morphology or cytoskeletal changes (Dudet et al., 1996; Lacerda et al., 1996), radiolabel incorporation into DNA synthesis (Higgins et al., 1992; Rozengurt et al., 1990; Wilson et al., 2000) or into total inositol phosphate pools (Murphy and Rozengurt, 1992; Orth et al., 2004; Staddon et al., 1991), and western blotting for activation of signaling pathways (Obreztchikova et al., 2006; Orth et al., 2007a; Orth et al., 2007b; Orth et al., 2005; Sabri et al., 2002; Seo et al., 2000; Wilson et al., 2000). However, all of these methods are cumbersome, time and labor intensive, and are difficult to adapt for high-throughput screening of toxin or toxin mutants for intracellular activity.
The NFAT-β-lactamase reporter assay system combines molecular and cell biology with a FRET-based real time detection method to create flexible, sensitive high-throughput screening assays for studying and quantifying numerous cellular processes in intact mammalian cells (Kunapuli et al., 2003). Use of black-wall, clear-bottom microtiter plates, which have low fluorescence background, enabled the adaptation of the blue/green assay for high-throughput screening using a fluorescence plate reader with bottom-read capabilities and appropriate filters. An advantage of using the NFAT-β-lactamase reporter gene assay in conjunction with the cell permeable fluorescent substrate, CCF4/AM, is the high detection sensitivity at the level of a single living cell for assessment of signaling activation as well as cellular morphology (see Fig. 1), and hence also its compatibility with flow cytometry or FACS analysis (Zlokarnik et al., 1998). The ability to observe the cellular response by fluorescence and to perform ratiometric measurements between two fluorescence intensities places this technology ahead of other reporter-gene assays by eliminating artifacts due to photobleaching or changes in focus (Hanson, 2006; Kunapuli et al., 2003; Oosterom et al., 2005). Use of the stable cell line NFAT-bla CHO-K1 bypasses the requirement for transient transfection with dual reporter gene vectors such as the dual luciferase reporter system, thereby avoiding problems with variable transfection efficiencies and further enhancing its high-throughput capabilities.
In this study, we utilized the NFAT-β-lactamase blue/green reporter assay as a functional readout to investigate PMT-induced activation of Ca2+ signaling. To demonstrate the utility of this method for screening toxin mutants, we generated a series of N-terminal and C-terminal deletion and site-specific point mutants of PMT and directly expressed them as GFP-fusion proteins in NFAT-bla CHO-K1 cells by using mammalian expression vectors. Results showed that regions or amino acids in PMT critical for intracellular activity could be identified using the NFAT-β-lactamase blue/green assay. Thus, this method can be used not only for studying the intracellular activity of PMT, but also for high-throughput screening of defective PMT mutants. This method holds promise for characterization of PMT-based vaccine candidates. Moreover, this method has potential application for screening and diagnosis of clinical isolates of toxinogenic P. multocida.
Acknowledgments
This work was supported in part by NIH/NIAID grant AI038396 (to B.A.W.). We thank Yuka Bannai and Leila Aminova for assistance with construction of some of the PMT mutants.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aminova LR, Wilson BA. Calcineurin-independent inhibition of 3T3-L1 adipogenesis by Pasteurella multocida toxin: suppression of Notch1, stabilization of beta-catenin and pre-adipocyte factor 1. Cell Microbiol. 2007;9:2485–96. doi: 10.1111/j.1462-5822.2007.00975.x. [DOI] [PubMed] [Google Scholar]
- Arashima Y, Kumasaka K. Pasteurellosis as zoonosis. Intern Med. 2005;44:692–3. doi: 10.2169/internalmedicine.44.692. [DOI] [PubMed] [Google Scholar]
- Bagley KC, Abdelwahab SF, Tuskan RG, Lewis GK. Pasteurella multocida toxin activates human monocyte-derived and murine bone marrow-derived dendritic cells in vitro but suppresses antibody production in vivo. Infect Immun. 2005;73:413–21. doi: 10.1128/IAI.73.1.413-421.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch C, Orth J, Djouder N, Aktories K. Biological activity of a C-terminal fragment of Pasteurella multocida toxin. Infect Immun. 2001;69:3628–34. doi: 10.1128/IAI.69.6.3628-3634.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudet LI, Chailler P, Dubreuil JD, Martineau-Doize B. Pasteurella multocida toxin stimulates mitogenesis and cytoskeleton reorganization in Swiss 3T3 fibroblasts. J Cell Physiol. 1996;168:173–82. doi: 10.1002/(SICI)1097-4652(199607)168:1<173::AID-JCP21>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Fisher CL, Pei GK. Modification of a PCR-based site-directed mutagenesis method. Biotechniques. 1997;23:570–1. 574. doi: 10.2144/97234bm01. [DOI] [PubMed] [Google Scholar]
- Foged NT. Pasteurella multocida toxin. The characterisation of the toxin and its significance in the diagnosis and prevention of progressive atrophic rhinitis in pigs. APMIS Suppl. 1992;25:1–56. [PubMed] [Google Scholar]
- Hanson BJ. Multiplexing Fluo-4 NW and a GeneBLAzer transcriptional assay for high-throughput screening of G-protein-coupled receptors. J Biomol Screen. 2006;11:644–51. doi: 10.1177/1087057106289982. [DOI] [PubMed] [Google Scholar]
- Harper M, Boyce JD, Adler B. Pasteurella multocida pathogenesis: 125 years after Pasteur. FEMS Microbiol Lett. 2006;265:1–10. doi: 10.1111/j.1574-6968.2006.00442.x. [DOI] [PubMed] [Google Scholar]
- Higgins TE, Murphy AC, Staddon JM, Lax AJ, Rozengurt E. Pasteurella multocida toxin is a potent inducer of anchorage- independent cell growth. Proc Natl Acad Sci U S A. 1992;89:4240–4. doi: 10.1073/pnas.89.10.4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunapuli P, Ransom R, Murphy KL, Pettibone D, Kerby J, Grimwood S, Zuck P, Hodder P, Lacson R, Hoffman I, Inglese J, Strulovici B. Development of an intact cell reporter gene beta-lactamase assay for G protein-coupled receptors for high-throughput screening. Anal Biochem. 2003;314:16–29. doi: 10.1016/s0003-2697(02)00587-0. [DOI] [PubMed] [Google Scholar]
- Lacerda HM, Lax AJ, Rozengurt E. Pasteurella multocida toxin, a potent intracellularly acting mitogen, induces p125FAK and paxillin tyrosine phosphorylation, actin stress fiber formation, and focal contact assembly in Swiss 3T3 cells. J Biol Chem. 1996;271:439–45. doi: 10.1074/jbc.271.1.439. [DOI] [PubMed] [Google Scholar]
- Liao CM, Huang C, Hsuan SL, Chen ZW, Lee WC, Liu CI, Winton JR, Chien MS. Immunogenicity and efficacy of three recombinant subunit Pasteurella multocida toxin vaccines against progressive atrophic rhinitis in pigs. Vaccine. 2006;24:27–35. doi: 10.1016/j.vaccine.2005.07.079. [DOI] [PubMed] [Google Scholar]
- Murphy AC, Rozengurt E. Pasteurella multocida toxin selectively facilitates phosphatidylinositol 4,5-bisphosphate hydrolysis by bombesin, vasopressin, and endothelin. Requirement for a functional G protein. J Biol Chem. 1992;267:25296–303. [PubMed] [Google Scholar]
- Obreztchikova M, Elouardighi H, Ho M, Wilson BA, Gertsberg Z, Steinberg SF. Distinct signaling functions for Shc isoforms in the heart. J Biol Chem. 2006;281:20197–204. doi: 10.1074/jbc.M601859200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oosterom J, van Doornmalen EJ, Lobregt S, Blomenrohr M, Zaman GJ. High-throughput screening using beta-lactamase reporter-gene technology for identification of low-molecular-weight antagonists of the human gonadotropin releasing hormone receptor. Assay Drug Dev Technol. 2005;3:143–54. doi: 10.1089/adt.2005.3.143. [DOI] [PubMed] [Google Scholar]
- Orth JH, Aktories K, Kubatzky KF. Modulation of host cell gene expression through activation of STAT transcription factors by Pasteurella multocida toxin. J Biol Chem. 2007a;282:3050–7. doi: 10.1074/jbc.M609018200. [DOI] [PubMed] [Google Scholar]
- Orth JH, Blocker D, Aktories K. His1205 and His1223 are essential for the activity of the mitogenic Pasteurella multocida toxin. Biochemistry. 2003;42:4971–7. doi: 10.1021/bi0272959. [DOI] [PubMed] [Google Scholar]
- Orth JH, Lang S, Aktories K. Action of Pasteurella multocida toxin depends on the helical domain of Galphaq. J Biol Chem. 2004;279:34150–5. doi: 10.1074/jbc.M405353200. [DOI] [PubMed] [Google Scholar]
- Orth JH, Lang S, Preuss I, Milligan G, Aktories K. Action of Pasteurella multocida toxin on Galpha(q) is persistent and independent of interaction with G-protein-coupled receptors. Cell Signal. 2007b;19:2174–2182. doi: 10.1016/j.cellsig.2007.06.016. [DOI] [PubMed] [Google Scholar]
- Orth JH, Lang S, Taniguchi M, Aktories K. Pasteurella multocida toxin-induced activation of RhoA is mediated via two families of G{alpha} proteins, G{alpha}q and G{alpha}12/13. J Biol Chem. 2005;280:36701–7. doi: 10.1074/jbc.M507203200. [DOI] [PubMed] [Google Scholar]
- Pennings AM, Storm PK. A test in vero cell monolayers for toxin production by strains of Pasteurella multocida isolated from pigs suspected of having atrophic rhinitis. Vet Microbiol. 1984;9:503–8. doi: 10.1016/0378-1135(84)90071-3. [DOI] [PubMed] [Google Scholar]
- Petersen SK, Foged NT, Bording A, Nielsen JP, Riemann HK, Frandsen PL. Recombinant derivatives of Pasteurella multocida toxin: candidates for a vaccine against progressive atrophic rhinitis. Infect Immun. 1991;59:1387–93. doi: 10.1128/iai.59.4.1387-1393.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettit RK, Ackermann MR, Rimler RB. Receptor-mediated binding of Pasteurella multocida dermonecrotic toxin to canine osteosarcoma and monkey kidney (vero) cells. Lab Invest. 1993;69:94–100. [PubMed] [Google Scholar]
- Pullinger GD, Sowdhamini R, Lax AJ. Localization of functional domains of the mitogenic toxin of Pasteurella multocida. Infect Immun. 2001;69:7839–50. doi: 10.1128/IAI.69.12.7839-7850.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Register KB, Sacco RE, Brockmeier SL. Immune response in mice and swine to DNA vaccines derived from the Pasteurella multocida toxin gene. Vaccine. 2007;25:6118–28. doi: 10.1016/j.vaccine.2007.05.028. [DOI] [PubMed] [Google Scholar]
- Riising HJ, van Empel P, Witvliet M. Protection of piglets against atrophic rhinitis by vaccinating the sow with a vaccine against Pasteurella multocida and Bordetella bronchiseptica. Vet Rec. 2002;150:569–71. doi: 10.1136/vr.150.18.569. [DOI] [PubMed] [Google Scholar]
- Rozengurt E, Higgins T, Chanter N, Lax AJ, Staddon JM. Pasteurella multocida toxin: potent mitogen for cultured fibroblasts. Proc Natl Acad Sci U S A. 1990;87:123–7. doi: 10.1073/pnas.87.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabri A, Pak E, Alcott SA, Wilson BA, Steinberg SF. Coupling function of endogenous alpha(1)- and beta-adrenergic receptors in mouse cardiomyocytes. Circ Res. 2000;86:1047–53. doi: 10.1161/01.res.86.10.1047. [DOI] [PubMed] [Google Scholar]
- Sabri A, Wilson BA, Steinberg SF. Dual actions of the Galpha(q) agonist Pasteurella multocida toxin to promote cardiomyocyte hypertrophy and enhance apoptosis susceptibility. Circ Res. 2002;90:850–7. doi: 10.1161/01.RES.0000016165.23795.1F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo B, Choy EW, Maudsley S, Miller WE, Wilson BA, Luttrell LM. Pasteurella multocida toxin stimulates mitogen-activated protein kinase via G(q/11)-dependent transactivation of the epidermal growth factor receptor. J Biol Chem. 2000;275:2239–45. doi: 10.1074/jbc.275.3.2239. [DOI] [PubMed] [Google Scholar]
- Staddon JM, Barker CJ, Murphy AC, Chanter N, Lax AJ, Michell RH, Rozengurt E. Pasteurella multocida toxin, a potent mitogen, increases inositol 1,4,5-trisphosphate and mobilizes Ca2+ in Swiss 3T3 cells. J Biol Chem. 1991;266:4840–7. [PubMed] [Google Scholar]
- Ward PN, Miles AJ, Sumner IG, Thomas LH, Lax AJ. Activity of the mitogenic Pasteurella multocida toxin requires an essential C-terminal residue. Infect Immun. 1998;66:5636–42. doi: 10.1128/iai.66.12.5636-5642.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson BA, Aminova LR, Ponferrada VG, Ho M. Differential modulation and subsequent blockade of mitogenic signaling and cell cycle progression by Pasteurella multocida toxin. Infect Immun. 2000;68:4531–8. doi: 10.1128/iai.68.8.4531-4538.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson BA, Ho M. Pasteurella multocida toxin as a tool for studying Gq signal transduction. Rev Physiol Biochem Pharmacol. 2004;152:93–109. doi: 10.1007/s10254-004-0032-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson BA, Ho M. Pasteurella multocida toxin. In: Alouf JE, Popoff MR, editors. The Comprehensive Sourcebook of Bacterial Protein Toxins. Burlington, MA: Academic Press; 2006. pp. 430–447. [Google Scholar]
- Wilson BA, Ponferrada VG, Vallance JE, Ho M. Localization of the intracellular activity domain of Pasteurella multocida toxin to the N terminus. Infect Immun. 1999;67:80–7. doi: 10.1128/iai.67.1.80-87.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson BA, Zhu X, Ho M, Lu L. Pasteurella multocida toxin activates the inositol triphosphate signaling pathway in Xenopus oocytes via G(q)alpha-coupled phospholipase C-beta1. J Biol Chem. 1997;272:1268–75. doi: 10.1074/jbc.272.2.1268. [DOI] [PubMed] [Google Scholar]
- Zlokarnik G. Fusions to beta-lactamase as a reporter for gene expression in live mammalian cells. Methods Enzymol. 2000;326:221–44. doi: 10.1016/s0076-6879(00)26057-6. [DOI] [PubMed] [Google Scholar]
- Zlokarnik G, Negulescu PA, Knapp TE, Mere L, Burres N, Feng L, Whitney M, Roemer K, Tsien RY. Quantitation of transcription and clonal selection of single living cells with beta-lactamase as reporter. Science. 1998;279:84–8. doi: 10.1126/science.279.5347.84. [DOI] [PubMed] [Google Scholar]
- Zywietz A, Gohla A, Schmelz M, Schultz G, Offermanns S. Pleiotropic effects of Pasteurella multocida toxin are mediated by Gq-dependent and -independent mechanisms. involvement of Gq but not G11. J Biol Chem. 2001;276:3840–5. doi: 10.1074/jbc.M007819200. [DOI] [PubMed] [Google Scholar]