

Abstract
Tetracyclines and tetracycline analogs are prepared by a convergent, single-step Michael–Claisen condensation of the AB precursors 1 or 2 with D-ring precursors of wide structural variability, followed by removal of protective groups (typically in two steps). A number of procedural variants of the key C-ring-forming reaction are illustrated in multiple examples. These include stepwise deprotonation of a D-ring precursor followed by addition of 1 or 2, in situ deprotonation of a D-ring precursor in mixture with 1 or 2, and in situ lithium-halogen exchange of a benzylic bromide D-ring precursor in the presence of 1 or 2, followed by warming. The AB plus D strategy for tetracycline synthesis by C-ring construction is shown to be robust across a range of different carbocyclic and heterocyclic D-ring precursors, proceeding reliably and with a high degree of stereochemical control. Evidence suggests that Michael addition of the benzylic anion derived from a given D-ring precursor to enones 1 or 2 is quite rapid at −78 °C, while Claisen cyclization of the enolate produced is rate-determining, typically occurring upon warming to 0 °C. The AB plus D coupling strategy is also shown to be useful for the construction of tetracycline precursors that are diversifiable by latter-stage transformations, subsequent to cyclization to form the C ring. Results of antibacterial assays and preliminary data obtained from a murine septicemia model show that many of the novel tetracyclines synthesized have potent antibiotic activities, both in bacterial cell culture and in vivo. The platform for tetracycline synthesis described gives access to a broad range of molecules that would be inaccessible by semi-synthetic methods (presently the only means of tetracycline production), and provides a powerful engine for the discovery and, perhaps, development of new tetracycline antibiotics.
Introduction
In prior research we showed that cyclohexenones 1 and 2 can be transformed into 6-deoxytetracycline antibiotics using a sequence of as few as three chemical steps.1
 |
The first and key step of the sequence forms the C ring of the tetracyclines by a Michael–Claisen cyclization reaction, a potentially general means for constructing tetracycline analogs widely variant in the left-hand or D-ring portion of the molecule.2 Here, we expand upon our original findings, describing the synthesis of more than fifty different tetracyclines and tetracycline analogs–many active in inhibiting the growth of cultured Gram-positive and Gram-negative bacteria, including tetracycline-resistant strains. We provide detailed protocols for the key cyclization reaction in its various forms and discuss features of stereochemistry, chemical efficiency, and mechanism. We also report minimum inhibitory concentration values for selected analogs in a number of different bacterial strains, as well as preliminary in vivo data obtained in a mouse septicemia model using a tetracycline-sensitive strain of Staphylococcus aureus.
Background
The first tetracycline antibiotic was discovered in 1948 when Benjamin Duggar of Lederle Laboratories isolated the natural product chlorotetracycline (Aureomycin®, 3) from the culture broth of a novel species of Streptomyces.3 Within two years a research team from Chas. Pfizer and Co. had isolated a second natural tetracycline, oxytetracycline (Terramycin®, 4),4 and in 1953 tetracycline itself (5) was prepared from chlorotetracycline by catalytic hydrogenolysis of the carbon-chlorine
 |
bond, a transformation discovered by Lloyd Conover of Pfizer.5 Subsequently, tetracycline was found to be a natural product and,6 later still, Lederle researchers isolated 6-demethyltetracyclines (see structure 6) from culture broths of a mutant strain of Streptomyces.7 Conover has provided detailed and insightful accounts of research efforts leading to new tetracyclines specifically and antibiotics more generally.8 All tetracyclines approved for human or veterinary use are fermentation products or are derived from fermentation products by semisynthesis. This is also true of most beta-lactam and all macrolide antibiotics. Tracing the paths of human efforts to produce new antibiotics from natural products not accessible by synthesis reveals an evolutionary process marked by specific, impactful discoveries. In the case of the tetracyclines, Pfizer scientists achieved a major enabling advance approximately 10 years after the class had been identified when they demonstrated that the C6-hydroxyl group of the natural products oxytetracycline (4), tetracycline (5) and 6-demethyltetracycline (6) could be removed reductively.9 The 6-deoxytetracyclines produced, including 6-deoxytetracycline itself (7), were found to be more stable than the parent compounds, yet retained broad-spectrum antibacterial activity. The important and now generic antibiotics doxycycline (Pfizer, 1967, 8) and minocycline (Lederle, 1972, 9) followed as a consequence, the latter arising from the additional discovery that electrophilic aromatic substitution at C7 becomes possible when the more stable 6-deoxytetracyclines are used as substrates.10 Decades later, a team of Wyeth scientists led by Frank Tally synthesized 7,9-disubstituted tetracycline derivatives, leading to the discovery of the antibiotic tigecycline (Tigacyl®, US approval 2005, 10).11
Tigacyl is one of only three new antibiotics to be brought to market in the United States in the past three years, and the only broad-spectrum agent.12 Diminishing economic incentives and increasing regulatory hurdles have led many pharmaceutical companies to discontinue efforts to develop new antibiotics, raising concern among public health officials, particularly with the emergence of antibiotic-resistant bacterial strains in community settings. While careful management of the use of existing antibiotics in society is warranted, it would seem unwise to abandon the search for new agents, given the diversity of bacteria and their capacity to evolve rapidly. Attempts to develop antibacterial agents with novel targets have met with little success.13 As a consequence, many research programs seeking to discover new antibiotics have been refocused toward the modification of agents in proven classes, such as the tetracyclines, with an emphasis on overcoming bacterial resistance. While the innovations of chemists seeking to modify the structure of naturally-occurring tetracyclines have been extraordinary, the slowing pace of discovery in this area is evident.
From the time that the structures of the tetracycline antibiotics were first revealed by Woodward and collaborators,14 many laboratories have sought to develop a practical route for their synthesis. In 2003, an expert opinion of the patent literature summarized the state of the art, concluding, “the original effort of Woodward has survived as the basic strategy for the total synthesis of this series and at greater than 25 steps is clearly not to be considered as practical.... we believe there is ample justification to explore new total synthetic and convergent synthetic routes that take full advantage of the body of chemistry that has become available since Woodward's original effort.”15 Among the many developments since Woodward and co-workers first synthesized sancycline (6-deoxy-6-demethyltetracycline) in 1962,16 one of particular note, and relevance to the results described herein, is Stork and Hagedorn's strategy for protection of the vinylogous carbamic acid group of the A ring of the tetracyclines as a 3-benzyloxyisoxazole group; subsequent deprotection occurs under mild (hydrogenolytic) conditions.17
Strategically, the original route developed by Woodward and collaborators for the synthesis of sancycline employed a “left-to-right” or D→A mode of construction. The Shemyakin and Muxfeldt research groups adopted a similar directionality in their remarkable syntheses of tetracycline (5, 1967) and terramycin (4, 1968), respectively, using a bicyclic CD-ring precursor as starting material.18,19 With the benefit of more than 50 years of structure-activity relationship data, as well as X-ray crystal structures of tetracycline bound to the bacterial ribosome (its putative target),20 the left-to-right mode of construction used in these pioneering synthetic efforts can be seen to present a practical disadvantage to the discovery of new tetracycline antibiotics, for the D ring has emerged as one of the most promising sites for structural variation. This was a primary consideration guiding our initial retrosynthetic analysis of the tetracycline class, leading us to focus upon disconnection of the C ring. Thus, we envisioned assembling tetracyclines by a convergent coupling of D- and AB-ring precursors. Although model studies suggested that both Diels–Alder and Michael–Claisen cyclization reactions might be used to form the C ring,21 only the latter proved successful with the AB precursors that we later targeted and synthesized (1 and 2).
Michael–Claisen and Michael–Dieckmann reaction sequences have been widely employed to construct naphthalene derivatives since 1978, when three different cyclization protocols were introduced by independent research groups. Hauser and Rhee used a sulfoxide-stabilized o-toluate ester anion as the nucleophilic component in a Michael–Dieckmann cyclization reaction with methyl crotonate (eq 1). In this case, aromatization occurred upon thermal elimination of phenylsulfenic acid.22 The use of phthalide and cyanophthalide anions as nucleophilic components was described by Broom and Sammes (eq 2),23 and Kraus and Sugimoto (eq 3),24 respectively. Formal loss of water and hydrogen cyanide, respectively, led to naphthoate ester products in these procedures. In 1979, the Weinreb and Staunton research groups first reported that simple o-toluate ester anions (unsubstituted at the benzylic position) undergo Michael–Claisen cyclization reactions with β-methoxycyclohexenones and γ-pyrones to form naphthyl ketones (see eq 4 for one example), a sequence sometimes referred to as the Staunton–Weinreb annulation.25 Aromatization in these cases occurs by elimination of alkoxide following cyclization.
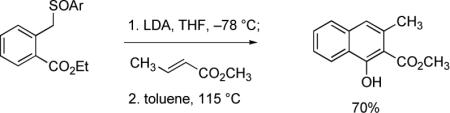 |
(1) |
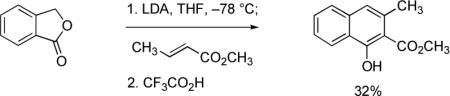 |
(2) |
 |
(3) |
 |
(4) |
There is also precedence for the formation of non-aromatic 6-membered rings by Michael–Claisen and Michael–Dieckmann reaction sequences.23,26,27 With few exceptions,27 stereochemical features of these cyclization reactions have not been discussed, frequently because they were of little consequence (aromatization followed cyclization). The Michael–Claisen cyclizations detailed below are unusual in their stereochemical complexity, stereocontrol and efficiency. In 2000, while our studies were in progress, Tatsuta and co-workers reported a synthesis of (−)-tetracycline (34 steps, 0.002% yield) that employed an early-stage Michael–Claisen cyclization reaction to form an aromatic C-ring precursor, which was dearomatized later in the sequence.28 Since 2005, our laboratory has reported two different routes to synthesize the AB precursor 1 in optically active form; the more recent of these was scaled to prepare >40 g of crystalline product in one batch.29 Here we provide details of the different protocols that can be used to construct the C ring of the tetracyclines and exemplify these protocols with the preparation of a number of novel substances with antibacterial properties.
Results
A critical early experiment in our attempts to assemble the C ring of the tetracyclines by a Michael–Claisen cyclization reaction provided both direction and mechanistic insight. As depicted in Scheme 1, treatment of a solution of organostannane 1130 in tetrahydrofuran (THF) with n-butyllithium at −78 °C, followed by transfer of this mixture to a solution of enone 1, also at −78 °C, and subsequent quenching with tert-butyldimethylsilyl triflate (TBSOTf) afforded the Michael addition product 12 as a single stereoisomer in 98% yield.
Scheme 1.

Michael addition of a benzylic anion (generated by tin-lithium exchange) to enone 1, followed by trapping of the resultant enolate with tert-butyldimethylsilyl triflate.
Crystallization of the chromatographically purified product from hexanes at −30 °C provided a single crystal suitable for X-ray analysis (mp 146 °C). Inspection of the crystal structure (Figure 1) revealed that the stereochemistry of positions “C5a” and “C6” of the adduct (12) corresponded to that of 6-deoxytetracycline (7) and doxycycline (8), which was fortuitous given that as many as four diastereomers could have been formed in the Michael addition reaction. The product that is formed apparently arises from selective addition of the nucleophile to the concave face of the enone. This selectivity may arise as a consequence of stereoelectronic factors (pseudoaxial addition to the enone) and/or steric effects (addition of the nucleophile to the π-face opposite the tert-butyldimethylsilyloxy substituent, which is also axial; see the space-filling model of enone 1 depicted in Figure 1).
Figure 1.

Crystal structures of the Michael addition product 12 and the enone 1, presented as ball and stick and space-filling models, respectively.
Directed by this key finding, we proceeded to strategically modify the D-ring precursor, with three objectives: (1) to activate the ester toward Claisen cyclization (which we did not observe with the methyl ester substrate 11), (2) to obviate the use of organotin intermediates, and (3) to mask the C10 phenoxy substituent with a protective group more labile than methyl. These objectives were achieved using the tert-butoxycarbonyl-protected phenyl ester substrate 13 (Scheme 2 below).31,32
Scheme 2.

Synthesis of 6-deoxytetracycline (7) by Michael–Claisen cyclization using the tert-butoxycarbonyl-protected phenyl ester 13 as the D-ring precursor and a stepwise protocol for addition of the base and enone 1, followed by deprotection.
Deprotonation of 13 (3 equiv) with lithium diisopropylamide (LDA, 4 equiv) at −78 °C in the presence of N,N,N′,N′-tetramethylethylenediamine (TMEDA, 4 equiv) afforded a deep red solution of the corresponding o-toluate ester anion; addition of a solution of the enone 1 (1 equiv) and slow warming of the resulting mixture to 0 °C over 3 h provided the Michael–Claisen cyclization product 14 in 81% yield in diastereomerically pure form after isolation by reverse-phase high-performance liquid chromatography (rp-HPLC). A minor diastereomer (<4%), believed to be epimeric at C6, was isolated separately.
Thus, Michael addition occurs with >20:1 stereoselectivity at C6, in the sense indicated in Scheme 2, and appears to proceed with complete stereocontrol at C5a (attack upon a single diastereoface of the enone). These observations have held consistently in more than 40 different C-ring-forming cyclization reactions examined to date, with two (closely related) exceptions, discussed below.
There is little question that the transformation of enone 1 to the 6-deoxytetracycline precursor 14 proceeds by a stepwise mechanism, involving sequential Michael and Claisen reactions, for the intermediate Michael adduct can be intercepted by protonation with acetic acid at −78 °C to give the keto ester 16 in 88% yield (eq 5).33 Indeed, we have observed across a range
 |
(5) |
of different D-ring precursors that Michael addition is relatively rapid at −78 °C, while Claisen cyclization proceeds more slowly, typically upon warming to 0 °C. Thus, we view Claisen cyclization and not Michael addition as rate-determining. It is evident from these observations that C-ring formation cannot occur by Diels–Alder cycloaddition of an o-quinonemethide intermediate, which is hypothetically a mechanistic alternative to Michael–Claisen cyclization.
With the establishment of an effective protocol for construction of the C ring, a two-step deprotection sequence was employed to transform the cyclization product 14 into 6-deoxytetracycline (Scheme 2 above). The tert-butoxycarbonyl and tertbutyldimethylsilyl protective groups were removed upon treatment of 14 with hydrofluoric acid in acetonitrile at 23 °C (2 days). Hydrogenolysis of the crude reaction product (15) in the presence of a palladium catalyst under an atmosphere of hydrogen in methanol-dioxane at 23 °C and subsequent purification by rp-HPLC then afforded 6-deoxytetracycline (7) in 85% yield.17 As the examples below will demonstrate, this two-step deprotection protocol has been found to be widely applicable, though in some instances it is advantageous to invert the ordering of the two steps. In general, the tert-butoxycarbonyl group is cleaved relatively rapidly in the deprotection step employing hydrofluoric acid, while the tertiary tert-butyldimethylsilyl ether undergoes protodesilylation more slowly.
The conditions developed for the cyclization reaction depicted in Scheme 2, sequential deprotonation of a D-ring precursor followed by addition of the enone 1, have been effective for the synthesis of a number of known tetracyclines as well as novel tetracycline analogs variant within or near the D ring. For example, treatment of phenyl ester 1734 (3 equiv, Scheme 3 below) with LDA (3 equiv) in the presence of TMEDA (6 equiv) at −78 °C, followed by addition of enone 1 (1 equiv) and warming of the resulting mixture to −10 °C provided the Michael–Claisen cyclization product 18 in 83% yield after purification by rp-HPLC. Two-step deprotection and purification by rp-HPLC then afforded minocycline (9) in 74% yield.
Scheme 3.

Synthesis of minocycline (9) by Michael–Claisen cyclization using the tert-butoxycarbonyl-protected phenyl ester 17 as the D-ring precursor and a stepwise protocol for addition of the base and enone 1, followed by deprotection.
Among the modified D-ring precursors we have investigated, substrates with benzylic anion-stabilizing groups were found to undergo particularly efficient cyclization reactions. For example, addition of enone 1 (1 equiv) to a solution of the stabilized anion derived from substrate 19 (3 equiv, Scheme 4 below),32 followed by warming of the resulting mixture to −15 °C, provided the Michael–Claisen cyclization product 20 in 97% yield after purification by rp-HPLC. Standard deprotection of 20 then afforded the novel tetracycline analog 6-(S)-phenylsancycline (21), which was found to effectively inhibit the growth of a number of Gram-positive bacteria, including tetracycline-resistant strains (vide infra), prompting the synthesis of a series of 6-aryl-substituted tetracyclines (see Table 1).35 It is worthy of note that more than half of the cyclization reactions presented in the tables below were attempted only once.
Scheme 4.

Synthesis of 6-(S)-phenylsancycline (21) by Michael–Claisen cyclization using the tert-butoxycarbonyl-protected phenyl ester 19 as the D-ring precursor and a stepwise protocol for addition of the base and enone 1, followed by deprotection.
Table 1.
Synthesis of 6-substituted tetracyclines by Michael—Claisen cyclization using D-ring precursors with anion-stabilizing substituents in the benzylic position and a stepwise protocol for addition of the base and enone 1, followed by deprotection [(i) HF (aq), CH3CN; (ii) H2, Pd/C (or Pd black), CH3OH-dioxane].
| D-ring Precursor | Cyclization Conditions | Cyclization Product | Cyclization Yield | Tetracycline | Deprotection Yield |
|---|---|---|---|---|---|
 |
LDA; 1 −78 → −10 °C |
 |
84% |  |
71% |
 |
LDA; 1 −78 → −5 °C |
 |
57% |  |
70% |
 |
LDA; 1 −78 → −10 °C |
 |
79% |  |
72% |
 |
LDA; 1 −78 → −10 °C |
 |
79% |  |
83% |
 |
LDA, DMPU; 1 −78 → −10 °C |
 |
81% | 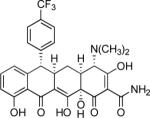 |
72% |
 |
LDA, DMPU; 1 −78 → −10 °C |
 |
37% |  |
59% |
 |
LDA, DMPU; 1 −78 → −10 °C |
 |
78% |  |
79% |
 |
LDA; 1 −78 → −5 °C |
 |
74% |  |
64% |
 |
LHMDS, HMPA; 1 −78 → −10 °C |
 |
49% |  |
79% |
Other notable features of the cyclization reactions of D-ring precursors with benzylic anion-stabilizing groups include the fact that additives such as TMEDA were typically not required, and that stoichiometries of just over one equivalent of a given D-ring precursor frequently sufficed to achieve a high yield of cyclization product. We also found that benzylic deprotonation could be conducted with the weaker base lithium bis(trimethylsilyl)amide (LHMDS) in lieu of the standard base LDA and, using N-imidazoyl substrate 22 (Scheme 5 below), the important observation was made that condensation could be achieved with high efficiency by an in situ deprotonation protocol, which is to say by addition of base to the D-ring precursor in the presence of enone 1. Thus, treatment of a mixture of phenyl ester 22 (1.3 equiv) and enone 1 (1 equiv) with an excess of LHMDS (3.3 equiv) at −78 °C, followed by warming of the resulting mixture to −30 °C over 90 min, provided the cyclization product 23 in 94% yield (Scheme 5); removal of protective groups then provided 6-(R)-N-imidazoylsancycline (24, 42% yield over two steps).
Scheme 5.
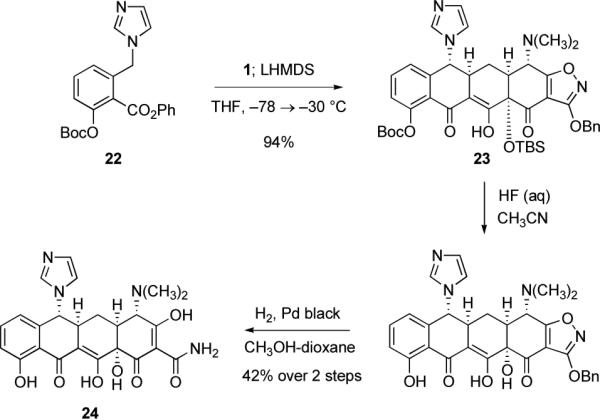
Synthesis of 6-(R)-N-imidazoylsancycline (24) by Michael–Claisen cyclization using the tert-butoxycarbonyl-protected phenyl ester 22 and enone 1 as substrates, with deprotonation in situ, followed by deprotection.
In situ deprotonation with LHMDS was also effective in bringing about cyclization of the methyl phenyl diester substrate 25 with enone 1, as well as cyclization of the corresponding benzyl phenyl diester substrate 27, as depicted in Scheme 6 below. In both cases, the major product of cyclization was epimeric at C6 relative to the major products of all other cyclization reactions we have studied (these providing the two exceptions referred to above), which we attribute to epimerization at C6 post cyclization. We have not yet conducted the rigorous studies necessary to establish if the product ratios were kinetically or thermodynamically determined in these examples.
Scheme 6.

Synthesis of 6-(S)-carbomethoxysancycline (26) by Michael–Claisen cyclization using the methyl phenyl diester 25 and enone 1 as substrates, with deprotonation in situ, followed by deprotection; in one experiment conducted with the corresponding benzyl phenyl diester 27 as the D-ring precursor, a diastereomeric mixture of cyclization products was obtained.
The in situ deprotonation protocol has proven to be effective for the Michael–Claisen cyclization reactions of a number of different D-ring precursors containing benzylic anion-stabilizing substituents (see Table 2 below); because of its greater experimental convenience this has become the preferred method for the synthesis of tetracycline analogs substituted at the C6-position.
Table 2.
Synthesis of 6-substituted tetracyclines by Michael—Claisen cyclization using D-ring precursors with anion-stabilizing substituents in the benzylic position and enone 1 as substrates, with deprotonation in situ, followed by deprotection [(i) HF (aq), CH3CN; (ii) H2, Pd/C, CH3OH-dioxane].37
| D-ring Precursor | Cyclization Conditions | Cyclization Product | Cyclization Yield | Tetracycline | Deprotection Yield |
|---|---|---|---|---|---|
 |
1; LDA −78 → −10 °C |
 |
58% |  |
58% |
 |
1; LDA −78 → −10 °C |
 |
72% |  |
60% |
 |
1; LDA −78 → −10 °C |
 |
44% |  |
23% |
 |
1; LDA −78 → −10 °C |
 |
83% |  |
98% |
 |
1; LDA, TMEDA −78 → −10 °C |
 |
25% |  |
64% |
 |
1; LHMDS −78 → −10 °C |
 |
72% |  |
43% |
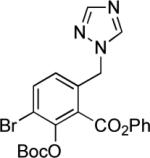 |
1; LHMDS −78 → −10 °C |
 |
38% |  |
65% |
D-ring heterocyclic analogs of tetracyclines had not been made before, so far as we are aware, and their construction by semisynthesis cannot be easily imagined. In targeting analogs of this type, we initially chose to explore the synthesis of the D-ring pyridine derivative 7-aza-10-deoxysancycline (30, Scheme 7 below). As we previously reported, in situ deprotonation of phenyl ester 28 (4 equiv) with LDA (5 equiv) at −95 °C in the presence of enone 1 (1 equiv) and hexamethylphosphoramide (HMPA, 10 equiv), followed by warming to −50 °C, afforded the Michael–Claisen cyclization product 29 in 76% yield (Scheme 7).1 Claisen ring-closure was notably more facile in this example than in others we have studied, proceeding to completion in less than 1 h upon warming to −50 °C. 7-Aza-10-deoxysancycline (30) was then obtained in 79% yield after removal of protective groups.
Scheme 7.

Synthesis of 7-aza-10-deoxysancycline (30) by Michael–Claisen cyclization using phenyl ester 28 and enone 1 as substrates, with deprotonation in situ, followed by deprotection.
Another novel class of tetracyclines that we have explored is the pentacyclines (Scheme 8 below). This required that we develop chemistry suitable for Michael–Claisen cyclization of naphthoate ester DE-ring precursors,22-24 as the protocols that we had used to this point were found to be largely ineffective with these substrates.
Scheme 8.

Synthesis of the pentacycline 33 by Michael–Claisen cyclization using the phenyl bromomethylnaphthoic acid ester 31 and enone 1 as substrates, and an in situ protocol for lithium-halogen exchange, followed by deprotection.
Lithium-halogen exchange is rarely employed to form benzyllithium reagents in organic synthesis, due to the propensity of the benzyl halide substrates to engage in Wurtz-type coupling reactions in the presence of an alkyllithium reagent.36 In a surprising transformation, treatment of a solution of phenyl bromomethylnaphthoic acid ester 31 (4 equiv) and enone 1 (1 equiv) with n-butyllithium (4 equiv) at −100 °C, followed by warming to 0 °C, provided the Michael–Claisen cyclization product 32 in 75% yield (Scheme 8).1 A three-step deprotection sequence then afforded pentacycline 33 in 74% yield.
The in situ lithium-halogen exchange protocol developed for the synthesis of the pentacycline 33 has proven to be generally effective for the synthesis of a number of quite different tetracycline analogs. In many cases we have adopted the procedural modification of substituting the less reactive reagent phenyllithium for n-butyllithium in the lithium-halogen exchange reaction. For example, addition of phenyllithium to a solution of pyrazole 34 (3 equiv) and enone 1 (1 equiv) containing HMPA (6 equiv) at −90 °C, followed by warming to 0 °C over 2 h, provided the Michael–Claisen cyclization product 35 in 81% yield (Scheme 9 below). Removal of the protective groups led to concomitant cleavage of the heteroaryl carbon-chlorine bond during hydrogenolysis, affording the tetracycline analog 36 containing a D-ring pyrazole (87% yield over two steps).
Scheme 9.

Synthesis of a pyrazole analog (36) by Michael–Claisen cyclization using the benzylic bromide 34 and enone 1 as substrates, and an in situ protocol for lithium-halogen exchange, followed by deprotection.
A very similar procedure produced 8-fluorosancycline (38) from the benzyl bromide 37 (Scheme 10). Like the D-ring pyrazole analog 36, the 8-fluorotetracycline analog 38 would have been difficult if not impossible to prepare by semi-synthetic methods.
Scheme 10.

Synthesis of 8-fluorosancycline (38) by Michael–Claisen cyclization using benzylic bromide 37 and enone 1 as substrates, and an in situ protocol for lithium-halogen exchange, followed by deprotection.
Lithium-halogen exchange was also employed for the synthesis of 10-deoxysancycline (41, Scheme 11 below). As we reported previously, treatment of a solution of phenyl 2-(bromomethyl)benzoate 39 (4 equiv) and enone 1 (1 equiv) with n-butyllithium (4 equiv) at −100 °C, followed by warming to 0 °C over 30 min, provided the Michael–Claisen cyclization product 40 in 81% yield.1,38 The typical two-step deprotection sequence then transformed the cyclized product 40 into 10-deoxysancycline (41) in 84% yield.
Scheme 11.

Synthesis of 10-deoxysancycline (41) by Michael–Claisen cyclization using phenyl 2-(bromomethyl)benzoate 39 and enone 1 as substrates, with deprotonation in situ, followed by deprotection.
Bromomethylquinoline derivatives39 were also employed as DE-ring precursors in Michael–Claisen cyclization reactions with enone 1, providing pentacycline precursors with heterocyclic E rings (Scheme 12). The typical two-step deprotection sequence afforded tetrahydroquinoline products in these examples, while the use of modified conditions led to deprotection without reduction of the pyridine E-ring. Cyclization reactions of quinoline substrates with 6-aryl substituents were also investigated,35b using stepwise deprotonation conditions (Scheme 13).
Scheme 12.

Synthesis of pentacyclines with heterocyclic E rings by Michael–Claisen cyclizations using bromomethylquinolines and enone 1 as substrates, and an in situ protocol for lithium-halogen exchange, followed by deprotection.
Scheme 13.

Synthesis of 6-aryl-substituted heterocyclic pentacyclines by Michael-Claisen cyclizations using benzylically-substituted quinolines as DE-ring precursors and a stepwise protocol for addition of the base and enone 1, followed by deprotection.
Thus far, each different cyclization reaction described has produced a single tetracycline analog. It is worth noting explicitly that the convergent coupling strategy employed to prepare individual tetracycline analogs can also be used to target structures that serve as branch-points to large numbers of analogs, versatile structures such as the aryl bromide 43, or the corresponding aldehyde 44 (Scheme 14). Both of these pentacycline precursors were targeted for synthesis.
Scheme 14.

Synthesis of the diversifiable pentacycline precursors 43 and 44 by Michael–Claisen cyclization using the benzylic bromide 42 and enone 1 as substrates, with an in situ protocol for selective lithium-halogen exchange. Further transformation of 44 by a reductive amination reaction provides a route to alkylaminomethylpentacyclines, as illustrated by the synthesis of the tert-butylaminomethylpentacycline 45.
Michael–Claisen cyclization of the benzylic bromide 4240 with enone 1 was successfully achieved by in situ lithium-halogen exchange using phenyllithium, but not n-butyllithium. Thus, treatment of a mixture of the (bis) bromide 42 (3 equiv) and enone 1 (1 equiv) with phenyllithium (3 equiv) at −100 °C, followed by addition of LHMDS (1 equiv) and warming of the resulting mixture to −10 °C provided the cyclization product 43 in 44% yield (Scheme 14 above).41 Although the yield of product in this cyclization reaction was moderate, it remained consistent during scale-up and allowed for sufficient quantities of 43 to be produced to explore late-stage diversification. The aryl bromide intermediate 43 was treated sequentially with phenyllithium and n-butyllithium to effect deprotonation and lithium-halogen exchange, respectively, and the resulting dianion was formylated with N,N-dimethylformamide (DMF) to give the aldehyde 44; reductive amination with tert-butylamine and subsequent deprotection afforded tert-butylaminomethylpentacycline 45 in 61% yield after purification by rp-HPLC. The use of a number of different amines in the reductive amination reaction led to the synthesis of various alkylaminomethylpentacycline analogs from the common intermediate 44 (Chart 1).
Chart 1.

Alkylaminomethylpentacyclines synthesized from aldehyde 44 by reductive amination followed by deprotection (see Supporting Information for experimental details).42
We also successfully pursued the synthesis of a parallel series of diversifiable pentacycline precursors containing a dimethylamino substituent at C7, as in minocycline (9) and tigecycline (10, see Scheme 15 below). In the case of the dimethylamino-substituted naphthoate ester 46, it was possible to conduct cyclization by in situ deprotonation, in the presence of enone 1, forming the dimethylamino-substituted aryl bromide 47 in 57% yield. Subjection of 47 to deprotonation and lithium-halogen exchange, and formylation of the resulting dianion with DMF gave the pentacycline precursor aldehyde 48 in 80% yield; reductive amination with azetidine and subsequent deprotection afforded azetidinylmethylpentacycline 49 in 74% yield after purification by rp-HPLC (Scheme 15). As with aldehyde 44 above, reductive amination of 48 could be conducted using a number of different amines, providing various 7-dimethylamino-alkylaminomethylpentacyclines upon deprotection (see Chart 2 below).
Scheme 15.
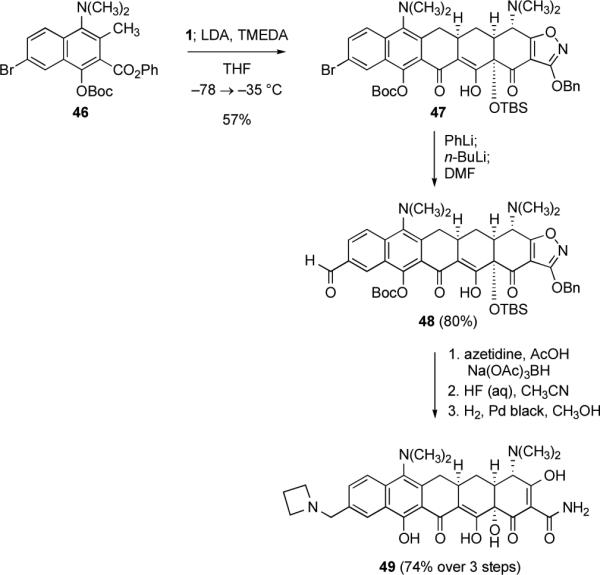
Synthesis of the diversifiable pentacycline precursors 47 and 48 by Michael–Claisen cyclization using the phenyl naphthoate ester 46 and enone 1 as substrates, with deprotonation in situ. Further transformation of 48 by a reductive amination reaction provides a route to 7-dimethylamino-alkylaminomethylpentacyclines, as illustrated by the synthesis of the 7-dimethylamino-azetidinylmethylpentacycline 49.
Chart 2.

7-Dimethylamino-alkylaminomethylpentacyclines synthesized from aldehyde 48 by reductive amination followed by deprotection.43
An alternative approach to diversification involved reduction of aldehyde 44 with sodium triacetoxyborohydride, mesylation of the resulting primary alcohol with methanesulfonic anhydride, and then nucleophilic displacement (see Scheme 16 below). When imidazole was used as the nucleophile, substitution afforded the N-imidazoylmethyl product 50 (59% yield over two steps); removal of protective groups afforded the corresponding pentacycline analog 51 in 40% yield.
Scheme 16.
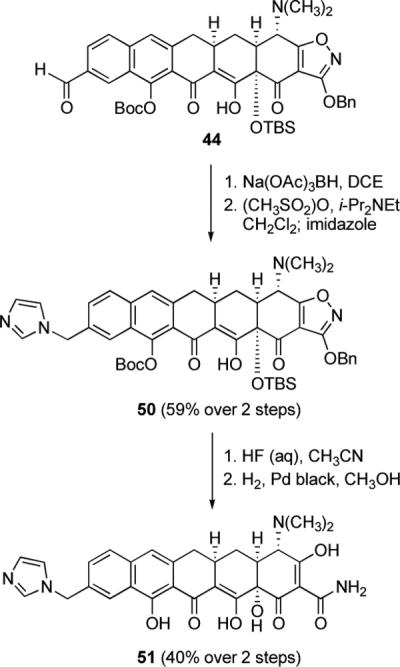
An alternative route to diversification of the aldehyde 44, involving reduction, activation, and nucleophilic displacement, as exemplified by the synthesis of the N-imidazoylmethylpentacycline 51.
As we have previously shown, by variation of our original synthetic route to the enone 1, the corresponding C5-hydroxylated substrate, protected as a benzyl carbonate (enone 2, see introductory paragraph), can be prepared in gram amounts.1 This in turn has enabled the synthesis of a number of C5-hydroxytetracyclines (Scheme 17 and Table 3 below). Like the corresponding cyclization reactions with enone 1, Michael–Claisen condensations with enone 2 proceed with uniformly high stereoselectivity. For example, addition of enone 2 (1 equiv) to a solution of the o-toluate ester anion derived from phenyl ester 13 (4.5 equiv) at −78 °C, followed by warming of the resulting mixture to 0 °C over 2 h, provided the Michael–Claisen cyclization product 52 in 79% yield in diastereomerically pure form after purification by rp-HPLC (Scheme 17). A minor diastereomer, believed to be 6-epi-52, was isolated separately (<7% yield). Removal of the protective groups of 52 and purification by rp-HPLC afforded doxycycline (8) in 90% yield.1
Scheme 17.

Synthesis of doxycycline (8) by Michael–Claisen cyclization using the tert-butoxycarbonyl-protected phenyl ester 13 as the D-ring precursor and a stepwise protocol for addition of the base and enone 2, followed by deprotection.
Table 3.
Synthesis of 5-hydroxytetracyclines by Michael—Claisen cyclization using a number of different D-ring precursors and enone 2 as substrates, followed by deprotection.
| D-ring Precursor | Cyclization Conditions | Cyclization Product | Cyclization Yield | Deprotection Conditions | Tetracycline | Deprotection Yield |
|---|---|---|---|---|---|---|
 |
2; PhLi, LHMDS −78 → 0 °C |
 |
88% | B |  |
58% |
 |
LDA, TMEDA; 2 −78 → −10 °C |
 |
62% | C |  |
50% |
 |
LDA; 2 −78 → −10 °C |
 |
39% | A |  |
82% |
 |
LDA; 2 −78 → −10 °C |
 |
88% | A |  |
73% |
 |
LDA; 2 −78 → 0 °C |
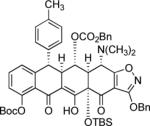 |
66% | A |  |
65% |
Deprotection Conditions: A (i) HF (aq), CH3CN; (ii) H2, Pd black, CH3OH-dioxane. B (i) HF (aq), CH3CN; (ii) H2, Pd/C, CH3OH-dioxane. C (i) H2, Pd black, CH3OH-dioxane; (ii) HF (aq), CH3CN.
Antibacterial Activities
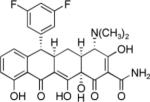
Minimum inhibitory concentrations (MICs) were determined in whole-cell antimicrobial assays using a panel of tetracycline-sensitive and tetracycline-resistant Gram-positive and Gram-negative bacteria. The results for selected compounds are shown in Table 4. Two promising new series of tetracycline antibiotics emerge from this study: 6-aryl tetracyclines, which are active in both tetracycline-sensitive and tetracycline-resistant Gram-positive strains, and alkylaminomethylpentacyclines, which show good activity in both tetracycline-sensitive and tetracycline-resistant Gram-positive and Gram-negative organisms.
Table 4.
Minimum inhibitory concentration (MIC) values for selected tetracycline analogs (μg/mL).
| |
SA 100 |
SA 2147 |
SA 757 |
SA 2011 |
EF 708 |
EF 1092 |
SP 1195 |
SP 911 |
EC 102 |
EC 2271 |
EC 119 |
EC 121 |
AB 1630 |
PA 103 |
HI 1224 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Resistance: | M | M, T | M, T | V, T | P, T | T | T | Mult | A, T | ||||||
 |
1 | 0.5 | 64 | 32 | 0.5 | >64 | 0.25 | 64 | 1 | 64 | 2 | 64 | 1 | 32 | 16 |
 |
0.25 | 0.12 | 0.5 | 0.12 | 0.12 | 0.5 | <0.06 | <0.06 | >16 | >16 | >16 | 8 | 0.25 | >16 | 2 |
 |
0.25 | 0.25 | 0.5 | 0.25 | 0.5 | 0.5 | 0.25 | 0.25 | >32 | >32 | ND | ND | 8 | >32 | >32 |
 |
0.25 | 0.25 | 0.5 | 0.25 | 0.25 | 0.5 | 0.25 | 0.25 | >32 | >32 | ND | ND | 2 | >32 | 64 |
 |
1 | 1 | 1 | 0.5 | 0.5 | 0.5 | 0.25 | 0.25 | >32 | >32 | >32 | >32 | 8 | >32 | 0.5 |
| 1 | 0.5 | 2 | 0.5 | 0.12 | 2 | <0.06 | 0.5 | 0.5 | 1 | 1 | 0.25 | 2 | >32 | 0.06 | |
| 0.5 | 0.25 | 2 | 0.5 | <0.06 | 1 | <0.06 | 0.5 | 0.25 | 1 | 1 | 0.25 | 0.5 | >32 | 0.06 | |
| 0.5 | 0.25 | 1 | 0.25 | <0.06 | 0.5 | <0.06 | 0.5 | 0.5 | 1 | 2 | 0.12 | 1 | 32 | 0.25 |
Organisms in red are antibiotic-resistant. Abbreviations: A, ampicillin; M, methicillin; P, penicillin; V, vancomycin; T, tetracycline; Mult, multiple. Organisms: SA, S. aureus; EF, E. faecium; SP, S. Pneumoniae; EC, E. coli; AB, A. baumanii; PA, P. aeruginosa; HI, H. influenzae.
Several compounds were selected for further study in a mouse septicemia model to determine efficacy in vivo (Table 5 below). Mice were inoculated intraperitoneally with an LD90−100 bolus of S. aureus (Smith). One hour later, tetracycline analogs were administered intravenously at dose levels ranging from 0.3 mg/kg to 30 mg/kg. Mortality was monitored once daily for 7 days. All mice survived at the highest level of dosing (30 mg/kg) for each of the compounds tested, indicating that the threshold of acute toxicity in mice is greater than this value. The 6-phenyl tetracycline analog showed some efficacy in this murine model (ED50 10.7 mg/kg), while the alkylaminomethylpentacyclines were more potent, with efficacy similar to that of tetracycline (ED50 1.7−2.8 mg/kg).
Table 5.
In vivo efficacy of selected tetracycline analogs in a tetracycline-sensitive strain of S. aureus.
| Compound | Number of Mice | Number of Deaths at Dose of |
ED50 (mg/kg) | ||||
|---|---|---|---|---|---|---|---|
| 30 mg/kg | 10 mg/kg | 3 mg/kg | 1 mg/kg | 0.3 mg/kg | |||
 |
6 | 0 | 0 | 2 | 6 | 0.97 | |
 |
6 | 0 | 5 | 6 | 6 | 6 | 10.71 |
| 6 | 0 | 0 | 1 | 6 | 6 | 2.80 | |
| 6 | 0 | 0 | 1 | 5 | 6 | 1.73 | |
| 6 | 0 | 0 | 1 | 5 | 6 | 1.73 | |
Mice were inoculated intraperitoneally with an LD90−100 bolus of Staphylococcus aureus (Smith) (3.5 × 105 CFU/mouse) in 0.5 mL of BHI broth containing 5% mucin. Tetracycline analogs were administered intravenously to test animals at one hour after bacterial inoculation. Mortality was monitored once daily for 7 days.
Conclusion
The results described illustrate the application of a general AB plus D strategy for the synthesis of more than fifty tetracyclines and tetracycline analogs. C-ring formation was achieved by a stereocontrolled Michael–Claisen cyclization reaction employing one or more of the protocols detailed herein. The examples discussed demonstrate that structural modification of both AB and D-ring components allows for the preparation of individual tetracyclines, as well as for the synthesis of tetracycline precursors that are readily diversified by late-stage transformations. In many cases a single cyclization attempt provided, after deprotection, sufficient material for antibacterial screening against a panel of Gram-positive and Gram-negative organisms. For compounds of interest, reactions could then be readily scaled to provide amounts necessary for further evaluation in assays such as a murine septicemia model. A number of novel structural classes were explored, including D-heteroaryl tetracyclines, pentacyclines, and E-heteroaryl pentacyclines. The platform for tetracycline synthesis described gives access to a broad range of molecules that would be inaccessible by semi-synthetic methods (presently the only means of tetracycline production), and provides a powerful engine for the discovery and, perhaps, development of new tetracycline antibiotics.
Supplementary Material
Acknowledgement
This work was generously supported by the National Institutes of Health (AI048825). We thank the NSF for predoctoral graduate fellowship support (JDB and MGC) and the Deutscher Akademischer Austausch Dienst for postdoctoral fellowship support (CDL). We acknowledge Vivisource for execution of in vivo studies, Micromyx, LLC for execution of in vitro studies, and Tetraphase Pharmaceuticals for sponsorship of these. We thank Dr. Richard Staples and Dr. Andrew Haidle for conducting the X-ray crystallographic analyses.
Footnotes
Supporting Information Available: Detailed experimental procedures for Michael-Claisen cyclization reactions and deprotection sequences, and characterization data for all tetracycline analogs (120 pages).
References
- 1.Charest MG, Lerner CD, Brubaker JD, Siegel DR, Myers AG. Science. 2005;308:395–398. doi: 10.1126/science.1109755. [DOI] [PubMed] [Google Scholar]
- 2.As pointed out by a reviewer, the second step of the key C-ring-forming reaction sequence is more properly designated as an internal Claisen reaction, rather than a Dieckmann reaction, as we previously described it [for discussion, see: Hauser CR, Swamer FW, Adams JT. Org. React. (N.Y) 1954;8:59.Schaefer JP, Bloomfield JJ. Org. React. (N.Y) 1967;15:1.]. For brevity, this sequence is referred to here as a Michael–Claisen cyclization.
- 3.a Duggar BM. Ann. N. Y. Acad. Sci. 1948;51:177–181. doi: 10.1111/j.1749-6632.1948.tb27262.x. [DOI] [PubMed] [Google Scholar]; b Duggar BM. Aureomycin and Preparation of Same. U.S. Patent 2,482,055. 1949 Sept 13;
- 4.a Finlay AC, Hobby GL, P'an SY, Regna PP, Routien JB, Seeley DB, Shull GM, Sobin BA, Solomons IA, Vinson JW, Kane JH.Science 19501118515400447 [Google Scholar]; b Sobin BA, Finlay AC. Terramycin and its Production. U.S. Patent 2,516,080. 1950 July 18;
- 5.a Booth JH, Morton J, Petisi JP, Wilkinson RG, Williams JH. J. Am. Chem. Soc. 1953;75:4621. [Google Scholar]; b Conover LH, Moreland WT, English AR, Stephens CR, Pilgrim FJ. J. Am. Chem. Soc. 1953;75:4622–4623. [Google Scholar]
- 6.Minieri PP, Sokol H, Firman MC. Process for the Preparation of Tetracycline and Chlorotetracycline. U. S. Patent 2,734,018. 1956 Feb 7;
- 7.McCormick JRD, Sjolander NO, Hirsch U, Jensen ER, Doerschuk AP. J. Am. Chem. Soc. 1957;79:4561–4563. [Google Scholar]
- 8.a Conover LH. Discovering Tetracycline. Research Management. 1984 Sept-Oct;:17–22. [Google Scholar]; b Conover LH. Advances in Chemistry Series. 1971;108:33–80. [Google Scholar]
- 9.a Stephens CR, Murai K, Rennhard HH, Conover LH, Brunings KJ. J. Am. Chem. Soc. 1958;80:5324–5325. [Google Scholar]; b McCormick JRD, Jensen ER, Miller PA, Doerschuk AP. J. Am. Chem. Soc. 1960;82:3381–3386. [Google Scholar]; c Stephens CR, Beereboom JJ, Rennhard HH, Gordon PN, Murai K, Blackwood RK, Schach von Wittenau M. J. Am. Chem. Soc. 1963;85:2643–2652. [Google Scholar]; d Blackwood RK, Stephens CR. J. Am. Chem. Soc. 1962;84:4157–4159. [Google Scholar]
- 10.a Martell MJ, Boothe JH. J. Med. Chem. 1967;10:44–46. doi: 10.1021/jm00313a009. [DOI] [PubMed] [Google Scholar]; b Church RFR, Schaue RE, Weiss MJ. J. Org. Chem. 1971;36:723–725. doi: 10.1021/jo00804a025. [DOI] [PubMed] [Google Scholar]; c Spencer JL, Hlavka JJ, Petisi J, Krazinski HM, Boothe JH. J. Med. Chem. 1963;6:405–407. doi: 10.1021/jm00340a014. [DOI] [PubMed] [Google Scholar]; d Zambrano RT. U.S. Patent 3,483,251. 1969 Dec 9;
- 11.a Sum P-E, Lee VJ, Testa RT, Hlavka JJ, Ellestad GA, Bloom JD, Gluzman Y, Tally FP. J. Med. Chem. 1994;37:184–188. doi: 10.1021/jm00027a023. [DOI] [PubMed] [Google Scholar]; b Sum P-E, Petersen P. Bioorg. Med. Chem. Lett. 1999;9:1459–1462. doi: 10.1016/s0960-894x(99)00216-4. [DOI] [PubMed] [Google Scholar]
- 12. [August 2008];Listing of Newly Approved Drug Therapies. 2008 http://www.centerwatch.com/patient/drugs/druglist.html.
- 13.Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. Nat. Rev. Drug Discovery. 2007;6:29–40. doi: 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- 14.a Hochstein FA, Stephens CR, Conover LH, Regna PP, Pasternack R, Gordon PN, Pilgrim FJ, Brunings KJ, Woodward RB. J. Am. Chem. Soc. 1953;75:5455–5475. [Google Scholar]; b Stephens CR, Conover LH, Hochstein FA, Regna PP, Pilgrim FJ, Brunings KJ. J. Am. Chem. Soc. 1952;74:4976–4977. [Google Scholar]
- 15.Podlogar BL, Ohemeng KA, Barrett JF. Expert Opin. Ther. Patents. 2003;13:467–478. [Google Scholar]
- 16.a Conover LH, Butler K, Johnston JD, Korst JJ, Woodward RB. J. Am. Chem. Soc. 1962;84:3222–3224. [Google Scholar]; b Woodward RB. Pure Appl. Chem. 1963;6:561–573. [Google Scholar]; c Korst JJ, Johnston JD, Butler K, Bianco EJ, Conover LH, Woodward RB. J. Am. Chem. Soc. 1968;90:439–457. [Google Scholar]
- 17.Stork G, Hagedorn AA., III J. Am. Chem. Soc. 1978;100:3609–3611. [Google Scholar]
- 18.a Gurevich AI, Karapetyan MG, Kolosov MN, Korobko VG, Onoprienko VV, Popravko SA, Shemyakin MM. Tetrahedron Lett. 1967;8:131–134. doi: 10.1016/s0040-4039(00)90501-x. [DOI] [PubMed] [Google Scholar]; b Kolosov MN, Popravko SA, Shemyakin MM. Lieb. Ann. 1963;668:86–91. [Google Scholar]
- 19.a Muxfeldt H, Hardtmann G, Kathawala F, Vedejs E, Mooberry JB. J. Am. Chem. Soc. 1968;90:6534–6536. doi: 10.1021/ja01025a063. [DOI] [PubMed] [Google Scholar]; b Muxfeldt H, Haas G, Hardtmann G, Kathawala F, Mooberry JB, Vedejs E. J. Am. Chem. Soc. 1979;101:689–701. doi: 10.1021/ja01025a063. [DOI] [PubMed] [Google Scholar]
- 20.a Brodersen DE, Clemons WM, Jr., Carter AP, Morgan-Warren RJ, Wimberly BT, Ramakrishnan V. Cell. 2000;103:1143–1154. doi: 10.1016/s0092-8674(00)00216-6. [DOI] [PubMed] [Google Scholar]; b Pioletti M, Schlünzen F, Harms J, Zarivach R, Glühmann M, Avila H, Bashan A, Bartels H, Auerbach T, Jacobi C, Hartsch T, Yonath A, Franceschi F. EMBO J. 2001;20:1829–1839. doi: 10.1093/emboj/20.8.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Parrish CA. Ph.D. Thesis. California Institute of Technology; Pasadena, CA: 1999. [Google Scholar]
- 22.Hauser FM, Rhee RP. J. Org. Chem. 1978;43:178–180. [Google Scholar]
- 23.Broom NJP, Sammes PG. J. Chem. Soc., Chem. Comm. 1978:162–164. [Google Scholar]
- 24.Kraus GA, Sugimoto H. Tetrahedron Lett. 1978;26:2263–2266. [Google Scholar]
- 25.a Dodd JH, Weinreb SM. Tetrahedron Lett. 1979;38:3593–3596. [Google Scholar]; b Dodd JH, Starrett JE, Weinreb SM. J. Am. Chem. Soc. 1984;106:1811–1812. [Google Scholar]; c Leeper FJ, Staunton J. J. Chem. Soc., Chem. Comm. 1979;5:206–207. [Google Scholar]; d Leeper FJ, Staunton J. J. Chem. Soc. Perkin Trans. 1. 1984:1053–1059. [Google Scholar]
- 26.a Tarnchompoo B, Thebtaranonth C, Thebtaranonth Y. Synthesis. 1986;9:785–786. [Google Scholar]; b Boger DL, Zhang M. J. Org. Chem. 1992;57:3974–3977. [Google Scholar]; c Nishizuka T, Hirosawa S, Kondo S, Ikeda D, Takeuchi T. J. Antibiotics. 1997;50:755–764. doi: 10.7164/antibiotics.50.755. [DOI] [PubMed] [Google Scholar]; d Hill B, Rodrigo R. Org. Lett. 2005;7:5223–5225. doi: 10.1021/ol052058u. [DOI] [PubMed] [Google Scholar]
- 27.For examples of stereocontrol in Michael–Claisen and Michael–Dieckmann cyclization reactions, see: Franck RW, Bhat V, Subramaniam CS. J. Am. Chem. Soc. 1986;108:2455–2457. doi: 10.1021/ja00269a059.Tatsuta K, Yamazaki T, Mase T, Yoshimoto T. Tetrahedron Lett. 1998;39:1771–1772.White JD, Demnitz FWJ, Qing X, Martin WHC. Org. Lett. 2008;10:2833–2836. doi: 10.1021/ol8009732.
- 28.Tatsuta K, Yoshimoto T, Gunji H, Okado Y, Takahashi M. Chem. Lett. 2000:646–647. [Google Scholar]
- 29.Brubaker JD, Myers AG. Org. Lett. 2007;9:3523–3525. doi: 10.1021/ol071377d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carpenter TA, Evans GE, Leeper FJ, Staunton J, Wilkinson MR. J. Chem. Soc. Perkin Trans. I. 1984:1043–1051. [Google Scholar]
- 31.Phenyl ester activation in toluate ester condensations is precedented: White JD, Nolen EG, Jr., Miller CH. J. Org. Chem. 1986;51:1150–1152. (see also reference 27c).
- 32.Bennetau B, Mortier J, Moyroud J, Guesnet J-L. J. Chem. Soc. Perkin Trans. 1. 1995:1265–1271. [Google Scholar]
- 33.For other examples of the isolation of uncyclized Michael adducts, see references 23, 25c, 25d and 26b.
- 34.Phenyl ester 17 was synthesized in 6 steps from ethyl 6-methylsalicylate; see: Olah GA, Malhotra R, Narang SC. Nitration, Methods and Mechanisms. VCH Publishers Inc.; New York: 1980. Bellamy FD, Ou K. Tetrahedron Lett. 1984;26:839–842.Abdel-Magid AF, Carson KG, Harris BD, Maryanoff CA, Shah RD. J. Org. Chem. 1996;61:3849–3862. doi: 10.1021/jo960057x..
- 35.Leading references for the synthesis of D-ring precursors to 6-aryl-substituted tetracyclines: Miyaura N, Suzuki A. Chem. Rev. 1995;95:2457–2483.Kotha S, Mandal K. Eur. J. Org. Chem. 2006;23:5387–5393.Wright SW, Hageman DL, McClure LD. J. Org. Chem. 1994;59:6095–6097.Osyanin VA, Purygin PP, Belousova ZP. Russ. J. Gen. Chem. 2005;75:111–117.Appukkuttan P, Dehaen W, Fokin VV, Van der Eycken E. Org. Lett. 2004;6:4223–4225. doi: 10.1021/ol048341v..
- 36.a Parham WE, Jones LD, Sayed YA. J. Org. Chem. 1978;41:1184–1186. [Google Scholar]; b Berk SC, Yeh MCP, Jeong N, Knochel P. Organometallics. 1990;9:3053–3064. [Google Scholar]
- 37.>The use of a methyl ether protecting group in the penultimate entry of Table 2 required one additional deprotection step (BBr3, CH2Cl2, −78 °C → 23 °C) following the typical two-step sequence.
- 38.Both the stepwise and in situ protocols for deprotonation-cyclization were also effective for the synthesis of the Michael–Claisen cyclization product 40 from phenyl o-toluate and enone 1, though yields were lower.
- 39.Van Leusen AM, Terpstra JW. Tetrahedron Lett. 1981;22:5097–5100. [Google Scholar]
- 40.Synthesized from 6-bromophthalide and methyl crotonate: Broom NJP, Sammes PG. J. Chem. Soc. Perkin Trans. 1. 1981:465–470.
- 41.The yield of the Michael–Claisen cyclization product 43 was somewhat lower (28%) when LHMDS was omitted from the reaction.
- 42.The final compound in Chart 1 was prepared by N-acetylation after reductive amination with methylamine.
- 43.7-Dimethylamino-alkylaminomethylpentacyclines were initially synthesized from a DE-ring precursor in which the phenolic hydroxyl group was protected as a methyl ether (necessitating a three-step deprotection sequence); it was later found that the corresponding DE-ring precursor containing a tert-butoxycarbonyl protecting group (compound 46) was also an effective cyclization substrate (allowing for standard two-step deprotection, see Supporting Information for details).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.