

Abstract
The mammalian central nervous system (CNS) is populated very early in development by tissue macrophages referred to as microglia. By adulthood, this CNS-resident population is found in all regions of the brain and spinal cord. Despite nearly a century of study, the in vivo function of microglia and the extent that they contribute to the onset, progression and recovery from neuroinflammatory disorders is still a subject of debate. Partly, the debate of whether activated microglia promote neuroprotection or neurodegeneration is fueled by the contrasting results derived from the different models used to assay microglial function. Here we discuss the strengths, weaknesses and utility of some of the most commonly used in vivo and in vitro models.
Introduction
Microglia are the resident tissue macrophage of the CNS (Fig. 1) (reviewed in [1–3]). They are readily detected within the CNS during early prenatal development and are found in all regions of the healthy adult brain and spinal cord. Because microglial activation is one of the earliest and most prominent features of nearly all neurodegenerative diseases, microglia have alternatively been credited with either promoting.
Figure 1.
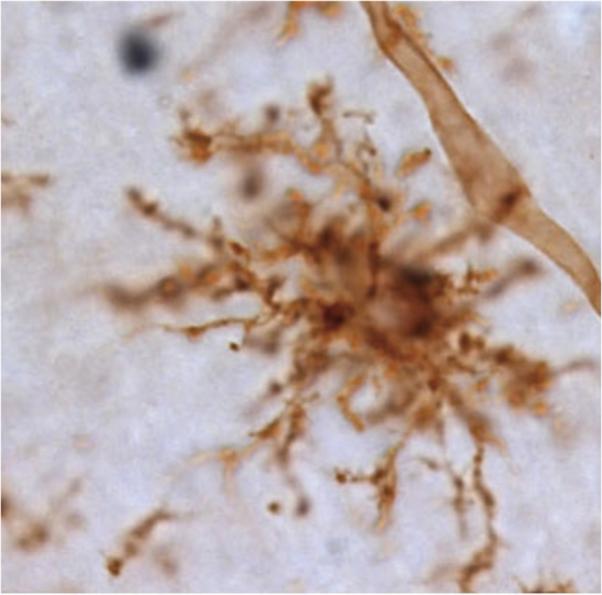
Microglia in the intact CNS. Microglial processes extend to all elements of their environment. A microglia and blood vessel are visualized in brown using tomato lectin.
(a) the transition from acute injury to chronic neurodegenerative disease
or
(b) the protection and regeneration of injured neurons (reviewed in [1–3]).
Partly, the inability to resolve this debate is due to the very different predictions of microglial function generated by the different in vitro and in vivo models of microglia being used. However, both possibilities suggest that microglia could be attractive therapeutic targets to treat a wide array of CNS disorders from mechanical/ischemic injury (spinal cord injury, stroke) to chronic neurodegenerative disorders (ALS, Parkinson's, Alzheimer's disease) to neuroinflammatory disorders (multiple sclerosis, AIDs dementia). The focus of this review will not be to resolve this debate. Rather the focus of this review will be to discuss the strengths, weaknesses and specific utility of the models most commonly used to probe microglial biology and to predict their contribution toward CNS pathophysiology.
The use and interpretation of in vitro and in vivo models of microglial function
In vitro models: overview
Microglia comprise ∼15% of the total number of cells within the CNS but comprise less than 0.1% of the total mass and RNA of the CNS RNA [1,2]. Therefore, characterization of microglial phenotype and function on a molecular level has been difficult to separate from that of CNS neurons, oligodendrocytes and astrocytes within the healthy and inflamed CNS. To help address this issue, several in vitro models of microglia have been developed. The most commonly used in vitro models are
Immortalized microglial cell lines (most commonly used rodent lines are the spontaneously immortalized EOC cell lines and the oncogene transformed BV-2 and N9 cell lines).
Primary microglia derived from established embryonic or neonatal mixed glial cultures.
Primary microglia acutely isolated from adult CNS tissue.
In vitro models: where are the neurons?
Although the purpose of these models is to study microglial phenotype and function in the absence of other contaminating CNS cell types, the absence of other types of CNS cells also introduces a major limitation in their predictive power for in vivo biology. Like all tissue macrophages, microglial phenotype is highly plastic and is continually being determined by the sum of their external cues [1,2]. Most notably, microglia express functional receptors for many neuronally expressed molecules including CD200, fractalkine and most CNS neurotransmitters [1,2]. In addition, unlike non-CNS macrophages, they also express inward rectifying potassium channels. Therefore, it is not unexpected that when microglia are cultured extensively in the absence of neuronal cues their phenotype begins to significantly diverge from that of microglia in vivo.
The scale of this problem is dramatically revealed by gene profiling studies contrasting primary microglia derived from established neonatal mixed glial cultures with peripheral macrophages. In these studies, several molecules were identified as being highly expressed in unactivated cultured microglia [1,2,4]. For example, in vitro nearly all cultured microglia (primary neonatal microglia and many immortalized microglial cell lines) express high levels of CXCL14, a molecule that is chemoattractive for macrophages [1,2,4]. Furthermore, in vitro CXCL14 expression is repressed ∼10-fold by treating cultured microglia with LPS/IFNg overnight. However, when examined by in situ hybridization, in vivo GABAnergic interneurons not microglia were found to be the cells expressing high levels of CXL14 [1,2,4]. Detection of very low levels of CXL14 could only be detected in adult murine microglia when real-time PCR methods were used to assess RNA purified from microglia immediately after isolating these cells from the healthy adult CNS [1,2,4].
Conversely, in the adult rodent, all peripheral macrophage populations express very high but similar levels of CD45, a protein tyrosine phosphatase [5,6]. Although the function of CD45 is not fully understood, it is does act as an inhibitory receptor on cells of myeloid origin. Microglia are unique in being the only macrophage population that expresses a log order lower level of CD45 than that measured on any other macrophages population [5–8]. Upon activation microglial CD45 levels can increase dramatically but tend to remain lower than the levels measured on unactivated peripheral macrophage populations [5–8]. By contrast, ‘unactivated’ microglia maintained in culture in the absence of neurons express CD45 levels that are higher than those measured on highly activated microglia in vivo [7,8]!
The influence of external cues on microglial phenotype is further demonstrated by contrasting the largely uniform phenotype of microglia in vitro with the diversity of phenotypes observed in vivo. For example, SPARC and Trem2 are two molecules expressed at relatively high levels by nearly all microglia in vitro, both immortalized cell lines and primary microglia from neonatal mixed glial cultures [1,2,4]. By contrast, in the adult murine CNS, SPARC is only highly expressed by hindbrain microglia. Forebrain microglia of the cortex, striatum and olfactory bulb express low to negligible levels of SPARC in the adult mouse CNS [1,2,4,9]. Similarly, in the adult murine CNS, the percentage of Trem2 expressing microglia and the level of Trem2 per microglia varies by brain region [1,2,4,9]. The highest levels and percentage of Trem2 expressing microglia are found in the cingulate cortex, corpus collosum, fimbiria and entorhinal cortex, whereas the lowest are found in the circumventricular organs and the laterodorsal thalamic nucleus. Strikingly, at birth through the first week of murine life, all microglia express fairly uniform levels of Trem2 [10]. It is not until after mice open their eyes (∼post-natal day 10) that heterogeneous expression of Trem2 begins to become detectable. These data suggest that alterations in neuronal activity and/or other coincident CNS developmental events directly modulate microglial phenotype and function.
From this discussion, three obvious questions are raised:
Do these differences in gene expression between microglia in vitro and in vivo preclude the use of cultured microglia as useful tools to elucidate microglial function and/or define potential points of therapeutic intervention?
Would it be more appropriate to use organotypic brain slice cultures, in which all major classes of CNS cells are represented?
If pure populations of microglia are required to test cell-type responses to specific molecules or pharmacologic substances, is it mandatory to only use defined populations acutely isolated from adult CNS tissue?
Affirmative answers to these three questions would represent an extreme viewpoint that ignores many of the practical advantages of using well-characterized cultured microglial models (Table 1).
Table 1.
Matching the model to the experimental question.
| In vitro models |
In vivo models |
|||||
|---|---|---|---|---|---|---|
| Immortalized cell lines | Primary microglia derived from mixed glial cultures | Ex vivo adult microglia | Organotypic slice cultures | Microglial deficient mice | Chimeric mice | |
|
Pros
|
Large quantities of homogeneous cells economically generated |
Large quantities of primary cells are readily obtained; cells differentiated in presence of CNS astrocytes and oligodendrocytes |
Differentiated in the presence of intact, functional CNS |
Retention of three-dimensional orientation of differentiated CNS neurons, microglia and microglia, easy delivery of cells and molecules |
Intact CNS environment |
Intact CNS environment, separate manipulation of CNS-resident microglia and peripheral immune cells |
|
Cons
|
Polarized phenotypes, different lines might yield different assays |
Differentiated in the absence of functional neurons, hyperresponsive to pathogenic stimuli |
Difficult to obtain sufficient numbers of cells for many assays |
Slice ‘wound’ introduces ill-defined inflammatory stimuli to culture system |
Incomplete deletion/depletion of only subsets of microglia; difficult to directly measure microglial function |
Chimeric mouse methodologies alters vasculature and circulating immune system, difficult to directly measure microglial function |
|
Best use of model
|
High throughput and first screen assays |
Signal transduction assays, molecular manipulation of gene expression and functional consequences of mutational |
Assays requiring purified microglia, final assays confirming key observations made in high throughput |
Controlled manipulations and measurements difficult to perform in the intact animal (addition of exogenous cells, compounds) |
Test of function within intact CNS and in presence of interacting endocrine and immune systems |
Test of function within intact CNS and in presence of interacting endocrine and immune systems |
|
How to get access
|
ATCC |
Prepare from fetal or neonatal CNS tissue |
Prepare from adult CNS tissue |
Prepare from intact CNS |
Various animal vendors and research groups |
Generate from rodent strains on hand |
| Refs | [16-18] | [7,8] | [5-7] | [14,15] | [21,22,26-28] | [23-25], Reviewed in [9] |
First, the differences in gene expression between in vitro and in vivo microglia can also be utilized as biosensors/biomeasures to test and quantify the regulatory effects of CNS derived cues if comprehensively documented. For example, because Trem2 expression was uniformly high on microglia cultured in the absence of neurons, we tested and confirmed that in vivo insults such as ischemic injury or kainic acid induced excitotocity that cause neuronal death and decreased neuronal activity dramatically increased local microglial expression of Trem2 ([2], Carson unpublished results). Similarly, CNS neurons in vivo constitutively express the ligands for at least three inhibitory receptors (CD45, CD200R, CXCR3) expressed by microglia (reviewed in [1,2,9]). In the absence of these neuronal ligands, cultured microglia become hyperresponsive to a wide variety of activating stimuli including pathogens or even apoptopic cell debris. Knockout and transgenic overexpression mice have confirmed in vivo the significance of these neuronal–microglial interactions for limiting neurotoxic microglial activation states and promoting the resolution of acute neuroinflammatory responses in the CNS [11–13] Thus, once these receptor–ligand interactions are documented, cultured microglia become a powerful resource to test pharmacological manipulation of these pathways in the context of known CNS pathogens and/or CNS toxins.
Second, organotypic brain slice cultures do have many advantages in that the relative orientation and cells present in a brain region are retained in a cultured slice of brain tissue [14]. Pharmacologic agents, pathogens, toxins and even cells are readily introduced into this integrated three-dimensional structure [14]. Indeed, adding primary cultured microglia to slice cultures has been of utility in demonstrating the robust ability of the CNS microenvironment to regulate microglial morphology and adhesion molecule expression. In addition, slice cultures offer a window into endogenous CNS inflammatory responses occurring in the absence of infiltrating peripheral immune cells or blood-derived products leaking into the CNS via damaged blood vessels [14]. For example, microglial engulfment of activated neutorphils was conclusively imaged when neutorphils were added to brain slice cultures [14]. The authors hypothesize that this might be a previously unknown mechanism by which CNS-microglia limit peripheral inflammation during acute CNS injury. However, many problems also exist with brain slice cultures. Neuronal activity and vascular dynamics are disrupted, while at the same time both microgliosis and astrogliosis are induced by the slice ‘wound’. The release of neuronal cell debris in these cultures has been demonstrated to exacerbate neuronal responses to activated microglia and astrocytes [15]. Thus, the organotypic slice culture can play a useful role in providing a readily manipulated CNS microenvironment to study microglial biology, but as yet the effects of slice ‘wounding’ on the regulatory ability of the CNS microenvironment are not yet fully characterized.
Third, collecting sufficient numbers of microglia from adult murine CNS to run high throughput assays sufficient to characterize the effects of pharmacological compound libraries is impractical. Testing even a single compound for its ability to modulate the kinetics, magnitude and type of inflammatory response to even a single inflammatory stimulus requires multiple time points, multiple doses and multiple replicates. Even in highly optimized assays of phagocytosis, antigen-presentation, production of growth factors, cytokines, chemokines and reactive oxygen species, 10s of thousands of cells are required per data sample. Unfortunately, standard methodologies for isolating microglia from adult rodent CNS and separating them from CNS-infiltrating macrophages not only require the availability of flow cytometric cell sorting, the yields of microglia are of notoriously of low. Typically ∼500 000−750 000 microglia are isolated from the pooled healthy adult mouse brain and spinal cord [7,8]. An additional complication is the relatively small mass of an adult microglia as compared with other CNS cells. Microglia produce only 10% of the RNA produced by CNS astrocytes [4,7,8]. Therefore, it is essential to quantify expression of astrocyte specific gene expression in ‘pure’ microglial samples because even minor astrocytic contamination will have large effects on measurements of RNA and protein expression. However, the number of cells that can be isolated from an adult murine CNS are sufficient for real-time PCR analysis and we have used flow cytometry and this approach to determine that CNS microglia express nearly 10-fold higher levels of Trem2 than CNS-infiltrating macrophages.
Lastly, in vivo microglia are a heterogenous collection of a diverse array of phenotypes specialized to specific brain regions and brain activities. In vitro, within a single culture of microglia, only a limited array of phenotypes is observed. In primary microglia maintained in mixed glial culture, microglial phenotype varies as a function of the differentiation state of the astrocytes and oligodendrocytes present in the culture [7,8]. Upon purifying microglia way from the astrocytes and oligodendrocytes, also promotes a uniform change in microglial phenotype. Therefore, this model system is a highly useful tool to identify microglial regulatory cues provided by glia. In addition, mixed glial cultures have high utility in generating preparative quantities of primary microglia. Upon harvesting and depleting microglia from mixed glial cultures, the few remaining microglia rapidly proliferate and repopulate the mixed glial population. In practice, four murine pup equivalents generate ∼1 million primary microglia every two weeks without the addition of exogenous macrophage growth factors [Carson, unpublished observations].
When even greater numbers and more uniform phenotype are required or when cell lines with well-characterized and/or stable genetic manipulations are required, this is when advantages of using immortalized microglial cell lines begin to outweigh their disadvantages. Two primary problems must be addressed when choosing specific microglial cell lines: broad diversity of phenotypes between microglial cell lines and the degree of accumulated genomic mutations and. For example, BV-2 cells and N9 cells are both oncogene transformed microglial cell lines derived from standard neonatal mixed glial cultures [16,17]. These cells differ in morphology, phagocytic activity, propensity to produce pro-inflammatory molecules (cytokines, proteases, nitric oxide) and to proceed toward activation induced cell death. It is possible that these differences reflect their different genetic backgrounds. However, phenotypic differences are observed even in the panel of CSF-1 dependent EOC microglial cell lines all derived from the same standard non-transformed murine neonatal mixed glial cultures [18]. Although the phenotype of each EOC cell line was relatively stable, there was great variation in their ability to process and present antigens to CD4+ and CD8+ T lymphocytes. An additional concern for using immortalized cell lines. Cloning and sequencing commonly expressed immunomodulatory molecules from BV-2 cells reveals a high degree of mutations, some with potential functional consequences [Carson, unpublished observations]. As with all long-term cell lines, it is crucial to keep track of the passage number and mutation-induced changes in cell function.
Despite these concerns, the uniform phenotype of immortalized cell lines has provided a consistent background to quantify the functional consequences of overexpressing and knocking down microglial expression of Trem2. Using these methods, we and others have been able to demonstrate that Trem2 expression promotes microglial phagocytosis of aggregated amyloid, cell debris, while limiting microglial-mediated activation of T cell cytokine production [1,4,10,19,20]. Considered together with our ex vivo assays of Trem2 expression in adult microglia and CNS-infiltrating macrophages (discussed above), these functional assays suggest that CNS-resident microglia play a fundamentally different role than CNS-infiltrating macrophages. While CNS-infiltrating macrophages display primarily pro-inflammatory functions, decreased neuronal activity induces a neuroprotective state in activated CNS-resident microglia.
In vivo models: the overview
To test for function, traditional experimental approaches involve quantifying the consequences of removing, altering and adding back the element being tested. Ideally it would be useful to deplete or remove microglia from the CNS to define and reveal the consequences of microglial function in normal and abnormal CNS physiology. As yet, there is no spontaneous viable mammalian mutant that lacks CNS-resident microglia. Even the op/op mouse, a spontaneous deletion mutation resulting in loss of CSF-1, a necessary macrophage growth factor, show only a modest reduction in CNS-microglia [21,22]. Interestingly, the reduction is only detectable in selected brain regions and is much less than that the reduction observed in other peripheral macrophage populations. In addition, by 6 months of age, microglial numbers have dramatically increased. Considered together these data strongly suggest that microglia play a non-redundant role in maintaining CNS function. Furthermore, microglia differ from all other tissue macrophages in that they are largely a long lived, self-renewing population that is only rarely replaced by bone marrow derived cells.
In vivo models: manipulating microglial genotype and function
By contrast, most tissue macrophages are short lived and are replaced by bone marrow derived cells every few weeks (reviewed in [9]). Therefore, the function of most tissue macrophages can be tested or even genetically modulated in two steps using a bone marrow donor and a bone marrow recipient. First the bone marrow of the recipient is killed (usually with irradiation). Second, following carefully dosed irradiation, the recipient receives bone marrow from a recipient. Without donor bone marrow, the recipient would die (no bone marrow equals no new red blood cells). The donor bone marrow can be modified genetically to allow for easy detection (for example, transgenic expression of green fluorescent protein). Within approximately 12 weeks, the entire peripheral immune system of the recipient has been replaced by the donor bone marrow, with one major exception: microglia remain recipient derived [9,23]. It is important to note that the dose of irradiation must be carefully monitored. If too high of a dose is used, non-specific radiation induced inflammation is observed throughout the body including the CNS. Interestingly, these results have been confirmed in humans receiving bone marrow transplants from gender-mismatched donors. When examined upon autopsy years after bone marrow transplant, CNS microglia are largely of the recipient gender genotype, while all the peripheral immune cells are of the donor's gender genotype [24].
Irradiation is known to cause long lasting changes to the vasculature; therefore, it was striking that the same types of observations were made in a very different model of parabiosis [25]. In this model, the vasculature of two genetically distinct mice are linked. Without any other manipulation, the replacement of tissue macrophages in each mouse by cells with the genotype of the surgically linked mouse can be histologically monitored. In these mice, CNS-resident microglia were not replaced with cells from the surgically linked mouse. These data confirm that the CNS is not routinely infiltrated by blood borne cells. These data also confirm that in the healthy CNS, in vivo microglia are not routinely exposed to serum components, in contrast to cultured microglia that are usually cultured in the presence of serum!
These data also illustrate the strengths and weaknesses of using chimeric mice. On the one hand, chimeric mice provide a method to genetically manipulate the peripheral immune system without altering CNS microglia. Using this approach, a large number of investigators have demonstrated that while microglia are not as effective as other peripheral immune cells in activating pro-inflammatory T cell responses (reviewed in [9]). In addition, using this approach, the unique ability of microglia to drive neuroprotective T cell responses was revealed. Furthermore, Neumann and colleagues have begun to dissect the molecular basis of this difference. In EAE, CNS-infiltrating myeloid cells express low or negligible levels of Trem2 [19,20]. By transfecting bone marrow with Trem2 overexpression constructs, Neumann and colleagues were able to confer ‘microglial-like neuroprotection’ to the peripheral immune cells. EAE severity was significantly reduced in chimeric mice receiving Trem2 expressing bone marrow.
On the other hand, chimeric mice demonstrate the difficulty of introducing modifications into the microglial compartment to test regulatory function of a specific molecule. Several approaches have been taken to attempt to remove or deplete microglia from the CNS. At least two groups have transgenically expressed mutant thymidine kinase (TK) under the control of macrophage promoters [26,27]. Treating these transgenic mice with ganciclovir kills all proliferating macrophages in the body. Therefore, irradiation chimeric approaches are used to restrict TK expression to CNS-resident microglia. Using these approaches, ganciclovir has little effect on microglia in the healthy CNS indicating that these cells are not routinely undergoing cell proliferation. However, in response to injury or pathogen exposure, microglia rapidly proliferate and become ganciclovir sensitive. Using these types of approaches, several groups have demonstrated the neuroprotective effects of activated microglia by demonstrating increased neuronal death in the presence of ganciclovir [26,28]. Most recently, this approach was used to reveal proliferating microglia as a key pool of IGF-1 following ischemic injury [28].
Model comparison and translation to humans
To this point, only animal-based models have been discussed. However the goal is to identify and/or develop models predictive of human biology. To this end, all of the strengths and weaknesses discussed for cultured rodent microglia apply to cultured human microglia, with two additional concerns. Human microglia are generally isolated from abortion/miscarriage fetal tissue or from human biopsy material (reviewed in [9]). With human fetal material, undisclosed or undefined genetic, viral or other ill-defined insults might lead to fundamental abnormalities in the phenotype and function of cultured microglia. Similarly, human CNS biopsy tissue is usually derived from tumor or severe epileptic tissue. Studies from rodents and other animal models suggest that microglia from these sources are not only heterogeneous, they are also not representative of microglia found in normal CNS tissue. Therefore, it is only by using all of the discussed animal models and by building molecular profiles associated with each functional phenotype that we will be able to define the functional consequences of precise molecular manipulation of microglia.
Once these molecular definitions of microglia are used to identify potential therapeutic targets in humans, it will be essential to develop real-time imaging of microglial activation. In the past, analysis of microglial function was based on indirect inference from clinical measures or was limited to static analysis of histological sections (biopsy/autopsy). More recently, two-photon microscopy has been used to image transgenic fluorescent microglia at extremely high resolution in rodent CNS [29,30]. This new approach dramatically altered the perspective of how many viewed microglial function in vivo. Before these studies, many assumed that in the absence of injury or pathogens, microglia are quiescent and inactive. By contrast, two photon studies demonstrated that microglia were constantly active sampling their environment. Recent calculations indicate that all microglial processes move and touch all elements in their environment every 6 h [29,30]. The ability to image microglia in vivo opens new possibilities to monitor their real time responses to changes in neuronal activity, metabolism or to pathogenic insults. Unfortunately, 2-photon microscopy is limited in the depth of tissue that can be imaged. Current technology only allows imaging in rodents of cortical areas in areas in which the skull has been removed or thinned [29,30,32]. Microglial activation has also been demonstrated to be caused by these surgical manipulations to the skull. Despite these caveats, 2-photon imaging is now opening a window on long-term imaging of microglia in CNS pathology and is confirming observations implied from histological analysis of chimeric mice. Most notably, long-term recurrent imaging of microglia surrounding amyloid plaques in transgenic mice reveals that individual microglia do not leave the plaque but can remain in situ over months phagocytosing Abeta [31].
2-Photon imaging while powerful to analyze living animal models, is not readily applicable to human patients. An alternative approach to imaging microglial activation in vivo is positron emission tomography (PET) imaging of peripheral benzodiazepine receptor complex [32]. Although both microglia and astrocytes are reported to express this receptor complex, most studies suggest that PET imaging of radiolabeled ligands for this receptor complex is monitoring microglial activation [32]. Although this imaging method is of very low resolution, PET imaging of peripheral benzodiazepine receptor complex in human patients has already begun to provide real-time information concerning the sites and kinetics of microglial activation in relationship to clinical/behavioral measurements [32].
Conclusion
Microglia are well suited to act as biosensors of CNS physiology on the basis of their highly plastic phenotype and their demonstrated ability to actively survey their local environment. This same phenotypic plasticity has also hampered interpretation of the extent that specific in vitro models are predictive of in vivo biology. In vivo models are hampered by the difficulty of manipulating the microglial compartment within the CNS. However, by characterizing the molecular profile of microglia in each model, contrasting the assayed functions and testing whether that function can be transferred into other macrophages compartments should reveal therapeutic points of intervention able to modulate the tipping point between neuroprotective and neurotoxic inflammation.
References
- 1.Carson MJ, et al. A rose by any other name: the potential consequences of microglial heterogeneity during CNS health and disease. Neurotherapeutics. 2007;4:571–579. doi: 10.1016/j.nurt.2007.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Melchior B, et al. Microglia and the control of autoreactive T cell responses. Neurochem. Int. 2006;49:145–153. doi: 10.1016/j.neuint.2006.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Butovsky O, et al. Induction and blockage of oligodendrogenesis by differently activated microglia in an animal model of multiple sclerosis. J. Clin. Investig. 2006;116:905–915. doi: 10.1172/JCI26836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schmid CD, et al. Heterogeneous expression of the triggering receptor expressed on myeloid cells-2 on adult murine microglia. J. Neurochem. 2002;83:1309–1320. doi: 10.1046/j.1471-4159.2002.01243.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sedgwick JD, et al. Isolation and direct characterization of resident microglial cells from the normal and inflamed central nervous system. PNAS. 1991;88:7438–7442. doi: 10.1073/pnas.88.16.7438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Renno T, et al. TNF-alpha expression by resident microglia and infiltrating leukocytes in the central nervous system of mice with experimental allergic encephalomyelitis. Regulation by Th1 cytokines. J. Immunol. 1995;154:944–953. [PubMed] [Google Scholar]
- 7.Carson MJ, et al. Mature microglia resemble immature antigen-presenting cells. GLIA. 1998;22:72–85. doi: 10.1002/(sici)1098-1136(199801)22:1<72::aid-glia7>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 8.Carson MJ, et al. Microglia stimulate naive T-cell differentiation without stimulating T-cell proliferation. J. Neurosci. Res. 1999;55:127–134. doi: 10.1002/(SICI)1097-4547(19990101)55:1<127::AID-JNR14>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 9.Carson MJ, et al. CNS immune privilege: hiding in plain sight. Imunol Rev. 2006;213:48–65. doi: 10.1111/j.1600-065X.2006.00441.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thrash, et al. Developmental regulation of TREM2 and DAP12 expression in the murine CNS: implications for Nasu-Hakola disease. Neurochemical Research. 2008 doi: 10.1007/s11064-008-9657-1. PMID: 18404378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chitnis T. Elevated neuronal expression of CD200 protects Wlds mice from inflammation-mediated neurodegeneration. Am. J. Pathol. 2007;170:1695–1712. doi: 10.2353/ajpath.2007.060677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deckert M. Regulation of microglial cell responses in murine Toxoplasma encephalitis by CD200/CD200 receptor interaction. Acta Neuropathol. 2006;111:548–558. doi: 10.1007/s00401-006-0062-z. [DOI] [PubMed] [Google Scholar]
- 13.Cardona AE. Control of microglial neurotoxicity by the fractalkine receptor. Nat. Neurosci. 2006;9:917–924. doi: 10.1038/nn1715. [DOI] [PubMed] [Google Scholar]
- 14.Neumann J. Microglia cells protect neurons by direct engulfment of invading neutrophil granulocytes: a new mechanism of CNS immune privilege. J. Neurosci. 2008;28:5965–5975. doi: 10.1523/JNEUROSCI.0060-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bernardino L, et al. Inflammatory events in hippocampal slice cultures prime neuronal susceptibility to excitotoxic injury: a crucial role of P2X7 receptor-mediated IL-1beta release. J. Neurochem. 2008;106:271–280. doi: 10.1111/j.1471-4159.2008.05387.x. [DOI] [PubMed] [Google Scholar]
- 16.Bocchini V, et al. An immortalized cell line expressed properties of activated microglial cells. J. Neurosci. Res. 1992;31:616–621. doi: 10.1002/jnr.490310405. [DOI] [PubMed] [Google Scholar]
- 17.Righi M, et al. myc-Immortalized microglial cells express a functional platelet-activating factor receptor. J. Neurochem. 1995;64:121–129. doi: 10.1046/j.1471-4159.1995.64010121.x. [DOI] [PubMed] [Google Scholar]
- 18.Walker WS, et al. Mouse microglial cell lines differing in constitutive and interferon-gamma-inducible antigen-presenting activities for naive and memory CD4+ and CD8+ T cells. J. Neuroimmunol. 1995;63:163–174. doi: 10.1016/0165-5728(95)00146-8. [DOI] [PubMed] [Google Scholar]
- 19.Takahashi K, et al. TREM2-transduced myeloid precursors mediate nervous tissue debris clearance and facilitate recovery in an animal model of multiple sclerosis. PLOS Med. 2007;4:e124. doi: 10.1371/journal.pmed.0040124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Neumann H. Essential role of the microglial triggering receptor expressed on myeloid cells-2 (TREM2) for central nervous tissue immune homeostasis. J. Neuroimmunol. 2007;184:92–99. doi: 10.1016/j.jneuroim.2006.11.032. [DOI] [PubMed] [Google Scholar]
- 21.Kondo, et al. Osteopetrotic (op/op) mice have reduced microglia, no Abeta deposition, and no changes in dopaminergic neurons. J. Neuroinflamm. 2007;4:31. doi: 10.1186/1742-2094-4-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sasaki, et al. Effects of macrophage-colony-stimulating factor deficiency on the maturation of microglia and brain macrophages and on their expression of scavenger receptor. Neuropathology. 2000;20:134–142. doi: 10.1046/j.1440-1789.2000.00286.x. [DOI] [PubMed] [Google Scholar]
- 23.Byram SC, et al. CD4-positive T cell-mediated neuroprotection requires dual compartment antigen presentation. J. Neurosci. 2004;24:4333–4339. doi: 10.1523/JNEUROSCI.5276-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Unger ER, et al. Male donor-derived cells in the brains of female sex-mismatched bone marrow transplant recipients: a Y-chromosome specific in situ hybridization study. J. Neuropathol. Exp. Neurol. 1993;52:460–470. doi: 10.1097/00005072-199309000-00004. [DOI] [PubMed] [Google Scholar]
- 25.Ajami B, et al. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat. Neurosci. 2008;10:1538–1543. doi: 10.1038/nn2014. [DOI] [PubMed] [Google Scholar]
- 26.Gowing G, et al. Mouse model for ablation of proliferating microglia in acute CNS injuries. GLIA. 2006;53:331–337. doi: 10.1002/glia.20288. [DOI] [PubMed] [Google Scholar]
- 27.Heppner FL, et al. Experimental autoimmune encephalomyelitis repressed by microglial paralysis. Nat. Med. 2005;11:146–152. doi: 10.1038/nm1177. [DOI] [PubMed] [Google Scholar]
- 28.Lalancette-Hébert M. Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J. Neurosci. 2007;27:2596–2605. doi: 10.1523/JNEUROSCI.5360-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Davolos D, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 2005;8:752–758. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- 30.Nimmerjahn, et al. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- 31.Grutzendler J. Two-photon imaging of synaptic plasticity and pathology in the living mouse brain. NeuroRx. 2008;3:489–496. doi: 10.1016/j.nurx.2006.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cagnin A. Positron emission tomography imaging of neuroinflammation. Neurotherapeutics. 2007;4:443–452. doi: 10.1016/j.nurt.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]