

Abstract
Previously, we showed that Abl family tyrosine kinases are activated by growth factors, and c-Abl is required for transition from G1 to S phase during PDGF-mediated proliferation. Here, we show that the SHP-2 tyrosine phosphatase, which acts to promote proliferation in response to cytokines and growth factors, is a novel substrate of endogenous Abl kinases during growth factor-mediated cellular proliferation. Using a pharmacological inhibitor and RNAi, we show that endogenous Abl kinases phosphorylate SHP-2 on Y580, and induce sustained activation of ERK kinases in response to growth factor stimulation in fibroblasts. Consistent with these data, SHP-2 is required for Abl-dependent PDGF-mediated proliferation since expression of an activated form of SHP-2 rescues the ability of c-Abl/Arg null fibroblasts to transit from G1 to S-phase, while inhibition of SHP-2 signaling reduces the ability of Abl kinases to rescue the proliferation defect. Abl kinases also indirectly mediate phosphorylation of SHP-2 on Y63 and Y279, which are frequent sites of germline mutation in two cancer susceptibility syndromes. Significantly, we demonstrate that phosphorylation of SHP-2 on Y279 downregulates growth factor-induced sustained ERK activation and proliferation, supporting a role for Abl kinases not only in potentiating growth factor-mediated SHP-2 signaling, but also in negative feedback regulation.
Keywords: c-Abl, Arg, SHP-2, proliferation, ERK
INTRODUCTION
The mammalian Abl family of nonreceptor tyrosine kinases (Abl kinases) includes two tightly regulated proteins, c-Abl and Abl-Related Gene (Arg) (Pendergast, 2002). Abl kinases have highly homologous catalytic, SH2, and SH3 domains in their amino-termini, but are more divergent in their carboxy-termini (Pendergast, 2002). Translocation of c-Abl next to BCR results in constitutively active BCR-Abl fusion proteins that drive the development of several forms of human leukemia (Pendergast, 2001). BCR-Abl transforms hematopoietic cells by phosphorylating a variety of proteins involved in cell proliferation, survival, adhesion, and motility (Pendergast, 2001). One of these proteins, the tyrosine phosphatase SHP-2, binds BCR-Abl, is tyrosine phosphorylated in cells transformed by BCR-Abl, and is required for BCR-Abl-mediated transformation in vitro and leukemogenesis in vivo (Chen et al., 2007; Sattler et al., 1997; Tauchi et al., 1994).
Although the role of BCR-Abl in leukemogenesis has been extensively studied, the function of endogenous Abl kinases has remained more elusive. In earlier studies, we demonstrated that platelet-derived growth factor (PDGF) and epidermal growth factor (EGF) induce activation of the cytoplasmic/membrane pool of Abl kinases in fibroblasts, and PDGF-induced activation of c-Abl requires Src family kinases and PLCγ1 (Plattner et al., 2003; Plattner et al., 1999; Plattner et al., 2004). Activation of c-Abl is required for PDGF-mediated proliferation, membrane ruffling, and PLCγ1-induced migration (Plattner et al., 2003; Plattner et al., 1999; Plattner and Pendergast, 2003). Endogenous c-Abl also promotes proliferation by accelerating the G1->S transition (Furstoss et al., 2002; Plattner et al., 1999; Plattner and Pendergast, 2003), and activates a Rac-dependent mitogenic pathway in fibroblasts (Boureux et al., 2005); however, it is not clear whether this pathway is the exclusive mechanism by which Abl kinases promote proliferation.
Mammalian SHP-2, encoded by the Ptpn11 gene, contains two SH2 domains and a protein tyrosine phosphatase domain (PTP)(Feng, 1999). SHP-2 catalytic activity is essential for signaling downstream of growth factor and cytokine receptors and integrins, and is required for proliferation, survival, adhesion, and migration (Feng, 1999). Intramolecular interactions between N-SH2 and PTP domains maintains SHP-2 in an inactive conformation, and binding of the N-SH2 domain to phosphotyrosine residues releases the inhibition and activates SHP-2 (Feng, 1999). SHP-2 binds the adapter protein Gab1, and this complex is required for Ras-extracellular-signal-regulated kinase (ERK) signaling in response to growth factor stimulation (Huang et al., 2002).
SHP-2 is tyrosine phosphorylated on two C-terminal residues (Y542, Y580) following growth factor stimulation. Y542 is phosphorylated by PDGFR-β, while the tyrosine kinase that phosphorylates Y580 has remained elusive (Bennett et al., 1994). The role of C-terminal tyrosine phosphorylation on SHP-2 activity and function is controversial. Early studies suggested that Y542/Y580 phosphorylation performs an adapter function by recruiting Grb2/SOS complexes to growth factor receptors, thereby increasing Ras activation, since Y542 and Y580 are consensus Grb2 binding sites (Bennett et al., 1994). However, the catalytic activity of SHP-2 clearly is required for activation of the Ras/ERK pathway (Yamauchi et al., 1995; Yart et al., 2001), and association of SHP-2 with Grb2 is not sufficient to promote full ERK activation (Araki et al., 2003). Some suggest that phosphorylation of Y542 and Y580 stimulates the catalytic activity of SHP-2 (Lu et al., 2001), while others show that phosphorylation of Y542 and Y580 is required for sustained activation of the Ras/ERK pathway in response to PDGF (Araki et al., 2003).
Somatic SHP-2 mutations have been identified in childhood leukemias and in solid tumors, and germline mutations have been identified in two cancer susceptibility syndromes: LEOPARD syndrome (multiple Lentigines, Electro-cardiographic conduction abnormalities, Occular hypertelorism, Pulmonary stenosis, Abnormal genitalia, Retardation of growth, sensorineural Deafness; LS), and Noonan syndrome (NS) (Bentires-Alj et al., 2004; Tartaglia and Gelb, 2005). Mutant forms of SHP-2 associated with Noonan syndrome and leukemia have increased activity and/or ability to signal, whereas mutant forms associated with LEOPARD syndrome are inactive and inhibit wild-type SHP-2 function (Kontaridis et al., 2006).
Since Abl kinases and SHP-2 are both activated downstream of growth factor receptors and are involved in growth factor-mediated cellular processes, and BCR-Abl induces SHP-2 phosphorylation in hematopoietic cells, we hypothesized that SHP-2 may be a target of endogenous Abl kinases during growth factor-induced proliferation. In the present report, we demonstrate that in response to EGF and PDGF stimulation, endogenous Abl kinases phosphorylate SHP-2 on Y580, and Abl kinases also indirectly mediate SHP-2 phosphorylation on two other sites (Y63, Y279). Additionally, we show that Abl kinase-mediated phosphorylation of SHP-2 on Y580 increases sustained ERK activation in response to PDGF, and Abl kinases promote PDGF-mediated proliferation at least, in part, by phosphorylating SHP-2. Our data are significant because we are the first to demonstrate that: 1) Abl kinases are the elusive kinases that phosphorylate SHP-2 onY580 in response to growth factor stimulation; 2) Endogenous Abl kinases mediate sustained PDGF-induced activation of ERK kinases; 3) SHP-2 is tyrosine phosphorylated on residues other than Y580 and Y542 (Y63, Y279) in an Abl kinase-dependent manner; and 4) tyrosine phosphorylation of Y63 and Y279 alters the ability of SHP-2 to signal and affect cellular proliferation.
RESULTS
Abl kinases tyrosine phosphorylate SHP-2 and promote EGF-dependent SHP-2 phosphorylation
To test whether SHP-2 is a downstream target of Abl kinases, we expressed wild-type c-Abl, constitutively active c-Abl or Arg (Abl-PP, Arg-PP), which have mutation of two interlinker proline residues, and wild-type SHP-2 in 293T cells and assessed the phosphorylation status of SHP-2. We found that active Abl kinases induced SHP-2 tyrosine phosphorylation, while kinase-inactive forms had no effect (Fig. 1A). Wild-type c-Abl induced SHP-2 phosphorylation because overexpression of c-Abl at high levels in 293T cells activates its kinase activity (Pendergast et al., 1991). Wild-type Arg did not induce SHP-2 tyrosine phosphorylation, most likely because expression levels were not high enough to activate Arg (Fig. 1A). Abl kinases also phosphorylated soluble SHP-2, in vitro (Fig. 1B), which demonstrates that Abl kinases directly phosphorylate SHP-2. Although wild-type Arg did not induce tyrosine phosphorylation of SHP-2 in cells (Fig. 1A), the immunoprecipitated, partially purified, wild-type protein was capable of phosphorylating SHP-2 in vitro (Fig. 1B), most likely because cellular inhibitors were washed away during immunoprecipitation (Pendergast et al., 1991).
Figure 1. Abl kinases phosphorylate SHP-2.

(A) SHP-2 was immunoprecipitated from 293T cells transfected with SHP-2 and kinase-inactive (KR), wild-type (WT), or constitutively active forms (PP) of c-Abl or Arg, and blotted with phosphotyrosine antibody (4G10/PY99) (top). The blot was stripped and reprobed with SHP-2 antibody (middle). Results are representative of three independent experiments. (B) Immunoprecipitated Abl kinases were incubated in a “cold” kinase assay with GST, GST-SHP-2, or GST-Crk, and probed with anti-phosphotyrosine antibody. Arg-WT was expressed to a greater extent than Arg-PP (data not shown). Data are representative of three independent experiments. (C) Serum-starved 10T1/2-EGFR cells were pretreated with STI571 (10 μM) or vehicle (water) for 4 hours, stimulated with EGF (100 ng/ml), SHP-2 was immunoprecipitated from the lysates, and probed with anti-phosphotyrosine antibody (top). Percent phosphorylation is relative to total immunoprecipitated SHP-2 protein, and is expressed as a percent of phosphorylation observed in untreated cells. (D) SHP-2 phosphorylation (relative to total protein levels) from STI571-treated cells (C) was compared to untreated cells and expressed as a percentage of untreated. Results from three independent experiments are shown (mean ± s.e.m). *p≤0.05 using a ratio paired t-test. (E) 10T1/2-EGFR cells, transfected with c-Abl or Arg siRNAs (40 nM), were starved and stimulated with EGF for 30 minutes, SHP-2 was immunoprecipitated and blotted with phosphotyrosine antibody. The blot was stripped and reprobed with SHP-2 antibody. SHP-2 phosphorylation (relative to total protein levels) from c-Abl or Arg siRNA-transfected cells was compared to scrambled control-transfected cells and expressed as a percentage of scrambled control. Since Arg antibodies do not work well for western blots, an in vitro kinase assay was utilized to determine knockdown efficiency (Srinivasan and Plattner, 2006). “Percentage knockdown” indicates the decrease in protein expression or activity relative to the scrambled control. (F) SHP-2 phosphorylation (relative to total protein levels) from c-Abl or Arg siRNA-transfected cells (E) was compared to scrambled control-transfected cells and expressed as a percentage of scrambled. Results from three independent experiments are shown (mean ± s.e.m). *p≤0.05, **p<0.005 using a one-way ANOVA followed by a Bonferroni post-hoc test.
To determine whether endogenous Abl kinases, activated by growth factors, can phosphorylate SHP-2, we assayed the effect of inhibiting c-Abl and Arg activities on growth factor-induced SHP-2 phosphorylation, using a pharmacological inhibitor of the Abl kinases, STI571 (Gleevec, imatinib). A concentration of 10 μM STI571 was utilized in these experiments because we and others found that this concentration is required to efficiently eliminate (75-90%) endogenous Abl kinase phosphorylation and activity (Burton et al., 2003; Srinivasan and Plattner, 2006; Zipfel et al., 2004), and this concentration has no effect on the activities of similar proteins such as the highly related Src kinases (Buchdunger et al., 2001; Srinivasan and Plattner, 2006). Serum-starved 10T1/2 fibroblasts that express the EGF receptor (10T1/2-EGFR) were pretreated with STI571 or vehicle for four hours prior to EGF stimulation, and SHP-2 phosphorylation was assessed by immunoprecipitation followed by western blotting. We found that EGF-induced tyrosine phosphorylation of SHP-2 was significantly decreased in STI571-treated cells (Fig. 1C,D). STI571 has no effect on the activity of EGFR but does inhibit PDGF receptors (Druker et al., 1996). Therefore, to rule out that the effect of STI571 on SHP-2 phosphorylation was not due to inhibition of PDGFR, which may affect EGFR activation via receptor crosstalk, we assessed whether STI571 inhibited EGF-induced EGFR activation. We found that phosphorylation of EGFR on Y1173, a measure of EGFR activation, was not altered in STI571-treated cells (Fig. 1C), which indicates that the effect of STI571 on SHP-2 phosphorylation is likely mediated by Abl kinase inhibition. A small amount of constitutive SHP-2 phosphorylation was observed in unstimulated cells, due to low-level constitutive activation of EGFR, and STI571-treatment also reduced constitutive SHP-2 phosphorylation (Fig. S1-top). To confirm the data obtained with STI571, we silenced c-Abl or Arg with siRNAs, starved and stimulated the cells with EGF for 30 minutes, and assessed SHP-2 phosphorylation by immunoprecipitation followed by phosphotyrosine blotting. Knockdown of either c-Abl or Arg also significantly reduced EGF-mediated SHP-2 phosphorylation (Fig. 1E,F). Since c-Abl and Arg are highly homologous, expression of the Arg siRNA partially inhibited c-Abl expression and vice versa. However, as we will show in subsequent figures, we do not believe that the modest cross-inhibition of the siRNAs has significant biological effects. Since STI571 inhibits the activities of PDGF receptors (Druker et al., 1996), it was not possible to assess the effect of STI571 on PDGF-induced SHP-2 phosphorylation.
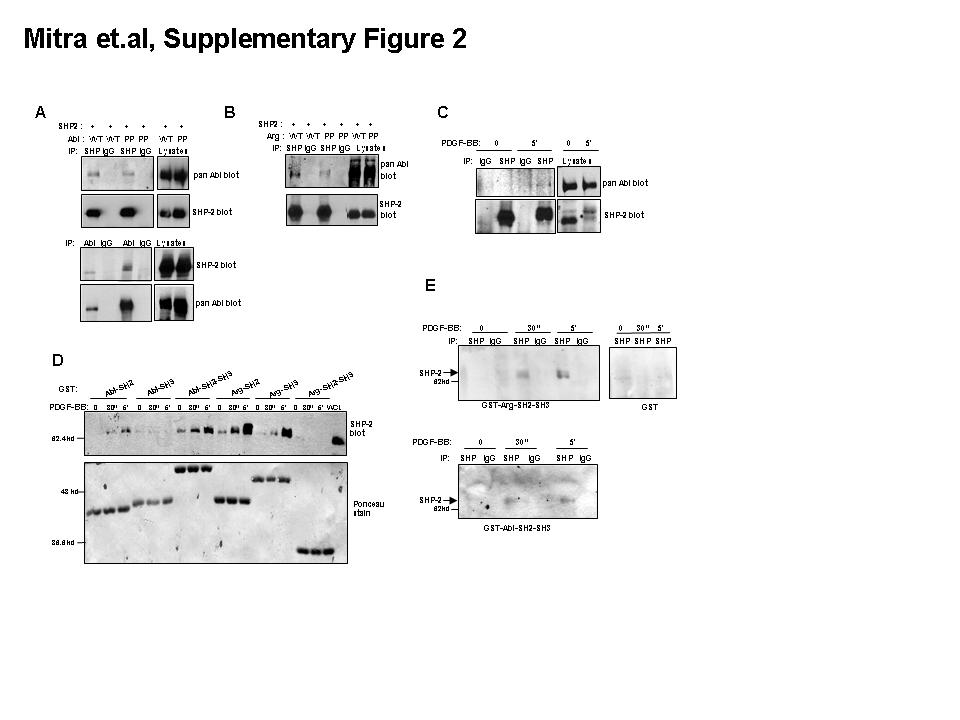
Consistent with its possible role as an Abl kinase substrate, SHP-2 coimmunoprecipitated with Abl kinases when coexpressed in 293T cells (Fig. S2A,B); endogenous SHP-2 formed a PDGF-inducible complex with endogenous Abl kinases in fibroblasts (Fig. S2C); GST-pulldown assays demonstrated that the interaction was mediated by c-Abl and Arg SH2 domains (Fig. S2D); and far western analyses indicated that the c-Abl/Arg-SHP-2 interactions were direct and didn’t require bridging proteins (Fig. S2E). Interestingly, Arg SH2 domains consistently interacted more strongly with SHP-2 immunoprecipitated from PDGF-stimulated NIH3T3 cells in GST-pulldown and far western assays, indicating that Arg may bind SHP-2 more strongly than c-Abl.
Abl kinases directly phosphorylate SHP-2 on Y580, and induce phosphorylation of Y63 and Y279
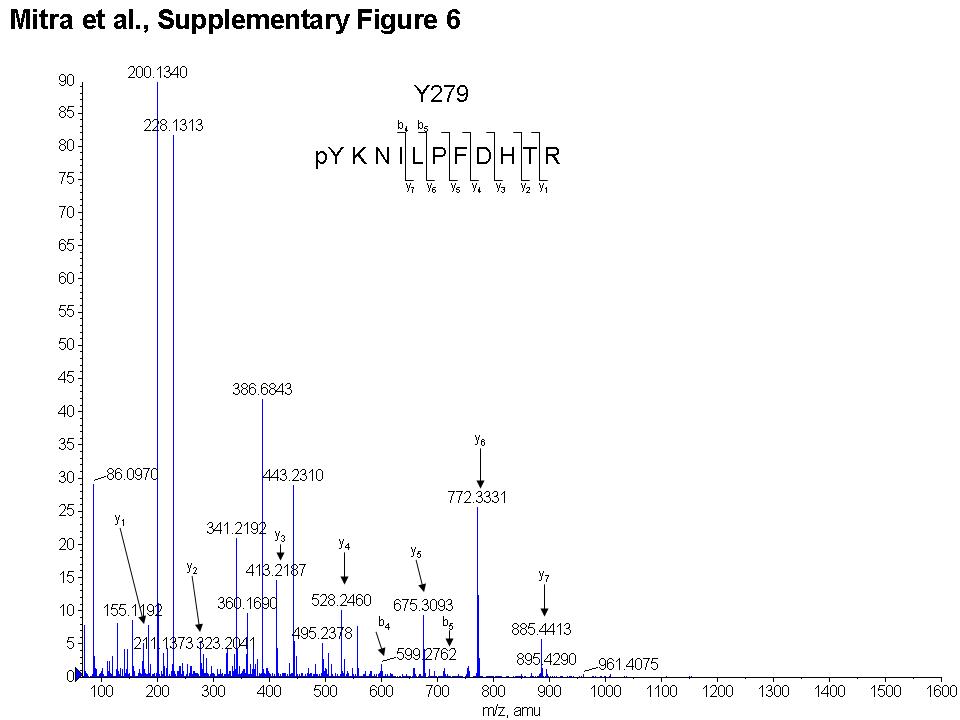
Mass spectrometry was utilized to identify the sites on SHP-2 that are phosphorylated by Abl kinases. We identified two residues on SHP-2 that were potentially phosphorylated by wild-type c-Abl, in vitro (Y542 and Y580), while Y580 was identified as the sole phosphorylation site induced by Abl-PP and Arg-PP, in vitro (Figs. 2A, S3, 4). Phosphorylation sites induced by wild-type Arg could not be detected by mass spectrometry (data not shown). Mutation of Y580 to phenylalanine dramatically reduced SHP-2 tyrosine phosphorylation induced by wild type c-Abl, Abl-PP and Arg-PP, while mutation of Y542 to phenylalanine did not decrease SHP-2 phosphorylation, indicating that Y580 and not Y542 is the true phosphorylation site (Fig. 2B-left). Mass spectrometry analysis performed on immunoprecipitated SHP-2 isolated from 293T cells expressing Arg-PP and wild-type SHP-2 led to the identification of two additional, unique putative phosphorylation sites (Y63 and Y279)(Fig. 2A, S5, 6). Y63 is located in the N-SH2 domain while Y279 is in the phosphatase (PTP) domain (Fig. 2C). Mutation of Y63 or Y279 to phenylalanine reduced SHP-2 phosphorylation induced by Arg-PP, while mutation of Y63 but not Y279 reduced phosphorylation induced by c-Abl and Abl-PP (Fig. 2B-right). These results indicate that c-Abl and Arg induce phosphorylation of Y63 while only Arg induces phosphorylation of Y279. Phosphorylation of Y580 is likely to precede phosphorylation of Y63 or Y279 since mutation of Y580 completely abolishes Abl-dependent phosphorylation of SHP-2 even though Y63 and Y279 sites are still present in the Y580F protein.
Figure 2. Abl kinases phosphorylate SHP-2 on Y580 and induce phosphorylation of SHP-2 on Y63 and Y279.

(A) GST-SHP-2 was phosphorylated by Abl kinases in vitro, and analyzed by mass spectrometry. For identification of SHP-2 phosphorylation sites in cells, c-Abl and Arg were coexpressed with SHP-2 in 293T cells, and immunoprecipitated SHP-2 was analyzed by mass spectrometry. Coverage is the percent of protein recovered as peptides. ND=not done. (B) Wild-type or mutant forms of SHP-2 and c-Abl or Arg were coexpressed in 293T cells, SHP-2 was immunoprecipitated and probed with phosphotyrosine antibody (top). Percentage phosphorylation is relative to total immunoprecipitated SHP-2 protein obtained from reprobed blots. Results shown are representative of three independent experiments. (C) Structure of SHP-2 and putative Abl kinase phosphorylation sites. (D) Immunoprecipitated Abl kinases were incubated in an in vitro kinase assay with wild-type or mutant forms of GST-SHP-2. Coomassie blue staining showed that SHP-2 proteins were equivalent (data not shown). Results shown are representative of three independent experiments.
Figure 4. SHP-2 phosphorylation mutants have altered activities towards phosphotyrosine-containing substrates.

(A) Wild-type or mutant forms of SHP-2 were immunoprecipitated from serum-starved, overexpressing 293T cells, incubated with pNPP substrate and supernatant A405 was measured. Some error bars are too small to be visualized. (B) Wild-type or mutant forms of SHP-2 were immunoprecipitated from serum-starved overexpressing 293T cells, incubated with 250 μM substrate (R-R-L-I-E-D-A-E-pY-A-A-R-G) for 30 minutes at 37°C, and phosphate release into the supernatant was quantitated by incubation with Malachite Green, and measurement of A620. Absorbance values were compared to a standard phosphate curve to determine pmoles of phosphate released. Values obtained for mutant forms of SHP-2 were expressed as a percentage of wild-type with mean ± s.e.m. from three independent experiments. ***p≤0.001 using one-way ANOVAs followed by Bonferroni post-hoc tests. Immunoprecipitates were blotted with SHP-2 antibody (representative experiments are shown). (C) SHP-2 immunoprecipitates from expressing 293T cells (B) were incubated in a phosphatase assay with increasing concentrations of peptide substrate. Velocity values were converted to pmol/min/pmol of enzyme utilized. A representative experiment is shown. (D) Velocity values from two independent experiments (C) were standardized by converting the values to percent of the maximum obtained in each experiment (mean ± s.e.m.). Some error bars are too small to be visualized.
Figure 6. Expression of SHP-2-Y279F increases PDGF-induced sustained ERK phosphorylation and proliferation in fibroblasts.

(A,B) Lysates from serum-starved, PDGF-stimulated NIH3T3 cells, infected with wild-type or mutant SHP-2 retroviruses, were blotted with phospho-ERK1/2 antibody. Changes in ERK1/2 phosphorylation shown are relative to total ERK1/2 levels obtained from reprobed blots. Different exposures are shown for each time point, so that differences between mutants can easily be visualized. Results are expressed as a percentage of wild-type. (B) Results from three independent experiments (A) with mean ± s.e.m. *p≤0.05, **p≤0.02, ***p≤0.005 using t-tests. (C,D) Tritiated thymidine incorporation was assessed in infected NIH3T3 cells labeled with tritiated thymidine for 24 hours. One representative experiment (C) (mean ± s.d.), and a composite of three independent experiments in which tritiated thymidine incorporation of cells expressing SHP-2 mutants compared to cells expressing wild-type SHP-2 is shown (mean ± s.e.m.). Some error bars are too small to be visualized. (D). Aliquots of infected cells were lysed and probed with SHP-2 antibody (C). *p≤0.05, ***p≤0.001 using a one-way ANOVA followed by a Bonferroni post-hoc test.
To determine whether active Abl kinases directly phosphorylate Y580, Y63 or Y279, soluble forms of the mutant SHP-2 proteins were incubated with immunoprecipitated Abl kinases in an in vitro kinase assay. Mutation of Y580 to phenylalanine abrogated c-Abl-and Arg-mediated phosphorylation of SHP-2 in vitro (Fig. 2D), which demonstrates that Y580 is directly phosphorylated by Abl kinases. Conversely, Abl kinases efficiently phosphorylated GST-tagged SHP-2 proteins lacking Y63 and Y279 (Y63F, Y279F; Fig. 2D), which indicates that Abl kinases activate tyrosine kinase intermediates that phosphorylate Y63 and Y279.
To determine whether endogenous Abl kinases phosphorylate SHP-2 on Y580 following activation by growth factors, we assayed the effect of inhibiting endogenous c-Abl and Arg on growth factor-mediated SHP-2 phosphorylation on Y580. Pretreatment of 10T1/2-EGFR cells with STI571 for 4 hours prior to EGF stimulation, significantly reduced EGF-dependent SHP-2 phosphorylation on Y580 (Fig. 3A,B), while phosphorylation of Y542 was not altered (Fig. 3A). Interestingly, the effect of STI571 increased with increasing times of EGF stimulation (Fig. 3B), indicating that phosphorylation of SHP-2 by Abl kinases is maximum 10-30 minutes after EGF stimulation in this cell type. A small amount of constitutive Y580 phosphorylation was observed in unstimulated cells, due to low-level activation of EGFR, and this phosphorylation also was decreased in cells treated with STI571 (Fig. S1, bottom). To confirm the results obtained with STI571, we silenced Abl kinases with siRNAs and assessed the effect on growth factor-induced SHP-2-Y580 phosphorylation. Significantly, silencing either c-Abl or Arg reduced EGF-induced SHP-2 phosphorylation on Y580 in 10T1/2-EGFR cells, although silencing c-Abl had a more dramatic effect (Fig. 3C-left, D). Conversely, in NIH3T3 cells, silencing Arg dramatically reduced SHP-2-Y580 phosphorylation in response to PDGF, and also slightly decreased SHP-2 protein levels, whereas the effect of silencing c-Abl on SHP-2-Y580 phosphorylation was not significant over three experiments (Fig. 3C-right, D). No basal phosphorylation of SHP-2 on Y580 was observed in NIH3T3 cells, and transfection of Abl siRNAs did not affect the low level basal phosphorylation of SHP-2 on Y580 in 10T1/2-EGFR cells (data not shown). Taken together our data definitively demonstrate that endogenous Abl kinases are required for efficient phosphorylation of SHP-2 on Y580 in response to growth factor stimulation.
Figure 3. Endogenous Abl kinases phosphorylate SHP-2 on Y580, and are required for sustained ERK activation in response to PDGF.

(A) Serum-starved 10T1/2-EGFR cells were pretreated with STI571 (10 μM), stimulated with EGF, and lysates were probed with phospho-specific SHP-2 antibodies (Y580, Y542). Percentage phosphorylation is relative to total SHP-2 protein levels obtained from reprobed blots. (B) Graphical representation of mean ± s.e.m. for three independent experiments for lysates probed with phospho-SHP-2-Y580 antibody (A). *p≤0.05 using a ratio paired t-test. (C) 10T1/2-EGFR or NIH3T3 cells, transfected with c-Abl or Arg siRNAs (40nM), were serum-starved, stimulated with EGF (100ng/ml; 10T1/2-EGFR) or PDGF-BB (12.5ng/ml; NIH3T3) for 30 or 10 minutes respectively, and lysates were probed with the indicated antibodies. SHP-2-Y580 phosphorylation in c-Abl or Arg siRNA-transfected cells was expressed as a percentage of SHP-2-Y580 phosphorylation observed in scrambled control-transfected cells and is relative to total SHP-2 protein levels obtained from reprobed blots. (D) Graphical representation of mean ± s.e.m. from three independent experiments (C). *p≤0.05, ***p≤0.005 using a one-way ANOVA followed by a Bonferroni post-hoc test. (E) NIH3T3 cells, transfected with siRNAs, were serum-starved, stimulated with PDGF-BB (12.5ng/ml) for 30 minutes, and phosphorylation of ERK1/2 was assessed by western blotting with a phospho-specific antibody (pTEpY). ERK phosphorylation shown is relative to total ERK protein levels obtained from reprobed blots, and is expressed as a percentage of phosphorylation observed in scrambled control-transfected cells. (F) Graphical representation of three independent experiments (E). *p≤0.05, **p≤0.005, ***p≤0.001 using a one-way ANOVA followed by a Bonferroni post-hoc test.
Since c-Abl and Arg are highly homologous, expression of the Arg siRNA partially inhibited c-Abl expression and vice versa. However, although the c-Abl siRNA modestly inhibited Arg activity in NIH3T3 cells, there was no significant effect of the Abl siRNA on SHP-2-Y580 phosphorylation (Fig. 3D), which demonstrates that the modest cross-inhibition of the siRNAs has little biological effect. Phospho-specific antibodies were not available for Y63 and Y279, and thus we were unable to determine whether endogenous Abl kinases mediate phosphorylation of these sites.
Since Abl kinases phosphorylate SHP-2 on Y580, and Y580 phosphorylation was previously shown to be required for sustained Ras/ERK activation in response to PDGF (Araki et al., 2003), we tested whether Abl kinases promote sustained ERK phosphorylation following PDGF stimulation. Indeed, silencing either c-Abl or Arg significantly reduced ERK activation after 20 minutes (data not shown) or 30 minutes PDGF stimulation (Fig. 3E, F), whereas Abl kinase siRNAs had no effect on basal ERK phosphorylation (data not shown). Therefore, in PDGF-stimulated NIH3T3 cells, silencing Arg significantly reduced SHP-2-Y580 phosphorylation and ERK activation, while silencing c-Abl only inhibited ERK activation and had little effect on SHP-2-Y580 phosphorylation. These data indicate that c-Abl may induce phosphorylation of SHP-2 on another residue, or c-Abl may mediate sustained ERK activation in a SHP-2-independent manner.
Mutation of Y63 or Y279 to phenylalanine affects the activity of SHP-2 towards phosphotyrosine-containing substrates
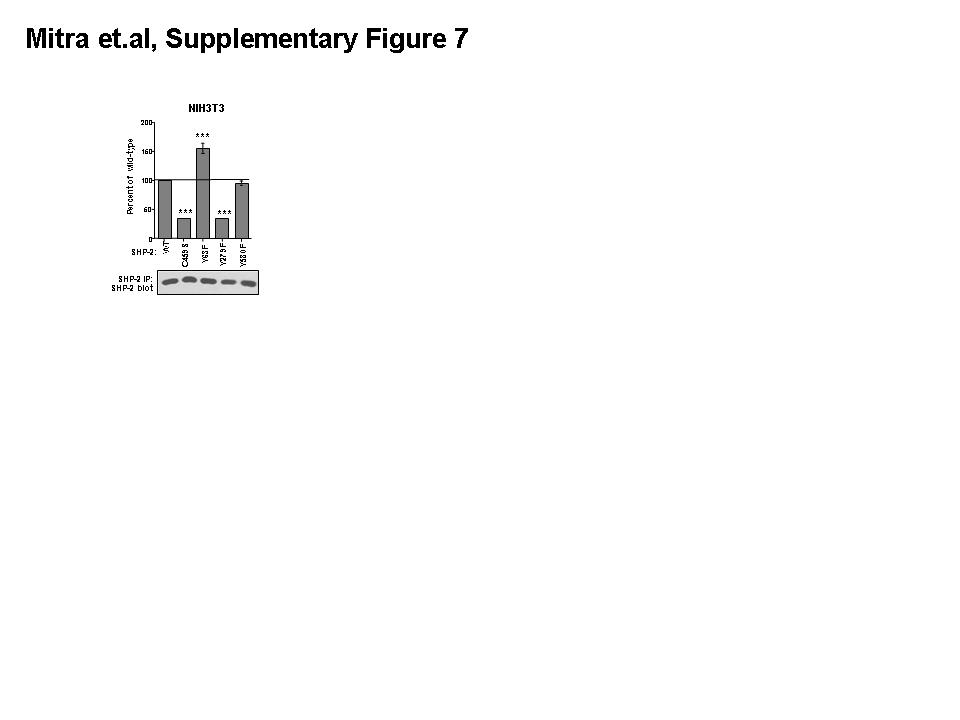
To determine whether phosphorylation of SHP-2 by Abl kinases affects SHP-2 catalytic activity or phosphotyrosine-induced activation, we assessed whether phosphorylation mutants have altered catalytic activity. Basal activities of mutant SHP-2 proteins were assessed by incubating immunoprecipitated SHP-2 proteins, isolated from serum-starved overexpressing 293T cells, in in vitro phosphatase assays using p-nitrophenyl phosphate (pNPP) as substrate. In agreement with a previous report (Kontaridis et al., 2006), wild-type SHP-2 had little to no catalytic activity towards pNPP, whereas E76K, a constitutively active form, had very high basal activity (Fig. 4A). Like wild-type SHP-2, catalytically-inactive (C459S), Y63F, Y279F, and Y580 forms of SHP-2 had little to no basal activities, indicating that none of these mutants is constitutive active (Fig. 4A). To determine whether mutant SHP-2 proteins have altered abilities to be activated by phosphotyrosine-containing substrates, phosphatase assays were performed on SHP-2 immunoprecipitates from serum-starved 293T and NIH3T3 cells overexpressing various forms of SHP-2 using a standard phosphopeptide, R-R-L-I-E-D-A-E-pY-A-A-R-G (derived from Src) as substrate. A constitutively active mutant (E76K) had an increased ability to dephosphorylate the substrate, while a catalytically inactive form (C459S) demonstrated very low-level activation (Fig. 4B, S7). SHP-2-Y63F had higher activity towards the phosphotyrosine-containing substrate than wild-type SHP-2, Y279F had dramatically decreased activity, and Y580F had approximately the same activity as wild-type (Fig. 4B, S7). Similar results were obtained for Y63F, Y279F, and Y580F mutants using a different phosphopeptide substrate, T-S-T-E-P-Q-pY-Q-P-G-E-N-L (Src529)(data not shown). To determine whether F63 and F279 have an altered affinity for the phosphopeptide substrate, velocity assays were performed. The velocity curve for F63 was shifted to the left of wild-type indicating that this mutant has a lower Km, whereas the curve corresponding to F279 was shifted to the right of wild-type, indicating it has a higher Km (Fig. 4C,D). In addition, maximum velocity values for F279 did not approach those obtained by wild-type or F63 SHP-2, suggesting that the catalytic activity (Vmax) also was significantly lower for this mutant. It was not possible to obtain high enough substrate concentrations to accurately determine Km and Vmax values.
Abl-dependent phosphorylation of SHP-2 affects ERK phosphorylation and proliferation
To identify the biological consequences of Abl-dependent SHP-2 phosphorylation, we assessed the ability of mutant SHP-2 proteins, lacking c-Abl and/or Arg phosphorylation sites, to induce ERK phosphorylation. Previous reports showed that SHP-2 activity and ability to signal downstream can be assessed by overexpressing mutant SHP-2 proteins together with the scaffold adaptor protein Gab1 and HA-tagged ERK in 293T cells, and assessing the level of growth factor-induced HA-ERK phosphorylation (Kontaridis et al., 2006). Therefore, 293T cells were transfected with wild-type or mutant forms of SHP-2, Gab1, and HA-tagged ERK2, cells were serum-starved, and HA-ERK2 phosphorylation was assessed in unstimulated or EGF-stimulated cellular lysates. In the absence of growth factor stimulation, expression of E76K, Y63F, and Y279F forms of SHP-2 increased HA-ERK2 phosphorylation as compared to wild-type SHP-2, while expression of C459S and Y580F forms decreased ERK phosphorylation in unstimulated cells (Fig. 5A,B). Stimulation of cells with EGF for 5 minutes induced similar levels of phosphorylated HA-ERK2 in 293T cells, and only the catalytically-inactive SHP-2 mutant (C459S) was unable to induce full HA-ERK phosphorylation (Fig. 5A). However, at later time points (20′, 60′) cells expressing E76K, Y63F, and Y279F had sustained levels of phosphorylated HA-ERK (Fig. 5C,D). In agreement with a previous report, expression of Y580F had little effect on EGF-induced ERK phosphorylation (Fig. 5C,D)(Araki et al., 2003).
Figure 5. Expression of SHP-2 Y63F and Y279F mutants increases EGF-induced sustained ERK phosphorylation in 293T cells.

(A,C) 293T cells were transfected with plasmids encoding wild-type or mutant forms of SHP-2 (10 μg), Gab1 (4 μg), and HA-tagged ERK2 (1 μg), cells were serum-starved, stimulated with EGF for the indicated times, and lysates were probed with phospho-ERK1/2 antibody. Only HA-tagged ERK2, which migrates slower than endogenous ERK1/2, is shown. Blots were stripped and reprobed with HA antibody. (B,D) Graphical representations of three unstimulated (A) or four EGF-stimulation experiments (C). Levels of phosphorylated HA-ERK2 relative to total HA-ERK2 for cells expressing SHP-2 mutants were expressed as a percentage of HA-ERK2 phosphorylation observed in cells expressing wild-type SHP-2. Results are mean ±s.e.m. *p≤0.05, **p≤0.03, ***p≤0.001 using t-tests.
To rule out the possibility that the effects observed in 293T cells were cell-type specific, or specific for the growth factor, EGF, we performed a related assay in fibroblasts. Wild-type or mutant forms of SHP-2 were expressed in NIH3T3 cells and the effect on endogenous ERK1/2 phosphorylation was assessed. Cells expressing Y279F had sustained phospho-ERK1/2 levels at 20 and 60 minute time points (Fig. 6A,B), similar to the results obtained for 293T cells (Fig. 5C,D). Cells expressing Y63F also had significantly increased ERK phosphorylation at 20 minutes, but it was not sustained for 60 minutes (Fig. 6A,B). In agreement with a previous report, expression of SHP-2-Y580F decreased sustained ERK phosphorylation (60′) in response to PDGF (Fig. 6A,B)(Araki et al., 2003).
To examine whether Abl-dependent phosphorylation of SHP-2 affects the ability of SHP-2 to promote cellular proliferation, we assessed the proliferative capability of NIH3T3 cells expressing wild-type or mutant forms of SHP-2. Expression of C459S decreased cellular proliferation, as evidenced by decreased uptake of tritiated thymidine (Fig. 6C,D) and induced a flat morphology reminiscent of SHP-2 null fibroblasts (data not shown)(Yu et al., 1998). Expression of Y63F significantly inhibited cell proliferation (p≤0.001)(Fig. 6C,D), while expression of Y279F increased the ability of cells to incorporate tritiated thymidine (p≤0.05)(Fig. 6C,D). These results are significant because effects on proliferation usually are difficult to observe in asynchronous cell populations unless the changes are fairly large. Therefore, our results indicate that phosphorylation of Y63 acts to potentiate SHP-2-mediated proliferation, while phosphorylation of Y279 turns off the pathway and inhibits proliferation.
SHP-2 is a target of endogenous Abl kinases during PDGF-mediated mitogenesis
Endogenous Abl kinases phosphorylate SHP-2 on Y580 and induce sustained ERK phosphorylation in response to PDGF. These data suggest that Abl kinases may promote PDGF-mediated proliferation by phosphorylating SHP-2. To test this hypothesis, we examined whether SHP-2 function is required for Abl-mediated mitogenesis. Previously, we and others showed that c-Abl null fibroblasts enter S-phase in response to PDGF stimulation at a slower rate than the same cells reconstituted with c-Abl (Furstoss et al., 2002; Plattner and Pendergast, 2003). c-Abl/Arg double null fibroblasts also are defective in their ability to enter S-phase following PDGF stimulation, and reintroduction of c-Abl and Arg into these cells increases their ability to respond to PDGF-BB (12.5ng/ml)(Fig. 7A,B). Expression of a constitutively active form of SHP-2 (E76K) completely rescued the ability of c-Abl/Arg null fibroblasts to proliferate in response to PDGF, and inhibition of SHP-2 signaling by expression of a catalytically-inactive, dominant-negative form of SHP-2 (C459S) significantly decreased the ability of c-Abl and Arg to rescue PDGF-mediated mitogenesis (Fig. 7A,B). Taken together, our data indicate that endogenous Abl kinases induce PDGF-mediated proliferation, at least in part, by activating a SHP-2-dependent pathway.
Figure 7. SHP-2 lies downstream of Abl kinases during PDGF-mediated mitogenesis.

c-Abl/Arg double null fibroblasts, infected with vectors (Migr1, MigCD4, PK1), c-Abl and Arg (MigCD4-Abl, PK1-Arg), and/or wild-type or mutant forms of SHP-2 (Migr1-SHP2-C459S, Migr1-SHP-2-E76K), were serum-starved, stimulated with PDGF-BB (12.5ng/ml) for 16-20 hours, pulsed with tritiated thymidine, and harvested. (A) Results from one representative experiment (mean ± s.d). Some error bars are too small to be visualized. Lysates from infected cells were probed with the indicated antibodies. (B) Composite of three independent experiments (mean ± s.e.m). Tritiated thymidine incorporation is expressed as a percentage of vector-transfected cells. *p≤0.05, **p≤0.01, ***p≤0.001 using a one-way ANOVA followed by a Bonferroni post-hoc test.
DISCUSSION
In this report, we provide strong evidence that endogenous SHP-2 is a substrate of endogenous Abl kinases: 1) PDGF- and EGF-mediated SHP-2 phosphorylation on Y580 is reduced in cells expressing Abl kinase siRNAs; 2) PDGF-induced sustained activation of ERK kinases requires Abl kinases; 3) expression of active SHP-2 rescues the ability of c-Abl/Arg null fibroblasts to respond to PDGF; and 4) inhibition of SHP-2 function reduces the ability of c-Abl and Arg to rescue the mitogenic defect observed in c-Abl/Arg null fibroblasts. c-Abl previously was shown to promote proliferation by activating a Rac/JNK/Nox pathway, since Rac activation in response to PDGF was reduced in c-Abl/Arg null fibroblasts, and expression of a constitutively active form of Rac (Rac-V12) rescued the ability of c-Abl/Arg null fibroblasts to transit from G1->S phase in response to PDGF (Boureux et al., 2005). However, the authors did not also show that inhibition of Rac function blocks Abl kinases from rescuing the proliferation defect (Boureux et al., 2005), which leaves open the possibility that c-Abl and Rac act in parallel pathways. In addition, it is unclear whether other Abl-dependent mitogenic pathways also exist. Here, we show that SHP-2 is an important mediator of Abl-dependent growth factor-induced proliferation. Since expression of a SHP-2 dominant-negative did not completely inhibit mitogenic rescue by c-Abl and Arg, the Abl-SHP-2 pathway may represent only one way by which Abl kinases promote proliferation, and another pathway, perhaps Rac/JNK/Nox, is likely also to contribute to Abl-dependent growth factor-mediated proliferation. Alternatively, Abl kinases, SHP-2, Rac, and JNK may lie in the same pathway since SHP-2/Gab2 complexes are required for activation of Rac/JNK downstream of c-Kit during mast cell proliferation (Yu et al., 2006)(Fig. 8).
Figure 8. Model of Abl-dependent proliferation in fibroblasts.
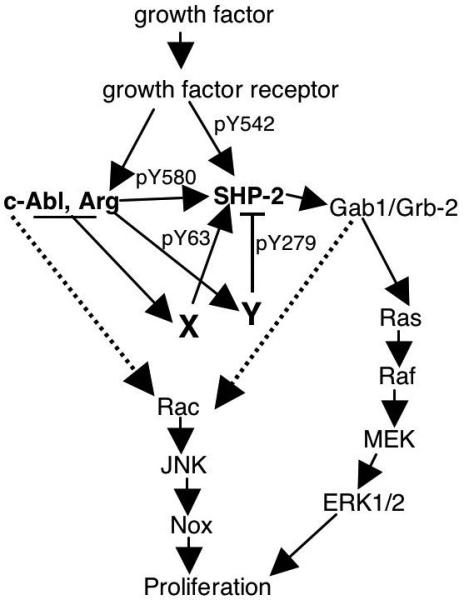
Endogenous Abl kinases directly phosphorylate SHP-2 on Y580, which increases sustained ERK activation in response to PDGF (Figs. 3,6)(Araki et al., 2003). Abl kinases also induce phosphorylation of Y63 and Y279 (Fig. 2B) presumably by activating unknown tyrosine kinases (X, Y). Phosphorylation of Y63 potentiates mitogenic signaling in fibroblasts (Fig. 6C,D), while phosphorylation of Y279 negatively regulates ERK activation (Fig. 6A,B) and proliferation (Fig. 6C,D). Abl kinases also activate a Rac/JNK pathway (Boureux et al., 2005), which may be dependent or independent of SHP-2 (Yu et al., 2006)(dotted lines).
Significantly, we are the first to demonstrate that Abl kinases are the elusive tyrosine kinases that phosphorylate SHP-2 on Y580, and we and others show that Y580 phosphorylation is required for sustained SHP-2 signaling in response to PDGF (Araki et al., 2003). Both c-Abl and Arg phosphorylate SHP-2 on Y580 in response to EGF stimulation in 10T/12-EGFR cells, while only Arg phosphorylates SHP-2 on Y580 following PDGF stimulation of NIH3T3 cells. The reason for this discrepancy is unclear; however there are several possibilities. 1) Expression of c-Abl may be lower in NIH3T3 cells than in 10T1/2-EGFR cells. However, this is not the case as c-Abl actually is expressed more highly in NIH3T3 cells (2-fold) than in 10T1/2-EGFR cells (data not shown). 2) PDGF stimulation may recruit Arg more efficiently than c-Abl to SHP-2 complexes whereas EGF stimulation may recruit both c-Abl and Arg, due to differences in binding sites on the receptors for Abl kinases or due to stronger binding of Arg to SHP-2. Consistent with this hypothesis, Arg SH2 domains interacted more strongly with SHP-2 immunoprecipitates isolated from PDGF-stimulated NIH3T3 cells (Fig. S2D, E). 3) There may be differential expression of other proteins in the two cell types, which are responsible for linking c-Abl to the SHP-2-containing complex.
In addition to directly phosphorylating Y580, Abl kinases also indirectly induce tyrosine phosphorylation of SHP-2 on two residues (Y63, Y279) that are important for SHP-2 function since they reside in the N-SH2 and PTP domains, respectively, which are important for keeping SHP-2 in an inactive state. Interestingly, phosphorylation of Y279 is Arg but not c-Abl dependent, while phosphorylation of Y63 is both Arg and c-Abl dependent. Therefore, it is likely that two different tyrosine kinase intermediates mediate these phosphorylation events, one that is activated by c-Abl and Arg and phosphorylates Y63, and another that is only activated by Arg and phosphorylates Y279 (Fig. 8).
Y63 and Y279 are frequently mutated in several disease states including somatic mutation in leukemia (Y63C), and germline mutation in Noonan (Y63C) and LEOPARD (Y279C) syndromes, which demonstrates that these residues are important for SHP-2 function (Kontaridis et al., 2006; Tartaglia and Gelb, 2005). In fact, Y279 is the most common mutated residue in LEOPARD Syndrome (Kontaridis et al., 2006). Y279 is located in the catalytic cleft of the phosphatase domain, which confers specificity for phosphotyrosine-containing substrates (Kontaridis et al., 2006). Mutation of Y279 to cysteine decreases SHP-2 activity toward phosphotyrosine substrates, and inhibits EGF-induced ERK phosphorylation in 293T cells (Kontaridis et al., 2006). Interestingly, we show that mutation of Y279 to phenylalanine also dramatically decreases SHP-2 activity towards phosphotyrosine substrates. It is possible that mutation of the tyrosine residue to phenylalanine changes the strength of the interaction between N-SH2- and PTP domains. However, since phenylalanine substitutions are unlikely to alter the depth of the catalytic cleft or orientation of the catalytic cysteine 459, as is predicted to occur for Y279C (Kontaridis et al., 2006), and mutation to phenylalanine has the identical effect on SHP-2 activation as mutation to cysteine, it is also possible that phosphorylation of Y279 contributes to phosphotyrosine-induced SHP-2 activation. Significantly, unlike Y279C, expression of Y279F does not inhibit signaling as it increases constitutive ERK activation in 293T cells, induces sustained EGF-induced ERK phosphorylation in 293T cells, sustained PDGF-induced ERK phosphorylation in fibroblasts, and increases fibroblast proliferation. There are several possible explanations for these contrasting results. Expression of Y279F may increase SHP-2 signaling in a phosphatase-independent manner by preventing the binding of a protein involved in negatively regulating ERK phosphorylation and proliferation. Catalytic-independent functions involved in promoting proliferation have been previously described for SHP-2 (Chen et al., 2007; Yu et al., 1998). Alternatively, mutation of Y279 to phenylalanine may alter substrate specificity; this could result in decreased affinity of SHP-2 towards some substrates (e.g. peptide substrates used in the phosphatase assays) and increased affinity towards others (e.g. substrate(s) involved in ERK activation and proliferation). Indeed, PTP mutants have been described that have altered substrate specificity, which results in more effective dephosphorylation of a subset of target proteins (Keilhack et al., 2005), and F279 velocity curves demonstrate decreased affinity for the Src peptide substrate (Fig. 4C,D). In either event, phosphorylation of Y279 clearly acts to downregulate SHP-2-dependent ERK signaling and proliferation.
Mutation of Y63 to cysteine occurs in patients with chronic myelomonocytic leukemia (CMML) and Noonan syndrome, and is an “activating” mutation (Bentires-Alj et al., 2004; Tartaglia and Gelb, 2005). Y63, located in the N-SH2 domain, is directly involved in N-SH2-PTP domain interactions, and mutation of Y63 to cysteine disrupts auto-inhibition (Tartaglia and Gelb, 2005). Interestingly, like Y63C, Y63F also increases phosphotyrosine-induced activation. These data suggest that phosphorylation of Y63 may play a role in maintaining SHP-2 in an inactive state. However, mutation of Y63 to glutamate also increases phosphotyrosine-induced activation (data not shown), which suggests that mutation of the tyrosine residue to phenylalanine changes the strength of the interaction between N-SH2- and PTP domains. Consistent with these data, overexpression of Y63F in 293T cells results in increased and sustained ERK activation in response to EGF. However, in fibroblasts, expression of Y63F only induces sustained ERK phosphorylation for 20 minutes following PDGF stimulation, and ERK phosphorylation is not maintained for 60 minutes as is observed for cells expressing Y279F. In addition, NIH3T3 cells expressing Y63F have a decreased capacity to proliferate. There are several possible explanations for these contrasting results. Perhaps in fibroblasts, mutation of Y63 prevents the binding of a protein(s) to phosphorylated Y63, which is involved in promoting proliferation. Alternatively, the Y63F mutant, although able to activate the ERK pathway, may be unable to activate other SHP-2-dependent proliferative pathways (such as the Rac/JNK pathway). Finally, it is possible that expression of interacting downstream signaling proteins is different in different cell types, and the recruitment of these proteins may differ in response to diverse extracellular stimuli (ie. EGF, PDGF, serum).
Clearly, SHP-2 signaling is a complicated process, as evidenced by the following: 1) “activating” and “inhibitory” mutants of SHP-2 both cause two highly related cancer-susceptibility syndromes (Noonan and LEOPARD); 2) some SHP-2 mutants identified in disease states do not appear to affect catalytic activity; 3) the transforming capability of SHP-2 leukemic mutants is not proportional to their catalytic activities; and 4) transformation by leukemic mutants requires catalytic activity, one C-terminal tyrosine residue (Y580 or Y542), and both SH2 domains must be able to bind phosphotyrosine-containing proteins (Keilhack et al., 2005; Kontaridis et al., 2006; Mohi and Neel, 2007; Tartaglia and Gelb, 2005). Future work clearly will be required to understand exactly how phosphorylation of Y63 and Y279 affects SHP-2 activity and signaling, as well as to identify the tyrosine kinases that phosphorylate Y63 and Y279. The fact that Y63 and Y279 are mutated in various disease states lends further credence to our finding that these sites are important for regulating growth factor-induced SHP-2 signaling.
In conclusion, we show that SHP-2 is a downstream substrate of Abl kinases during cell proliferation. Direct phosphorylation of SHP-2 by Abl kinases (Y580) promotes sustained activation of SHP-2 signaling and proliferation, and c-Abl/Arg-dependent activation of tyrosine kinase X further potentiates SHP-2 signaling by phosphorylating SHP-2 on Y63. Finally, Arg-dependent activation of tyrosine kinase Y acts as a novel negative feedback mechanism, turning-off the signaling pathway by phosphorylating SHP-2 on Y279 (Fig. 8).
MATERIALS AND METHODS
Cell lines and reagents
10T1/2 fibroblasts expressing EGFR (Sally Parsons, University of Virginia), c-Abl/Arg -/-fibroblasts (Anthony Koleske, Yale University), c-Abl/Arg-/-fibroblasts reconstituted with c-Abl and Arg, NIH3T3 and 293T cells, Arg antibody, and c-Abl and Arg GST-fusion proteins were previously described (Plattner et al., 1999; Plattner et al., 2004). 293T and NIH3T3 cells were serum-starved in medium containing 0.1% fetal bovine serum or 0.1% calf serum, respectively for 16-24 hours. STI571, (Novartis Pharmaceuticals; Basel, Switzerland), was dissolved in water (10 mM) and stored at -80°C. The following antibodies were obtained commercially: c-Abl (immunoprecipitation; K12), GST, SHP-2, α-tubulin, pan-ERK (K23) and phosphotyrosine (PY99) antibodies (Santa Cruz Biotechnology; Santa Cruz, CA); c-Abl (western blotting; 8E9) and EGFR (BD Biosciences; Chicago, IL); HA (Roche Diagnostics Corp.; Indianapolis, IN); pERK1/2 (Promega; Madison, WI); phosphotyrosine (4G10), pEGFR, Gab1 (Upstate Biotechnology, Lake Placid, NY); and pSHP-2 (Cell Signaling Technology; Danvers, MA). EGF was purchased from Roche Corp. (Indianapolis, IN), PDGF-BB from Upstate Biotechnology (Lake Placid, NY), and tritiated-thymidine from Perkin Elmer (Waltham, MA). Mouse siRNAs (Abl: GGGAGGGUGUACCACUACA; Arg: GGCACUGAAUGAAGCGAUC) were obtained from Ambion (Austin, TX).
Statistics
Statistics were performed using SigmaStat for Windows (Systat Software, Inc.; San Jose, CA). Ratio t-tests were performed using GraphPad Prism (San Diego, CA).
Plasmids and in vitro mutagenesis
c-Abl and Arg constructs were previously described (Plattner et al., 2003; Plattner et al., 1999; Plattner et al., 2004). Murine SHP-2 in pcDNA (Yu et al., 1998) was transferred to Migr1 and pBabepuro retroviral vectors (Plattner et al., 2003; Plattner et al., 1999; Plattner et al., 2004) by blunting the HindIII/Xba insert and cloning into the blunted EcoR1 site of Migr1 and into the SnaB1 site in pBabepuro. SHP-2 was mutagenized using Quikchange (Stratagene, La Jolla, CA). Mutagenesis primers were:
Y542F-forward-AGAAAAGGACATGAATTTACCAATATTAAGTAT
Y542-reverse-ATACTTAATATTGGTAAATTCATGTCCTTTTCT
Y580F-forward-GACAGCGCCCGAGTCTTTGAGAACGTGGGC
Y580F-reverse-GCCCACGTTCTCAAAGACTCGGGCGCTGTC
Y63F-forward--CACTGGGGACTACTTTGACCTCTATGGTGG
Y63F-reverse-CCACCATAGAGGTCAAAGTAGTCCCCAGTG
Y279F-forward--CAAAAACAGATTCAAAAACATCC
Y279F-reverse--GGATGTTTTTGAATCTGTTTTTG
E76K-forward--GAAGTTTGCCACTTTGGCTAAACTGGTTCAGTATTACATGG
E76K-reverse--CCATGTAATACTGAACCAGTTTAGCCAAAGTGGCAAACTTC.
Constructs were sequenced to confirm that no additional mutations were introduced.
Mass Spectrometry
Abl kinases were immunoprecipitated from expressing 293T cells and incubated with soluble GST-SHP-2 in the presence of 1 mM ATP, at 37°C for 30′ (in vitro)(Plattner et al., 2003). SHP-2 also was immunoprecipitated from 293T cells cotransfected with Abl kinases and wild-type SHP-2 (in cells). Kinase reactions and immunoprecipitates were run on SDS-PAGE gels, SHP-2 was cut from the Coommassie-stained gels, washed with 50 mM NH4HCO3/50% CH3CN, reduced with 10 mM DTT at 57° C for 30′, alkylated with 50 mM iodoacetamide for 30′, and digested with trypsin (10ng/μl) for 18 hours at 37°C. Peptides were extracted with 0.1% formic acid followed by 50% acetonitrile/0.1% formic acid. Nano-flow reverse phase LC-MS/MS was performed on extracted peptides using a capillary HPLC system (LC Packings, Amsterdam, Netherlands) coupled with a QSTAR XL quadrupole time-of-flight mass spectrometer (ABI/MDS Sciex) through a nanoelectrospray ionization source (Protana). Analyst QS software was used for system control and data collection. Peptide solutions were desalted on a C18 trap column, separated by a C18 reverse phase column, and introduced into the mass spectrometer. Each cycle consisted of a 1-s TOF MS survey from 400 to 1600 (m/z) and two 2-s MS/MS scans with mass range of 65-1600 (m/z). The LC-MS/MS data were submitted to a local MASCOT server for MS/MS ions search.
Transfections
293T cells were transfected for 5-8 hours using calcium phosphate (15 μg DNA, 62 μl calcium chloride, 500 μl Hepes Buffered Saline, and 438 μl water per 60 mm dish) in medium containing 0.1 mM chloroquine. pcDNA3 constructs were utilized for 293T transfections, and Migr1 or pBabepuro constructs were used for retroviral infection (Plattner et al., 1999; Yu et al., 1998). siRNAs (40nM) were transfected with Lipofectamine 2000 (Invitrogen, La Jolla, CA).
Immunoprecipitations, GST-pulldowns, kinase assays, immunoblotting, and far western analyses
Procedures were previously described (Plattner et al., 1999; Plattner et al., 2004; Srinivasan and Plattner, 2006). Cell lysis and immunoprecipitations for kinase assays, GST-pulldowns, and far western blots, cells were performed in kinase lysis buffer (50mM HEPES pH7, 150 mM NaCl, 10% glycerol, 1% triton-X-100, 1.5 mM MgCl2, 1mM EGTA, and inhibitors (1mM PMSF, 10 μg/ml leupeptin, aprotinin, pepstatin)), and RIPA buffer (50 mM Tris pH 7.5, 150 mM NaCl, 1% triton-X 100, 0.1% SDS, 1% sodium deoxycholate, inhibitors) was used for to prepare lysates for phospho-blots. For kinase assays, c-Abl or Arg immunoprecipitates were washed twice in RIPA buffer, twice in NaCl buffer (10 mM Tris, pH7.4, 5 mM EDTA, 1% triton-X-100, 100 mM NaCl, inhibitors), twice in the previous buffer lacking NaCl, and twice in kinase buffer (20 mM Tris pH7.4, 10 mM MgCl2, 1 mM DTT), and incubated for 40′ at room temperature in kinase buffer containing 1 μM cold ATP, 5 μCi 32P-γ-ATP and 1 μg substrate. In some instances, cold kinase assays were used to demonstrate that phosphorylation observed occurred on tyrosine residues. For these assays, kinase reactions lacked radioactive ATP, and were probed with phospho-tyrosine antibody. For GST-pulldowns, 1 μg of GST-tagged fusion proteins were incubated with cellular lysate and glutathione-sepharose, and precipitates were probed with anti-SHP-2 antibody. For far westerns, blots containing SHP-2 immunoprecipitates, were blocked in 5% BSA in 50mM Tris-HCl, pH 7.4, 150mM NaCl, were incubated with GST-fusion proteins (2μg/ml) in 20 mM Hepes, pH 7.2, 150 mM NaCl, 0.1% Triton-X 100, 10% glycerol, 2% BSA, and GST binding was assessed by western blot with anti-GST antibody. Western blots were quantitated by analyzing scanned blots with ImageQuant software (Molecular Dynamics, GE Healthcare; Piscataway, NJ). Changes in phosphoprotein levels were assessed by comparing to total protein levels obtained from stripped blots. Kinase assays were quantitated on a Storm phosphoimager (Molecular Dynamics, GE Healthcare; Piscataway, NJ).
Tritiated thymidine assays
293T cells were transfected with expression plasmids and pSVψ2 to produce retrovirus as previously described (Plattner et al., 1999). c-Abl/Arg double null fibroblasts or NIH3T3 cells were infected with retroviruses containing 4 μg/ml polybrene in 6-well dishes, by spinning at 2300 rpm for 2 hours. Two days after infection, c-Abl/Arg null cells were replated in 12-well dishes (5 × 104/well) in triplicate, serum-starved 5 hours later, stimulated with PDGF-BB (12.5 ng/ml) the following day, and labeled with tritiated thymidine (5 μCi) for two hours. NIH3T3 cells were infected with retrovirus obtained from transfection of 293T cells with pBabepuro contructs, cells were replated in 12-wells (2.5 ×104cells/well) in triplicate, and labeled with tritiated thymidine for 24 hours. Cells were harvested by washing with phosphate-buffered saline, 10% trichloroacetic acid (TCA), incubating in 10% TCA for 45′, solubilizing radioactivity in 0.2N NaOH, and reading on a scintillation counter.
Phosphatase Assays
293T cells were transfected with mutant or wild-type SHP-2 constructs (pcDNA) and NIH3T3 cells were infected with retroviruses (Migr1), cells were serum starved the following day in basal medium for 24 hours. Cells were washed with ice-cold Hepes/NaCl (25mM Hepes, pH7.4, 150mM NaCl), lysed in phosphatase lysis buffer (25mM Hepes, pH7.4, 150mM NaCl, 2mM EDTA, 0.5% Triton-X-100, 1mM PMSF, 10 μg/ml leupeptin, aprotinin, pepstatin), SHP-2 proteins were immunoprecipitated, washed twice in lysis buffer and twice in phosphatase assay buffer (25mM Hepes, pH7.5, 5% glycerol, 2mM EDTA, 1mM DTT), and incubated with phosphopeptide substrate R-R-L-I-E-D-A-E-pY-A-A-R-G for 30′ at 37°C (#12-217; kit #17-125; Upstate Biotechnology, Lake Placid, NY). Phosphate release into the supernatant was detected by addition of Malachite Green to the supernatant, and measurement of absorbance at 620 nm (Upstate protocol). For some experiments T-S-T-E-P-Q-pY-Q-P-G-E-N-L (#12-218) substrate was utilized. For velocity assays, immunoprecipitates were incubated with increasing concentrations of substrate. Velocity (pmol/min) values were divided by pmol of enzyme utilized, which was assessed by comparing SHP-2 immunoprecipitate bands to bovine serum albumin standards on SDS-PAGE gels. Assays were linear with respect to time and immunoprecipitated protein. To determine basal phosphatase activity, 5% BSA (1.7μl) and phosphatase assay buffer (27μl) were added to immunoprecipitates, incubated for 15′ at 37°C, pNPP (para-nitrophenyl phosphate; 40μl of 1.5mg/ml) was added, and incubated 15′ at 37°C (#17-125; Upstate Biotechnology, Lake Placid, NY). Supernatant absorbance was measured at 405 nm. Immunoprecipitates were blotted with SHP-2 antibody.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
ACKNOWLEDGEMENTS
10T1/2-EGFR cells were provided by Dr. Sally Parsons (University of Virginia), and c-Abl/Arg null fibroblasts by Dr. Anthony Koleske (Yale University). This work was supported by a Concern Foundation Young Investigator Award, American Cancer Society Pilot Grant #85-001-16 IRG, NIH Grant P20 RR20171 from the National Center for Research Resources, and NIH/NCI Grants 1R01CA116784 to R.P. and R01CA078606 to G.S.F. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of NIH. Mass spectrometric analysis was performed at the University of Kentucky, Center for Structural Biology Protein Core Facility, supported in part by funds from NIH National Center for Research Resources (NCRR) grant P20 RR020171. We thank Divya Srinivasan for reading the manuscript, and Dr. David Rodgers and Dr. Lou Hersh for structural biology and phosphatase velocity consultation, respectively.
REFERENCES
- Araki T, Nawa H, Neel BG. Tyrosyl phosphorylation of Shp2 is required for normal ERK activation in response to some, but not all, growth factors. J Biol Chem. 2003;278:41677–41684. doi: 10.1074/jbc.M306461200. [DOI] [PubMed] [Google Scholar]
- Bennett AM, Tang TL, Sugimoto S, Walsh CT, Neel BG. Protein-tyrosine-phosphatase SHPTP2 couples platelet-derived growth factor receptor beta to Ras. Proc Natl Acad Sci U S A. 1994;91:7335–7339. doi: 10.1073/pnas.91.15.7335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentires-Alj M, Paez JG, David FS, Keilhack H, Halmos B, Naoki K, Maris JM, Richardson A, Bardelli A, Sugarbaker DJ, et al. Activating mutations of the noonan syndrome-associated SHP2/PTPN11 gene in human solid tumors and adult acute myelogenous leukemia. Cancer Res. 2004;64:8816–8820. doi: 10.1158/0008-5472.CAN-04-1923. [DOI] [PubMed] [Google Scholar]
- Boureux A, Furstoss O, Simon V, Roche S. Abl tyrosine kinase regulates a Rac/JNK and a Rac/Nox pathway for DNA synthesis and Myc expression induced by growth factors. J Cell Sci. 2005;118:3717–3726. doi: 10.1242/jcs.02491. [DOI] [PubMed] [Google Scholar]
- Buchdunger E, Matter A, Druker BJ. Bcr-Abl inhibition as a modality of CML therapeutics. Biochim Biophys Acta. 2001;1551:M11–18. doi: 10.1016/s0304-419x(01)00022-1. [DOI] [PubMed] [Google Scholar]
- Burton EA, Plattner R, Pendergast AM. Abl tyrosine kinases are required for infection by Shigella flexneri. EMBO J. 2003;22:5471–5479. doi: 10.1093/emboj/cdg512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Yu WM, Daino H, Broxmeyer HE, Druker BJ, Qu CK. SHP-2 phosphatase is required for hematopoietic cell transformation by Bcr-Abl. Blood. 2007;109:778–785. doi: 10.1182/blood-2006-04-019141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, Zimmermann J, Lydon NB. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med. 1996;2:561–566. doi: 10.1038/nm0596-561. [DOI] [PubMed] [Google Scholar]
- Feng GS. Shp-2 tyrosine phosphatase: signaling one cell or many. Exp Cell Res. 1999;253:47–54. doi: 10.1006/excr.1999.4668. [DOI] [PubMed] [Google Scholar]
- Furstoss O, Dorey K, Simon V, Barila D, Superti-Furga G, Roche S. c-Abl is an effector of Src for growth factor-induced c-myc expression and DNA synthesis. EMBO J. 2002;21:514–524. doi: 10.1093/emboj/21.4.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Q, Lerner-Marmarosh N, Che W, Ohta S, Osawa M, Yoshizumi M, Glassman M, Yan C, Berk BC, Abe J. The novel role of the C-terminal region of SHP-2. Involvement of Gab1 and SHP-2 phosphatase activity in Elk-1 activation. J Biol Chem. 2002;277:29330–29341. doi: 10.1074/jbc.M112450200. [DOI] [PubMed] [Google Scholar]
- Keilhack H, David FS, McGregor M, Cantley LC, Neel BG. Diverse biochemical properties of Shp2 mutants. Implications for disease phenotypes. J Biol Chem. 2005;280:30984–30993. doi: 10.1074/jbc.M504699200. [DOI] [PubMed] [Google Scholar]
- Kontaridis MI, Swanson KD, David FS, Barford D, Neel BG. PTPN11 (Shp2) mutations in LEOPARD syndrome have dominant negative, not activating, effects. J Biol Chem. 2006;281:6785–6792. doi: 10.1074/jbc.M513068200. [DOI] [PubMed] [Google Scholar]
- Lu W, Gong D, Bar-Sagi D, Cole PA. Site-specific incorporation of a phosphotyrosine mimetic reveals a role for tyrosine phosphorylation of SHP-2 in cell signaling. Mol Cell. 2001;8:759–769. doi: 10.1016/s1097-2765(01)00369-0. [DOI] [PubMed] [Google Scholar]
- Mohi MG, Neel BG. The role of Shp2 (PTPN11) in cancer. Curr Opin Genet Dev. 2007;17:23–30. doi: 10.1016/j.gde.2006.12.011. [DOI] [PubMed] [Google Scholar]
- Pendergast AM. BCR-ABL protein domain, function, and signaling. In: Carella AM, Daley GQ, Eaves CJ, Goldman JM, Helmann R, editors. Chronic Myeloid Leukaemia: Biology and Treatment. Martin Dunitz, Lt.; London: 2001. pp. 19–39. [Google Scholar]
- Pendergast AM. The Abl family kinases: mechanisms of regulation and signaling. Adv Cancer Res. 2002;85:51–100. doi: 10.1016/s0065-230x(02)85003-5. [DOI] [PubMed] [Google Scholar]
- Pendergast AM, Muller AJ, Havlik MH, Clark R, McCormick F, Witte ON. Evidence for regulation of the human Abl tyrosine kinase by a cellular inhibitor. Proc Natl Acad Sci USA. 1991;88:5927–5931. doi: 10.1073/pnas.88.13.5927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plattner R, Irvin BJ, Guo S, Blackburn K, Kazlauskas A, Abraham RT, York JD, Pendergast AM. A New Link Between the c-Abl Tyrosine Kinase and Phosphoinositide Signaling via PLC-γ1. Nat Cell Biol. 2003;5:309–319. doi: 10.1038/ncb949. [DOI] [PubMed] [Google Scholar]
- Plattner R, Kadlec L, DeMali KA, Kazlauskas A, Pendergast AM. c-Abl is activated by growth factors and Src family kinases and has a role in the cellular response to PDGF. Genes Dev. 1999;13:2400–2411. doi: 10.1101/gad.13.18.2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plattner R, Koleske AJ, Kazlauskas A, Pendergast AM. Bidirectional Signaling Links the Abelson Kinases to the Platelet-Derived Growth Factor Receptor. Mol Cell Biol. 2004;24:2573–2583. doi: 10.1128/MCB.24.6.2573-2583.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plattner R, Pendergast AM. Activation and signaling of the Abl tyrosine kinase: bidirectional link with phosphoinositide signaling. Cell Cycle. 2003;2:273–274. [PubMed] [Google Scholar]
- Sattler M, Salgia R, Shrikhande G, Verma S, Choi JL, Rohrschneider LR, Griffin JD. The phosphatidylinositol polyphosphate 5-phosphatase SHIP and the protein tyrosine phosphatase SHP-2 form a complex in hematopoietic cells which can be regulated by BCR/ABL and growth factors. Oncogene. 1997;15:2379–2384. doi: 10.1038/sj.onc.1201422. [DOI] [PubMed] [Google Scholar]
- Srinivasan D, Plattner R. Activation of abl tyrosine kinases promotes invasion of aggressive breast cancer cells. Cancer Res. 2006;66:5648–5655. doi: 10.1158/0008-5472.CAN-06-0734. [DOI] [PubMed] [Google Scholar]
- Tartaglia M, Gelb BD. Noonan syndrome and related disorders: genetics and pathogenesis. Annu Rev Genomics Hum Genet. 2005;6:45–68. doi: 10.1146/annurev.genom.6.080604.162305. [DOI] [PubMed] [Google Scholar]
- Tauchi T, Feng GS, Shen R, Song HY, Donner D, Pawson T, Broxmeyer HE. SH2-containing phosphotyrosine phosphatase Syp is a target of p21bcr-abl tyrosine kinase. J Biol Chem. 1994;269:15381–15287. [PubMed] [Google Scholar]
- Yamauchi K, Milarski KL, Saltiel AR, Pessin JE. Protein-tyrosine-phosphatase SHPTP2 is a required positive effector for insulin downstream signaling. Proc Natl Acad Sci U S A. 1995;92:664–668. doi: 10.1073/pnas.92.3.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yart A, Laffargue M, Mayeux P, Chretien S, Peres C, Tonks N, Roche S, Payrastre B, Chap H, Raynal P. A critical role for phosphoinositide 3-kinase upstream of Gab1 and SHP2 in the activation of ras and mitogen-activated protein kinases by epidermal growth factor. J Biol Chem. 2001;276:8856–8864. doi: 10.1074/jbc.M006966200. [DOI] [PubMed] [Google Scholar]
- Yu DH, Qu CK, Henegariu O, Lu X, Feng GS. Protein-tyrosine phosphatase Shp-2 regulates cell spreading, migration, and focal adhesion. J Biol Chem. 1998;273:21125–21131. doi: 10.1074/jbc.273.33.21125. [DOI] [PubMed] [Google Scholar]
- Yu M, Luo J, Yang W, Wang Y, Mizuki M, Kanakura Y, Besmer P, Neel BG, Gu H. The scaffolding adapter Gab2, via Shp-2, regulates kit-evoked mast cell proliferation by activating the Rac/JNK pathway. J Biol Chem. 2006;281:28615–28626. doi: 10.1074/jbc.M603742200. [DOI] [PubMed] [Google Scholar]
- Zipfel PA, Zhang W, Quiroz M, Pendergast AM. Requirement for Abl kinases in T cell receptor signaling. Curr Biol. 2004;14:1222–1231. doi: 10.1016/j.cub.2004.07.021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.