

Abstract
Insulin signaling is dysfunctional in obesity and diabetes. Moreover, central glucose-sensing mechanisms are impaired in these diseases. This is associated with abnormalities in hypothalamic glucose-sensing neurons. Glucose-sensing neurons reside in key areas of the brain involved in glucose and energy homeostasis, such as the ventromedial hypothalamus (VMH). Our results indicate that insulin opens the KATP channel on VMH GE neurons in 5, 2.5, and 0.1 mM glucose. Furthermore, insulin reduced the sensitivity of VMH GE neurons to a decrease in extracellular glucose level from 2.5 to 0.1 mM. This change in the glucose sensitivity in the presence of insulin was reversed by the phosphatidylinositol 3-kinase (PI3K) inhibitor wortmannin (10 nM) but not by the mitogen-activated kinase (MAPK) inhibitor PD-98059 (PD; 50 μM). Finally, neither the AMPK inhibitor compound C nor the AMPK activator AICAR altered the activity of VMH GE neurons. These data suggest that insulin attenuates the ability of VMH GE neurons to sense decreased glucose via the PI3K signaling pathway. Furthermore, these data are consistent with the role of insulin as a satiety factor. That is, in the presence of insulin, glucose levels must decline further before GE neurons respond. Thus, the set point for detection of glucose deficit and initiation of compensatory mechanisms would be lowered.
Keywords: adenosine 5′-triphosphate-sensitive K+ channel, adenosine 5′-monophosphate-activated protein kinase, phosphatidylinositol 3-kinase, electrophysiology
insulin receptors located in the ventromedial hypothalamus (VMH) regulate energy homeostasis (3, 13, 33). Mice with neuron-specific disruption of the insulin receptor gene (NIRKO mice) exhibit hyperphagia, increased adiposity, hyperinsulinemia, and insulin resistance, consistent with type 2 diabetes mellitus (5, 30). Similar effects are noted in VMH-lesioned rats (14, 15). Moreover, microinjection of insulin directly into the VMH significantly reduces the firing rate of sympathetic nerves that supply interscapular brown adipose tissue. Destruction of VMH neurons via kainic acid injection blocks the inhibitory effects of insulin on sympathetic firing rate (31a, 32). These results suggest that dysfunctional VMH insulin signaling impairs sympathetic nervous system activity and is correlated with the development of type 2 diabetes mellitus.
The central effects of insulin on food intake and energy balance are mediated by the phosphoinositol 3-kinase (PI3K) signaling pathway. The PI3K inhibitors wortmannin and LY-294002 block the inhibitory effect of intracerebroventricular insulin on food intake and body weight gain in rats (45). The PI3K signaling pathway also mediates the effects of insulin on VMH glucose-sensing neurons (40). That is, wortmannin blocks insulin-induced activation of the ATP-sensitive K+ (KATP) channel and subsequent inhibition of neuronal activity of VMH glucose-excited (GE) neurons exposed to 10 mM glucose. These data suggest that VMH glucose-sensing neurons may play a role in the central effects of insulin on energy homeostasis.
The VMH possesses two subtypes of glucose-sensing neurons that directly sense changes in extracellular glucose. GE neurons increase and glucose-inhibited (GI) neurons decrease their action potential frequency, membrane potential, and input resistance as glucose increases (38, 39). Glucose closes the KATP channel on GE neurons, whereas it opens a chloride channel on GI neurons (38). Both GE and GI neurons respond to hormonal signals of peripheral energy homeostasis [e.g., insulin and leptin (6, 43)]. Previous studies of VMH glucose-sensing neurons utilized extracellular glucose concentrations (e.g., 10 mM or higher) that exceed physiological brain glucose levels. Thus, it has been difficult to ascertain the physiological role of VMH glucose-sensing neurons. However, work from our laboratory has established that VMH GE and GI neurons in brain slices are sensitive to changes in glucose between 0.1 and 2.5 mM (38, 43), suggesting that they are physiologically relevant brain glucose sensors.
Insulin also opens the KATP channel on VMH GE neurons in 2.5 mM glucose. However, in contrast to what was observed in 10 mM glucose, insulin-induced KATP channel activation in 2.5 mM glucose was not sufficient to alter neuronal activity (43). The interaction between insulin and glucose concentration is potentially relevant. That is, insulin may not have a profound effect on action potential frequency at a particular glucose concentration within the physiological range. However, insulin-induced activation of the KATP channel may blunt the effect of lowering glucose on KATP channel activity and thus on neuronal activity. We hypothesize that this will reduce the ability of GE neurons to sense decreased glucose. A reduction in the sensitivity of the brain to small glucose decreases is consistent with the role of insulin as a central satiety factor. Thus, this study will test the hypothesis that insulin blunts the response of VMH GE neurons in brain slices to physiological glucose decreases as a result of KATP channel activation.
METHODS
Preparation of brain slices.
Male 14- to 21-day-old Sprague-Dawley rats were housed at the New Jersey Medical School animal facility, with their dams in a controlled temperature of 22–23°C and on a 12:12-h light-dark cycle. Animals were maintained on Purina Rat Chow 5001 and water ad libitum. On the day of the experiment, rats were anesthetized with 50 mg/kg pentobarbital and transcardially perfused. Brains were then rapidly removed and placed in an ice-cold slushy oxygenated perfusion solution. Sections (350 μm) through the hypothalamus were maintained at 32°C in oxygenated artificial cerebrospinal fluid (ACSF) composed of the following (in mM): 126 NaCl, 1.9 KCl, 1.2 KH2PO4, 26 NaHCO3, 2.5 glucose, 9 MgCl2, and 0.3 CaCl2; osmolarity was adjusted to ∼300 mOsm with sucrose, pH 7.4. Slices were then transferred to normal oxygenated ACSF (2.4 mM CaCl2, 1.3 mM MgCl2) for the remainder of the experiment. All procedures were approved by the Institutional Animal Care and Use Committee at the New Jersey Medical School.
Electrophysiology: whole cell recordings in brain slices.
Viable neurons were visualized and studied under infrared differential-interference contrast microscopy using a Leica DMLFS microscope equipped with a ×40 long-working distance water-immersion objective. Current- and voltage-clamp recordings from VMH neurons were performed using an Axopatch-1D amplifier (Axon Instruments, Foster City, CA). Data were simultaneously digitized at 5 kHz (Digidata 1320A; Axon Instruments) and analyzed using pCLAMP 9.2 software. During recording, brain slices were perfused at 6 ml/min with normal oxygenated ACSF. Borosilicate pipettes (1.4–3.5 MΩ; Sutter Instruments, Novato, CA) were filled with an intracellular solution containing (in mmol/l) 128 K-gluconate, 10 KCl, 4 KOH, 10 HEPES, 4 MgCl2, 0.5 CaCl2, and 5 EGTA, pH 7.2. Osmolarity was adjusted to 290–300 mOsm with sucrose. Amphotericin B was included in the patch pipette to establish the perforated patch recording configuration (final concentration 240 μg/ml, stock 60 mg/ml DMSO). The junction potential between the bath and the patch pipette was nulled before the formation of a gigaohm seal. Membrane potential and action potential frequency were allowed to stabilize for 10–15 min after formation of a gigaohm seal to allow for the amphotericin B to perforate the membrane. Neurons with access resistance that exceeds 40 MΩ were rejected. Input resistance was calculated from the change in membrane potential in response to small, 500-ms hyperpolarizing pulses (−10 to −20 pA) given every 3 s, as described previously. The reversal potential was calculated from the change in membrane current in response to voltage steps from −120 to +60 mV from a holding potential of −60 mV. Steady-state currents were determined by measuring data points within the last 5 ms of the 200-ms voltage pulses. GE neurons are identified as those that decrease their action potential frequency, membrane potential, and input resistance with a decrease in extracellular glucose from 2.5 to 0.1 mM; GI neurons do the converse. Experimental manipulation of extracellular glucose levels and chemicals added to the ACSF are described in the figure legends. All chemicals were obtained from Sigma (St. Louis, MO).
Statistical analysis.
All data were expressed as means ± SE. Statistical analysis was performed using Student's t-test or one-way analysis of variance. P < 0.05 was considered statistically significant.
RESULTS
Localization of VMH GE neurons.
Whole cell patch clamp recordings were used to characterize 359 VMH neurons with regard to their glucose and insulin sensitivity. When recordings were made predominately in the lateral arcuate nucleus (ARC) within the VMH, 3.2% (4 of 124 neurons) were GE neurons, whereas none were GI neurons. In contrast, when recordings were made along the ventrolateral border of the ventromedial nucleus (VL-VMN) within the VMH, 19.6% (46 of 235 neurons) were GE neurons. GI neurons made up <1% of the neurons in the VL-VMN (1 of 235 neurons; Fig. 1). These studies indicate that the densest population of GE neurons is found in the VL-VMN. For this reason, we focused on VL-VMN GE neurons for the present study.
Fig. 1.
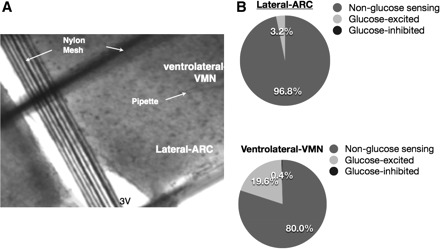
Localization of glucose-sensing neurons in the ventrolateral border of the ventromedial nucleus (VL-VMN) compared with the lateral arcuate nucleus (ARC) in the rat. A: direct-interference infrared (DIC-IR) image of a 350-μm hypothalamic section through the ventromedial hypothalamus (×40). The pipette tip is located in the region where glucose-excited (GE) neurons are concentrated. B: pie charts representing the relative percentage of GE, glucose-inhibited (GI), and non-glucose-sensing neurons recorded from the lateral ARC vs. the VL-VMN.
Evaluation of the insulin concentration-response relationship.
When insulin (5 nM) was added to 5 mM glucose, the action potential frequency of VL-VMN GE neurons decreased by 55 ± 10% (n = 9, P < 0.05). However, the addition of insulin to 2.5 or 0.1 mM glucose had no significant effect on the action potential frequency of VL-VMN GE neurons (2.5 mM glucose: 6 ± 3%, n = 15, P > 0.05; 0.1 mM glucose: 0 ± 0%, n = 6, P > 0.05). In contrast, insulin significantly decreased membrane potential and input resistance of GE neurons in all three glucose concentrations (Fig. 2). However, the inhibitory effect of insulin on membrane potential and input resistance of GE neurons in 2.5 and 0.1 mM glucose was significantly attenuated compared with that in 5 mM glucose (P < 0.05; Fig. 2). The effects of insulin persisted during the whole treatment period and were readily reversible with removal of insulin from the perfusion solution.
Fig. 2.
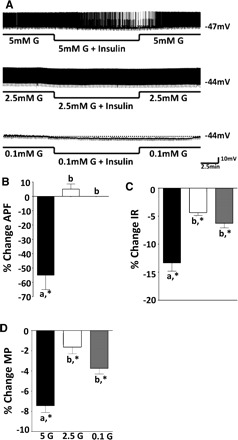
A: consecutive whole cell current clamp recordings of spontaneous electrical activity in a VL-VMN GE neuron. Resting membrane potential is indicated by the horizontal dotted line and noted at the right of each trace. Downward deflections represent the membrane voltage response to a constant hyperpolarizing pulse. The membrane voltage response is directly proportional to the change in input resistance (IR). Insulin causes significant hyperpolarization and decreases in action potential frequency (APF) in 5 (n = 9) but not 2.5 (n = 25) or 0.1 mM glucose (G) (n = 6; A and B). However, insulin significantly decreased IR and membrane potential (MP) in all 3 glucose concentrations (C and D). The effect of insulin on IR and MP in 5 mM glucose was greater than that in 2.5 or 0.1 mM glucose (columns with different letters are statistically different; *significantly different from 0% change, P < 0.05).
Previous studies have shown that the effects of insulin (0.5–500 nM) on VMH GE neurons were independent of insulin concentration (40, 43). Therefore, we evaluated lower insulin concentrations to determine the threshold for the inhibitory effect of insulin on GE neurons in physiological brain glucose concentrations. Because insulin had no effect on action potential frequency in 2.5 and 0.1 mM glucose, we calculated the insulin concentration-response relationship using the percent change in input resistance. Input resistance is also a sensitive measure of neuronal excitability and reflects changes in ion channel activity. There was no significant difference between the effect of insulin on the input resistance of GE neurons in 2.5 mM glucose and that in 0.1 mM glucose (Fig. 2); thus, these data were combined to generate the concentration-response relationship for insulin. Using insulin concentrations from 5 pM to 300 nM, we found that the half-maximal effective concentration (EC50) for insulin on input resistance of VL-VMN GE neurons in 2.5 or 0.1 mM glucose was 3.3 nM (Fig. 3).
Fig. 3.
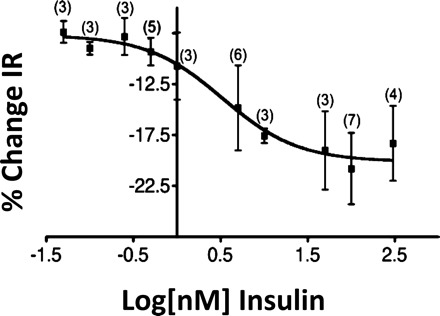
Insulin concentration-response relationship for %change in IR in VL-VMN GE neurons in brain slices. There was no significant difference in the effect of 5 pM to 300 nM insulin on IR in 2.5 or 0.1 mM glucose; thus, these data points were combined. The EC50 for insulin was 3.3 nM.
KATP channel activation mediates insulin's effects on VL-VMN GE neurons.
Current-voltage relationships show that the insulin-induced current in VL-VMN GE neurons reverses at approximately −96 ± 5.9 mV (adjusted to a −20-mV junction potential; n = 9) in all three glucose concentrations (Fig. 4). This is near the theoretical K+ equilibrium potential of −99 mV in our solutions. In addition, the KATP channel blocker tolbutamide abolished insulin's effects in 5, 2.5, and 0.1 mM glucose (Fig. 5, A–C). Current-voltage relationships for the effects of insulin in the presence and absence of tolbutamide reversed at the theoretical K+ equilibrium potential (Fig. 5D). These data are consistent with earlier reports that insulin opens the KATP channel on GE neurons in 10 and 2.5 mM glucose (40, 43). Finally, tolbutamide inhibited the effects of both 50 pM and 5 nM insulin (data not shown), suggesting that KATP channel activation mediates the effects of insulin on VL-VMN GE neurons in all insulin concentrations.
Fig. 4.
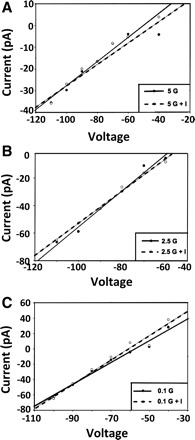
Current-voltage relationships indicate that the insulin-sensitive conductance reverses near the theoretical K+ equilibrium potential (−99 mV) in 5 (A), 2.5 (B), and 0.1 mM glucose (C). I, insulin.
Fig. 5.
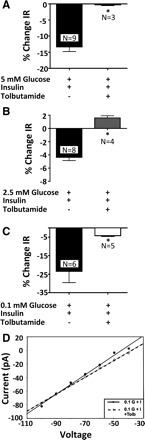
Tolbutamide (tolb; 100 μM) blocks the effects of I on the IR of VL-VMN GE neurons in 5 (A), 2.5 (B), and 0.1 mM G (C). D: current-voltage relationship for the effect of insulin in 0.1 mM G in the presence and absence of tolbutamide reverses near the theoretical K+ equilibrium potential (*P < 0.05).
Insulin suppresses the ability of VL-VMN GE neurons to sense decreased glucose.
Decreasing extracellular glucose from 2.5 to 0.1 mM decreased the action potential frequency of VL-VMN GE neurons by 71 ± 5% (n = 19, P < 0.05; Fig. 6). In contrast, when glucose was lowered in the presence of 5 nM insulin, action potential frequency was decreased by only 34 ± 8% (n = 9, P < 0.05; Fig. 6). As shown in Fig. 7, the decrease in input resistance and membrane potential in response to decreased glucose was similarly attenuated in the presence of insulin.
Fig. 6.
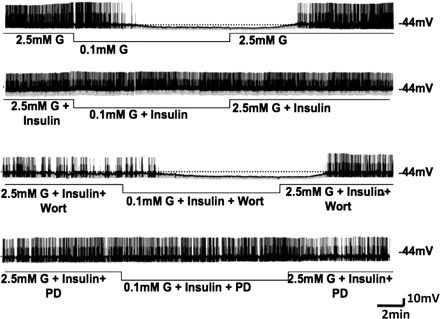
Consecutive whole cell current clamp recordings of spontaneous electrical activity in a VL-VMN GE neuron. Resting MP is indicated by the horizontal dotted line and note at the right of each trace. Downward deflections represent the membrane voltage response to a constant hyperpolarizing pulse. The membrane voltage response is directly proportional to the change in IR. Decreasing extracellular G levels from 2.5 to 0.1 mM causes hyperpolarization, decreased IR, and decreased APF (top trace). The response to decreased glucose is reduced in the presence of 5 nM insulin (upper middle trace). The phosphatidylinositol 3-kinase (PI3K) inhibitor wortmannin (wort; 10 nM) blocks the effects of insulin (lower middle trace); however, the MAPK inhibitor PD-98039 (PD; 50 μM) does not (bottom trace).
Fig. 7.
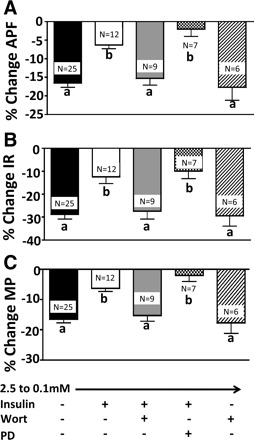
%Change in APF (A), IR (B), and MP (C) in VL-VMN GE neurons in response to decreased glucose from 2.5 to 0.1 mM in the presence of insulin and inhibitors of insulin signaling. The PI3K inhibitor wort (10 nM) alone had no effect on glucose sensitivity; however, wort blocked the effects of insulin, whereas the MAPK inhibitor PD-98039 (50 μM) did not. Columns with different letters are statistically different (P < 0.05).
The effect of insulin on the glucose sensitivity of VL-VMN GE neurons was reversed in the presence of the PI3K inhibitor wortmannin (10 nM). That is, when glucose was lowered from 2.5 to 0.1 mM in the presence of insulin and wortmannin, action potential frequency was decreased by 69 ± 9% (n = 8, P < 0.05; Fig. 6, lower middle trace). This is not significantly different from that observed in response to a decrease in extracellular glucose alone (P > 0.05). Similar results were noted for membrane potential and input resistance (Fig. 7). The response to decreasing extracellular glucose in the presence of wortmannin was not significantly different compared with a decrease in glucose alone (Fig. 7).
In contrast to the effects of wortmannin, the MAPK inhibitor PD-98059 (50 μM) did not alter the effect of insulin on the glucose sensitivity of VL-VMN GE neurons (Figs. 6 and 7). That is, decreasing glucose from 2.5 to 0.1 mM in the presence of insulin decreased action potential frequency to a similar extent independent of the presence of PD-98059 in the perfusion solution (insulin, −34 ± 8%; insulin + PD-98059, −20 ± 11%, n = 6, P > 0.05; Fig. 6; upper middle and bottom traces, respectively). As shown in Fig. 7, there was no significant difference in input resistance and membrane potential in response to decreased extracellular glucose with insulin in the presence and absence of PD-98059.
AMP-activated protein kinase (AMPK) signaling does not appear to play a role in glucose sensing in VL-VMN GE neurons. Decreasing glucose from 2.5 to 0.1 mM in the presence of the specific AMPK inhibitor compound C (10 μM; Ref. 18) decreased action potential frequency by 78 ± 9% (n = 4, P < 0.05; Fig. 8A, middle trace). This was not significantly different from the decrease in action potential frequency when glucose was decreased in the absence of compound C (71 ± 5%, n = 19, P < 0.05; Fig. 8A, top trace). As shown in Fig. 8, B and C, top, there was no significant difference in input resistance and membrane potential in response to decreased glucose in the presence and absence of compound C. Furthermore, there was no significant difference in the action potential frequency of VL-VMN GE neurons in the presence and absence of the AMPK activator 5-aminoimidazole-4-carboxamide (0.5 mM) in 2.5 mM glucose (Fig. 8A, bottom trace). Similar results were seen for input resistance and membrane potential (Fig. 8, B and C, bottom).
Fig. 8.
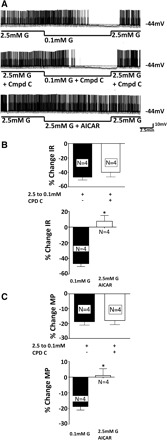
A: consecutive whole cell current clamp recordings of spontaneous electrical activity in a VL-VMN GE neuron. Resting MP is indicated by the horizontal dotted line and noted at the right of each trace. Downward deflections represent the membrane voltage response to a constant hyperpolarizing pulse. The membrane voltage response is directly proportional to the change in IR. Decreasing extracellular glucose levels from 2.5 to 0.1 mM causes hyperpolarization, decreased IR, and decreased APF (top trace). The effect of decreased glucose is not altered in the presence of the AMPK inhibitor compound C (CPD C; 10 μM, middle trace). The AMPK activator 5-aminoimidazole-4-carboxamide (AICAR) did not decrease the APF of VL-VMN GE neurons (bottom trace). B: the change in IR and MP in response to decreased glucose from 2.5 to 0.1 was the same in the presence and absence of CPD C. C: the administration of 0.5 mM AICAR to 2.5 mM glucose did not mimic the effects of decreased glucose on IR and MP.
The effects of insulin on VL-VMN GE neurons were direct, as indicated by the persistence of response in the presence of the selective voltage-dependent Na+ channel blocker tetrodotoxin (TTX; 300 nM, n = 5; Fig. 9). Finally, insulin had no significant effect on input resistance, action potential frequency, or membrane potential in non-glucose-sensing neurons in the VL-VMN (n = 22; data not shown).
Fig. 9.
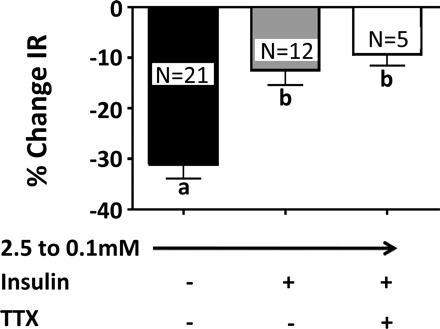
%Change in IR of VL-VMN GE neurons in response to decreased glucose from 2.5 to 0.1 mM in the presence or absence of insulin and the Na+ channel blocker tetrodotoxin (TTX; 300 nM). The effect of insulin on VL-VMN GE neurons persists in the presence of TTX (columns with different letters are statistically different, P < 0.05).
DISCUSSION
Previous studies have shown that peripheral signals of energy balance, such as insulin and leptin, regulate the activity of hypothalamic glucose-sensing neurons (40, 41, 43). We show here that insulin blunts the response of GE neurons to glucose decreases. These data are consistent with the role of insulin as a central satiety factor (34, 35). That is, if VL-VMN GE neurons are important for initiating and/or maintaining signals of energy sufficiency, it would be inappropriate for these neurons to become inhibited by small decreases in glucose associated with meal-to-meal oscillations. Rather, in the presence of normal insulin levels, VL-VMN GE neurons are inhibited only during severe glucose deficits that would compromise brain function.
Numerous studies indicate that the VL-VMN is necessary for signals of energy sufficiency to be correctly interpreted by the brain (21). VMH lesions have been associated with obesity (14). In particular, specific lesions, which included the VL-VMN and the capsule surrounding the VMN, cause the most profound obesity (15, 21). Later studies further support a critical role for this region in the regulation of energy balance. The neurotoxin gold thioglucose, which causes obesity, destroys neurons specifically in the VL-VMN and the “cell-poor” region between the VMN and ARC (22). Moreover, many VMH-efferent projections to other brain regions involved in metabolic regulation arise from the VL-VMN (21). The data above suggest that the VL-VMN supports catabolic processes. It is noteworthy that we find the greatest concentration of GE neurons in the VL-VMN. The regulation of VL-VMN GE neurons by insulin and glucose is consistent with the hypothesis that the VL-VMN is important for the integration of signals of energy sufficiency. Moreover, our data showing that insulin regulates the glucose sensitivity of VL-VMN GE neurons support a role for these neurons in the maintenance of energy homeostasis.
Our observation that insulin did not change the action potential frequency of VL-VMN GE neurons in 2.5 mM glucose appears to contradict the present literature. Spanswick and colleagues (40, 41) demonstrated that insulin abolished action potentials and caused profound inhibition of VMH GE neurons by opening the KATP channel. However, Spanswick and colleagues (40, 41), like many others (1, 2, 25, 27, 28, 29), evaluated GE neurons in 10 mM glucose (or higher). These extracellular glucose concentrations were based on the assumption that brain glucose levels were equal to those found in the periphery. Since these earlier experiments were completed, a number of studies have been carried out that indicate that brain glucose levels are much lower than those in the blood (8, 23, 36, 37). In fact, during peripheral energy sufficiency, VMH glucose levels range from 1.5 to 2.5 m (8, 36, 37). Thus, it was necessary to reevaluate the effects of insulin on GE neurons using physiological glucose concentrations.
To this end, we evaluated the effects of insulin in glucose concentrations that would be found in the brain during peripheral euglycemia (2.5 mM), hypoglycemia (0.1 mM), and hyperglycemia (5 mM) (36, 37). As Spanswick et al. (40) observed in 10 mM glucose, we found that insulin also decreased the membrane potential and action potential frequency of VL-VMN GE neurons in 5 mM glucose. This was associated with a decrease in input resistance, suggesting that an ion channel opened. The insulin-activated current reversed at the K+ equilibrium potential and was blocked by the KATP channel blocker tolbutamide. Thus, insulin inhibited VL-VMN GE neurons in 5 mM glucose by opening the KATP channel, as occurs in 10 mM glucose (40). Insulin also opened the KATP channel on VL-VMN GE neurons in 2.5 and 0.1 mM glucose, as indicated by decreased input resistance, reversal at the K+ equilibrium potential, and blockade with tolbutamide. Thus, our data show that insulin opens the KATP channel on VL-VMN GE neurons regardless of glucose concentration.
However, the magnitude of the effect of insulin on input resistance of VL-VMN GE neurons was inversely correlated with glucose concentration. That is, the insulin-induced decrease in input resistance was significantly attenuated in 2.5 and 0.1 mM glucose compared with that in 5 mM glucose. This indicates that the effect of insulin on the KATP channel current was diminished in lower glucose concentrations. In fact, insulin-induced KATP channel activation in 2.5 and 0.1 mM glucose was insufficient to alter action potential frequency. Although insulin does not modify action potentials in physiological glucose, its effects on input resistance and KATP channel activity are highly significant in terms of regulating the function of VL-VMN GE neurons. One of the primary functions of “nonsynaptic” K+ channels is to regulate the excitability or responsiveness of neurons to other stimuli (16). Thus, in the presence of insulin, the cellular (input) resistance of GE neurons is lower. When cellular resistance is lower, the magnitude of the voltage change in response to a given presynaptic input must be greater to exert the same effect on membrane potential and action potential frequency as would be observed when cellular resistance is higher. These data suggest that, in the presence of insulin, VL-VMN GE neurons are less responsive to presynaptic input, whether it is excitatory or inhibitory.
Alterations in glucose sensitivity are another very important consequence of the effect of insulin on VL-VMN GE neurons in physiological glucose concentration. We show here that insulin blunts the response of VL-VMN GE neurons to glucose decreases. This hypothesis is further supported by the observation that there was no significant difference in the effect of insulin in 2.5 and 0.1 mM glucose. We hypothesize that these data indicate that the effect of insulin is mediated by two distinct populations of KATP channels on GE neurons, those that are sensitive to physiological changes in extracellular glucose and those that are not (Fig. 10). This hypothesis is based on our observation that insulin significantly increases KATP channel activation in 0.1 mM glucose. This suggests that some KATP channels on VL-VMN GE neurons are sensitive to insulin but not to changes in extracellular glucose. If all KATP channels were sensitive to physiologically relevant changes in extracellular glucose levels, they would be maximally open in 0.1 mM glucose and thus insensitive to further activation by insulin. This hypothesis is further supported by our observation that insulin activated KATP channels on VL-VMN GE neurons in 5 mM glucose to a greater extent than in 0.1 mM. These results are consistent with previously published data showing that lactate administration closes KATP channels on GE neurons even when increases in extracellular glucose cause no further effect (39). Colocalization of KATP channels with glycolytic enzymes at the plasma membrane, as seen in cardiac and intestinal epithelial cells (9, 44), may account for the existence of multiple populations of KATP channels. Such colocalization could render these KATP channels sensitive to changes in extracellular glucose, since cytosolic ATP levels are well buffered (9).
Fig. 10.
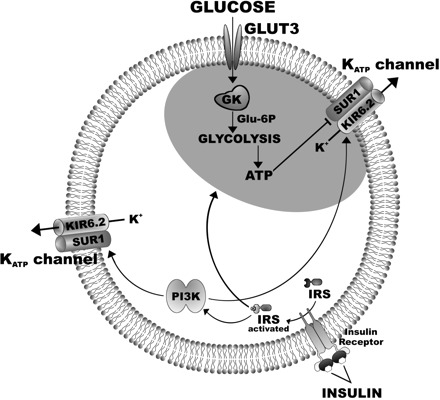
Hypothetical model of the effects of glucose and insulin on ATP-sensitive K+ (KATP) channels on GE neurons. Two subpopulations of KATP channels exist, one that is colocalized with glycolytic enzymes and thus sensitive to changes in extracellular glucose and another that is not. Glucose is transported into neurons via the glucose transporter 3 (GLUT3). Glucokinase (GK) catalyzes the first step in glycolysis in GE neurons (19). Decreased extracellular glucose results in decreased levels of intracellular ATP. If glycolytic enzymes are compartmentalized adjacent to KATP channels, decreased ATP levels will open these “glucose-sensitive” KATP channels, allow potassium (K) efflux, and inhibit GE neurons. Insulin may have opposing effects on glucose-sensitive KATP channels. First, insulin binds to its receptor, phosphorylates the insulin receptor subunit (IRS), activates PI3K, and opens both the glucose-sensitive and -insensitive populations of KATP channels. However, in addition to direct effects on KATP channel activation, insulin may also close glucose-sensitive KATP channels by increasing neuronal glucose uptake (11, 42) and stimulating glycolysis (18). The net effect of insulin on VL-VMN GE neurons is to blunt the response to decreased glucose. SUR1, sulfonylurea receptor; Kir6.2, pore-forming subunit of the KATP channel; Glu-6-P, glucose 6-phosphate.
Multiple KATP channel populations raise the possibility that the effects of insulin and decreased glucose on KATP channels are additive. If so, it suggests that insulin attenuates the effect of decreased glucose by “washing out” the effect of decreased glucose on KATP channel activation. Our data do not support this hypothesis for the following reasons. The effect of decreased glucose from 2.5 to 0.1 mM on input resistance and membrane potential of VL-VMN GE neurons (Fig. 7) is greater than the effect of insulin in 5 mM glucose (Fig. 2). This might be a consequence of the use of the EC50 for insulin rather than a maximal concentration. However, the effect of decreasing glucose from 2.5 to 0.1 mM on input resistance is also greater than the maximal effect of insulin in 2.5 mM glucose (Fig. 3). Moreover, the decreased membrane potential and input resistance observed with insulin in 2.5 mM glucose combined with the further decrease in these parameters when glucose is subsequently lowered to 0.1 mM in the presence of insulin is still smaller than observed when glucose is lowered from 2.5 to 0.1 mM in the absence of insulin. Taken together, our data point toward an interaction between insulin and glucose metabolism. Reports that insulin increases neuronal glucose transporter 3 (42) and hypothalamic glucose transporter 4 translocation (11) as well as pyruvate dehydrogenase activity (18) support this hypothesis. Thus, insulin would have opposing effects on KATP channel activation (Fig. 10). On the one hand, insulin directly opens both populations of KATP channels; however, insulin also increases ATP production, leading to closure of glucose-sensitive KATP channels. The latter effect of insulin may explain the attenuated the response of GE neurons to decreased glucose in the presence of insulin. Future studies are required to test this hypothesis.
Our results showing that PI3K mediates insulin activation of the KATP channel are consistent with the literature concerning the role of the PI3K pathway in insulin signaling in the regulation of energy status (34, 35, 45). Insulin is also linked to the MAPK and AMPK signaling pathways. For example, inhibition of the MAPK pathway blocked insulin-induced sympathoactivation in brown adipose tissue (31). However, like Spanswick et al. (40), our results show that administration of the MAPK inhibitor PD-98059 had no effect on insulin action in VL-VMN GE neurons. Insulin has also been reported to decrease hypothalamic AMPK activity (24). However, we found that neither AMPK activation nor inhibition altered the activity or glucose sensitivity of VL-VMN GE neurons. Thus, it is highly unlikely that AMPK inhibition mediates the effect of insulin on VL-VMN GE neurons. These data suggest that insulin alters the activity of VL-VMN GE neurons via PI3K but not MAPK- or AMPK-dependent mechanisms.
Finally, although we evaluated glucose sensitivity of VL-VMN GE neurons in the presence and absence of insulin, these neurons are unlikely to be exposed to insulin absence in vivo. The studies herein are a necessary first step to determine the effects of insulin on the glucose sensitivity of VL-VMN GE neurons. This enables comparison with previous studies of glucose-sensing neurons in which glucose-sensing neurons are evaluated in the absence of basal hormonal and nutrient signals of peripheral energy homeostasis (e.g., insulin, leptin, free fatty acids). However, it is now critical to begin to evaluate these neurons in an in vitro “estimate” of the physiological extracellular brain milieu. For example, it will be important to evaluate the effects of leptin and free fatty acids, first singly and then in combination on the glucose sensitivity of VL-VMN GE neurons. It will also be important to determine how pre- to postprandial fluctuations in insulin levels affect the glucose sensitivity of VL-VMN GE neurons, although this will be a difficult task since physiological brain insulin levels are not yet known.
In conclusion, insulin regulates not only the activity but the glucose sensitivity of VL-VMN GE neurons. Our data suggest that insulin blunts the response of VL-VMN GE neurons to physiological glucose decreases. These data are consistent with the role of insulin as a satiety factor. That is, inhibition of the VL-VMN leads to a shift in body energy balance that favors decreased energy expenditure. During energy sufficiency, it would not be efficient for neurons in the VL-VMN to be inhibited by small decreases in extracellular glucose associated with meal-to-meal oscillations. However, when peripheral energy stores are low, insulin levels decline. Decreased insulin levels would enhance the response of VL-VMN GE neurons to glucose decreases. Thus, VL-VMN GE neurons would be more likely to be inhibited during daily glucose fluctuations, leading to an overall decrease in the activity of the VL-VMN. Our data lead to the hypothesis that insulin resistance during obesity and type 2 diabetes mellitus enhances the response of VL-VMN GE neurons to decreased glucose. This would lead to signals of glucose or energy deficit during energy sufficiency, which could contribute to the development of these metabolic diseases.
GRANTS
This study was supported in part by National Institute of Diabetes and Digestive and Kidney Diseases Grant 2-RO1-DK-55619 and National Institutes of Health Research Service Award 5F31NS05681.
Acknowledgments
We thank Kurt Fakira for assistance with the production of Fig. 10.
REFERENCES
- 1.Ashford ML, Boden PR, Treherne JM. Tolbutamide excites rat glucoreceptive ventromedial hypothalamic neurones by indirect inhibition of ATP-K+ channels. Br J Pharmacol 101: 531–540, 1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ashford ML, Boden PR, Treherne JM. Glucose-induced excitation of hypothalamic neurones is mediated by ATP-sensitive K+ channels. Pflugers Arch 415: 479–483, 1990 [DOI] [PubMed] [Google Scholar]
- 3.Baskin DG, Figlewicz-Lattemann D, Seeley RJ, Woods SC, Porte D Jr, Schwartz MW. Insulin and leptin: dual adiposity signals to the brain for the regulation of food intake and body weight. Brain Res 848: 114–123, 1999 [DOI] [PubMed] [Google Scholar]
- 5.Brüning J, Gautam D, Burks DJ, Gillette J, Schubert M, Orban PC, Klein R, Krone W, Muller-Wieland D, Kahn CR. Role of brain insulin receptor in control of body weight and reproduction. Science 289: 2122–2125, 2000 [DOI] [PubMed] [Google Scholar]
- 6.Canabal DD, Song Z, Potian JG, Beuve A, McArdle JJ, Routh VH. Glucose, insulin, and leptin signaling pathways modulate nitric oxide synthesis in glucose-inhibited neurons in the ventromedial hypothalamus. Am J Physiol Regul Integr Comp Physiol 292: R1418–R1428, 2007 [DOI] [PubMed] [Google Scholar]
- 7.Claret M, Smith MA, Batterham RL, Selman C, Choudhury AI, Fryer LG, Clements M, Al-Qassab H, Heffron H, Xu AW, Speakman JR, Barsh GS, Viollet B, Vaulont S, Ashford ML, Carling D, Withers DJ. AMPK is essential for energy homeostasis regulation and glucose sensing by POMC and AgRP neurons. J Clin Invest 117: 2325–2336, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Vries M, Arseneau LM, Lawson ME, Beverly JL. Extracellular glucose in rat ventromedial hypothalamus during acute and recurrent hypoglycemia. Diabetes 52: 2767–2773, 2003 [DOI] [PubMed] [Google Scholar]
- 9.Dubinsky WP, Mayorga-Wark O, Schultz SG. Colocalization of glycolytic enzyme activity and KATP channels in basolateral membrane of Necturus enterocytes. Am J Physiol Cell Physiol 275: C1653–C1659, 1998 [DOI] [PubMed] [Google Scholar]
- 10.Fioramonti X, Contié S, Song Z, Routh VH, Lorsignol A, Pénicaud L. Characterization of glucosensing neuron subpopulations in the arcuate nucleus: integration in neuropeptide Y and pro-opio melanocortin networks. Diabetes 56: 1219–1227, 2007 [DOI] [PubMed] [Google Scholar]
- 11.Grillo CA, Tamashiro KL, Piroli GG, Melhorn S, Gass JT, Newsom RJ, Reznikov A, Smith SP, Wilson RR, Sakai RR, Reagan LP. Lentivirus-mediated downregulation of hypothalamic insulin receptor expression. Physiol Behav 92: 691–701, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hakansson M, Brown H, Ghilardi N, Skoda R, Meister B. Leptin receptor immunoreactivity in chemically defined target neurons of the hypothalamus. J Neurosci 18: 559–572, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Havrankova J, Roth J, Brownstein M. Insulin receptors are widely distributed in the central nervous system of the rat. Nature 272: 827–829, 1978 [DOI] [PubMed] [Google Scholar]
- 14.Hetherington AW, Ranson SW. Hypothalamic lesions and adiposity in the rat. Anat Rec 78: 149–172, 1940 [Google Scholar]
- 15.Hetherington AW, Ranson SW. The relation of various hypothalamic lesions to adiposity in the rat. J Comp Neurol 76: 475–499, 1942 [Google Scholar]
- 16.Hille B. Ion Channels of Excitable Membranes (3rd ed.). Sunderland, MA: Sinauer Associates, 2001, p. 72–85
- 17.Ibrahim N, Bosch MA, Smart JL, Qiu J, Rubinstein M, Rønnekleiv O, Low MJ, Kelly M. Hypothalamic proopiomelanocortin neurons are glucose responsive and express K(ATP) channels. Endocrinology 144: 1331–1340, 2003 [DOI] [PubMed] [Google Scholar]
- 18.Johnson SA, Denton RM. Insulin stimulation of pyruvate dehydrogenase in adipocyte involves two distinct signaling pathways. Biochem J 369: 351–356, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kang L, Sanders NM, Dunn-Meynell AA, Gaspers LD, Routh VH, Thomas AP, Levin BE. Prior hypoglycemia enhances glucose responsiveness in some ventromedial hypothalamic glucosensing neurons. Am J Physiol Regul Integr Comp Physiol 294: R784–R792, 2008 [DOI] [PubMed] [Google Scholar]
- 20.Kefas BA, Cai Y, Kerckhofs K, Ling Z, Martens G, Heimberg H, Pipeleers D, Van de Casteele M. Metformin-induced stimulation of AMP-activated protein kinase in beta-cells impairs their glucose responsiveness and can lead to apoptosis. Biochem Pharmacol 68: 409–411, 2004 [DOI] [PubMed] [Google Scholar]
- 21.King B. The rise, fall, and resurrection of the ventromedial hypothalamus in the regulation of feeding behavior and body weight. Physiol Behav 87: 221–244, 2006 [DOI] [PubMed] [Google Scholar]
- 22.Kishi E, Takahashi A, Ishimaru H, Ikarashi Y, Maruyama Y. Development of obesity and neurochemical backing in aurothioglucose-treated mice. Auton Neurosci 92: 21–27, 2001 [DOI] [PubMed] [Google Scholar]
- 23.McNay EC, Gold PE. Extracellular glucose concentrations in the rat hippocampus measured by zero-net-flux: effects of microdialysis flow rate, strain, and age. J Neurochem 72: 785–790, 1999 [DOI] [PubMed] [Google Scholar]
- 24.Minokoshi Y, Alquier T, Furukawa N, Kim YB, Lee A, Xue B, Mu J, Foufelle F, Ferré P, Birnbaum MJ, Stuck BJ, Kahn BB. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature 92: 21–27, 2004 [DOI] [PubMed] [Google Scholar]
- 25.Mizuno Y, Oomura Y. Glucose responding neurons in the nucleus tractus solitarius of the rat: in vitro study. Brain Res 307: 109–116, 1984 [DOI] [PubMed] [Google Scholar]
- 26.Nakano Y, Oomura Y, Lenard L, Nishino H, Aou S, Yamamoto T, Aoyai K. Feeding related activity of glucose and morphine-sensitive neurons in the monkey amygala. Brain Res 399: 167–172, 1986 [DOI] [PubMed] [Google Scholar]
- 27.Oomura Y. Glucose as a regulator of neuronal activity. Adv Metab Disord 10: 31–65, 1983 [DOI] [PubMed] [Google Scholar]
- 28.Oomura Y, Ono T, Ooyama H, Wayner MJ. Glucose and osmosensitive neurons of the rat hypothalamus. Nature 222: 282–284, 1969 [DOI] [PubMed] [Google Scholar]
- 29.Oomura Y, Kimura K, Ooyama H, Maeo T, Iki M, Kuniyoshi N. Reciprocal activities of the ventromedial and lateral hypothalamic area of cats. Science 143: 484–485, 1964 [DOI] [PubMed] [Google Scholar]
- 30.Orgel SC, Hansen B, Shafrir E. Insulin Resistance and Insulin Resistance Syndrome. New York: Informa Healthcare, 2002, p. 126
- 31.Rahmouni K, Morgan DA, Morgan GM, Liu X, Sigmund CD, Mark A, Haynes WG. Hypothalamic PI3K and MAPK differentially mediate regional sympathetic activation to insulin. J Clin Invest 114: 652–658, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31a.Sakaguchi T, Bray GA. Intrahypothalamic injection of insulin decreases firing rate of sympathetic nerves. Proc Natl Acad Sci USA 84: 2012–2014, 1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sakaguchi T, Takahashi M, Bray GA. Lateral hypothalamus and sympathetic firing rate. Am J Physiol Regul Integr Comp Physiol 255: R507–R512, 1988 [DOI] [PubMed] [Google Scholar]
- 33.Schwartz MW, Sipols AJ, Marks JL, Sanacora G, White JD, Scheurink A, Kahn SE, Baskin DG, Woods SC, Figlewicz DP. Inhibition of hypothalamic neuropeptide Y gene expression by insulin. Endocrinology 130: 3608–3616, 1992 [DOI] [PubMed] [Google Scholar]
- 34.Schwartz MW, Woods SC, Porte D Jr, Seeley RJ, Baskin DG. Central nervous system control of food intake. Nature 404: 661–671, 2000 [DOI] [PubMed] [Google Scholar]
- 35.Schwartz MW, Niswender KD. Insulin and leptin revisited: adiposity signals with overlapping physiological and intracellular signaling capabilities. Front Neuroendocrinol 21: 1–10, 2003 [DOI] [PubMed] [Google Scholar]
- 36.Silver I, Erencinska M. Glucose-induced intracellular ion changes in sugar-sensitive hypothalamic neurons. J Neurophysiol 79: 1733–1745, 1998 [DOI] [PubMed] [Google Scholar]
- 37.Silver I, Erencinska M. Extracellular glucose concentration in mammalian brain: continuous monitoring of changes during increased neuronal activity and upon limitation in oxygen supply in normo-, hypo-, and hyperglycemic animals. J Neurosci 14: 5068–5076, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Song Z, Levin B, McArdle JJ, Bakhos N, Routh VH. Convergence of pre- and postsynaptic influences on glucosensing neurons in the ventromedial hypothalamic nucleus. Diabetes 50: 2673–2681, 2001 [DOI] [PubMed] [Google Scholar]
- 39.Song Z, Routh VH. Differential effects of glucose and lactate on glucosensing neurons in the ventromedial hypothalamic nucleus. Diabetes 54: 15–22, 2005 [DOI] [PubMed] [Google Scholar]
- 40.Spanswick D, Mirshamsi S, Routh VH, Ashford ML. Insulin activates ATP -sensitive K+ channels in hypothalamic neurons of lean, but not obese rats. Nat Neurosci 3: 757–758, 2000 [DOI] [PubMed] [Google Scholar]
- 41.Spanswick D, Mirshamsi S, Groppi VE, Logan SD, Ashford ML. Leptin inhibits hypothalamic neurons by activation of ATP-sensitive potassium channels. Nature 390: 521–525, 1997 [DOI] [PubMed] [Google Scholar]
- 42.Uemura E, Greenlee HW. Insulin regulates neuronal glucose uptake by promoting translocation of glucose transporter GLUT3. Exp Neurol 198: 48–53, 2006 [DOI] [PubMed] [Google Scholar]
- 43.Wang R, Liu X, Hentges ST, Dunn-Meynell AA, Levin BE, Wang W, Routh VH. The regulation of glucose-excited neurons in the hypothalamic arcuate nucleus by glucose and feeding-relevant peptides. Diabetes 53: 1959–1965, 2004 [DOI] [PubMed] [Google Scholar]
- 44.Weiss J, Lamp ST. Metabolic regulation of cardiac ATP-sensitive K+ channels. Cardiovasc Drugs Ther 7: 499–505, 1993 [DOI] [PubMed] [Google Scholar]
- 45.Woods SC, Lotter EC, McKay LD, Porte D Jr. Chronic intracerebroventricular infusion of insulin reduces food intake and body weight of baboons. Nature 282: 503–505, 1979 [DOI] [PubMed] [Google Scholar]