

Abstract
One of the unanswered questions in muscle hypertrophy is how new contractile units are inserted into a stable existing cytoskeletal meshwork. Regulation of actin capping by CapZ may play a role in remodeling processes, therefore, CapZ dynamics are determined during rapid growth of cardiac cells in vitro. Neonatal rat ventricular myocytes were infected with adenovirus expressing green fluorescent protein-CapZ β1 and responded normally to hypertrophic stimuli. CapZ dynamics were analyzed by fluorescence recovery after photobleaching in cultured myocytes treated with endothelin-1 (100 nM) or phenylephrine (10 μM). Recovery by 30 s was greater with endothelin treatment. Analysis 30 min postbleach showed CapZ-infected cells treated with endothelin recovered more completely than controls (77 ± 9% vs. 50 ± 6%, P < 0.001). Similar results were found with phenylephrine (77 ± 5%, P < 0.05). A potential mechanism for phosphatidylinositol bisphosphate (PIP2) mediation of increased CapZ exchange in endothelin- and phenylephrine-treated cells was tested. PIP2 sequestration with neomycin (500 μM) blocked both endothelin- (43 ± 6%, P < 0.001) and phenylephrine (36 ± 4%, P < 0.001)-mediated recovery. The protein kinase C inhibitor chelerythrine chloride (10 μM) also blocked endothelin- (53 ± 10%, P < 0.001) and phenylephrine (42 ± 3%, P < 0.001)-mediated recovery. This study demonstrates for the first time that endothelin and phenylephrine alter CapZ dynamics through PIP2- and PKC-dependent pathways, which might destabilize the existing framework and permit sarcomeric remodelling to proceed.
Keywords: sarcomere remodeling, heart hypertrophy, thin filament assembly, actin binding, Z-disk
cardiac hypertrophy is an adaptive response to increased mechanical load. Cells elongate in response to increased ventricular filling by adding sarcomeres in series, and they thicken in response to increased systolic pressure by adding filaments in parallel (24). However, the precise way in which this occurs within an existing sarcomeric meshwork is still under investigation. Endothelin-1 (ET) and phenylephrine (PE) elicit both hypertrophy and cardiac remodeling through activation of Gq, phospholipase C (PLC-β), and small G proteins such as Ras, Rac, and Rho via downstream activation of mitogen-activated protein kinase signaling pathways (6, 7, 14). In experiments reported here, we tested the hypothesis that the downstream signaling involves altered dynamics of interactions of the capping protein CapZ.
The cap for the barbed end of actin filaments found in all eukaryotes is a heterodimer of α- and β-subunits. The β2 isoform is localized to focal adhesions, whereas β1, the predominant isoform expressed in striated muscle, is at the Z-disk and is thus named CapZ (26). The COOH-terminal extension is important for actin capping (35). In most cells actin filaments are capped after they have formed, but in muscle, CapZ is first localized to the Z-disk, after which it nucleates filament formation (27). A point mutation in the β1-subunit amino acid 262 from leucine to arginine occurs in the COOH-terminal extension and is thought to confer a 12-fold decrease in affinity for actin. Transgenic mice overexpressing this point mutation showed myofibrillar disarray, cardiac hypertrophy, and early death (13).
The role of CapZ in cardiac hypertrophy and remodeling may involve phosphoinositide signaling. Studies have used fluorescence recovery after photobleaching (FRAP) to interrogate cellular remodeling and the regulatory role of phosphoinositide on the dynamics of α-actinin and other actin binding proteins (10, 28, 31). Actin filament nucleation assays show a half-life for a cap to remain on the filament of 28 min, whereas the half-life to remain uncapped is about 0.2 s (28), demonstrating in homeostatic conditions that spontaneous uncapping of a filament and destabilization of the cytoskeleton is rare. Studies of myofibrillogenesis using FRAP demonstrated an importance for the binding affinity between Z-disk constituents in sarcomere formation (31). Phosphatidylinositol bisphosphate (PIP2) binding sites in actin capping protein have also been proposed based on crystallographic and total internal reflection fluorescence microscopy studies (17, 35). PIP2 binding of CapZ results in a reduction in binding affinity of the COOH-terminal extensions of the CapZ dimer subunits for the actin filament (21). Phosphoinositide binding mutants of α-actinin green fluorescent protein (GFP) fusion proteins demonstrated a dependence on phosphoinositide for disassociation of α-actinin from actin stress-fibers (11). In addition to modulation of actin capping by PIP2, there is also good evidence that ET and PE share in common the initial activation of Gq and phospholipase C (PLC-β) and have direct effects on the δ- and ɛ-isoforms of PKC (5).
The present study uses adenoviruses to express GFP and a GFP-CapZ fusion protein to examine the role of CapZ in agonist-induced hypertrophy and cytoskeletal remodeling. In addition, the role PIP2 and PKC, potential regulators of CapZ, are also evaluated. Our FRAP data indicate that signaling through endothelin and PE leading to hypertrophy involves an alteration in CapZ dynamics through PIP2 and PKC, which might destabilize the existing framework and permit sarcomeric remodelling to proceed.
EXPERIMENTAL PROCEDURES
Cell culture.
Primary heart cultures were obtained from neonatal rats according to Institutional Animal Care and Use Committee and National Institutes of Health guidelines for the care and use of laboratory animals. Hearts were removed and cells isolated from 1- to 2-day-old neonatal Sprague-Dawley rats as previously described (3). The cells were resuspended, filtered through a metal sieve to remove large material, and preplated, and enriched neonatal rat ventricular myocytes (NRVM) were finally plated at high density (1,000 cells/mm2) on fibronectin-coated glass coverslips in PC-1 medium (BioWhitaker/Cambrex). After 24 h PC-1 was replaced by Dulbecco's Modified Eagle's Medium (DMEM) (Sigma Aldrich) in a 4-to-1 ratio with M199 (GIBCO).
Adenovirus.
Recombinant adenoviruses were constructed utilizing AdEasy XL adenoviral vector system (Stratagene). The plasmid containing the cDNA of NH2-terminal enhanced GFP fusion of full-length murine CapZ β1 was generously provided by Dr. Dorothy Schafer (University of Virginia) and subcloned into pShuttle-cytomeglavirus (CMV). Resultant adenovirus was confirmed by restriction digest, sequencing, and Western analysis. Adenovirus was amplified and titered by standard techniques. Cells were infected after 24 h of culture at a multiplicity of infection of 20. Adenoviral infection was allowed to proceed for 50 min, at which time the virus containing media was aspirated off and 4:1 DMEM-M199 was added. Cells were allowed to express the transgene for 24–48 h before assaying.
Cell fixation, immunostaining, and confocal microscopy.
Cells were washed with PBS, fixed with 4% paraformaldehyde (Sigma Aldrich) for 10 min, placed in cold 70% ethanol, and stored at −20°C until immunostaining. Cells were rehydrated in phosphate-buffered saline (PBS) for 10 min and then incubated on a shaker table in 1% BSA in PBS for 1 h at 25°C. Primary α-actinin (AbCam) antibody was diluted (1:250) in 1% BSA in PBS, incubated on a shaker table at 4°C overnight, rinsed in PBS at 25°C, and blocked in 1% BSA in PBS for 1 h at 25°C. Cells were incubated for 1 h at 25°C with the secondary antibody (Molecular Probes) diluted at a ratio of 1:500 in 1% BSA in PBS. After the cells were washed in PBS, anti-fade reagent with DAPI (Molecular Probes) was added, and coverslips were mounted on glass slides for imaging by a confocal microscope (Zeiss LSM 510).
Surface area.
Cells were plated at lower density (50 cells/mm2) to visualize individual cell boundaries on glass coverslips in PC-1 media for 24 h. After 48 h of infection in PC-1, cells were either left untreated or were treated with 100 nM ET for 24 h. For determination of cell surface area, NRVM were analyzed by using 2′,7′-bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein, acetoxymethyl ester as previously described (15).
FRAP.
Cells infected with GFP-CapZ were incubated for 48 h and subsequently subjected to photobleaching (Zeiss LSM Meta). One to five beating, well-striated cells per culture were randomly selected for FRAP studies. Ar/Kr-laser power (excitation at 488 nm) was increased to 100% and GFP bleached in a square region of interest (ROI) of uniform area 5.76 μm2. The time course to recovery was followed by standard methods. Images were analyzed by Zeiss Imaging Browser software. Results were compared with an unbleached control area in the same cell. Intensities were determined according to the following equations (subscripts indicate the data point number):
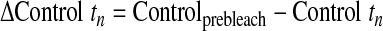 |
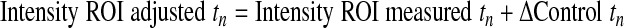 |
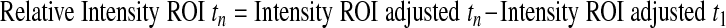 |
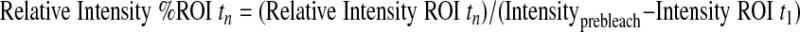 |
The relative intensities account for changes in cellular fluorescence levels during the 30-min time course of the experiment and are given as a percentage of the difference between the initial prebleach intensity and the adjusted postbleach intensity of the ROI. Curves were fit to the two-phase exponential association curve I = I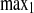 ·[1 − e
·[1 − e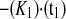 ] + I
] + I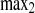 ·[1 − e
·[1 − e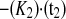 ], where I is intensity expressed as a percentage, K1 and K2 are the binding affinity constants for the first and second recovery phases expressed as inverse seconds, and t1 and t2 are the half-times to full recovery for the first and second phases expressed in seconds. K is the ratio of Koff/Kon (where Koff is the protein off-rate constant and Kon is the protein on-rate constant).
], where I is intensity expressed as a percentage, K1 and K2 are the binding affinity constants for the first and second recovery phases expressed as inverse seconds, and t1 and t2 are the half-times to full recovery for the first and second phases expressed in seconds. K is the ratio of Koff/Kon (where Koff is the protein off-rate constant and Kon is the protein on-rate constant).
Hypertrophic stimuli PIP2 and PKC inhibition.
Cells were either left unstimulated before photobleaching or were treated with either PE (Sigma-Aldrich) or ET (Sigma-Aldrich). PE was administered at a concentration of 10 μM for 24 h before photobleaching (48 h after initial plating). ET was given at a concentration of 100 nM for 4 h before FRAP analysis (68 h after plating). At least 4 h of hypertrophic stimulation was used so that maximal sarcomere remodeling could be seen, as indicated by previous work from our lab (19). PIP2 inhibition was initiated 4 h before FRAP analysis with 500 μM neomycin (Sigma-Aldrich) with the dose selected for effectiveness (1), thus added simultaneously to ET-treated cultures and 18 h following the start of PE treatment so that PIP2 inhibition would be initiated. Chelerythrine chloride (10 μM) was chosen as a broad-spectrum PKC blocker (4, 20).
Statistics.
Data are presented as means ± SE. Sample number (n) for surface area assays were defined as individual cells isolated from three different cultures done on different days. For FRAP assays sample number was defined as individual cells, of which 1–5 cells were analyzed per culture, with at least 3 separate cultures were analyzed per experimental condition. Statistical significance was determined for FRAP curves by two-way ANOVA analysis without secondary tests. Curves were analyzed as single events occurring over time to demonstrate that each curve is its own analysis condition (P < 0.0001). Additional analyses were performed on these curves at individual time points and were performed as one-way ANOVA analyses. Secondary Newman-Keuls multiple comparison tests were performed to compare individual curves. Significance was taken at P < 0.05.
RESULTS
Localization of GFP-CapZ fusion protein in vitro.
Addition of GFP to the NH2 terminus of CapZ did not interfere with its expected cellular localization at the end of the actin filament in the Z-disk of the myocyte (Fig. 1A). Moreover, CapZ colocalized with α-actinin (Fig. 1B), which is expected because CapZ and α-actinin are known binding partners at the Z-disk (21).
Fig. 1.

Wild-type (WT) GFP-CapZ fusion proteins localize appropriately to Z-disk. Cells were cultured for 24 h, infected for 1 h, and cultured for another 24 h at which point they were fixed and subjected to either staining with phalloidin for actin (A) or immunostaining for antibody for α-actinin (B). Cells stained for actin and infected with CapZ adenovirus demonstrate proper localization of CapZ at the Z-disk and actin localization adjacent to Z-disks in the I-band. Inset, higher magnification images of area delineated by the white box in the lower magnification image. Scale bar = 10 μm.
Hypertrophic response of cultured neonatal rat ventricular cardiac myocytes expressing GFP-CapZ-fusion protein.
To eliminate concerns that the virus interfered in the hypertrophic pathway, cell area was assessed after ET treatment. Surface area increased with ET treatment (878 ± 32 μm2) above control with GFP alone (704 ± 20 μm2) (P < 0.01) and for CapZ ET treatment (759 ± 25 μm2) versus control was (601 ± 17 μm2) (P < 0.01).
Hypertrophic stimulation with ET alters binding kinetics of CapZ fusion protein.
Cultured myocytes were infected with GFP or GFP-CapZ adenoviruses and exposed to ET. FRAP recovery necessitates dissociation of bound, bleached protein within the ROI, movement of unbleached protein from outside into the ROI, and finally association of this pool of protein to its binding partner(s). Bleaching of the ROI does not remove the cap but only quenches the signal. When the cap disassociates the filament is then recapped by unbleached GFP-CapZ from the cytosol. Hence, if the recovery is faster the implication is the filament was uncapped more quickly given that Koff dominates Kon (32). These events were quantified by plotting the intensity of fluorescence signal over time. GFP alone demonstrated that 80% of the bleached area is recovered in a 13-s time frame under normal conditions and with increased contractility (data not shown). In FRAP experiments with CapZ fusion protein, this early phase was characterized by a filling of the bleached ROI with a green fluorescent signal that lacks specificity (data not shown). Cells treated with PE did not initially recover as quickly as those treated with ET, suggesting that increased mixing of cellular contents due to increased cellular contraction rate is not responsible for the increase in early recovery seen in the ET-treated cells. By 30 min, recovery of ET and PE is similar, suggesting the initial rates are not the most relevant factors in determining absolute recovery and the dynamic protein exchange that is occurring in each treatment condition. These data support observations that ET-mediated signaling activation and protein synthesis are faster than that for PE treatment (9). Further credence to this is given as recovery of signal in cells treated with PE and either neomycin or chelerythrine chloride was slower than all other conditions and more specifically was slower than cells treated with ET and either of these inhibitory drugs.
The FRAP recovery profile was altered dramatically by 4 h treatment with ET (Fig. 2A). FRAP recovery of ET-treated CapZ at 30 min was 77 ± 9%, which is significantly different from the 50 ± 6% recovery reported for untreated CapZ (P < 0.001, n = 7), indicating altered binding affinity of CapZ for actin.
Fig. 2.

Fluorescence recovery after photobleaching (FRAP) recovery curves for WT GFP-CapZ fusion protein treated with endothelin (ET). FRAP analysis was performed on cells expressing WT green fluorescence protein (GFP)-tagged CapZ protein. Recovery percentages were reported as a percentage of prebleach intensity. A: cells were cultured for 24 h, infected for 1 h, and cultured for an additional 44 h at which time they were either left untreated for 4 h, treated with ET for 4 h, treated with ET and neomycin (Neo), an inhibitor of PIP2 signaling, for 4 h, or treated with ET and chelerythrine chloride (CC), an inhibitor of PKC signaling, for 4 h. B: confocal images of cells analyzed in each of the experimental conditions in A at time points immediately before photobleaching, immediately after photobleaching, and 30 min postphotobleaching.
PIP2 inhibition abrogates ET-induced alterations in CapZ binding kinetics.
The hypothesis that the ET-mediated effects on capping may in part be mediated by a PIP2-dependent process was tested with FRAP analysis using a known PIP2 scavenger neomycin (Fig. 2A). Neomycin has a high affinity for PIP2 and readily sequesters any available PIP2 in the cellular milieu, rendering it unavailable for its normal physiological roles (1). NRVM exposed to ET and neomycin for 4 h showed no ET-mediated increase in FRAP recovery rate (Fig. 2A). At 30 min the recovery of the neomycin-treated cells was 43 ± 6%, which was significantly less than the recovery of cells treated with ET alone (77 ± 9%) (P < 0.001, n = 7) and untreated cells (50 ± 6%) (P < 0.001, n = 7).
PKC blockade abrogates ET-induced alterations in CapZ binding kinetics.
The effects of PKC blockade on the FRAP recovery profile following ET treatment was assessed. Cells treated simultaneously with ET and chelerythrine chloride, a known PKC inhibitor, for 4 h demonstrated no increase in recovery rate (53 ± 10%) when compared with untreated (50 ± 6%). A decreased amount of recovery at 30 min was observed compared with ET-treated alone (P < 0.001, n = 6) (Fig. 2A). Representative images of all FRAP recovery conditions are shown, illustrating fluorescence immediately prebleach, immediately postbleach, and 30 min postbleach (Fig. 2B).
PE hypertrophic stimulation alters binding kinetics of CapZ fusion protein.
Treatment of CapZ-infected cells with PE (Fig . 3A) demonstrated FRAP recovery similar to that of the ET-treated cells (Fig. 2A). This recovery was significantly different from untreated (P < 0.05, n = 8). Yet, when compared with ET recovery, the initial rate of recovery of PE-treated myocytes was less rapid as can be seen at the 10-min time point. This may be explained by a slower time course for signaling activation and protein synthesis following PE treatment compared with that for ET (9, 22).
Fig. 3.

FRAP recovery curves for WT GFP-CapZ fusion protein treated with phenylephrine (PE). FRAP analysis was performed on cells expressing GFP-tagged CapZ protein. Recovery percentages were reported as a percentage of prebleach intensity. A: cells were cultured for 24 h, infected for 1 h, and cultured for an additional 44 h at which time they were left untreated for 4 h, treated with PE for 24 h, treated with PE for 24 h and Neo for 4 h, or treated with PE for 24 h and CC for 4 h. B: confocal images of cells analyzed in each of the experimental conditions in A at time points immediately before photobleaching, immediately after photobleaching, and 30 min postphotobleaching.
PIP2 inhibition abrogates PE-induced alterations in CapZ binding kinetics.
The hypothesis that the PE-mediated effects on capping may be partly due to PIP2 was tested by FRAP. CapZ recovery was decreased with the PE effects due to PIP2 blocked by 4 h neomycin treatment (Fig. 3A). NRVM treated with both neomycin and PE showed a 36 ± 4% recovery after 30 min; a significant decline compared with cells treated with PE alone (P < 0.001, n = 11) and untreated CapZ cells (P < 0.001, n = 8).
PKC blockade abrogates PE-induced alterations in CapZ binding kinetics.
FRAP analysis showed that the enhanced CapZ binding affinity demonstrated by PE treatment is also mediated by PKC, since with PKC blockade recovery is (42 ± 3%) compared with NRVM treated with PE alone (P < 0.001, n = 10) and untreated (P < 0.001, n = 8) (Fig. 3A). Representative images of FRAP recovery conditions are shown in Fig. 3B and illustrate fluorescence immediately prebleach, immediately postbleach, and 30 min postbleach.
DISCUSSION
One of the unanswered questions in muscle hypertrophy is how new contractile units are inserted into a stable existing cytoskeletal meshwork. This study demonstrates for the first time that treatment of NRVM with hypertrophic agonists results in altered steady-state equilibrium of CapZ dynamics. Decreases in binding affinity for CapZ to actin and other proteins following both ET and PE treatment are presumably dominated by changes in Koff with its fourfold order of magnitude difference in wild-type capping protein (32). Furthermore, ET and PE elicited increased exchange of CapZ protein via PIP2- and PKC-dependent pathways. It should be pointed out, however, that addition of PE or ET to the culture medium of low-density NRVM cultures can elicit calcium oscillations and “spontaneous” contractile activity. This induction of intracellular Ca2+ concentration ([Ca2+]i) transients and/or mechanical activity may have directly or indirectly contributed to the alteration in CapZ dynamics. Electrical pacing-induced calcium transients, and beating activity are known to induce PKC-δ and PKC-ɛ translocation and to increase sarcomeric assembly and hypertrophic growth independently of G protein-coupled receptor activation (29). Thus some of the effects of PE and ET treatment on CapZ dynamics may be a consequence of alterations in [Ca2+]i and mechanical activity. Nevertheless, modulation of actin capping by CapZ dynamics may play a role in sarcomere remodeling whereby the stability of the existing sarcomeric framework is altered by agonist or mechanical stimulation to allow insertion of filament proteins during the induction of cellular hypertrophy.
Myocytes contain phosphatidylinositol-4-phosphate-5-OH-kinase that localizes with α-actinin at the sarcomeric Z-disk that overlay with titin T12 close to the Z-disk (36).
PIP2 is thought to regulate α-actinin incorporation and actin stress fiber formation during hypertrophy (8–12, 36), and mutations in PIP2 binding regions of α-actinin cause abnormal actin filament formation. In platelets, the half-life for a capped filament is 28 min and the half-life for a filament to remain uncapped is 0.2 s, but with the addition of PIP2 the half-life of the capped filament in stimulated platelets is reduced to 46 s (28). A recent study suggests that PIP2 effects on capping do not occur through a direct PIP2-to-CapZ interaction but through an interaction of PIP2 and actin-related protein permitting a more complex filament bundling to occur (18). This fits nicely with previous studies suggesting that sarcomerogenesis is accomplished by insertion of new sarcomeres (37). PIP2 may bind CapZ at conserved basic groups facilitating disassociation of capping protein by preventing the CapZ α-subunit interaction with actin, leaving the wobble of the β-subunit (17). COOH-terminal extensions of Cap Z interact with adjacent actin monomers allowing β-subunit and α-subunit release by interaction with PIP2 (16, 17, 32, 33). PIP2 may inhibit unbound CapZ from capping existing filaments, but Schafer et al. (28) explicitly states kinetic simulations do not support this and instead support a model where PIP2 binding of actin-bound capping protein promotes its disassociation from the filament. In other cell types agonist stimulation elicits PIP2-mediated alterations in actin binding protein affinity for actin permitting regulation of shape, which is dynamically regulated, and other cellular functions (2). The present study with agonist stimulation shows that a similar phenomenon may occur in cardiac myocyte hypertrophy, whereby PIP2 alters the dynamic protein exchange of the sarcomeric cytoskeletal protein CapZ β1. Increasing the off-rate of CapZ may destabilize the Z-disk permitting filament remodeling.
PKC phosphorylates several sarcomeric proteins and activates multiple transcriptional pathways in response to hypertrophic stimuli of ET and PE (7). The present study suggests a role for PKC whereby it decreases affinity of CapZ for the actin filament. This is supported by the observation that blockade of PKC following agonist treatment slows the disassociation of CapZ from actin compared with agonist treatment alone. Although chelerythrine may have actions other than PKC inhibition, in a previous study (20), we compared the effectiveness of chelerythrine to calphostin C in inhibiting PKC. Both inhibitors gave the same result. Muscle bundles with reduced CapZ levels have increased calcium sensitivity, reduced PKC-β, and in the presence of ET and PE, decreased PKC-ɛ at the myofilament suggesting interplay between PKC activity levels and CapZ function (22). CapZ is a factor in anchoring PKC-β to the myofilament (23). Disassociation of CapZ from the filament could perhaps explain how PKC-β overexpression leads to cardiac hypertrophy and failure (30). Results here complement those studies, demonstrating the importance of CapZ in cellular remodeling, sarcomere assembly, and the hypertrophic response via a PKC-dependent process.
Previous work by our group has focused on the cellular processes and signaling cascades that occur during the sarcomeric remodeling steps characteristic of both physiological and pathological muscle hypertrophy. We previously demonstrated a requirement for focal adhesion kinase and PKC in the length remodeling process (19). The present study suggests a complementary process by which myocyte exposure to ET and PE lead to hypertrophy by altering the binding affinity of capping protein for the actin filament, destabilizing the existing framework and allowing insertion of proteins-an alteration conducive to the remodeling process perhaps involving the dynamic nature of Z-band proteins (25).
GRANTS
This work was supported by National Heart, Lung, and Blood Institute HL-62426.
Acknowledgments
We thank Dr. Dorothy Schafer for the gift of the CapZ DNA constructs and Dr. Mei-Ling Chen for technical assistance with the FRAP experiments.
REFERENCES
- 1.Anderson KE, Dart AM, Woodcock EA. Inositol phosphate release and metabolism during myocardial ischemia and reperfusion in rat heart. Circ Res 76 261–268, 1995. [DOI] [PubMed] [Google Scholar]
- 2.Barkalow K, Witte W, Kwiatkowski DJ, Hartwig JH. Coordinated regulation of platelet actin filament barbed ends by gelsolin and capping protein. J Cell Biol 134: 389–399, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boateng SY, Hartman TJ, Ahluwalia N, Vidula H, Desai TA, Russell B. Inhibition of fibroblast proliferation in cardiac myocyte cultures by surface microtopography. Am J Physiol Cell Physiol 285: C171–C182, 2003. [DOI] [PubMed] [Google Scholar]
- 4.Chmura SJ, Dolan ME, Cha A, Mauceri HJ, Kufe DW, Weichselbaum RR. In vitro and in vivo activity of protein kinase C inhibitor chelerythrine chloride induces tumor cell toxicity and growth delay in vivo. Clin Can Res 6: 737–742, 2000. [PubMed] [Google Scholar]
- 5.Clerk A, Bogoyevitch MA, Andersson MB, Sugden PH. Differential activation of protein kinase C isoforms by endothelin-1 and phenylephrine and subsequent stimulation of p42 and p44 mitogen-activated protein kinases in ventricular myocytes cultured from neonatal rat hearts. J Biol Chem 269: 32845–32857, 1994. [PubMed] [Google Scholar]
- 6.Clerk A, Kemp TJ, Harrison JG, Mullen AJ, Barton PJR, Sugden PH. Up-regulation of c-jun mRNA in cardiac myocytes requires the extracellular signal-regulated kinase cascade, but c-Jun N-terminal kinases are required for efficient up-regulation of c-Jun protein. Biochem J 368: 101–110, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dorn GW, Force T. Free in PMC protein kinase cascades in the regulation of cardiac hypertrophy. J Clin Invest 115: 527–537, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eble DM, Qi M, Waldschmidt S, Lucchesi PA, Byron KL, Samarel AM. Contractile activity is required for sarcomeric assembly in phenylephrine-induced cardiac myocyte hypertrophy. Am J Physiol Cell Physiol 274: C1226–C1237, 1998. [DOI] [PubMed] [Google Scholar]
- 9.Eskildsen-Helmond Y, Bezstarosti K, Dekkers DH, van Heugten HA, Lamers JM. Cross-talk between receptor-mediated phospholipase C-beta and D via protein kinase C as intracellular signal possibly leading to hypertrophy in serum-free cultured cardiomyocytes. J Mol Cell Cardiol 29: 2545–2559, 1997. [DOI] [PubMed] [Google Scholar]
- 10.Fraley TS, Pereira CB, Tran TC, Singleton CA, Greenwood JA. Phosphoinositide binding regulates alpha-actinin dynamics: mechanism for modulating cytoskeletal remodeling. J Biol Chem 280: 15479–15482, 2005. [DOI] [PubMed] [Google Scholar]
- 11.Fukami K, Endo T, Imamura M, Takenawa T. Alpha-Actinin and vinculin are PIP2-binding proteins involved in signaling by tyrosine kinase. J Biol Chem 269: 1518–1522, 1994. [PubMed] [Google Scholar]
- 12.Fukami K, Sawada N, Endo T, Takenawa T. Identification of a phosphatidylinositol 4,5-bisphosphate-binding site in chicken skeletal muscle alpha-actinin. J Biol Chem 271: 2646–2650, 1996. [DOI] [PubMed] [Google Scholar]
- 13.Hart MC, Cooper JA. Vertebrate isoforms of actin capping protein beta have distinct functions in vivo. J Cell Biol 147: 1287–1298, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heidkamp MC, Bayer AL, Scully BT, Eble DM, Samarel AM. Activation of focal adhesion kinase by protein kinase C epsilon in neonatal rat ventricular myocytes. Am J Physiol Heart Circ Physiol 285: H1684–H1696, 2003. [DOI] [PubMed] [Google Scholar]
- 15.Heidkamp MC, Iyengar R, Szotek EL, Cribbs LL, Samarel AM. Protein kinase cepsilon-dependent MARCKS phosphorylation in neonatal and adult rat ventricular myocytes. J Mol Cell Cardiol 42: 422–431, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim K, Yamashita A, Wear MA, Maeda Y, Cooper JA. Capping protein binding to actin in yeast: biochemical mechanism and physiological relevance. J Cell Biol 164: 567–580, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim K, McCully ME, Bhattacharya N, Butler B, Sept D, Cooper JA. Structure/function analysis of the interaction of phosphatidylinositol 4,5-bisphosphate with actin-capping protein: implications for how capping protein binds the actin filament. J Biol Chem 282: 5871–5879, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuhn JR, Pollard TD. Single molecule kinetic analysis of actin filament capping. Polyphosphoinositides do not dissociate capping proteins. J Biol Chem 282: 28014–28024, 2007. [DOI] [PubMed] [Google Scholar]
- 19.Mansour H, de Tombe PP, Samarel AM, Russell B. Restoration of resting sarcomere length after uniaxial static strain is regulated by protein kinase Cepsilon and focal adhesion kinase. Circ Res 94: 642–649, 2004. [DOI] [PubMed] [Google Scholar]
- 20.Montgomery DE, Chandra M, Huang Q, Jin J, Solaro RJ. Transgenic incorporation of skeletal TnT into cardiac myofilaments blunts PKC-mediated depression of force. Am J Physiol Heart Circ Physiol 280: H1011–H1018, 2001. [DOI] [PubMed] [Google Scholar]
- 21.Papa I, Astier C, Kwiatek O, Raynaud F, Bonnal C, Leb art MC, Roustan C, Benyamin Y. Alpha actinin-CapZ, an anchoring complex for thin filaments in Z-line. J Muscle Res Cell Motil 20: 187–197, 1999. [DOI] [PubMed] [Google Scholar]
- 22.Pyle WG, Hart MC, Cooper JA, Sumandea MP, de Tombe PP, Solaro RJ. Actin capping protein: an essential element in protein kinase signaling to the myofilaments. Circ Res 90: 1299–1306, 2002. [DOI] [PubMed] [Google Scholar]
- 23.Pyle WG, La Rotta G, de Tombe PP, Sumandea MP, Solaro RJ. Control of cardiac myofilament activation and PKC-betaII signaling through the actin capping protein, CapZ. J Mol Cell Cardiol 41: 537–543, 2006. [DOI] [PubMed] [Google Scholar]
- 24.Russell B, Motlagh D, Ashley WW. Form follows function: how muscle shape is regulated by work. J Appl Physiol 88: 1127–1132, 2000. [DOI] [PubMed] [Google Scholar]
- 25.Sanger JM, Sanger JW. The dynamic Z bands of striated muscle cells. Sci Signal 1: pe37, 2008. [DOI] [PubMed]
- 26.Schafer DA, Korshunova YO, Schroer TA, Cooper JA. Differential localization and sequence analysis of capping protein beta-subunit isoforms of vertebrates. J Cell Biol 127: 453–465, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schafer DA, Hug C, Cooper JA. Inhibition of CapZ during myofibrillogenesis alters assembly of actin filaments. J Cell Biol 128: 61–70, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schafer DA, Jennings PB, Cooper JA. Dynamics of capping protein and actin assembly in vitro: uncapping barbed ends by polyphosphoinositides. J Cell Biol 135: 169–179, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Strait JB, Samarel AM,. Isoenzyme-specific protein kinase C and c-Jun N-terminal kinase activation by electrically stimulated contraction of neonatal rat ventricular myocytes. J Mol Cell Cardiol 32: 1553–1566, 2000. [DOI] [PubMed] [Google Scholar]
- 30.Wakasaki H, Koya D, Schoen FJ, Jirousek MR, Ways DK, Hoit BD, Walsh RA, King GL. Targeted overexpression of protein kinase C beta2 isoform in myocardium causes cardio myopathy. Proc Natl Acad Sci USA 94: 9320–9325, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Shaner J, N, Mittal B, Zhou Q, Chen J, Sanger JM, Sanger JW. Dynamics of Z-band based proteins in developing skeletal muscle cells. Cell Motil Cytoskeleton 61: 34–48, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wear MA, Yamashita A, Kim K, Maeda Y, Cooper JA. How capping protein binds the barbed end of the actin filament. Curr Biol 13: 1531–1537, 2003. [DOI] [PubMed] [Google Scholar]
- 33.Wear MA, Cooper JA. Capping protein: new insights into mechanism and regulation. Trends Biochem Sci 29: 418–428, 2004. [DOI] [PubMed] [Google Scholar]
- 34.Wear MA, Cooper JA. Capping protein binding to S100B: implications for the tentacle model for capping the actin filament barbed end. J Biol Chem 279: 14382–14390, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yamashita A, Maeda K, Maeda Y. Crystal structure of CapZ: structural basis for actin filament barbed end capping. EMBO J 22: 1529–1538, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Young P, Gautel M. The interaction of titin and alpha-actinin is controlled by a phospholipid-regulated intramolecular pseudoligand mechanism. EMBO J 19: 6331–6340, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu JG, Russell B. Cardiomyocyte remodeling and sarcomere addition after uniaxial static strain in vitro. J Histochem Cytochem 53: 839–844, 2005. [DOI] [PubMed] [Google Scholar]