

Abstract
Serine palmitoyltransferase (SPT) catalyzes the condensation of l-serine and palmitoyl-CoA, which is the rate-limiting step in the de novo synthesis of sphingolipids. SPT activity is commonly measured by monitoring the incorporation of radiolabeled l-serine into 3-ketodihydrosphingosine. In this article, we introduce several adaptations of the established protocol to improve sensitivity, reproducibility, and practicability of the assay. A significant improvement of this new protocol is the possibility to measure SPT activity in total cell lysate instead of microsomes. The assay is furthermore extended by the introduction of a nonradioactive, HPLC-based detection protocol. The suggested HPLC method offers several advantages, most importantly, a 20-fold lower detection limit compared with the radioactive assay and the possibility to use an internal standard to correct for variation in the extraction.
Keywords: sphingolipids, high-performance liquid chromatography, acyl-CoA thioesterases
Serine palmitoyltransferase (SPT) (EC 2.3.1.50) catalyzes the first, rate-limiting step in the de novo synthesis of sphingolipids: the condensation of palmitoyl-CoA and l-serine to 3-ketodihydrosphingosine (3KDS) (1, 2).
Sphingolipids are bioactive metabolites and modulate the activities of various enzymes, such as protein kinases, protein phosphatases, and phospholipases in cells or cell-free systems (3). They are involved in many cellular events, including proliferation, differentiation, senescence, apoptosis, and inflammatory responses (4). Abnormalities in the function of SPT cause clinical disorders, such as hereditary sensory neuropathy type I (5, 6). Recent studies also indicate that human plasma sphingolipid levels are an independent risk factor for coronary artery disease (7) and that lowering sphingolipid levels by pharmaceutical interventions can reduce the risk for atherogenesis in apolipoprotein E knockout mice (8). Other data indicate that sphingolipids are involved in the pathological aggregation of amyloid-β fragments, which is a key process in the development of Alzheimer's disease (9).
To address these issues on a biochemical level, a robust, sensitive, and reproducible SPT activity assay would be desirable. SPT activity is usually determined by the method described by Merrill (10) and Williams, Wang, and Merrill (11) using [3H] or [14C]l-serine as a substrate, or variants thereof.
In this work, we looked at various aspects of this method and suggest several adaptations for simplification and to improve assay precision and sensitivity. We furthermore provide a protocol for a standardized SPT activity assay to improve reproducibility and interassay comparability.
EXPERIMENTAL PROCEDURES
All chemicals, unless otherwise stated, were purchased from Sigma-Aldrich (St. Louis, MO). l-[U-14C]serine was from GE Healthcare (Little Chalfont, Buckinghamshire, UK). C17- and C20-sphingosine were from Avanti Polar Lipids (Alabaster, AL).
Photometric assays
Thioesterase activity was measured photometrically by monitoring the reaction of free CoA with DTNB at 412 nm in a spectrophotometer (12). Concentrations were determined with a molar absorption coefficient of 13.6 mM−1 cm−1 for the 2-nitrobenzoate anion. The assay typically contained 50 μM palmitoyl-CoA, 100 μM DTNB, and the corresponding amount of cells as stated in the text.
The influence of DTT on the release of free CoA from the substrate was determined enzymatically using pyruvate dehydrogenase. The assay contained 50 mM HEPES (pH 8), 50 μM palmitoyl-CoA, 1 mU pyruvate dehydrogenase, 1 mM sodium pyruvate, 2.5 mM thiamine pyrophosphate, 2.5 mM MgCl2, 2.5 mM NAD+, 100 μM NADH, 0.5 mg/ml BSA, 50 mM sodium oxamate (to inhibit the lactate dehydrogenase), and the corresponding amount of DTT. NADH oxidation was monitored spectrophotometrically at a wavelength of 340 nm using an absorption coefficient of 6.22 mM−1 cm−1. Background reactions were monitored in control samples without palmitoyl-CoA.
Photometric assays were usually performed in a 96-well polystyrene flat-bottom microtiter plate (Nunc, Roskilde, Denmark) and measured using a microplate spectrophotometer (PowerWave 340; BioTek, Winooski, VT).
Sample preparation, SPT reaction, and lipid extraction
Human Embryonic Kidney cells (HEK293) were grown in DMEM supplemented with 10% fetal calf serum, penicillin G (100 U/ml), and streptomycin (100 μg/ml). Cultures were maintained at 37°C in a 5% CO2 atmosphere at 100% humidity. Cell membrane microsomes were prepared as follows: A HEK293 cell monolayer in a 10-cm dish was washed twice with 3 ml of PBS and suspended in 500 μl of 50 mM HEPES (pH 8) and 1 mM EDTA. The cell suspension was sonicated for 15 s at 50% power and 50% pulsation (Sonopuls HD 2070; Bandelin, Berlin, Germany) and centrifuged at 2,500 g for 2 min. The supernatant was then centrifuged at 100,000 g for 30 min, and the pellet was resuspended in the desired buffer (usually 50 mM HEPES, pH 8, and 1 mM EDTA). Preparation of total cell lysate, SPT reaction, and lipid extraction were performed as described in the Appendix. Protein determinations were performed according to Bradford (13), using the protein assay kit from Bio-Rad (Hercules, CA).
For some experiments a prokaryotic, soluble form of SPT from Sphingomonas paucimobilis was used (14). The protein was expressed in Escherichia coli (BL21 star (DE3)) using the pET expression system (pET21b, Invitrogen, Carlsbad, CA) according to the manufacturer's protocol. Cells were lysed in 50 mM HEPES (pH 8), 5 mM EDTA by sonification and stored at -20°C in 50% glycerol.
Detection of radioactivity by liquid scintillation counting
Radioactivity was determined using Ultima Gold (Perkin-Elmer, Wellesley, MA) liquid scintillation cocktail and a Packard Tri-Carb 1900TR (Perkin-Elmer) scintillation counter.
Product detection by HPLC
The HPLC system consisted of a LC-10Ai solvent delivery module (Shimadzu, Kyoto, Japan), SIL-10ADvp automatic injector (Shimadzu), SCL-10Avp system controller (Shimadzu), and a 1046A fluorescence detector (Hewlett-Packard, Palo Alto, CA). Peaks were analyzed and quantified with VP-CLASS (version 6.14 SP4).
RESULTS AND DISCUSSION
Influence of DTT on palmitoyl-CoA stability
The addition of DTT is considered to have a beneficial effect on SPT activity by protecting active site Cys residues from oxidation. However, the presence of DTT might also influence the stability of palmitoyl-CoA due to the reduction of the thioester bond in activated fatty acids. To determine the effect of DTT on substrate stability, we incubated palmitoyl-CoA with DTT in concentrations from 0 to 10 mM. The DTT-dependent release of free CoA is illustrated in Fig. 1. DTT concentration of 5 mM, as it was suggested originally by Merrill (10) and Williams, Wang, and Merrill (11), did not significantly influence palmitoyl-CoA concentrations within 10 min of incubation. However, ∼50% of the palmitoyl-CoA was reduced after 60 min. This considerable reduction in substrate concentration has to be considered when performing the assay with longer incubation times. Although the presence of DTT might have a beneficial effect, we never observed a reduced SPT activity due to oxidation, even in samples that were stored up to 6 months at −20°C. Therefore, we believe that the presence of DTT in the assay is not essential to maintain SPT activity. However, if DTT is desired, the concentration should be limited to 0.1–0.5 mM.
Fig. 1.

Reduction of palmitoyl-CoA as a side reaction in the presence of different DTT concentrations. The release of free CoA was monitored via the pyruvate dehydrogenase reaction as described in the Experimental Procedures.
Sample preparation and acyl-CoA thioesterases
SPT activity is usually determined in microsomal preparations. This results in an improved specific activity of the enzyme since cytoplasmatic proteins are largely removed during the microsomal preparation. More important, the use of microsomes also reduces the number of interfering enzymes like acyl-CoA thioesterases or nonspecific hydrolases that compete with the SPT for the substrate palmitoyl-CoA. However, the preparation of microsomes is time-consuming, requires an ultracentrifuge, and needs rather large amounts of tissue and cells. Furthermore, the resolubilization of the pelleted microsomal membranes in the absence of a detergent is critical and results frequently in inhomogeneities of the membrane suspensions. The possibility to measure SPT activity directly in cell lysate would significantly simplify these preparative steps. However, the major obstacle for the use of cell lysate is the presence of highly active acyl-CoA thioesterases that results in a fast hydrolyzation of the substrate palmitoyl-CoA (11). Acyl-CoA thioesterases are a ubiquitously expressed family of enzymes that show high activities in the cytosol of most eukaryotic cells. Their exact physiological role is not fully clear, but probably important to maintain appropriate intracellular levels of acyl-CoA, free CoA, and free fatty acids (15).
To reduce this perturbing background activity, we aimed to block the thioesterase activity without affecting SPT activity. Thioesterase activity is influenced by several detergents, some lipids, and divalent ions like Mn2+ and Ni2+ (16–19). We investigated several substances in regard to their inhibitory effect on thioesterases and their interference with SPT activity. We found that sucrose monolaurate (SML) showed the best inhibitory effect on thioesterase activity without a detectable inhibitory effect on SPT activity (Table 1). The residual thioesterase activity in cell lysate with 0.1% SML was comparable to the thioesterase activity in microsomes (Fig. 2A). SPT activity in microsomes was 50 pmol/min/mg, which is in the range of previously reported results (2.6 to 127 pmol/min/mg in different rat tissues) (11, 20, 21). SPT activity in total cell lysate without any additions was about 6-fold lower. The addition of 0.1% SML significantly improved SPT activity, and the activity was comparable to microsomes. In the presence of 0.1% SML, the reaction was linear for up to 60 min (Fig. 2B). Therefore, the assay can be performed over a longer time span, which leads to more product and an improved signal-to-noise ratio. The addition of 0.1% SML to the microsomal preparation did not further increase SPT activity.
TABLE 1.
Specific SPT activity in various lysate preparations of HEK293 cells
| Cell preparation | Thioesterase Inhibitor | SPT Activity [pmol/min/mg Total Protein]a | |
|---|---|---|---|
| Total cell lysate | None | 2.4 ± 0.3b | 2.4 ± 0.3c |
| 0.1% SML | 12.4 ± 1.6b | 12.4 ± 1.6c | |
| Microsomes | None | 14.0 ± 2.8b | 50.3 ± 10.2c |
| 0.1% SML | 12.9 ± 1.2b | 46.5 ± 4.4c | |
| Supernatant (100,000 g) | None | Not assayed | Not assayed |
| 0.1% SML | 0.3 ± 0.4b | 0.5 ± 0.6c | |
Activities were determined using the radioactivity-based assay.
Mean ± standard deviation, n = 3.
SPT activity normalized to the total amount of extracted protein.
SPT activity normalized to protein amount of the assayed fraction.
Fig. 2.

A: SPT and acyl-CoA thioesterase activities in total lysate and microsomes prepared from HEK293 cells in the presence and absence of 0.1% SML. Low SPT activity and high thioesterase activities were seen in total lysate in the absence of SML. The addition of 0.1% SML increased SPT activity 6-fold and reduced acyl-CoA thioesterase activities by 90%. SPT activity in total lysate in the presence of 0.1% SML was similar to the activity in microsomes. The addition of 0.1% SML to the microsomal preparation did not further improve SPT activity. B: The SPT reaction in microsomes and total cell lysate + 0.1% SML was linear over 60 min. SPT activity in total cell lysate is reduced by 80% in the absence of SML. SPT activity was assayed using [14C]l-serine as described in the Appendix. For comparability, the activities were normalized to total amount of extracted protein. n = 3.
A comparison of SPT activity in the various preparations of HEK293 cells is summarized in Table 1.
Optimal substrate concentration
The Km for l-serine was reported to be in the range of 0.1 to 1.8 mM for the mammalian SPT (11, 22–24). We redetermined Km and Vmax with the new assay conditions (Fig. 3). l-Serine shows a regular Michaelis-Menten kinetics, while palmitoyl-CoA shows an inhibitory effect at higher substrate concentrations. This substrate inhibition was already reported earlier for the mammalian (11, 25, 26) and plant enzyme (24). The maximum of activity was seen at a concentration of ∼0.05–0.1 mM palmitoyl-CoA (Fig. 3B). A Km of 1.2 mM for l-serine was determined by an Eadie-Hofstee plot. This shows a rather low affinity of SPT toward its substrate l-serine. Commonly, the SPT assay is performed at l-serine concentrations between 0.5 and 1 mM. This is far from saturating conditions that are required to measure enzyme activity at its maximal velocity (Vmax). However, increasing l-serine concentrations in the assay bears the disadvantage that the amount of radioactive l-serine also must be increased to maintain the specific radioactivity of the substrate. This is often unfavorable due to cost, health, and security reasons. We therefore aimed to establish a nonradioactive, HPLC-based detection method to circumvent the need for radioactivity and to perform the assay under optimal substrate conditions. The HPLC detection is based on the method of Riley et al. (27). Because 3KDS, as the direct product of the SPT reaction, cannot be efficiently detected with this method, we converted the 3KDS chemically with sodium borohydride (NaBH4) to sphinganine. This reaction is not stereospecific and results in the formation of erythro- and threo-sphinganine. These two products can be derivatized with ortho-phthalaldehyde (OPA) and quantified by HPLC (Fig. 4). Total SPT activity is reflected by the sum of both diastereoisomers. In the negative control, SPT activity was specifically blocked by the addition of myriocin, a commonly used SPT inhibitor (28). Trace amounts of free sphinganine might also be found in the control extract, so that the net SPT activity is calculated by the difference between sample and control.
Fig. 3.

Influence of substrate concentration on SPT activity. SPT activity was determined using the HPLC-based assay as described in the Appendix. For A, palmitoyl-CoA concentrations were kept at 50 μM, and for B, l-serine concentrations were kept at 2 mM. Error bars indicate standard deviation. n = 3.
Fig. 4.

HPLC chromatograms of threo-sphinganine (thr-Sa), erythro-sphinganine (ery-Sa), and C20-sphingosine (C20). A: Standard solutions: 100 pmol threo-/erythro-sphinganine and 100 pmol C20-sphingosine. B: Chromatograms of a SPT activity assay with (dashed line) and without (solid line) the addition of NaBH4. C: Overview of the SPT reaction and the following reduction of 3KDS to sphinganine.
The detection by HPLC might be biased because the SPT product, 3KDS, also could potentially get acylated by the ceramide synthase. The diacyl form of 3KDS would not be visible in the HPLC because the primary amine is blocked by the N-linked fatty acid and cannot form a fluorescent derivative with OPA. To exclude this possibility, we performed the assay in the presence and absence of fumonisin B1, a specific inhibitor of ceramide synthase (29). We found that the presence of fumonisin B1 had no influence on the detected sphinganine levels (data not shown).
The limit of detection (LOD) for the radioactivity- and HPLC-based method was compared. The LOD was defined as the activity for which the standard deviation exceeds 20%. Under the chosen condition, the LOD for the HPLC-based detection was 0.1 pmol/min versus 2 pmol/min for the radioactive assay. This means that the detection limit of the HPLC-based method is ∼20 times lower compared with the radioactive method.
However, both methods showed a very close correlation (r2 = 0.9519) (Fig. 5B). This demonstrates that the product determination by HPLC is as specific as the radioactive assay and not biased by the presence of an underlying metabolite that might be misinterpreted in the HPLC chromatogram. A further important advantage of the HPLC-based detection is the possibility to add an internal standard to correct for variations in the preparative steps. We used C20-sphingosine or preferably C17-sphingosine, a nonnaturally occurring sphingoid base, as an internal standard. Although the HPLC-based method shows significant improvements in terms of sensitivity and variability, it is on the other hand more laborious and time-consuming in comparison to the radioactive assay.
Fig. 5.

LOD and correlation. A: For a maximal assay variance of 20% (dotted line), the HPLC method had a LOD of 0.1 pmol/min and the [14C]l-serine based assay a LOD of 2 pmol/min. The HPLC-based detection was therefore ∼20 times more sensitive compared with the radioactive assay. B: Correlation of the radioactivity- and HPLC-based detection method. Activity assay was performed according to the method described in the Appendix followed by the addition of NaBH4. The final chloroform phase was split and either analyzed on the scintillation counter or by the described HPLC method. Both methods showed a close linear correlation with a correlation coefficient of r2 = 0.9519.
CONCLUSIONS
In this work, we aimed to improve various aspects of the commonly used method to determine SPT activity. One of the major improvements is the possibility to measure activity directly in total cell lysate. We showed that the presence of 0.1% SML significantly inhibits the thioesterase background activity that interferes with the SPT activity measurements. Under these conditions, SPT activity in the total cell lysate was similar to that in microsomes. This adaptation allows it to perform the assay with significantly smaller amounts of tissue and cells. The addition of 0.1% SML also significantly improves the linearity of the reaction. In the presence of 0.1% SML, SPT activity was linear for up to 60 min, which resulted in an improved signal-to-noise ratio. We furthermore identified a negative effect of DTT on the stability of palmitoyl-CoA and suggested to reduce or omit the addition of DTT to the assay. Finally, we introduced a nonradioactive, HPLC-based detection method, which makes it possible to measure SPT activity under saturating substrate concentration. This HPLC-based method shows a 20-fold lower detection limit in comparison to the radioactive detection.
We think that these adaptations lead to significant improvements in the robustness, sensitivity, and reproducibility of the SPT activity assay.
Acknowledgments
The authors thank the Hartmann Müller Foundation, the Herzog-Egli Foundation, the EMDO Foundation, the Foundation for Scientific Research (University of Zurich), and the 6th Framework Programme of the European Commission (LSHM-CT-2006-037631) for financially supporting this work. We also thank H. Kagamiyama for providing us with the expression plasmid of the soluble SPT from S. paucimobilis.
Abbreviations
3KDS, 3-ketodihydrosphingosine
LOD, limit of detection
OPA, ortho-phthalaldehyde
SML, sucrose monolaurate
SPT, serine palmitoyltransferase
APPENDIX: SAMPLE PROTOCOL FOR AN IMPROVED SPT ACTIVITY ASSAY
This protocol describes an improved assay to determine SPT activity. Two different detection methods are suggested:
Radioactivity-based assay: [14C]labeled l-serine is incorporated into 3KDS, which is quantified by liquid scintillation counting.
HPLC-based assay: 3KDS is chemically reduced by NaBH4 to threo- and erythro-sphinganine, whose OPA derivatives are separated by HPLC and quantified by fluorescence detection.
Reaction conditions
Reaction buffer: 50 mM HEPES (pH 8) containing 1 mM EDTA and 0.1% (w/v) SML
5 mM l-serine (HPLC-based assay) or 0.5 mM l-serine (radioactivity-based assay)
50 μM palmitoyl-CoA
20 μM pyridoxal 5′-phosphate
1. General reagents
a. Stock solutions: l-serine (200 mM), palmitoyl-CoA (5 mM), pyridoxal 5′-phosphate (5 mM), HEPES-NaOH (0.5 M, pH 8), EDTA (0.5 M, pH 8), SML [10% (w/v)], myriocin solution (1 mg/ml in methanol), NH4OH (2 N).
b. Alkaline water: 0.1 ml NH4OH (2 N) in 100 ml of water.
c. Methanolic KOH: dissolve 0.7g KOH (platelets) in 100 ml of methanol.
d. Methanol/KOH:CHCl3 (4:1): mix 4 vol of methanolic KOH with 1 vol of CHCl3; prepare fresh immediately before use.
e. Lysis buffer: Mix 1 ml of HEPES-NaOH (0.5 M, pH 8), 20 μl of EDTA (0.5 M, pH 8), and 100 μl of SML [10% (w/v)]. Bring to a volume of 10 ml with water.
The following reagents are only required for the radioactivity-based assay:
f. l-[U-14C]serine (50 μCi/ml)
g. 20× [14C] assay mix (including [14C]l-serine): example for 250 μl (suitable for 25 samples):
Mix 12.5 μl of l-serine (200 mM), 20 μl of pyridoxal 5′-phosphate (5 mM), 50 μl of palmitoyl-CoA (5 mM), 100 μl of l-[U-14C]serine (50 μCi/ml), and 68 μl of water.
h. Liquid scintillation cocktail (e.g., Ultima Gold; Perkin-Elmer)
The following reagents are only required for the HPLC-based assay:
i. 20× HPLC assay mix: example for 250 μl (suitable for 25 samples):
Mix 125 μl of l-serine (200 mM), 20 μl of pyridoxal 5′-phosphate (5 mM), 50 μl of palmitoyl-CoA (5 mM), and 55 μl of water.
j. NaBH4 (5 mg/ml in water): prepare immediately before use.
k. Sphingolipid standard solutions: C17-sphingosine (1 mM in ethanol), threo-/erythro-sphinganine (1 mM in ethanol)
l. HPLC standard mix: mix 5 μl of C17-sphingosine (1 mM in ethanol) and 5 μl of threo-/erythro-sphinganine (1 mM in ethanol). Bring to a volume of 7.5 ml with methanol:ethanol:water (85:47.5:17.5).
m. Methanol:CHCl3:0.1 M KOH + C17-Sphingosine (internal standard): Add 0.2 μl of C17-sphingosine (1 mM in ethanol) per ml methanol/KOH:CHCl3. 0.5 ml is needed per sample. Prepare always fresh immediately before use.
n. Boric acid [3% (w/v), pH 10.5]: adjust pH with KOH as necessary. Check pH before use.
o. OPA (50 mg/ml in ethanol)
p. OPA reagent: mix 10 μl OPA (50 mg/ml in ethanol), 990 μl boric [3% (w/v), pH 10.5)], and 0.5 μl of 2-mercaptoethanol. Store in dark, 4°C. Prepare fresh daily.
q. Potassium phosphate buffer (5 mM, pH 7): mix 5 mM K2HPO4 (0.87 g in 1,000 ml of water) and 5 mM KH2PO4 (0.68 g in 1000 ml of water) in a ratio of 61:39 (v/v) and adjust pH as necessary.
r. Running buffer for HPLC: methanol:5 mM potassium phosphate buffer (pH 7) (90:10, v/v)
2. Preparation of total cell lysate
All steps at 4°C
a. Cell monolayer in a 10-cm dish (80–90% confluent)
b. After washing twice with PBS, suspend cells in 1 ml of lysis buffer.
c. Sonicate for 15 s at 50% power and 50% pulsation. Solution should be homogeneous. No clumps should be visible.
d. Centrifuge at 2,500 g for 2 min to remove debris.
e. Take supernatant and determine protein concentration. Total protein concentration should be ∼2 mg/ml. Dilute with lysis buffer if desired.
3. SPT activity assay
Do the reaction in a 2 ml polypropylene reaction tube.
a. Take 190 μl of total cell lysate (∼200–400 μg total protein) and 10 μl of the corresponding 20× assay mix ([14C] or HPLC). Vortex briefly and keep samples on ice. For negative control, add 2 μl of myriocin solution. To correct for a methanol effect, also add 2 μl of methanol to the samples.
b. To start the reaction, transfer tubes to a water bath. Incubate at 37°C for 60 min.
The following step is only necessary for the HPLC-based assay:
c. Add 50 μl of NaBH4 (5 mg/ml) and let react for 5 min at room temperature.
4. Lipid extraction and product detection
a. Add 0.5 ml of methanol/KOH:CHCl3 (for the radioactivity-based assay) or 0.5 ml of methanol:CHCl3:0.1 M KOH + C17-sphingosine (for the HPLC-based assay) and mix intensively (vortex).
b. Add 0.5 ml CHCl3.
c. Add 0.5 ml alkaline water and 100 μl of 2N NH4OH and mix intensively (vortex).
d. Centrifuge (12,000 g, 1 min at room temperature).
e. Remove the upper phase with gentle aspiration.
f. Wash the lower phase twice with 900 μl of alkaline water.
The following steps are only necessary for the radioactivity-based assay:
g. Transfer 400 μl of the lower phase to a polyethylene scintillation vial.
h. Let the CHCl3 evaporate under a stream of nitrogen.
i. Add 5 ml of liquid scintillation cocktail and count the incorporated radioactivity by a scintillation counter. For activity calculations, take into account that the transferred 400 μl CHCl3 are 2/3 of the total CHCl3 phase (600 μl).
The following steps are only necessary for the HPLC-based assay:
j. Transfer 400 μl of the lower phase to a new 2 ml polypropylene vial.
k. Let the CHCl3 evaporate under a stream of nitrogen.
l. Redissolve the residue in 150 μl of methanol:ethanol:H2O (85:47.5:17.5).
m. In an additional vial, add 150 μl of HPLC standard mix. This vial will provide the defined standard peaks.
n. Add 5 μl of OPA reagent to each vial and let react for 2 h in the dark.
o. Analyze samples by HPLC using the following conditions:
C18 reverse-phase column (100 Å pore size, 3 μm particle size, 4 × 125 mm column)
Injection volume: 50 μl
Flow rate: 1.0 ml/min
Pump program:
0–20 min: methanol:5 mM potassium phosphate buffer (pH 7) (90:10, v/v), isocratic
20–25 min: methanol (100%), isocratic (wash)
25–30 min: methanol:5 mM potassium phosphate buffer (pH 7) (90:10, v/v), isocratic
Measure OPA fluorescence at an excitation wavelength of 335 nm and an emission wavelength of 440 nm.
p. Retention times for C17-sphingosine, threo-, and erythro-sphinganine standards are ∼5.5, 8.5, and 9 min.
q. SPT activity is calculated using the following formula:
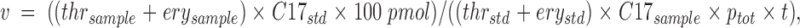 |
where v is the specific SPT activity in pmol/min/mg, thr and ery are the areas of threo- and erythro-sphinganine peaks, C17 is the area of C17-sphingosine peak, ptot is the total amount of protein in milligrams, and t is the reaction time in minutes. The subscripts sample and std stand for peaks from the samples and from the HPLC standard mix, respectively.
This work was supported by the Hartmann Müller Foundation, the Herzog-Egli Foundation, the EMDO Foundation, the Foundation for Scientific Research (University of Zurich), and the 6th Frame-work Programme of the European Commission (LSHM-CT-2006-037631).
Published, JLR Papers in Press, January 29, 2009.
References
- 1.Hanada K. 2003. Serine palmitoyltransferase, a key enzyme of sphingolipid metabolism. Biochim. Biophys. Acta. 1632 16–30. [DOI] [PubMed] [Google Scholar]
- 2.Merrill A. H., Jr., and E. Wang. 1986. Biosynthesis of long-chain (sphingoid) bases from serine by LM cells. Evidence for introduction of the 4-trans-double bond after de novo biosynthesis of N-acylsphinganine(s). J. Biol. Chem. 261 3764–3769. [PubMed] [Google Scholar]
- 3.Hannun Y. A., C. Luberto, and K. M. Argraves. 2001. Enzymes of sphingolipid metabolism: from modular to integrative signaling. Biochemistry. 40 4893–4903. [DOI] [PubMed] [Google Scholar]
- 4.Hannun Y. A., and L. M. Obeid. 2002. The ceramide-centric universe of lipid-mediated cell regulation: stress encounters of the lipid kind. J. Biol. Chem. 277 25847–25850. [DOI] [PubMed] [Google Scholar]
- 5.Bejaoui K., C. Wu, M. D. Scheffler, G. Haan, P. Ashby, L. Wu, P. de Jong, and R. H. Brown, Jr. 2001. SPTLC1 is mutated in hereditary sensory neuropathy, type 1. Nat. Genet. 27 261–262. [DOI] [PubMed] [Google Scholar]
- 6.Dawkins J. L., D. J. Hulme, S. B. Brahmbhatt, M. Auer-Grumbach, and G. A. Nicholson. 2001. Mutations in SPTLC1, encoding serine palmitoyltransferase, long chain base subunit-1, cause hereditary sensory neuropathy type I. Nat. Genet. 27 309–312. [DOI] [PubMed] [Google Scholar]
- 7.Jiang X. C., F. Paultre, T. A. Pearson, R. G. Reed, C. K. Francis, M. Lin, L. Berglund, and A. R. Tall. 2000. Plasma sphingomyelin level as a risk factor for coronary artery disease. Arterioscler. Thromb. Vasc. Biol. 20 2614–2618. [DOI] [PubMed] [Google Scholar]
- 8.Park T. S., R. L. Panek, S. B. Mueller, J. C. Hanselman, W. S. Rosebury, A. W. Robertson, E. K. Kindt, R. Homan, S. K. Karathanasis, and M. D. Rekhter. 2004. Inhibition of sphingomyelin synthesis reduces atherogenesis in apolipoprotein E-knockout mice. Circulation. 110 3465–3471. [DOI] [PubMed] [Google Scholar]
- 9.Grimm M. O., H. S. Grimm, A. J. Patzold, E. G. Zinser, R. Halonen, M. Duering, J. A. Tschape, B. De Strooper, U. Muller, J. Shen, et al. 2005. Regulation of cholesterol and sphingomyelin metabolism by amyloid-beta and presenilin. Nat. Cell Biol. 7 1118–1123. [DOI] [PubMed] [Google Scholar]
- 10.Merrill A. H., Jr. 1983. Characterization of serine palmitoyltransferase activity in Chinese hamster ovary cells. Biochim. Biophys. Acta. 754 284–291. [DOI] [PubMed] [Google Scholar]
- 11.Williams R. D., E. Wang, and A. H. Merrill, Jr. 1984. Enzymology of long-chain base synthesis by liver: characterization of serine palmitoyltransferase in rat liver microsomes. Arch. Biochem. Biophys. 228 282–291. [DOI] [PubMed] [Google Scholar]
- 12.Berge R. K., and B. Dossland. 1979. Differences between microsomal and mitochondrial-matrix palmitoyl-coenzyme A hydrolase, and palmitoyl-L-carnitine hydrolase from rat liver. Biochem. J. 181 119–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bradford M. M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72 248–254. [DOI] [PubMed] [Google Scholar]
- 14.Ikushiro H., H. Hayashi, and H. Kagamiyama. 2001. A water-soluble homodimeric serine palmitoyltransferase from Sphingomonas paucimobilis EY2395T strain. Purification, characterization, cloning, and overproduction. J. Biol. Chem. 276 18249–18256. [DOI] [PubMed] [Google Scholar]
- 15.Hunt M. C., and S. E. Alexson. 2002. The role Acyl-CoA thioesterases play in mediating intracellular lipid metabolism. Prog. Lipid Res. 41 99–130. [DOI] [PubMed] [Google Scholar]
- 16.Broustas C. G., and A. K. Hajra. 1995. Purification, properties, and specificity of rat brain cytosolic fatty acyl coenzyme A hydrolase. J. Neurochem. 64 2345–2353. [DOI] [PubMed] [Google Scholar]
- 17.Lee K. Y., and H. Schulz. 1979. Isolation, properties, and regulation of a mitochondrial acyl coenzyme A thioesterase from pig heart. J. Biol. Chem. 254 4516–4523. [PubMed] [Google Scholar]
- 18.Berge R. K., and M. Farstad. 1979. Dual localization of long-chain acyl-CoA hydrolase in rat liver: one in the microsomes and one in the mitochondrial matrix. Eur. J. Biochem. 95 89–97. [DOI] [PubMed] [Google Scholar]
- 19.Sanjanwala M., G. Y. Sun, and R. A. MacQuarrie. 1987. Purification of long-chain acyl-CoA hydrolase from bovine heart microsomes and regulation of activity by lysophospholipids. Arch. Biochem. Biophys. 258 299–306. [DOI] [PubMed] [Google Scholar]
- 20.He X., X. L. Guan, W. Y. Ong, A. A. Farooqui, and M. R. Wenk. 2007. Expression, activity, and role of serine palmitoyltransferase in the rat hippocampus after kainate injury. J. Neurosci. Res. 85 423–432. [DOI] [PubMed] [Google Scholar]
- 21.Merrill A. H., Jr., D. W. Nixon, and R. D. Williams. 1985. Activities of serine palmitoyltransferase (3-ketosphinganine synthase) in microsomes from different rat tissues. J. Lipid Res. 26 617–622. [PubMed] [Google Scholar]
- 22.Holleran W. M., M. L. Williams, W. N. Gao, and P. M. Elias. 1990. Serine-palmitoyl transferase activity in cultured human keratinocytes. J. Lipid Res. 31 1655–1661. [PubMed] [Google Scholar]
- 23.Merrill A. H., Jr., and E. Wang. 1992. Enzymes of ceramide biosynthesis. Methods Enzymol. 209 427–437. [DOI] [PubMed] [Google Scholar]
- 24.Lynch D. V., and S. R. Fairfield. 1993. Sphingolipid long-chain base synthesis in plants (characterization of serine palmitoyltransferase activity in squash fruit microsomes). Plant Physiol. 103 1421–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Braun P. E., and E. E. Snell. 1967. The biosynthesis of dihydrosphingosine in cell-free preparations of Hansenula ciferri. Proc. Natl. Acad. Sci. USA. 58 298–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Merrill A. H., Jr., and R. D. Williams. 1984. Utilization of different fatty acyl-CoA thioesters by serine palmitoyltransferase from rat brain. J. Lipid Res. 25 185–188. [PubMed] [Google Scholar]
- 27.Riley R. T., W. P. Norred, E. Wang, and A. H. Merrill. 1999. Alteration in sphingolipid metabolism: bioassays for fumonisin- and ISP-I-like activity in tissues, cells and other matrices. Nat. Toxins. 7 407–414. [DOI] [PubMed] [Google Scholar]
- 28.Miyake Y., Y. Kozutsumi, S. Nakamura, T. Fujita, and T. Kawasaki. 1995. Serine palmitoyltransferase is the primary target of a sphingosine-like immunosuppressant, ISP-1/myriocin. Biochem. Biophys. Res. Commun. 211 396–403. [DOI] [PubMed] [Google Scholar]
- 29.Wang E., W. P. Norred, C. W. Bacon, R. T. Riley, and A. H. Merrill, Jr. 1991. Inhibition of sphingolipid biosynthesis by fumonisins. Implications for diseases associated with Fusarium moniliforme. J. Biol. Chem. 266 14486–14490. [PubMed] [Google Scholar]