

Abstract
A diverse set of experimental systems has been developed to probe protein-lipid interactions. These include measurements with the headgroups of membrane lipids in solution, immobilized membrane lipids, and analysis of protein binding to membrane lipids reconstituted in liposomes. Each of these methodologies has strengths but also substantial limitations. For example, measurements between proteins and lipid headgroups or with immobilized membrane lipids do not probe interactions in their natural environment, the lipid bilayer. The use of liposomes, however, was so far mostly restricted to biochemical flotation experiments that do not provide quantitative and/or kinetic data. Here, we present a fast and sensitive flow cytometric method to detect protein-lipid interactions. This technique allows for quantitative measurements of interactions between multiple fluorescently labeled proteins and membrane lipids reconstituted in lipid bilayers. The assay can be used to quantify binding efficiencies and to determine kinetic constants. The method is further characterized by a short sampling time of only a few seconds that allows for high-content screening procedures. Finally, using light scatter measurements, the described method also allows for monitoring changes of membrane curvature as well as tethering of liposomes evoked by binding of proteins.
Keywords: method, quantitative, fluorescence-activated cell sorter, liposome, phosphoinositide, lipid binding, Pleckstrin Homology domain, Fibroblast Growth Factor 2, competitive binding, high-throughput
In recent years, a large number of protein domains have been identified that directly bind to membrane lipids. These include a variety of distinct lipid binding motifs, such as Pleckstrin Homology (PH), FYVE (Fab1, YOTB, Vac1, EEA1), PKC (Protein Kinase C), PX (Phox homology), and BAR (Bin, Amphiphysin, and Rvs) domains (1–4) as well as basic and/or hydrophobic clusters (5–7).
Alongside the discovery of these domains, the spectrum of methods used to study protein-lipid interactions has also been diversifying. These methods can be classified according to the state and background in which the lipid of interest is presented. Immobilized lipids spotted onto nitrocellulose membranes are convenient to analyze. On the other hand, the results are only qualitative and lipids are in a nonphysiological state and environment (8). Using surface plasmon resonance (SPR), lipids can be presented as bilayers (liposomes) to study interactions with immobilized proteins. This method allows for real-time measurements and results in quantitative data (9). However, a disadvantage of this technique is that either the protein or the membrane lipid in question must be immobilized and, therefore, is not presented in a physiologically relevant environment. SPR experiments are also inconvenient to analyze large numbers of individual samples, thus excluding high-content screening procedures. Additionally, competitive binding studies of multiple proteins cannot be performed.
A standard method to quantitatively analyze protein-lipid interactions in suspension is isothermal titration calorimetry (ITC) (10). Protein binding to both lipid headgroups and liposomes has successfully been measured (11). The main drawback remains the large amounts of protein (typically in the milligram range) being required for each individual measurement. Like for SPR experiments, ITC is not suited for competitive binding studies and high-content screening procedures.
Finally, a widespread method to probe interactions between proteins and chemically defined liposomes are biochemical flotation experiments. Again, this approach does not allow for analyzing large numbers of samples in parallel and is basically a qualitative method. This is because it involves a complicated and time-consuming multistep procedure with a potential loss of protein by degradation along with other possible issues.
The new experimental system described here is based on flow cytometry, which is a powerful tool to measure both light scatter and fluorescence properties of individual particles suspended in a continuous flow (12). Cells, membrane vesicles, or particles, such as liposomes, are carried through a flow cell one at a time where they are illuminated by one or more lasers. In general, the forward scatter of laser light is measured to determine the size of the particle as well as the sideward scatter, roughly defining the granularity and internal complexity of the sample. Emitted fluorescence passes through several optical filters to reach the appropriate detector channel, termed FL1, FL2, etc., each measuring a specific emission range. The generated signal intensity is linearly dependent on the amount of molecules present. Typically, 10,000 events are measured and depicted as population histograms or, when two channels are directly compared, as dot plots. An average population signal can be extracted for the comparison of different samples.
Flow cytometric analysis of liposomes has so far mostly been used to determine size and volume properties (13–15). Here, we made use of it as a quantitative method to detect interactions between membrane lipids and peripheral membrane proteins in suspension, i.e., both interactions partners are presented in their natural environment. In addition to measuring direct interactions on a quantitative basis, this method is also suited to detect membrane surface effects that may be the result of proteins binding to specific membrane lipids. This includes clustering of liposomes by multivalent interactions and changes of membrane curvature. The experimental system is a single-step assay without the need for extensive washing procedures. It does not require large amounts of purified proteins and, provided that different fluorescent tags are used, allows for competitive binding studies.
A general limitation in applications using liposomes is that membrane asymmetry between the two leaflets of a lipid bilayer cannot be reconstituted. Also, in the absence of membrane proteins, it is difficult to reconstitute microdomains with locally enriched membrane lipids. However, on the surface of liposomes, it is possible to mimic a certain leaflet, such as the cytoplasmic side of plasma membranes with a chemically defined composition of membrane lipids. Also, using a mixture of membrane lipids containing cholesterol and sphingomyelin, lipid microdomains can be reconstituted with local enrichments such as phosphoinositides. To establish the experimental method described here, we used liposomes with an overall lipid composition resembling plasma membranes of mammalian cells (16). Because we used phosphoinositide binding proteins as a paradigm, the lipid mixture was designed to support the formation of cholesterol-dependent lipid microdomains in which the phosphoinositide phosphatidylinositol-4,5-bisphosphate [PI(4,5)P2] is enriched (17, 18). In native membranes, PI(4,5)P2 is a component of the inner leaflet of plasma membranes and has been shown to function in a number of cellular processes, including signaling and protein targeting (17, 19, 20). To probe known interactions of proteins with PI(4,5)P2, we chose two peripheral membrane proteins that bind PI(4,5)P2 with two different kinds of recognition motifs for phosphoinositides. First, the PH domain of phospolipase Cδ1 (PH-PLCδ1) was included as a classical example of a phosphoinositide binding domain (11). Second, we used Fibroblast Growth Factor 2 (FGF-2), a recently described example of a protein with a polybasic cluster that binds to phosphoinositides (7).
METHODS
Generation of liposomes
A plasma membrane-like mixture of membrane lipids, including PI(4,5)P2 (see Table 1), was prepared from individual chloroform stock solutions (3 mM). With the exception of rhodamine-labeled phosphatidylethanolamine, PE-N-(Lissamine Rhodamine B Sulfonyl) (rhod-PE), all listed membrane lipids were natural extracts and were purchased from Avanti Polar Lipids. The respective order numbers are 840055 (phosphatidylcholine), 840032 (phosphatidylserine), 840042 (phosphatidylinositol), 840026 (phosphatidylethanolamine), 700000 (cholesterol), 860061 (sphingomyelin), 810158 (rhod-PE), and 840046 [PI(4,5)P2]. All stock solutions were stored under argon at −20°C and used for experiments within 1 month. The lipid mixtures were supplemented with 1 mol% rhod-PE to allow detection of liposomes by flow cytometry. Where indicated the mol% of PI(4,5)P2 was varied. The PI(4,5)P2 content was either increased at the cost of phosphatidylcholine or lowered by substitution with phosphatidylcholine. In this way, the relative molar ratios of all other membrane lipids stayed the same for all experiments.
TABLE 1.
Composition of plasma membrane-like liposomes
| Abbreviation | Lipid | Mol% | Source |
|---|---|---|---|
| PC | Phosphatidylcholine | 12.5 | Bovine liver |
| PS | Phosphatidylserine | 5.0 | Porcine brain |
| PI | Phosphatidylinositol | 5.0 | Bovine liver |
| PE | Phosphatidylethanolamine | 9.0 | Bovine liver |
| CL | Cholesterol | 50.0 | Ovine wool |
| SM | Sphingomyelin | 12.5 | Poultry eggs |
| rhod-PE | 16:0 diacyl PE-N-(Lissamine Rhodamine B Sulfonyl) | 1.0 | Synthetic |
| PI(4,5)P2 | Phosphatidylinositol-4,5-bisphosphate | 5.0 | Porcine brain |
A homogeneous lipid film was obtained by drying lipid mixtures in round bottom glass flasks under vacuum at 37°C for 30 min. The lipid film was rehydrated in reconstitution buffer (150 mM KCl and 25 mM HEPES, pH7.4) with 10% (w/v) sucrose (cleared for particles with 0.22 μm filters) at 37°C for 20 min under constant rotation. The total lipid concentration of the liposome suspension was 4 mM. Following 10 cycles of freezing (dry ice in isopropanol) and thawing (37°C; water bath), size extrusion using 1,000 nm filters was performed (mini-extruder; Avanti Polar Lipids). Where indicated, liposomes were loaded with green fluorescent protein (GFP) by supplementing reconstitution buffer with 100 μM recombinant GFP. Before analysis, liposomes were sedimented to remove nonincorporated GFP (16,000 g, 10 min).
The obtained liposomes were diluted to a final lipid concentration of 0.2 mM. Average diameters were measured by dynamic light scattering at 25°C (Zetasizer 1000HSA; Malvern Instruments; 90° angle, 633 nm). Liposome yield was controlled by rhod-PE-derived fluorescence comparing input signals to the final liposome suspension. For all liposome preparations, the yield was >70% with a consistent average diameter of ∼250 nm. Cryo-electron microscopy demonstrated that the resulting liposomes were mostly unilamellar structures (data not shown).
Protein purification
Recombinant GFP, FGF-2-GFP, and PH-PLCδ1-GFP were expressed in Escherichia coli using the expression vectors pGEX-2T (GE Healthcare) and pET28a (Novagen), respectively. Purification was as described previously (7, 21). The affinity tags were proteolytically cleaved off, and a further purification step using either heparin sepharose (in case of FGF-2-GFP) or gel filtration (in case of PH-PLCδ1-GFP and GFP) yielded homogeneous protein preparations. Purified proteins were stored at −80°C in reconstitution buffer. Concentrations were determined spectrophotometrically using a Nanodrop ND-1000 instrument (Peqlab).
Flow cytometry
Liposomes were analyzed with a fluorescence-activated cell sorter (FACS) Calibur instrument, and data were processed with CellQuest Pro software (Becton Dickinson). Forward scatter and sideward scatter of light were calibrated using size-defined latex beads (Sigma-Aldrich). The region in which the linear density was 100% defined the corresponding size gates. Measurement of 0.22 μm filtered reconstitution buffer defined the size region where noise was observed, which was subsequently deleted from further analysis. Settings for sideward scatter were optimized so the noise did not exceed the first log scale. Settings for forward scatter maximized the particle count within the detectable range.
Rhodamine-derived fluorescence (FL2-H, 585/42 nm) was calibrated by setting liposomes without rhodamine to a median of ±5 fluorescence units (F.U.), allowing for setting a gate in which <1% false positives occurred. GFP fluorescence settings (FL1-H, 530/30 nm) were defined by a median of ±2.5 F.U. for buffer-treated liposomes. Spectral overlap between GFP and rhodamine was compensated by rhodamine-containing GFP-free liposomes in one direction and GFP-loaded rhodamine-free liposomes in the other. The final values for the FL1 and FL2 channels used for analysis were the geometrical means 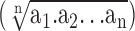 , a representative way of handling the logarithmic FACS data.
, a representative way of handling the logarithmic FACS data.
For FACS measurements, liposomes (total lipid concentration = 1 mM) were first blocked with 3% (w/v) fatty acid free BSA (Roche) for 1 h at 25°C, washed, pelleted, and resuspended again at 1 mM into the final protein solution. After 3–4 h incubation at 25°C, samples were washed and measured (30,000 events). All washing steps were performed with 0.22 μm filtered reconstitution buffer for 10 min at 16,000 g. In all experiments, binding of GFP fusion proteins was compared with GFP alone as a negative control. Both rhodamine and GFP-derived F.U. for each size gate were used for further analysis as described under data processing.
Data processing
Raw FACS data (in F.U.) for each sample containing defined liposomes and a given GFP fusion protein were corrected for background by subtracting GFP-derived signals from the negative control treated with recombinant GFP alone (Fig. 3A, B). In addition, as the rhodamine-derived signal has a linear dependency on the liposome surface area, a shape index factor was introduced based on rhodamine-derived fluorescence. This factor compensates for surface activity of the specific GFP fusion protein. The result, termed “corrected fluorescence units” (c.F.U.) can be obtained using the following formulas:
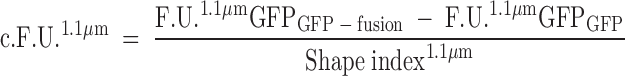 |
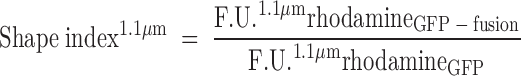 |
Fig. 3.

Detection of liposomes with bound GFP fusion proteins using flow cytometry. A: Dot plot of forward scatter to GFP-derived fluorescence, showing liposomes with 5 mol% PI(4,5)P2 treated with 5 μM FGF-2-GFP. Gating parameters are as described in Fig. 2B. Axes are in logarithmic scale. B: Histogram of FGF-2-GFP binding to liposomes with 5 mol% PI(4,5)P2 having a 0.6 μm diameter. The GFP-derived signal distribution of the 30,000 measured events as well as the population geo mean are depicted. Data were filtered through gates as described in Fig. 2B. The x axis is in logarithmic scale. C: Titration curve of liposomes with varying PI(4,5)P2 content treated with 5 μM FGF-2-GFP. Curves represent he different size gates defined in Fig. 2B. The c.F.U. values (y axis, log scale) are obtained by subtracting the geo mean of the background sample (GFP-treated) from the geo mean of the FGF-2-GFP-treated sample and dividing the outcome by a shape index, a correction factor for liposome aggregation and deformation. D, E: Standard curves for the conversion of GFP-derived F.U. to molecules of GFP (D) and of rhodamine-derived F.U. to molecules of PI(4,5)P2 (E). Axes depict the logarithmic values (see “data processing”). Equations and graphs represent the average of three independent experiments and the R2 exceed 0.99.
In these formulas, the subscripts define the specific sample and F.U.1.1 μmRhodamine or F.U.1.1 μmGFP correspond to the geometric means of the respective FACS channel (FL1-H or FL2-H) and size gate (e.g., 1.1 μm; see Fig. 1A). In case only GFP-treated liposomes were analyzed, the corrections were performed with signal from untreated liposomes. The c.F.U. is a correction of the raw FACS output to compensate for unspecific binding of the GFP part of the GFP fusion protein to liposomes and for any deviation the measured liposomes have from a reference spherical state (shape index). This deviation can result from either liposome aggregation (tethering) or from curvature (positive or negative) induced on the liposome bilayer by protein binding as described in detail under “shape alteration.”
Fig. 1.

Size gating of liposomes in FACS sideward to forward scatter. “Deleted events” corresponds to filtered background noise. The 0.1 μm (black), 0.3 μm (red), 0.6 μm (yellow), and 1.1 μm (blue) boxes correspond to size gates defined by measurement of beads with known sizes. Both axes are in logarithmic scale. A: Merge of the 100% linear density plots of 0.1 μm, 0.3 μm, 0.6 μm, and 1.1 μm latex beads. B: Density plot (100% linear) of liposomes generated by size extrusion. C: Dot plot corresponding to the liposomes in B, showing a broad size range. D: Average size distribution of liposomes (n = 6) as obtained by dynamic light scatter.
A data set was generated per size gate for each concentration of PH-PLCδ1-GFP or FGF-2-GFP. A total of 10 different concentrations ranging from 0 to 10 μM were analyzed. The c.F.U. (x axis) per size gate (n = 3) was plotted against the concentration of GFP fusion protein applied (in μM; y axis). The data were analyzed with GraphPad Prism 4 software and fitted for a one site binding model using the equation Y = Bmax*X/(KD+X). The constraint that dissociation constant (Kd) does not differ between liposome sizes was used. The initial binding curves were fitted for a linear model with the constraint of an intercept >0.
Since FACS data are only relative fluorescent values, we calibrated both the FL1 (GFP) and the FL2 (rhod-PE) channel using a Quantum R-PE (phycoerythrin) MESF and a Quantum FITC MESF kit, respectively (Polysciences; #826A and #827A). These systems allow for a conversion of F.U. to the number of fluorochrome molecules using beads with known amounts of coupled fluorochromes. Beads were measured using the same FACS settings (see “flow cytometry”) as the liposome samples according to the manufacturer's description. The resulting data were converted into two standard curves (see Fig. 3D, E). A coefficient for the conversion of phycoerythrin and FITC fluorescence to rhod-PE and GFP fluorescence, respectively, was determined by measuring concentrations of 25, 50, 100, 150, and 200 nM on a Jasco FP-6500 spectrofluormeter (488 nm excitation and 530/20 nm for FITC and GFP or 585/20 nm for phycoerythrin and rhod-PE emission). The settings matched the corresponding FACS channels. FITC and phycoerythrin were purchased from Sigma-Aldrich and measured in reconstitution buffer, as was GFP (see “protein purification”). Rhod-PE was measured in chloroform. Concentration versus fluorescence values were plotted, and the conversion coefficient was calculated from the two regression curves, indicating that the fluorescence intensity of FITC is 4.2 times that of GFP and the intensity of phycoerythrin is 17.4 that of rhod-PE. These experiments were performed in triplicate (R2 > 0.99) (data not shown). In addition, the level of self-quenching of rhod-PE upon insertion into liposomes was controlled by dissolving liposomes in 1% Triton X-100, as described previously (22). Self-quenching effects were found to be <15% and therefore disregarded in further analysis (data not shown).
RESULTS AND DISCUSSION
Liposome detection by flow cytometry
Flow cytometry allows for the analysis of single particles, such as liposomes, with regard to both fluorescent parameters and light scatter. In this study, liposomes could be detected in two ways. First, liposomes were selected on a size basis by forward and sideward scatter of light. Parallel measurements of size-defined beads were used to determine the diameter of various liposome populations (Fig. 1A). Reconstitution buffer, used to generate liposomes from dried lipid films, identified background signals that were subsequently excluded from the analysis (deleted events; Fig. 1A). These measurements defined size gates that were maintained for all subsequent measurements. Second, a fluorescent membrane lipid (rhod-PE) was included in the liposomal membrane and used to detect liposomes using rhodamine-derived fluorescence. Rhodamine can be excited by a 488 nm laser and detected in the FACS FL2 channel in parallel to GFP (FL1). A rhodamine positive gate was set so that <1% false positives occurred (Fig. 2A). Combining this gate setup with the already defined size gates allowed for a stringent detection and analysis of well-defined liposomes (Fig. 2B). All raw data were passed through these filters before further analysis.
Fig. 2.

Rhodamine-based gating of liposomes. A: Histogram overlay of liposomes without (black) and with 1 mol% rhod-PE (red), defining a cut-off value for rhodamine-positive liposomes where <1% false positives are detected. B: Dot plot of forward scatter to rhodamine-derived fluorescence showing the rhodamine gate (gated events) and the size gates as described in the legend to Fig. 1. Axes are in logarithmic scale.
Liposomes with a defined lipid composition (see Table 1) were generated by size extrusion. The use of a large pore size filter generated liposomes with a broad size range around 0.3 μm as shown by FACS measurements (Fig. 1B, C). Dynamic light scattering confirmed the obtained average size of about 270 nm with a range from ∼100 to 600 nm (Fig. 1D). The relatively broad size range of liposome populations and the possibility to analyze these populations individually allowed for the analysis of a potential role of membrane curvature in protein binding to membrane lipids.
Protein-lipid interactions analyzed by flow cytometry
To measure protein-lipid interactions in flow cytometry, a fluorescent marker is required. This can be achieved either by the detection of antigens with fluorescently labeled antibodies, protein conjugation with fluorophores, or by fusion of the protein of interest with fluorescent proteins. In this study, we used GFP as a fluorescent tag fused to the PH domain of PLC-δ1 and FGF-2, respectively. As a control, reference liposomes loaded with GFP were measured (data not shown), showing that the sensitivity of FACS is sufficient to detect these small amounts of fluorescent material. Liposomes were first blocked and then incubated with FGF-2-GFP (5 μM), a known PI(4,5)P2 binding protein (7). In Fig. 3, it is illustrated how raw FACS data (panels A and B) were converted into a titration curve (panel C) depicting the binding of FGF-2-GFP to liposomes with a rising concentration of PI(4,5)P2, again illustrating the sensitivity of the assay. Each size gate yields a mean GFP-derived fluorescence signal, which is corrected for background binding of the GFP moiety of FGF-2-GFP. Normalization for possible liposome aggregation or membrane deformation effects was achieved by dividing the corrected fluorescence signal by a shape index (see “shape alteration”). This final value was termed the corrected fluorescence units (c.F.U.).
A standard curve to determine the absolute number of fluorescent molecules was generated using calibration beads carrying phycoerythrin and FITC, respectively (see “data processing”). These fluorescence intensities were converted to rhod-PE and GFP fluorescence using a 4.2 and 17.4 conversion coefficient, respectively, yielding two final standard curves (Fig. 3D, E). Note that these curves are specific for the settings and the FACS machine used.
Kinetics of PH-PLCδ1 and FGF-2 binding to liposomes
The PH domain family is one of the largest and best studied lipid binding domain families (3). To characterize our newly established FACS-based assay, we chose the PH domain of PLCδ1 (PH-PLCδ1) that binds to PI(4,5)P2. Recombinant PH-PLCδ1-GFP was incubated with liposomes (Table 1) at 10 different concentrations. Binding was measured using the experimental settings described above. Data were collected for each size gate and corrected for the corresponding GFP-derived background as described under “data processing.” As shown in Fig. 4A, C, we observed linear and concentration-dependent binding at low protein levels. At higher protein concentrations, saturation of binding was attained (Fig. 4B, D). For liposomes containing 5 mol% PI(4,5)P2, a Kd of 2.69 ± 0.49 μM for PH-PLCδ1 and 2.89 ± 0.26 μM for FGF-2 was calculated, in line with previous data on binding properties of the PI(4,5)P2 headgroup to these proteins (7, 11, 23). The calculated Kd for PH-PLCδ1 is consistent with previously published data obtained with various independent methods: 1.7 ± 0.8, 1.4 ± 0.6 or 1.6 ± 0.4, and 2.1 μM for ITC, sedimentation, and SPR, respectively (11, 23–25). Compared with other suspension-based methods, the slightly higher Kd value calculated by the FACS-based method may be due to the ability to compensate for potential aggregation effects. These data corroborate the accuracy of the FACS-based method described here.
Fig. 4.

Titration curves of binding to liposomes of PH-PLCδ1 and FGF-2. A, C: Linear fitting (full line) of the initial binding slope to liposomes dependent on liposome size gating for PH-PLCδ1 (A) and FGF-2 (C). B, D: Nonlinear regression fit (full line) for binding of PH-PLCδ1 (B) and FGF-2 (D) to liposomes for each liposome size gate. The dotted line represents the 95% confidence band. c.F.U. values are derived as described under “data processing.”
Maximal binding (Bmax) per liposome was dependent on size as expected. Also, for both PH-PLCδ1 and FGF-2, the data did not indicate a preference in terms of binding to liposomes with a particular size, suggesting that both proteins do not bind membranes in a curvature-dependent manner. The stoichiometry of binding at saturation could be obtained by converting values for the bound protein maximum (Bmax) to amounts of protein molecules using the previously defined standard curves (Fig. 3D). This was done for all gates of liposome sizes (Table 2). Likewise, the rhodamine-derived fluorescence could be converted to the number of lipid molecules present in liposomes of the given size, considering that five times more PI(4,5)P2 (5 mol%) was present than rhod-PE (1 mol%). A stoichiometry of 0.83 for the binding of PH-PLCδ1 to PI(4,5)P2 was obtained under the assumption that 50% of the lipids is present on the extraluminal side and therefore available for binding. Note that data derived from the 0.1 μm size gate were excluded from analysis because the fluorescence intensity was below a reliable detection threshold.
TABLE 2.
Titration results of FGF-2 and PH-PLCδ1 binding to PI(4,5)P2
| F.U. |
Moleculesb |
Moleculesb |
Bmaxa |
Moleculesb |
Stoichiometryc |
Bmaxa |
Moleculesb |
Stoichiometryc |
|
|---|---|---|---|---|---|---|---|---|---|
| ∅liposomes | Rhod-PE | PI(4,5)P2 | PH-PLCδ1 | FGF-2 | |||||
| 0.1 μm | 134 ± 27 | n/a | n/a | 286 ± 400 | n/a | n/a | 59 ± 179 | n/a | n/a |
| 0.3 μm | 710 ± 171 | 0.28 ± 0.06 | 0.70 ± 0.15 | 2418 ± 424 | 0.77 ± 0.12 | 1.1 ± 0.17 | 2395 ± 193 | 0.76 ± 0.05 | 1.09 ± 0.07 |
| 0.6 μm | 2644 ± 701 | 1.19 ± 0.28 | 2.98 ± 0.70 | 8129 ± 643 | 2.78 ± 0.19 | 0.93 ± 0.06 | 6543 ± 263 | 2.21 ± 0.07 | 0.74 ± 0.02 |
| 1.1 μm | 7523 ± 1214 | 3.75 ± 0.51 | 9.38 ± 1.28 | 12520 ± 833 | 4.40 ± 0.25 | 0.47 ± 0.03 | 13178 ± 427 | 4.64 ± 0.12 | 0.50 ± 0.01 |
| Kd = 2.69 ± 0.49 μMa | n = 0.83 ± 0.09 | Kd = 2.89 ± 0.26 μMa | n = 0.78 ± 0.03 | ||||||
Values represent the average of three independent experiments ± standard deviation. n/a, data derived from liposomes with a 0.1 μm diameter were excluded from analysis because the obtained fluorescence values are below the reliable threshold.
Bmax (in c.F.U.) and Kd were obtained with GraphPad Prism 4 when fitting the respective titration curve to Y = Bmax*X/(KD+X), with Y the c.F.U. and X the protein concentration of the respective sample. Bmax represents the asymptotic maximum of the binding curve.
Amount of molecules in millions. The conversion from F.U. was performed using the standard curves in Fig. 3D, E. Liposomes contained five times more mol% PI(4,5)P2 than rhodamine, of which 50% was assumed to be available for binding.
The stoichiometry was calculated by dividing the amount of PH-PLCδ1 or FGF-2 by the amount of PI(4,5)P2 molecules.
Shape alteration
One of the benefits of the method described here is that it allows for monitoring both binding preferences based on different degrees of membrane curvature and changes in liposome shape. In the former case, curvature-dependent protein binding to liposomes would be revealed by differences in the stoichiometry as well as Bmax values (see Table 2) when size gates are compared. As shown in Table 2, as expected, this is not the case for PH-PLCδ1-GFP or FGF-2-GFP.
In the latter case, if protein binding changes the liposomal spherical shape, a liposome can shift to a different size class in the FACS size gating (Fig. 1C). Indeed, the reference size gates are defined for spherical particles and any major deviation thereof will result in a different scatter signal and hence size gate. Such a shifted liposome will differ in signal intensity of the fluorescent membrane dye (i.e., rhod-PE) from a spherical reference liposome. These membrane deformations can be summarized by defining a shape index based on rhodamine-derived fluorescence that illustrates a change in lipid molecules per liposome size (see “data processing”). As previously described, rhodamine-derived fluorescence directly correlates with the amount of lipid molecules in liposomes of a given size. If protein binding changes this value, this index will differ from 1. Values larger than 1 indicate that a liposome consists of more lipids than a reference spherical liposome of the same measured size; for example, negative curvature or liposome tethering has occurred. Conversely, a value smaller than 1 may indicate the induction of positive membrane curvature; for example, elongated liposomes would have a different surface-to-size ratio as reference spherical liposomes. The shape index of the entire sample is the average of the shape indexes derived for all measured size gates:
 |
For a definite conclusion on the nature of the shape change, events with a changed shape index can be enriched through a FACS-sorting module and subjected to further analysis employing for example confocal microscopy. Fig. 5 exemplifies this principle by the titration of PH-PLCδ1 and FGF-2 to liposomes, including GFP as a negative control. Although the overall effect is moderate, liposomes are slightly deformed at protein concentrations above 1 μM. This is consistent with the ability of PH-PLCδ1-GFP to insert into membranes, especially when containing high amounts of PI(4,5)P2, cholesterol, and phosphatidylethanolamine (26). Fig. 5 also suggests a similar ability for FGF-2-GFP. The slight increase in size at low protein concentrations could be explained as either an electrostatic effect (the negative liposome charge is somewhat shielded due to bound protein) or liposome tethering. The latter was confirmed in the case of FGF-2 by independent methods (data not shown). As a result, a few events in the FACS are no longer caused by a single liposome. These events have a higher surface-to-size ratio and hence a stronger rhodamine signal. In addition, a changed distribution of events is noticed between size gates, distinguishing the observed effect from changes in membrane curvature.
Fig. 5.

Presumed alterations of liposome shape upon protein binding. Liposomes were incubated with the proteins indicated and measured by flow cytometry using the gates defined in Fig. 2B. Shape index values (y axis) above 1 are indicative of an increase (negative membrane curvature or tethering), and values below 1 indicate a decrease (positive curvature) in rhodamine-derived signal averaged for all liposome size gates. Mean values of three independent experiments with standard deviations are shown.
Method application
Nowadays, researchers interested in lipid protein interactions can choose from a broad variety of methods, each of which comes with both advantages and disadvantages. The novel FACS-based assay described in this study represents a very strong alternative as it combines almost all of the advantages of previous experimental systems but eliminates most of their disadvantages.
Membrane lipids are presented in their natural environment as part of a typical bilayer rather than immobilized on some kind of a surface. A vast collection of commercially available membrane lipids (both natural and synthetic products) can be used to generate bilayers of a defined lipid composition to mimic lipid compositions of subcellular membranes on the exposed surface of liposomes. The new assay does not involve any centrifugation steps or flotation gradients. This prevents loss of material during multistep procedures and allows for accurate quantification of protein binding to liposomes.
Individual measurements are completed within a few seconds per sample. Thus, a flow cytometer equipped with a high-throughput sampler allows for the analysis of, for example, 96 samples in a multiwell format in just 15 min. With this kind of setup, high-throughput screens using, for example, small molecule libraries can be conducted to identify inhibitors of a given protein-lipid interaction. The other way around, large sets of proteins can be analyzed for potential lipid binding properties.
The amount of material required is minimized. For example, the whole procedure to titrate the interaction of PH-PLCδ1 with PI(4,5)P2-containing liposomes consumed only 200 μg PI(4,5)P2 and 0.5 mg of protein (n = 3; Fig. 4B). A single measurement required <3 μg PI(4,5)P2, an order of magnitude less than what is needed for individual measurements in ITC.
Although the method itself is qualitative in nature, an equilibrium-based titration can be conducted. From such measurements, dissociation constants can be calculated easily. By implementing the right FACS quantitative calibration kits, binding stoichiometries can be determined as well.
In this study, we measured just one protein at a time using GFP fusion proteins. Based on the use of additional fluorescent tags, such as mCherry (or chemical modifications to make proteins fluorescent), and the availability of multiple channels in flow cytometry, competitive binding studies on liposomal surfaces are possible. Additionally, using adequate fluorophores, such as yellow fluorescent protein and cyan fluorescent protein, hetero-oligomerization induced by binding to membranes could be measured by fluorescence resonance energy transfer using a flow cytometer. Conversely, distinct liposome populations containing different fluorescently labeled lipids (NBD, fluorescein, or others) suitable for FACS analysis are commercially available. A combination of two distinct liposome populations, each containing specific membrane lipids, could be implemented to reveal protein-mediated tethering of liposomes.
Due to the combination of fluorescent parameters from proteins and lipids as well as light scattering data, our assay has the potential to monitor the physical impact of protein binding to membranes, such as the induction of membrane curvature and membrane tethering. For further microscopic analysis, liposomes with specific properties can be enriched through the use of a FACS-sorting module.
In conclusion, we introduce a FACS-based system to directly monitor binding and surface activity of multiple proteins toward membrane lipids, without perturbing the equilibrium, suited for high-throughput applications and with only small requirements in terms of amounts of proteins and lipids. Using appropriate controls, the assay also allows for the calculation of apparent Kd values.
Acknowledgments
The authors thank Volker Haucke and Michael Krauß (Freie Universitāt Berlin, Germany) for providing the PH-PLCδ1-GFP construct and Özgen Deniz (Heidelberg University Biochemistry Center, Germany) for the purification of PH-PLCδ1-GFP protein.The authors are obliged to James Riches and John Briggs (EMBL Heidelberg) for the analysis of liposomes by cryo-electron microscopy and to Annabel Parret for critical reading of the manuscript.
Abbreviations
(c.)F.U., (corrected) fluorescence units
FACS, fluorescence-activated cell sorter
FGF-2, Fibroblast Growth Factor 2
GFP, green fluorescent protein
ITC, isothermal titration calorimetry
Kd, dissociation constant
PH-PLCδ1, Pleckstrin Homology domain of Phospolipase C-δ1
PI(4,5)P2, phosphatidylinositol-4,5-bisphosphate
rhod-PE, rhodamine-labeled phosphatidylethanolamine
SPR, surface plasmon resonance
This study was supported by the Collaborative Research Center 638 of the German Research Foundation.
Published, JLR Papers in Press, January 14, 2009.
References
- 1.Seet L. F., and W. Hong. 2006. The Phox (PX) domain proteins and membrane traffic. Biochim. Biophys. Acta. 1761 878–896. [DOI] [PubMed] [Google Scholar]
- 2.Hurley J. H. 2006. Membrane binding domains. Biochim. Biophys. Acta. 1761 805–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lemmon M. A. 2008. Membrane recognition by phospholipid-binding domains. Nat. Rev. Mol. Cell Biol. 9 99–111. [DOI] [PubMed] [Google Scholar]
- 4.Hayakawa A., S. Hayes, D. Leonard, D. Lambright, and S. Corvera. 2007. Evolutionarily conserved structural and functional roles of the FYVE domain. Biochem. Soc. Symp. 74 95–105. [DOI] [PubMed] [Google Scholar]
- 5.Heo W. D., T. Inoue, W. S. Park, M. L. Kim, B. O. Park, T. J. Wandless, and T. Meyer. 2006. PI(3,4,5)P3 and PI(4,5)P2 lipids target proteins with polybasic clusters to the plasma membrane. Science. 314 1458–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McLaughlin S., and D. Murray. 2005. Plasma membrane phosphoinositide organization by protein electrostatics. Nature. 438 605–611. [DOI] [PubMed] [Google Scholar]
- 7.Temmerman K., A. D. Ebert, H. M. Muller, I. Sinning, I. Tews, and W. Nickel. 2008. A direct role for phosphatidylinositol-4,5-bisphosphate in unconventional secretion of fibroblast growth factor 2. Traffic. 9 1204–1217. [DOI] [PubMed] [Google Scholar]
- 8.Dowler S., R. A. Currie, C. P. Downes, and D. R. Alessi. 1999. DAPP1: a dual adaptor for phosphotyrosine and 3-phosphoinositides. Biochem. J. 342 7–12. [PMC free article] [PubMed] [Google Scholar]
- 9.Narayan K., and M. A. Lemmon. 2006. Determining selectivity of phosphoinositide-binding domains. Methods. 39 122–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wiseman T., S. Williston, J. F. Brandts, and L. N. Lin. 1989. Rapid measurement of binding constants and heats of binding using a new titration calorimeter. Anal. Biochem. 179 131–137. [DOI] [PubMed] [Google Scholar]
- 11.Lemmon M. A., K. M. Ferguson, R. O'Brien, P. B. Sigler, and J. Schlessinger. 1995. Specific and high-affinity binding of inositol phosphates to an isolated pleckstrin homology domain. Proc. Natl. Acad. Sci. USA. 92 10472–10476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shapiro, H. M. 1995. Practical Flow Cytometry. Jon Wiley & Sons, New York.
- 13.Sato K., K. Obinata, T. Sugawara, I. Urabe, and T. Yomo. 2006. Quantification of structural properties of cell-sized individual liposomes by flow cytometry. J. Biosci. Bioeng. 102 171–178. [DOI] [PubMed] [Google Scholar]
- 14.Vorauer-Uhl K., A. Wagner, N. Borth, and H. Katinger. 2000. Determination of liposome size distribution by flow cytometry. Cytometry. 39 166–171. [PubMed] [Google Scholar]
- 15.Childers N. K., S. M. Michalek, J. H. Eldridge, F. R. Denys, A. K. Berry, and J. R. McGhee. 1989. Characterization of liposome suspensions by flow cytometry. J. Immunol. Methods. 119 135–143. [DOI] [PubMed] [Google Scholar]
- 16.van Meer G. 1998. Lipids of the Golgi membrane. Trends Cell Biol. 8 29–33. [DOI] [PubMed] [Google Scholar]
- 17.McLaughlin S., J. Wang, A. Gambhir, and D. Murray. 2002. PIP(2) and proteins: interactions, organization, and information flow. Annu. Rev. Biophys. Biomol. Struct. 31 151–175. [DOI] [PubMed] [Google Scholar]
- 18.Pike L. J., and J. M. Miller. 1998. Cholesterol depletion delocalizes phosphatidylinositol bisphosphate and inhibits hormone-stimulated phosphatidylinositol turnover. J. Biol. Chem. 273 22298–22304. [DOI] [PubMed] [Google Scholar]
- 19.Di Paolo G., and P. De Camilli. 2006. Phosphoinositides in cell regulation and membrane dynamics. Nature. 443 651–657. [DOI] [PubMed] [Google Scholar]
- 20.Martin T. F. 2001. PI(4,5)P(2) regulation of surface membrane traffic. Curr. Opin. Cell Biol. 13 493–499. [DOI] [PubMed] [Google Scholar]
- 21.Milosevic I., J. B. Sorensen, T. Lang, M. Krauss, G. Nagy, V. Haucke, R. Jahn, and E. Neher. 2005. Plasmalemmal phosphatidylinositol-4,5-bisphosphate level regulates the releasable vesicle pool size in chromaffin cells. J. Neurosci. 25 2557–2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.MacDonald R. I. 1990. Characteristics of self-quenching of the fluorescence of lipid-conjugated rhodamine in membranes. J. Biol. Chem. 265 13533–13539. [PubMed] [Google Scholar]
- 23.Garcia P., R. Gupta, S. Shah, A. J. Morris, S. A. Rudge, S. Scarlata, V. Petrova, S. McLaughlin, and M. J. Rebecchi. 1995. The pleckstrin homology domain of phospholipase C-delta 1 binds with high affinity to phosphatidylinositol 4,5-bisphosphate in bilayer membranes. Biochemistry. 34 16228–16234. [DOI] [PubMed] [Google Scholar]
- 24.Gambhir A., G. Hangyas-Mihalyne, I. Zaitseva, D. S. Cafiso, J. Wang, D. Murray, S. N. Pentyala, S. O. Smith, and S. McLaughlin. 2004. Electrostatic sequestration of PIP2 on phospholipid membranes by basic/aromatic regions of proteins. Biophys. J. 86 2188–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hirose K., S. Kadowaki, M. Tanabe, H. Takeshima, and M. Iino. 1999. Spatiotemporal dynamics of inositol 1,4,5-trisphosphate that underlies complex Ca2+ mobilization patterns. Science. 284 1527–1530. [DOI] [PubMed] [Google Scholar]
- 26.Flesch F. M., J. W. Yu, M. A. Lemmon, and K. N. Burger. 2005. Membrane activity of the phospholipase C-delta1 pleckstrin homology (PH) domain. Biochem. J. 389 435–441. [DOI] [PMC free article] [PubMed] [Google Scholar]