

Abstract
We describe a simple iterative approach to augment TCR affinity, which we studied using a myelin oligodendroglial glycoprotein (MOG)-specific TCR. We hypothesized that single amino acid modifications in T-cell receptor (TCR) CDR3 could enhance TCR sensitivity through focal interactions with antigenic peptide while minimizing the risk of cross-reactivity observed previously in TCRs more broadly mutagenized using in vitro evolution techniques. We show that this iterative method can indeed generate TCR with antigen sensitivity 100-fold greater than the parental receptor, and can endow TCR with co-receptor independence. However, we also find that single amino acid mutations in the CDR3 can alter TCR fine specificity, affecting recognition requirements for antigen residues over most of the length of the MHC binding groove. Further, minimal changes in surface-exposed CDR3 amino acids, even the addition of a single hydroxyl group or conversion of a methyl or sulfhydryl moiety to a hydroxyl, can confer modified antigen-specific TCR with new self reactivity. In vivo modeling of modified TCR through retroviral TCR gene transfer into Rag−/− mice confirmed the biological significance of these altered reactivities, though also demonstrated the feasibility of producing antigen-specific, positively selecting, co-receptor independent receptors with markedly increased antigen sensitivity. These results affirm the possibility of readily generating affinity-enhanced TCR for therapeutic purposes, but demonstrate that minimal changes in TCR CDR3 structure can promote self reactivity and thereby emphasize the importance of caution in validating receptors with even subtle alterations prior to clinical application.
Introduction
Limits on the T-cell receptor (TCR) repertoire imposed by developmental selection and peripheral tolerance restrict the availability of T-cells that can effectively target tumors or some pathogens. To overcome this, there has been growing interest in therapeutically skewing the T cell compartment through the enforced expression on T-cells of cloned antigen-specific TCR, thereby promoting desirable immune responses (1,2). One recent clinical trial, for example, showed that tumors in a subset of metastatic melanoma patients remitted after the infusion of CTL re-directed with retrovirally-transduced tumor antigen-specific TCR (3). Re-direction of regulatory T-lymphocytes with introduced autoantigen-specific TCR has similarly been proposed for use in the treatment of autoimmune conditions (4), and has been a subject of our interest. However, TCR gene therapy is currently limited by the availability within the natural TCR repertoire of antigen-specific receptors with desirable therapeutic properties. Both tumor-specific and autoantigen-specific TCR are often of low affinity. T-cells transduced with these frequently express low levels of receptor that must compete with endogenous TCR for expression (5). Target ligand may be weakly expressed, or expressed in the context of insufficient costimulation. Simple approaches to engineer TCR response characteristics, particularly affinity, are therefore desirable and may aid in the creation of receptors with optimized therapeutic potential.
Essentially two approaches have been used to manipulate TCR affinity, and studies of these have been largely performed with class I MHC-restricted TCR. Random mutagenesis of single-chain TCR expressed on yeast followed by multiple rounds of selection for peptide –MHC (pMHC) binding, or in vitro evolution, has allowed the generation of mutant TCR with even a 1,000-fold or greater increase in affinity (6-8). This approach, which generally produces TCR with multiple substitutions compared to the parental receptor, has been best studied in analyses of the well-characterized 1G4 and 2C TCRs. Interestingly, many of the mutants with the highest affinities were also stimulated with syngeneic antigen presenting cells (APCs) in the absence of cognate peptide, indicating that the TCR mutations either increased affinity for self MHC independently of peptide antigen or otherwise fostered a more degenerate recognition of antigen (9,10).
In vitro evolution studies of a class II MHC restricted TCR, the Ek-hemoglobin (Hb)-specific 3.L2 TCR, showed an up to 800-fold increase in affinity, increased degeneracy, yet no self specificity (11,12). As for class I MHC-restricted TCR, multiple mutations were typically present in TCR with enhanced affinity. It was hypothesized that the self-reactivity present in the mutated class I-restricted TCR could be receptor specific, or that the recognition behavior of class I- and class II-restricted TCR may differ in this regard.
A simpler approach to augment TCR affinity is to selectively manipulate just one or two TCR amino acids within the TCR α and β CDR 1, 2, and 3 loops (13). This was recently successfully tested with class I MHC-restricted NY-ESO-1 and MART-1 specific TCR. Candidate amino acids identified based on prior in vitro evolution studies were mutated, and TCRs were generated with modestly increased affinity, preserved specificity, and increased T cell sensitivity to Ag in in vitro analyses (14).
CDR 1 and 2 amino acids interface with cognate pMHC predominantly through the MHC rather than peptide. In contrast, the hypervariable CDR3 loops are typically oriented so as to primarily engage the peptide lying in the MHC's groove (13,15). The very high affinity 1G4 and 2C TCR mutants made to date, particularly those in which new self-specificity has been seen, have typically incorporated mutations in the CDR 1 and/or 2 regions that interact with MHC, as well as CDR3 (8,9). An enhanced generic specificity for MHC generated by substitutions in CDRs 1, 2 or other structural regions of the TCR will stabilize the TCR-MHC interface. This may augment the natural weak interactions between TCR and self pMHC that are important for T-cell positive selection and homeostasis, converting them into stimulatory high affinity interactions. We hypothesized that, in contrast, substitutions of surface exposed CDR3 amino acids, by focusing on peptide-specific rather than MHC-specific interactions, would be more likely to alter affinity for a defined peptide and less likely to broadly skew the selectivity of a TCR.
To test this, we performed iterative mutagenesis of CDR3 residues anticipated to contact antigenic peptide in an autoreactive myelin oligodendroglial glycoprotein (MOG)-specific, Ab-restricted TCR. We show that single amino acid CDR3 mutations in a class II MHC-restricted TCR can dramatically increase TCR sensitivity. However, subtle changes in the TCR CDR3, even the addition of a single hydroxyl, can also confer new, MOG-independent self-reactivity. Further, such changes can dramatically alter the fine specificity of TCR recognition of pMHC, even in regions distant to the anticipated binding site of the mutated CDR3 residue. Our studies demonstrate the feasibility of generating enhanced TCR through simple iterative mutagenesis. However, they also show that even minor changes in CDR3 structure can globally influence TCR recognition, the difficulty in maintaining specificity while manipulating TCR affinity, and the importance of caution as engineered TCR are clinically translated.
Materials and Methods
Mice
C57BL/6 (B6), B10.BR, NOD, and C57BL/6 Rag1−/− mice were obtained from The Jackson Laboratory and bred under specific pathogen free, including all detectable strains of helicobacter, conditions. C57BL/6 Ab−/− mice were generously provided by the laboratory of Dr. Peter Doherty. Experiments were performed in accordance with institutional animal care and use procedures.
Media, Reagents, Antibodies, and Flow Cytometry
T-cell culture conditions were as described (16). MOG35−55 peptide (MEVGWYRSPFSRVVHLYRNGK), alanine substitutions of this peptide, Sendai virus hemagglutinin-neuraminidase (HN)163−178 (VGEPYLSSDPKISLLPG), HN199−215 (SIGEAIYAYSSNLITQG), Sendai virus matrix protein (M)146−161 (IFNANKVALAPQCLPV), and Ovalbumin (OVA)326−338 (AVHAAHAEINEAGR) were synthesized and HPLC purified by the St. Jude Hartwell Center for Biotechnology. Monoclonal antibodies specific for CD4 (clone L3T4), CD8 (clone 53−6.7), CD3ε (2C11) and TCR Vβ8.1, 8.2 (clone MR5−2) were from BD Biosciences. Flow cytometry was performed on a FACSCalibur (Becton Dickinson), and flow cytometric sorting was performed on a MoFlo high-speed cell sorter (DakoCytomation).
TCR constructs and mutagenesis
The 1MOG9 TCR α and β chains, cloned into the MSCV-I-GFP murine stem cell virus-based retroviral vector, was previously described (17). Site directed TCR mutagenesis was performed using the Quickchange II Site Directed Mutagenesis kit (Stratagene, La Jolla, CA). All mutations were confirmed by DNA sequencing (St. Jude Hartwell Center).
Retroviral transduction of TCR
Retrovirus was produced as described (18,19) and used to infect surface TCR-deficient 4G4 T hybridoma cells (C. Janeway, Yale University, New Haven, CT) that had been transduced with mouse CD4-expressing retrovirus. To transduce primary T lymphocytes, freshly isolated C57BL/6 lymph node (LN) cells were sorted for either CD4 or CD8 and the absence of TRBV13−2, and stimulated with soluble CD3 and CD28 specific antibodies and 10 U/mL rhIL-2 (NCI BRB Repository, Frederick, MD). Cleared supernatant from retroviral producer cells with 8 μg/mL polybrene and rhIL-2 was added at day 1, and the cells spun at 1800 rpm for 90 minutes in a Jouan CR422 centrifuge. A second spin-infection cycle was repeated on day 2. Transduced T cells were flow cytometrically sorted on day 4−5 for expression of GFP and either CD4 or CD8, and expanded in EHAA medium (Invitrogen, Carlsbad, CA) / 10% FCS in the presence of rhIL-2 prior to analysis on day 7.
Generation of Retrogenic Mice
Retrogenic mice were generated as described (18,20). Briefly, bone marrow cells were harvested from the femurs of Rag1−/− mice 48 h after the administration of 0.15 mg 5-fluorouracil/g body weight. The pooled cells were cultured in complete Click's medium containing 20% FCS, IL-3 (20 ng ml−1), IL-6 (50 ng ml−1), and SCF (50 ng ml−1) for 48 h at 37°C / 5%CO2. The cells were then co-cultured for an additional 48 h with 1200 rad irradiated retroviral producer cells. The progenitor cells were harvested, washed with PBS, transduction confirmed by flow cytometry for GFP, and injected into sub-lethally irradiated (450 rad) Rag1−/− recipient mice at a ratio of 2 recipient mice per bone marrow donor. Mice were analyzed at 8−10 weeks of age.
Cytokine Analysis
1×105 4G4.CD4 cells were cultured in the presence of 3×105 APCs and the indicated stimulus. Culture supernatants were collected at 24 h and analyzed for IL-2 by sandwich ELISA (BD-Pharmingen) or Bio-Plex (Bio-Rad) assay. Alternatively, 5 × 104 flow cytometrically sorted transduced primary T cells, 7 d after prior stimulation and transduction, were re-stimulated with 3×105 irradiated splenic APCs and the indicated stimulus. After 24 h, cell free supernatant was harvested and IFN-γ content determined by sandwich ELISA (BD-Pharmingen).
Statistics
Error bars represent ± 1 standard deviation (s.d.). Two tailed student's t-tests were performed using Excel software (Microsoft). A p<0.05 was considered statistically significant.
Results
CDR3 mutations in a MOG-specific TCR
We previously described the 1MOG9 (TRAV3*01 TRAJ17*01, TRBV13−2*01 TRBJ2−4*01) TCR, which recognizes the immunodominant 35−55 epitope of MOG in association with Ab (17). Crystal structures of other TRBV13−2+ TCRs, including the D10, 172.10, 2W20, and YAe62 receptors, together with their cognate class II A MHC-peptide antigen show a conserved docking arrangement between the TCR and pMHC, aligning the antigenic peptide and TCR CDR3 (21-24). Considering this, we anticipated that the 1MOG9 CDR3 α and β would similarly align with antigenic peptide. We therefore performed alanine substitutions of surface-exposed CDR3 residues (Fig. 1). Mutated TCRs were retrovirally transduced into TCR-deficient 4G4.CD4 T-hybridoma cells and flow cytometrically purified based on co-expressed GFP. Expression levels of the wild-type TCR and its mutants were similar (Supp. Fig. 1). Because the transduced T-cells were essentially identical except for single amino acid mutations in their TCR, differences in their responsiveness to antigen should be primarily determined by differential affinity for pMHC ligand.
Figure 1. Alanine substitutions in the 1MOG9 TCR CDR3.

Sequences of the 1MOG9 CDR3α and β chains are shown. Predicted surface exposed residues mutated to alanine are indicated.
The TCR+ hybridomas were stimulated with graded amounts of MOG35−55 peptide (Fig. 2) and IL-2 production was measured as an indicator of response. As expected, control non-transduced cells were not stimulated by MOG35−55 whereas cells transduced with wild type 1MOG9 TCR responded well. TCR with substitutions in the CDR3α did not respond to Ag except for N103A, which showed an ∼10 fold decrease in sensitivity to MOG35−55 (Fig. 2a). This implies that the integrity of the CDR3α is crucial for pMHC recognition. A D101A substitution in the CDR3β also abrogated recognition, whereas all other CDR3β substitutions were more permissive, with some IL-2 production detected (Fig. 2b). Two substitutions increased responsiveness compared with the 1MOG9 TCR. A G99A substitution in CDR3β marginally enhanced activity (Fig. 2c). More interestingly, a neighboring E100A substitution led to a >10 fold increase in sensitivity and an approximate tripling of maximal response. Implicitly, E100, a large acidic residue, interferes with TCR binding to Ab-MOG35−55. These results show that structural conservation of many, but not all, external loop amino acids of the CDR3 is critical for 1MOG9 TCR stimulation by cognate Ag. They further support the utility of simple mutagenesis in engineering TCR with enhanced recognition properties.
Figure 2. Functional analysis of alanine substituted TCR.

Untransduced or TCR transduced 4G4.CD4 cells were stimulated in the absence of or with the indicated concentration of MOG35−55 peptide in the presence of irradiated C57BL/6 splenic APCs. IL-2 production was measured by Bio-plex after 24 h. Purified cells were also stimulated with soluble anti-CD3ε antibody (sαCD3), with responses ranging from 449 − 985 pg/ml IL-2 for the different TCR. (A) Results for cell lines with mutations in CDR3α. (B) Results for cell lines with mutations in CDR3β that show diminished sensitivity to MOG35−55 compared with cells transduced with the parental 1MOG9 receptor (filled diamond). (C) Results for cell lines with increased sensitivity to MOG35−55. Data in (A-C) is from a single representative experiment.
To better evaluate the role of E100 in MOG35−55-Ab recognition, we next mutated this TCR position to the remaining 18 amino acids. Again, retrovirus incorporating the modified TCR was produced and used to transduce 4G4.CD4 T cells. TCR expression levels were similar among hybridomas expressing the 20 amino acids at E100 (Supp. Fig. 2). Stimulation of the different TCR with MOG35−55 demonstrated distinct responsiveness among the E100 mutants (Fig. 3a-e). Surprisingly, a response was seen with all but 2 of the mutations. Therefore, flexibility in the 1MOG9 TCR interaction with Ab-MOG35−55 at E100 seemingly makes this location well suited for modulating TCR recognition properties.
Figure 3. Functional analysis of E100 mutant TCR.
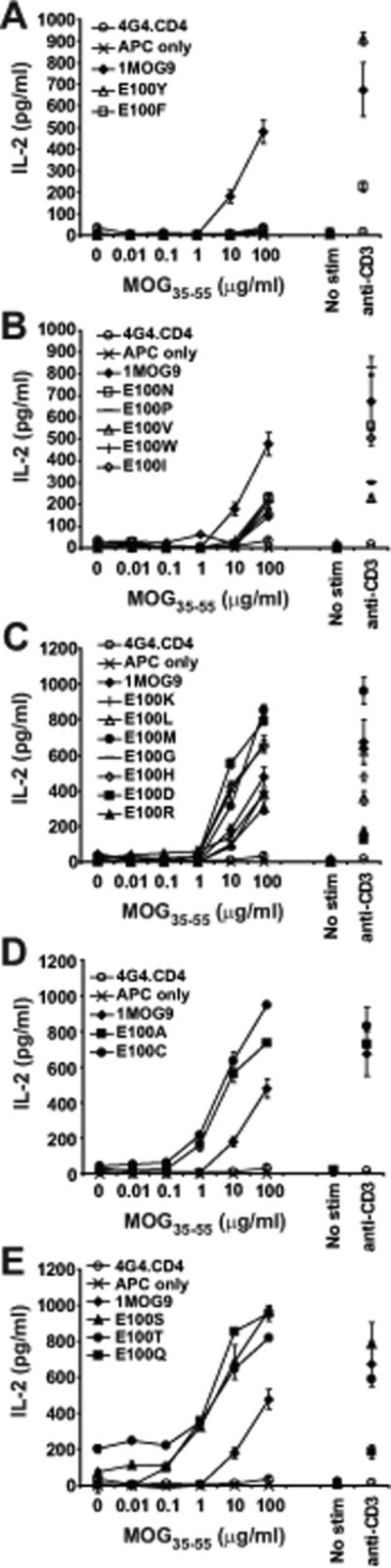
Purified untransduced or TCR transduced 4G4.CD4 cells were stimulated with the indicated concentration of MOG35−55 in the presence of irradiated splenic APC or with plate bound αCD3. IL-2 production was measured at 24 h by ELISA. (A) Results from mutant TCR-transduced cell lines demonstrating no response to MOG35−55 Ag compared with the parental 1MOG9 TCR. (B) Transduced cell lines with a ∼10 fold decreased sensitivity to Ag when compared with 1MOG9 transduced cells. (C) Transduced cells with a sensitivity for Ag similar to 1MOG9 transduced cells. (D) Transduced cells with a ∼10 fold increased sensitivity for Ag. (E) Transduced cells with a >10 fold increase in sensitivity for Ag. Data plotted in (A-E) are from a single representative experiment.
Responsiveness among the E100 mutants fell into distinct categories. Substitution of E100 with the bulky hydrophobic residues F or Y eliminated MOG35−55 reactivity (Fig. 3a). N, P, V, W, and I substitutions, consisting also largely of bulky hydrophobic residues, substantially decreased T-cell sensitivity (>1 log) to titrations of antigenic peptide compared with the parental 1MOG9 TCR (Fig. 3b). The sensitivity of D, G, H, K, L, M, and R substitutions was similar to that of the parental 1MOG9 receptor (Fig. 3c). A and C substitutions, residues with smaller side chains, showed an approximately 1 log increase in response sensitivity (Fig. 3d). Mutations of E100 to T, S, and Q showed the strongest response, with at least a 2 log increase in sensitivity to MOG35−55 (Fig. 3e). Therefore, single amino acid mutations in a class II-restricted TCR can dramatically enhance affinity. A change as small as the conversion of a glutamic acid carboxyl to an amide can increase the sensitivity of a TCR for pMHC by a factor of 100.
Two of the E100 mutations were of particular interest. E100S and E100T transduced cells demonstrated the highest sensitivity to titrations of antigenic peptide, with response seen even at the lowest Ag concentration tested. However, in each case some APC dependent, MOG-independent IL-2 production was seen. This response was readily apparent with the E100T transduced hybridoma cells (Fig. 3e). With the E100S transduced cells it was only recognizable from background by its consistent presence in multiple assays with independently transduced hybridomas. Therefore, as with class I-MHC restricted TCR, mutations of class II-restricted TCR can provoke new self reactivity. Because the E100A, E100C, and E100V mutations lacked this MOG-independent reactivity, our observations show that a change as subtle as the conversion of a CDR3 methyl or sulfhydryl to a hydroxyl group is capable of conferring new self-specificity on a TCR.
The MOG-independent reactivity of 1MOG9 E100T and E100S mutants is pMHC restricted
To determine whether the reactivity of the E100T and E100S mutated TCRs was Ab restricted, we tested reactivity against class II MHC deficient (C57BL/6 Ab−/−) APCs, or allogeneic Ak (B10.BR) or Ag7 (NOD) APCs (Fig. 4a and 6, and data not shown). Whereas the E100T and E100S T-cells responded to Ab expressing APC as above, they did not respond to any of these alternative stimuli. Therefore the MOG-independent reactivity of the hybridomas is selective for Ab and these mutations do not confer more generic specificity for class II A MHC, class I MHC, or other cell-surface molecules.
Figure 4. E100S and E100T TCR response to alternative APC and peptides.

(A) 1MOG9 or mutated TCR-transduced 4G4.CD4 cells were stimulated with irradiated C57BL/6, B10.BR, or NOD splenic APC, or with C57BL/6 APC and 100 μg/ml of MOG35−55 peptide. (B) 1MOG9 or mutated TCR transduced 4G4.CD4 cells were stimulated with T2-Ab APC previously pulsed with Ab-binding peptides, including MOG35−55, Sendai virus HN163−178, HN199−215, or M146−161, or OVA326−339 to establish whether the response of the E100S and E100T transduced T cells is peptide dependent. IL-2 production was measured at 24 h by ELISA. The stimulatory activity of the different assayed peptides was confirmed with peptide-specific T cells in separate control studies (data not shown).
Figure 6. Response of transduced primary T cells.

Flow cytometrically sorted CD4+TRBV13−2− or CD8+TRBV13−2− T cells were stimulated with anti-CD3/anti-CD28 and transduced with 1MOG9, E100A, E100S, or E100T TCR. The transduced cells were re-sorted for CD4 or CD8 and GFP, and expanded in IL-2. Expression of the transduced TRBV13−2+ TCR was confirmed by flow cytometry (not shown). Cells were analyzed 7 days after initial stimulation. (A) Transduced CD4+ T-cells were stimulated with titered MOG35−55 or with mitogen (ConA) in the presence of irradiated C57BL/6 APCs, or with control C57BL/6 Ab−/− APCs. IFN-γ production was measured in cell-free supernatant by ELISA 24 h after stimulation. (B) Analysis was performed as in (A) with simultaneously prepared TCR-modified CD8+ T lymphocytes.
Most of the binding energy linking a TCR to a pMHC complex results from interactions between the TCR and MHC rather than antigenic peptide (13,25). We therefore analyzed whether the altered structure of the E100S and E100T TCR allowed for peptide-independent stimulation by MHC. We pulsed T2 cells transfected with Ab (T2-Ab cells) with either MOG35−55 or 4 alternative, well characterized Ab-binding peptides; Sendai virus HN163−178 and HN199−215, Sendai virus M146−161, and OVA326−339 (26-28). Class II MHC molecules in T2 antigen-processing-defective cells are empty or associated with invariant chain or CLIP in the absence of exogenously supplied class II-binding peptides (29). Neither the E100S nor E100T hybridomas responded to T2-Ab cells, or to these cells pulsed with any of the 4 peptides (Fig. 4b). In contrast, strong responses were seen after pulsing T2-Ab cells with MOG35−55 peptide. Therefore, the E100S and E100T mutations do not bind empty, CLIP-bound, or random peptide associated Ab. This result argues against promiscuous MHC-directed binding and demonstrates peptide-specificity in the MOG-independent mutant TCR response.
Fine specificity of TCR interaction
We anticipated that single amino acid mutations in the CDR3 would selectively impact association of the mutant residue with juxtaposed MOG35−55 peptide residues. Yet the MOG-independent responses of the E100S and T mutants suggested the possibility of more substantial changes in fine specificity. To better define this, we compared the fine specificity of the 1MOG9 and E100S TCR for alanine-substituted MOG35−55 peptides. The S and not the T mutant was studied because E100S-modified T cells produced only very small amounts of IL-2 when cultured with unpulsed APCs, minimizing the confounding effect of MOG-independent activation in the analyses.
Stimulation of the 1MOG9-transduced 4G4.CD4 cells with peptide-pulsed APCs demonstrated a response pattern consistent with prior studies of other MOG35−55-specific T-cells (Fig. 5a, b) (30,31). Those studies identified R41, F44, R46, and V47 as primary TCR contact residues. Indeed, the 1MOG9 transduced cells showed significantly diminished responses to the corresponding R41A, F44A, R46A, and V47A substituted peptides. G38 and W39 have also been shown to interact with TCR, and the 1MOG9 TCR showed enhanced reactivity to the W39A peptide. S42 and P43, which neighbor other previously identified TCR-contact residues, also proved important to response. The fine-specificity of E100S-transduced T-cells showed some similarities with the 1MOG9-transduced cells. As for 1MOG9, there was significantly decreased response to S42A, F44A, and R46A peptides. However, the remaining 4 amino acids affecting 1MOG9 recognition, W39, R41, P43, and V47, did not significantly influence E100S recognition (Fig. 5a), demonstrating that the E100S and 1MOG9 TCR have distinct peptide contact requirements. When the data was normalized to the different T-cells’ specific response to unmutated MOG35−55, differences were seen between 1MOG9 and E100S in the recognition of 5 of the 21 MOG35−55 residues - W39, R41, S42, P43, and V47 (Fig. 5b). Response by the E100S T-cells to W39A substituted peptide was decreased compared with that of 1MOG9 T-cells. In contrast, response was relatively increased to R41A, S42A, P43A, and V47A peptides. Therefore, contradicting our initial prediction that local CDR3 mutations would only locally influence peptide recognition, the E100S mutation influences peptide recognition requirements across at least a 9 amino acid segment of antigenic peptide, from W39 to V47. Conceivably, an overall enhanced affinity of the E100S TCR obviates the need for some peptide interactions for effective pMHC engagement. However, increased overall affinity would be expected to only increase responses to mutant peptides, yet the E100S TCR response to the W39A mutant peptide is decreased compared with the parental 1MOG9 TCR. This suggests an alternative explanation, that the E100S mutation more fundamentally alters how the TCR associates with pMHC. Considering that models of MOG35−55 binding to Ab place Y40 in the p1 pocket of the MHC (31), our results imply that this CDR3β mutation is influencing recognition of residues, such as W39, in the peptide N-terminal end of bound pMHC, a region anticipated from other TRBV13−2 – class II A structures to associate predominantly with the TCR CDR3α chain (13,22-24). Therefore, even single amino acid mutations in surface exposed CDR3β residues may profoundly influence recognition requirements at distal TCR contact sites.
Figure 5. Fine specificity of TCR recognition of MOG35−55.

(A) 1MOG9, E100S, or untransduced 4G4.CD4 T cells were stimulated with 100 μg/ml MOG35−55 or the indicated alanine-substituted peptides in the presence of irradiated C57BL/6 APC. IL-2 production was measured at 24 h by ELISA. (B) Data in (A) was normalized to the response of the cell line to MOG35−55 and plotted to facilitate comparison of the 1MOG9 and E100S response. (C) Response of 1MOG9, E100S, and E100A transduced cells to a subset of relevant alanine-substituted peptides. Responses are normalized to the MOG35−55-specific response, which was 144±26, 308±2, and 257±8 for 1MOG9, E100S, and E100A transduced T cells respectively. In (A) and (C), results of t-tests comparing response with the same cells’ MOG35−55 specific response is shown. In (B) results of t-tests comparing 1MOG9 and E100S results for an individual peptide is shown. ‡, p<0.05 for MOG35−55 vs the indicated peptide for 1MOG9; ° for E100S; †, for E100A. *; p<0.05 for 1MOG9 vs E100S for the indicated peptide.
Notably, this broadly altered fine specificity of the E100S TCR did not seem to be directly associated with the acquisition of MOG-independent reactivity. The fine specificity of E100S resembled that of E100A, which is structurally similar and showed high Ag sensitivity but no MOG-independent self-reactivity (Fig. 5c). As for the E100S mutant, decreased response was observed to S42A, F44A, and R46A mutated Ag compared with wild type MOG35−55. As for E100S, the increased response to W39A and decreased response to R41A and V47A peptide present with 1MOG9 transduced T cells was not seen. The one distinction was a significantly decreased response to P43A in E100A but not E100S cells. Considering the similar fine specificity of the E100A and E100S mutant T-cells, their difference in MOG-independent self-reactivity may primarily reflect response magnitude rather than binding orientation.
Primary T cell reactivity and co-receptor independence of mutant TCR
To confirm that the MOG-independent reactivity and increased antigen sensitivity in the hybridomas was also present in primary T-cells, we transduced the E100S, T, and A mutants or wild type 1MOG9 TCR into primary T lymphocytes. CD4+ or CD8+ TRBV13−2− T lymphocytes were flow cytometrically purified, stimulated, transduced with retrovirus, and the CD4+ or CD8+ GFP+ T-cells flow cytometrically isolated. These were expanded with IL-2, then re-stimulated with APCs in the presence or absence of MOG35−55 7 days after initial stimulation. IFN-γ production was measured as an indicator of Ag sensitivity. Primary CD4 T cell responses resembled that of transduced 4G4.CD4 cells (Fig. 6a). The E100S and A mutants responded more strongly at lower doses of antigen than the parental 1MOG9 TCR, verifying the increased sensitivity of these receptors. The E100T-transduced cells responded strongly to APCs even in the absence of MOG35−55 peptide, with an incremental response observed when cultures were also supplemented with MOG35−55. A small amount of IFN-γ production was seen with the E100S mutant in response to APCs without MOG peptide. However, similar low levels of IFN-γ were produced by the 1MOG9 and E100A transduced primary T cells, possibly reflecting low-level Ag-independent responses to APCs by residual activated T cells, and it was not possible in these assays to identify a MOG-independent E100S response from control responses. Therefore, the response of primary transduced CD4 T-cells parallels that of the 4G4.CD4 hybridomas, though was not adequately sensitive to identify the acquisition of MOG-independent reactivity in E100S-transduced T-cells.
Transduced CD8+ T-cells showed a different response pattern. The MOG-independent reactivity of the CD4+ E100T cells was not seen, demonstrating CD4 dependence for this. Further, sensitivity to dilutions of MOG35−55 was diminished in transduced CD8+ T-cells (Fig. 6b) compared with simultaneously assayed CD4+ T-cells (Fig. 6a). Indeed, wild type 1MOG9 transduced CD8+ cells were wholly unresponsive to MOG, demonstrating co-receptor dependence for MOG recognition. CD8+ E100T, E100S, and E100A transduced cells showed an approximately 10−100-fold decreased sensitivity for MOG35−55 compared with corresponding CD4+ T cells. Therefore, the A, S, and T mutations in E100 can confer co-receptor independence in the primary MOG-specific response. However, this is associated with an ∼10−100 fold penalty in T-cell sensitivity.
Retrogenic modeling of 1MOG9 mutations
We previously used retroviral transgenic, or retrogenic, modeling to demonstrate that the 1MOG9 TCR is positively selected and functionally expressed on T-cells (17). Our results above suggest that E100T and possibly E100S expressing thymocytes will be negatively selected due to their self-reactivity. To test this, we retrovirally transduced the 1MOG9, E100A, E100S, and E100T TCR into Rag−/− hematopoietic progenitor cells and transplanted these into Rag−/− mice. Because the Rag−/− mice cannot rearrange endogenous TCR, developing T-cells will exclusively express the retrovirally introduced receptor. 1MOG9 retrogenic T cells engrafted well in the mice. Thymocytes developed predominantly into CD4+ T lymphocytes (Fig. 7a), and CD4+TRBV13−2+ T cells were virtually exclusively apparent in the spleen and LN (Fig. 7b). The E100A retrogenic mice also showed T cell positive selection into the CD4 lineage and good engraftment of CD4+ T-cells in lymphoid organs. However, increased numbers of mature CD8+ T cells were present compared with the 1MOG9 mice (Fig. 7c). This suggests increased positive selection or improved homeostatic expansion of CD8+ E100A T cells, which may be associated with enhanced co-receptor independent interactions with endogenous class II-peptide complexes. The in vitro, MOG-specific proliferative response of isolated retrogenic E100A T cells to MOG35−55 was also enhanced compared with 1MOG9 T cells, demonstrating increased Ag sensitivity similar to the transduced T-cell hybridomas and primary T-cells (data not shown).
Figure 7. Flow cytometric analysis of 1MOG9, E100A, E100S, and E100T retrogenic mice.

Groups of retrogenic mice including either 1MOG9 and E100A, or 1MOG9, E100S, and E100T were produced and analyzed separately. (A) Thymocytes from 1MOG9, E100A, and control C57BL/6 mice were stained with CD4 and CD8 and analyzed by flow cytometry (top row). Thymi of 1MOG9, E100S, and E100T mice were independently analyzed (bottom row). Values indicate percentage of cells within the indicated regions. CD4 (B) and CD8 (C) T cell engraftment in the LN and spleen of 1MOG9, E100A, or control C57BL/6 mice are plotted. (D) The absence of peripheral T cell engraftment in retrogenic E100S and E100T mice, compared with control 1MOG9 mice, is shown.
In contrast to the 1MOG9 and E100A TCRs, T-cell engraftment was not apparent in mice retrogenic for the E100S or E100T TCRs. Thymi from these showed good progression from double negative (DN) to double positive (DP) cells. However, these cells failed to advance to single positive (SP) T cells (Fig. 7a). Further, mature CD4+ or CD8+ TRBV13−2+ T cells were virtually absent from the periphery in the E100S and E100T retrogenic mice (Fig. 7d and not shown). This confirms that these modest changes in TCR structure can lead to the negative selection of thymocytes expressing the modified TCR.
Discussion
Antigen-specific T lymphocytes have proven a capable therapeutic for both cancer and infectious diseases (32-34). To overcome the difficulty in isolating and expanding autologous T lymphocytes, several laboratories have explored the re-direction of endogenous T cells with genetically introduced TCR (1,35). This permits the rapid production of large numbers of Ag-specific T lymphocytes. These TCR may be further engineered to desired specifications, facilitating the development of optimized cellular therapies.
TCR from many antigen-specific T cells, particularly those recognizing tumor or other self antigens, have suboptimal recognition characteristics. These lower affinity TCR may not adequately signal in TCR-modified T lymphocytes. This can lead to suboptimal induction of effector pathways or poor survival. In vitro evolution systems can dramatically enhance TCR affinity, as evidenced by work with the 2C, 1G4, and 3.L2 TCR (8-10,12). However, this does entail considerable effort, and generally leads to receptors with multiple mutations and therefore an increased risk of altered specificity. More directed mutations can also enhance affinity, though not to the levels observed with in vitro evolution (14). Yet affinities even 10−100 fold increased compared to a low affinity parental TCR may be more than adequate for therapeutic needs. The commonly accepted kinetic proofreading model of TCR signaling suggests that repetitive low affinity engagements between pMHC and TCR support T-cell activation (36). Indeed, T-cells expressing 3.L2 mutants with an ∼800 fold increase of affinity did not demonstrate a proportionately increased sensitivity for antigen when measured by functional responses (12). Similarly high affinity mutants of class I MHC-restricted TCR showed a threshold for optimal response at affinities of ∼1 μM; affinities in excess of this did not improve antigen sensitivity but did impart co-receptor independence (37). This suggests that the signal transduction machinery for T-cells is responsive to a limited range of TCR affinities, and there may be little benefit in increasing affinity outside of this realm.
Autoantigen-specific TCR may be useful in the cellular therapy of autoimmune diseases using TCR-modified regulatory T lymphocytes. We describe the functional impact of single amino acid mutations in the CDR3 of a TCR specific for the MOG autoantigen. We hypothesized that individual CDR3 mutations, by focusing on peptide-specific interactions, would be more likely to maintain specificity than broadly distributed TCR mutations. We present several new findings. First, we demonstrate that an iterative approach can indeed be used to identify “hot spots” and affinity mature TCR. This approach differs from recent analyses of class I-restricted TCR studying single or dual amino acid mutations in that it does not rely on prior knowledge from in vitro evolution systems of potentially advantageous changes (14). Alanine substitutions of the 1MOG9 CDR3 identified two locations in which responsiveness was increased, though this was only substantial for one of these, E100. This bulky acidic residue resides next to another similarly charged amino acid, D101. Interestingly, a D101A mutation was the only CDR3β mutation that wholly abrogated TCR recognition of MOG35−55. Alignment of 1MOG9 and pMHC based on the TCRBV13−2+ 172.10 TCR and Au-MBP1−9 structure (22) and a model of Ab-MOG35−55 (31) would place the negatively charged D101 in proximity with the positively charged R46 of the MOG35−55 peptide. Though speculative, it is possible that E100, by providing an unopposed additional negative charge, may adversely influence a D101-R46 interaction. Indeed, an E100Q mutation that neutralizes this negative charge substantially enhanced TCR sensitivity. More generally, some amino acids, such as E100 here, may be important precisely because they negatively influence TCR binding, and thereby ensure that a TCR's affinity is not too high for its cognate pMHC. Identification of those residues and substituting more structurally compatible amino acids through iterative mutagenesis can therefore generate a series of TCR with varying affinities.
Because the majority of the TCR interface engages the MHC portion of pMHC, random TCR mutagenesis followed by affinity-based selection will act primarily by enhancing TCR-MHC interactions. If additional free energy is provided by TCR-MHC engagements, less free energy must be contributed by TCR-peptide interactions to stimulate a T-cell. At the extreme, no peptide interactions would be needed to provide the binding energy needed for T cell stimulation. Peptide specificity will decrease and degenerate recognition will occur. To avoid this, we focused on CDR3 mutations that we anticipated would more likely affect the TCR-peptide interface and therefore less likely to promote new specificities. The E100S and E100T mutations, however, did lead to new self peptide-specific reactivity. Fine specificity analysis using alanine-substituted MOG35−55 peptides showed that alanine substitutions of some MOG amino acids critical for binding of the parental 1MOG9 TCR, such as R41 and V47, were altogether unimportant for mutant TCR binding. Alanine substitutions of other interspersed MOG residues, such as F44 and R46 were equally important for binding by both the parental and mutant TCR, and one residue, W39, enhanced binding to the wild type but not E100 mutant TCR. This suggests that the mutant TCR are binding peptide specifically and in a similar orientation, though in a manner distinct from the parental 1MOG9 TCR. Because the mutant TCR did not recognize unoccupied or random-peptide-occupied Ab, the MOG-independent binding was peptide-specific. Therefore, mutations in a MOG-Ab restricted TCR seem to lead to fine adjustments in pMHC recognition, and in contrast to studies of mutations in the CD4+ Hb-Ek-specific 3.L2 TCR (12), new self-specificities. Further, CDR3 mutations alone are adequate for this, and even minimal changes in exposed TCR residues, such as from an alanine in the MOG-selective E100A to a serine in the MOG-independent E100S TCR, can endow a TCR with new self-reactivity.
Whereas prior studies have been limited to analyzing mutant TCR in hybridomas or T cell lines in vitro, we further showed the utility of retroviral gene transfer to assess TCR in vivo. Our results validate, but also extend the findings with cultured T cells. Both the E100S and E100T mutants recognized APCs in the absence of cognate antigen, presaging their negative selection. The E100A mutation showed co-receptor independence; CD8+ T cells transduced with the receptor responded to MOG35−55-Ab, though with decreased sensitivity compared to transduced CD4+ T-cells. Similarly, a small number of CD8+ E100A T cells developed in the retrogenic mice. It is therefore interesting to speculate that the co-receptor independence of the TCR in transduced T cells in response to cognate antigen is mirrored in the interactions responsible for positive selection or homeostasis. Indeed, MOG is expressed at very low levels in the thymus (38) and it is possible that co-receptor independent reactivity to this small amount of Ag may facilitate CD8+ T-cell selection. Alternatively, the E100A TCR may be recognizing the same non-MOG self ligand or ligands recognized by the E100S TCR on APCs, but at lower affinity.
Class II A molecules on APC are estimated to present ∼650−2000 distinct peptides (39). Whereas it is convenient to examine whether a mutated TCR has acquired self-responsiveness using splenic APCs or peripheral blood mononuclear cells, tissue specific APCs may present a distinct array of peptides, and specificity for these may not be detectable by assaying response to lymphoid derived cells. Adoptive transfer of T-cells that have acquired specificity for unknown tissue-specific antigens may therefore have unintended and deleterious consequences. For this reason, thorough pre-clinical analyses of altered TCR are essential. Retrogenic or transgenic modeling may therefore be prudent before any clinical application of mutated TCRs. This can be performed using HLA Tg mice to assess human TCR with common HLA specificities.
In summary, we describe an iterative approach for enhancing TCR affinity. We demonstrate using this technique the feasibility of generating affinity-enhanced class II MHC-restricted TCR, and describe a series of MOG-specific TCRs with variable sensitivity for Ag. However, we also show that subtle modifications of TCR can have substantial effects both on TCR fine specificity and self-reactivity, and that in vivo modeling using TCR retrogenesis may serve as a means to pre-clinically characterize mutant TCRs and assure their safety.
Supplementary Material
Supplemental Figure 1. Expression of alanine substituted TCR. The indicated retrovirally encoded TCRs were transduced into TCRαβ-deficient 4G4.CD4 T cell hybridomas. Transduced hybridomas were flow cytometrically sorted based on co-expressed GFP, the sorted cells expanded, and stained with CD4 and TCR specific antibodies. Results for control untransduced cells are plotted in the upper left.
Supplemental Figure 2. Expression of TCR mutated at CDR3β E100. The 1MOG9 TCR was mutated at E100 to all 20 amino acids. 4G4.CD4 T cells were transduced, flow cytometrically purified based on GFP expression, expanded, stained with TCR-specific antibody, and analyzed. Histogram plots display results for untransduced cells or cells transduced with the different receptors.
Acknowlegements
We thank Richard Cross and Yuxia He for assistance with flow cytometric sorting, and Jennifer Smith for assistance with Bio-plex analyses.
This work was supported by the National Institutes of Health Grant R01 AI056153 (to TLG) and by the American Lebanese Syrian Associated Charities (ALSAC)/St. Jude Children's Research Hospital (to all authors).
Footnotes
Publisher's Disclaimer: This is an author-produced version of a manuscript accepted for publication in The Journal of Immunology (The JI). The American Association of Immunologists, Inc. (AAI), publisher of The JI, holds the copyright to this manuscript. This version of the manuscript has not yet been copyedited or subjected to editorial proofreading by The JI; hence, it may differ from the final version published in The JI (online and in print). AAI (The JI) is not liable for errors or omissions in this author-produced version of the manuscript or in any version derived from it by the U.S. National Institutes of Health or any other third party. The final, citable version of record can be found at www.jimmunol.org.
Reference List
- 1.Moss PA. Redirecting T cell specificity by TCR gene transfer. Nat. Immunol. 2001;2:900–901. doi: 10.1038/ni1001-900. [DOI] [PubMed] [Google Scholar]
- 2.Yang L, Qin XF, Baltimore D, Van PL. Generation of functional antigen-specific T cells in defined genetic backgrounds by retrovirus-mediated expression of TCR cDNAs in hematopoietic precursor cells. Proc. Natl. Acad. Sci. U. S. A. 2002;99:6204–6209. doi: 10.1073/pnas.092154599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, Royal RE, Topalian SL, Kammula US, Restifo NP, Zheng Z, Nahvi A, de Vries CR, Rogers-Freezer LJ, Mavroukakis SA, Rosenberg SA. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–129. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bluestone JA, Thomson AW, Shevach EM, Weiner HL. What does the future hold for cell-based tolerogenic therapy? Nat. Rev. Immunol. 2007;7:650–654. doi: 10.1038/nri2137. [DOI] [PubMed] [Google Scholar]
- 5.Heemskerk MH, Hagedoorn RS, van der Hoorn MA, d. van V, Hoogeboom M, Kester MG, Willemze R, Falkenburg JH. Efficiency of T-cell receptor expression in dual-specific T cells is controlled by the intrinsic qualities of the TCR chains within the TCR-CD3 complex. Blood. 2007;109:235–243. doi: 10.1182/blood-2006-03-013318. [DOI] [PubMed] [Google Scholar]
- 6.Richman SA, Kranz DM. Display, engineering, and applications of antigen-specific T cell receptors. Biomo. Eng. 2007;24:361–373. doi: 10.1016/j.bioeng.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 7.Holler PD, Holman PO, Shusta EV, O'Herrin S, Wittrup KD, Kranz DM. In vitro evolution of a T cell receptor with high affinity for peptide/MHC. Proc. Natl. Acad. Sci. U. S. A. 2000;97:5387–5392. doi: 10.1073/pnas.080078297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li Y, Moysey R, Molloy PE, Vuidepot AL, Mahon T, Baston E, Dunn S, Liddy N, Jacob J, Jakobsen BK, Boulter JM. Directed evolution of human T-cell receptors with picomolar affinities by phage display. Nat. Biotechnol. 2005;23:349–354. doi: 10.1038/nbt1070. [DOI] [PubMed] [Google Scholar]
- 9.Holler PD, Chlewicki LK, Kranz DM. TCRs with high affinity for foreign pMHC show self-reactivity. Nat. Immunol. 2003;4:55–62. doi: 10.1038/ni863. [DOI] [PubMed] [Google Scholar]
- 10.Zhao Y, Bennett AD, Zheng Z, Wang QJ, Robbins PF, Yu LY, Li Y, Molloy PE, Dunn SM, Jakobsen BK, Rosenberg SA, Morgan RA. High-affinity TCRs generated by phage display provide CD4+ T cells with the ability to recognize and kill tumor cell lines. J Immunol. 2007;179:5845–5854. doi: 10.4049/jimmunol.179.9.5845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weber KS, Donermeyer DL, Allen PM, Kranz DM. Class II-restricted T cell receptor engineered in vitro for higher affinity retains peptide specificity and function. Proc. Natl. Acad. Sci. U. S. A. 2005;102:19033–19038. doi: 10.1073/pnas.0507554102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Donermeyer DL, Weber KS, Kranz DM, Allen PM. The study of high-affinity TCRs reveals duality in T cell recognition of antigen: specificity and degeneracy. J Immunol. 2006;177:6911–6919. doi: 10.4049/jimmunol.177.10.6911. [DOI] [PubMed] [Google Scholar]
- 13.Rudolph MG, Stanfield RL, Wilson IA. How TCRs bind MHCs, peptides, and coreceptors. Annu. Rev. Immunol. 2006;24:419–466. doi: 10.1146/annurev.immunol.23.021704.115658. [DOI] [PubMed] [Google Scholar]
- 14.Robbins PF, Li YF, El-Gamil M, Zhao Y, Wargo JA, Zheng Z, Xu H, Morgan RA, Feldman SA, Johnson LA, Bennett AD, Dunn SM, Mahon TM, Jakobsen BK, Rosenberg SA. Single and dual amino acid substitutions in TCR CDRs can enhance antigen-specific T cell functions. J Immunol. 2008;180:6116–6131. doi: 10.4049/jimmunol.180.9.6116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garcia KC, Teyton L, Wilson IA. Structural basis of T cell recognition. Annu. Rev. Immunol. 1999;17:369–397. doi: 10.1146/annurev.immunol.17.1.369. [DOI] [PubMed] [Google Scholar]
- 16.Inaba H, Geiger TL. Defective cell cycle induction by IL-2 in naive T-cells antigen stimulated in the presence of refractory T-lymphocytes. Int. Immunol. 2006;18:1043–1054. doi: 10.1093/intimm/dxl038. [DOI] [PubMed] [Google Scholar]
- 17.Alli R, Nguyen P, Geiger TL. Retrogenic modeling of experimental allergic encephalomyelitis associates T cell frequency but not TCR functional affinity with pathogenicity. J Immunol. 2008;181:136–145. doi: 10.4049/jimmunol.181.1.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Holst J, Szymczak-Workman AL, Vignali KM, Burton AR, Workman CJ, Vignali DA. Generation of T-cell receptor retrogenic mice. Nat. Protoc. 2006;1:406–417. doi: 10.1038/nprot.2006.61. [DOI] [PubMed] [Google Scholar]
- 19.Moisini I, Nguyen P, Fugger L, Geiger TL. Redirecting Therapeutic T Cells against Myelin-Specific T Lymphocytes Using a Humanized Myelin Basic Protein-HLA-DR2-{zeta} Chimeric Receptor. J Immunol. 2008;180:3601–3611. doi: 10.4049/jimmunol.180.5.3601. [DOI] [PubMed] [Google Scholar]
- 20.Holst J, Vignali KM, Burton AR, Vignali DA. Rapid analysis of T-cell selection in vivo using T cell-receptor retrogenic mice. Nat. Methods. 2006;3:191–197. doi: 10.1038/nmeth858. [DOI] [PubMed] [Google Scholar]
- 21.Reinherz EL, Tan K, Tang L, Kern P, Liu J, Xiong Y, Hussey RE, Smolyar A, Hare B, Zhang R, Joachimiak A, Chang HC, Wagner G, Wang J. The crystal structure of a T cell receptor in complex with peptide and MHC class II. Science. 1999;286:1913–1921. doi: 10.1126/science.286.5446.1913. [DOI] [PubMed] [Google Scholar]
- 22.Maynard J, Petersson K, Wilson DH, Adams EJ, Blondelle SE, Boulanger MJ, Wilson DB, Garcia KC. Structure of an autoimmune T cell receptor complexed with class II peptide-MHC: insights into MHC bias and antigen specificity. Immunity. 2005;22:81–92. doi: 10.1016/j.immuni.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 23.Dai S, Huseby ES, Rubtsova K, Scott-Browne J, Crawford F, Macdonald WA, Marrack P, Kappler JW. Crossreactive T Cells spotlight the germline rules for alphabeta T cell-receptor interactions with MHC molecules. Immunity. 2008;28:324–334. doi: 10.1016/j.immuni.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huseby ES, White J, Crawford F, Vass T, Becker D, Pinilla C, Marrack P, Kappler JW. How the T cell repertoire becomes peptide and MHC specific. Cell. 2005;122:247–260. doi: 10.1016/j.cell.2005.05.013. [DOI] [PubMed] [Google Scholar]
- 25.Manning TC, Schlueter CJ, Brodnicki TC, Parke EA, Speir JA, Garcia KC, Teyton L, Wilson IA, Kranz DM. Alanine scanning mutagenesis of an alphabeta T cell receptor: mapping the energy of antigen recognition. Immunity. 1998;8:413–425. doi: 10.1016/s1074-7613(00)80547-6. [DOI] [PubMed] [Google Scholar]
- 26.Cole GA, Tao T, Hogg TL, Ryan KW, Woodland DL. Binding motifs predict major histocompatibility complex class II-restricted epitopes in the Sendai virus M protein. J Virol. 1995;69:8057–8060. doi: 10.1128/jvi.69.12.8057-8060.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cole GA, Katz JM, Hogg TL, Ryan KW, Portner A, Woodland DL. Analysis of the primary T-cell response to Sendai virus infection in C57BL/6 mice: CD4+ T-cell recognition is directed predominantly to the hemagglutinin-neuraminidase glycoprotein. J Virol. 1994;68:6863–6870. doi: 10.1128/jvi.68.11.6863-6870.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Robertson JM, Jensen PE, Evavold BD. DO11.10 and OT-II T cells recognize a C-terminal ovalbumin 323−339 epitope. J Immunol. 2000;164:4706–4712. doi: 10.4049/jimmunol.164.9.4706. [DOI] [PubMed] [Google Scholar]
- 29.Riberdy JM, Newcomb JR, Surman MJ, Barbosa JA, Cresswell P. HLA-DR molecules from an antigen-processing mutant cell line are associated with invariant chain peptides. Nature. 1992;360:474–477. doi: 10.1038/360474a0. [DOI] [PubMed] [Google Scholar]
- 30.Petersen TR, Bettelli E, Sidney J, Sette A, Kuchroo V, Backstrom BT. Characterization of MHC- and TCR-binding residues of the myelin oligodendrocyte glycoprotein 38−51 peptide. Eur. J Immunol. 2004;34:165–173. doi: 10.1002/eji.200324669. [DOI] [PubMed] [Google Scholar]
- 31.Ben-Nun A, Kerlero de RN, Kaushansky N, Eisenstein M, Cohen L, Kaye JF, Mendel I. Anatomy of T cell autoimmunity to myelin oligodendrocyte glycoprotein (MOG): prime role of MOG44F in selection and control of MOG-reactive T cells in H-2b mice. Eur. J Immunol. 2006;36:478–493. doi: 10.1002/eji.200535363. [DOI] [PubMed] [Google Scholar]
- 32.Heslop HE, Rooney CM. Adoptive cellular immunotherapy for EBV lymphoproliferative disease. Immunol Rev. 1997;157:217–222. doi: 10.1111/j.1600-065x.1997.tb00984.x. [DOI] [PubMed] [Google Scholar]
- 33.Gattinoni L, Powell DJ, Jr., Rosenberg SA, Restifo NP. Adoptive immunotherapy for cancer: building on success. Nat. Rev. Immunol. 2006;6:383–393. doi: 10.1038/nri1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dudley ME, Rosenberg SA. Adoptive cell transfer therapy. Semin. Oncol. 2007;34:524–531. doi: 10.1053/j.seminoncol.2007.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xue S, Gillmore R, Downs A, Tsallios A, Holler A, Gao L, Wong V, Morris E, Stauss HJ. Exploiting T cell receptor genes for cancer immunotherapy. Clin. Exp. Immunol. 2005;139:167–172. doi: 10.1111/j.1365-2249.2005.02715.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lanzavecchia A, Lezzi G, Viola A. From TCR engagement to T cell activation: a kinetic view of T cell behavior. Cell. 1999;96:1–4. doi: 10.1016/s0092-8674(00)80952-6. [DOI] [PubMed] [Google Scholar]
- 37.Holler PD, Kranz DM. Quantitative analysis of the contribution of TCR/pepMHC affinity and CD8 to T cell activation. Immunity. 2003;18:255–264. doi: 10.1016/s1074-7613(03)00019-0. [DOI] [PubMed] [Google Scholar]
- 38.Delarasse C, Daubas P, Mars LT, Vizler C, Litzenburger T, Iglesias A, Bauer J, Della GB, Schubart A, Decker L, Dimitri D, Roussel G, Dierich A, Amor S, Dautigny A, Liblau R, Pham-Dinh D. Myelin/oligodendrocyte glycoprotein-deficient (MOG-deficient) mice reveal lack of immune tolerance to MOG in wild-type mice. J Clin. Invest. 2003;112:544–553. doi: 10.1172/JCI15861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hunt DF, Michel H, Dickinson TA, Shabanowitz J, Cox AL, Sakaguchi K, Appella E, Grey HM, Sette A. Peptides presented to the immune system by the murine class II major histocompatibility complex molecule I-Ad. Science. 1992;256:1817–1820. doi: 10.1126/science.1319610. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Expression of alanine substituted TCR. The indicated retrovirally encoded TCRs were transduced into TCRαβ-deficient 4G4.CD4 T cell hybridomas. Transduced hybridomas were flow cytometrically sorted based on co-expressed GFP, the sorted cells expanded, and stained with CD4 and TCR specific antibodies. Results for control untransduced cells are plotted in the upper left.
Supplemental Figure 2. Expression of TCR mutated at CDR3β E100. The 1MOG9 TCR was mutated at E100 to all 20 amino acids. 4G4.CD4 T cells were transduced, flow cytometrically purified based on GFP expression, expanded, stained with TCR-specific antibody, and analyzed. Histogram plots display results for untransduced cells or cells transduced with the different receptors.