

Abstract
The question of the preferred substrate of glutamatergic neurons at high neural activity has been vibrantly debated for over a decade since the classical hypothesis (CH) of the primacy of glucose has been challenged by the astrocyte-neuron lactate shuttle hypothesis (ANLSH), which replaces the primacy of glucose with astrocyte produced lactate. We perform Bayesian Flux Balance Analysis (BFBA) with a new mathematical model of cellular brain energetics, comprising detailed biochemical pathways in and between astrocytes and glutamatergic neurons and partitioning of each cell type into cytosol and mitochondria. Supported by the results of our in silico studies, which are in remarkable agreement with previously published results, we posit the Glucose Shunt Hypothesis (GSH) that during high activity, the inhibition of the phosphofructokinase (PFK) enzyme in neuron impairs neuronal glycolysis, enabling the process by which lactate effluxed by astrocytes is taken up by glutamatergic neurons, whereas at low activity, glucose remains the preferred substrate for neurons. We postulate that the ANLS is a shunt utilized by glutamatergic neurons to bypass their glycolysis impaired by the inhibition of PFK in connection with increased oxidative phosphorylation at high neuronal activity.
INTRODUCTION
The brain is a highly oxidative organ that requires a continuous supply of nutrients and a disproportionate amount of energy compared with most of the other organs in the body. It is well established that under normal physiological conditions, glucose is the main obligatory fuel of the brain, (Siesjo 1978). The classical concept that glucose is the sole energy substrate used by the neurons in adult brain to sustain neuronal activity has dominated the field of brain energy metabolism for many years until the results of PET imaging (Fox and Raichle 1986) suggested a possible uncoupling between oxygen consumption and glucose utilization. This observation encouraged the consideration of alternative substrates, for example lactate, as the main fuel for glutamatergic neurons on activation and led to the formulation of the Astrocyte-Neuron Lactate Shuttle hypothesis (ANLSH). According to the ANLSH, anaerobic glycolysis in astrocytes, activated by increased glutamate level in the synapses of excited glutamatergic neurons, produces lactate, which is taken up by neurons to satisfy their energetic needs, effectively shifting the primacy of neuronal energy substrate from glucose toward lactate (Pellerin and Magistretti 1994).
Since the primacy of glucose was challenged, plenty of evidence has been provided to support both classical (Bak et al. 2006; Zielke et al. 2007) and ANLS hypotheses (Magistretti et al. 1993; McKenna et al. 1993), but the debate remains vibrant because obtaining in vivo and in situ data is difficult, and indirect observations may have a feasible interpretation in both frameworks (Chih and Roberts 2003; Chih et al. 2001). Recent two-photon fluorescence imaging studies of NADH in hippocampal slice preparations (Kasischke et al. 2004) have been interpreted as experimental evidence of the ANLS (Pellerin and Magistretti 2004), although it has been argued that none of the key components of the ANLS, i.e., lactate or pyruvate, were measured in this study (Gjedde 2007; Riera et al. 2008).
The ANLSH, featuring astrocytes as suppliers of lactate at high activity, does not fully explain why neurons should prefer lactate to glucose. Indeed, expression of glycolytic enzymes in neurons and abundance of the GLUT-3 glucose transporters in synaptic membranes (Leino et al. 1997), with higher affinity compared with the GLUT-1 transporters in astrocytes (Vannucci et al. 1997), would suggest the opposite.
One of the problems in settling the question of the lactate shuttling and quantifying the contribution of lactate generated within the brain to neuronal energetics is the difficulty of distinguishing in vivo the transfer of lactate among brain cells (Dienel and Cruz 2004). It has in fact been observed that the lactate shuttling is a cell-to-cell phenomenon (Dienel and Cruz 2004) not limited to the lactate exchange between astrocytes and neurons, but it can occur within populations of cells of the same type. As pointed out in Aubert et al. (2005), “interpretation of lactate kinetics in terms of cellular production, utilization, or disposal remains complex.”
In this article, we propose a mechanism capable of explaining the shift of the neuronal preference toward lactate at high activation. We hypothesize that increased oxidative metabolism of glutamatergic neurons at high neuronal activity exerts an inhibition on the glycolytic enzymes phosphofructokinase (PFK) and hexokinase (HK). Our computational results show that while the same mechanism occurs in astrocytes, their lower oxidative activity does not significantly inhibit glycolysis, therefore shifting their equilibrium toward lactate production. The astrocyte produced lactate, effluxed in extracellular space, is taken up by neurons to fuel their TCA cycle, thus bypassing the glycolytic quench, whereas at low activity, glucose remains the main energy substrate. In practice, we implement the glycolytic inhibition as a downregulation of PFK by the increased oxidative phosphorylation of the cell. The allosteric sensitivity of PFK to changing cytosolic conditions, e.g., the equilibrium state of adenosine phosphates (Gjedde 2007), variations in pH, redox states, and creatine kinase equilibrium (Sappey-Marinier et al. 1992), is known. This leaves several possibilities open to explain the regulatory mechanism.
The Glucose Shunt hypothesis (GSH) posits that neuronal uptake of astrocyte produced lactate is a glucose shunt at high activity, whereas at low activity, the neuron's own glycolysis is the main source of pyruvate entering the TCA. To test the feasibility of the GSH, we perform steady-state analysis with a new five-compartment model of the astrocyte-neuron cellular complex using Bayesian Flux Balance Analysis (BFBA) (Heino et al. 2007). Classical Flux Balance Analysis (FBA) is a methodology for studying steady-state configurations in metabolic networks starting from mass balance equations (Ramakrishna et al. 2001). The BFBA is a stochastic extension of FBA, where all unknown biochemical reaction fluxes and transport rates are modeled as random variables, the stoichiometric and physiological constraints of the system determining their joint probability distribution. The stochastic formulation allows the expression of the degree of uncertainty in both model and data, and the use of sampling methods for estimating the fluxes and transport rates of interest makes it possible to carry out this type of analysis even where the classical FBA would fail, e.g., when the model is not fully consistent or contains nonlinear constraints. This is the case for our model where the inhibition of PFK in connection with an increase in oxidative phosphorylation is implemented in the form of a nonlinear constraint between these two reaction fluxes.
The results of our BFBA confirm that a GSH is consistent with experimental steady-state data at high and low neuronal activities (Gruetter et al. 2001; Hyder et al. 2006; Shen et al. 1999) and conforms to the original formulation of the ANLS hypothesis in Pellerin and Magistretti (1994), recently revisited in Pellerin et al. (2007).
The present work makes a further contribution toward a unified framework for brain energy metabolism, suggesting a regulatory mechanism of the metabolic interaction between astrocytes and glutamatergic neurons that neither assumes nor excludes either the classical or the ANLS hypothesis. The use of mathematical models to create a coherent quantitative framework for energetics and substrate fluxes through the complex biochemical pathways in astrocytes and neurons has been previously advocated, e.g., by Aubert and coworkers (Aubert and Costalat 2005; Aubert et al. 2005, 2007) to study the dynamics of lactate and test the existence of the ANLSH between astrocyte and neuron under different physiological conditions.
METHODS
New five-compartment model of cellular brain energy metabolism
The new metabolic model for brain energetics that we propose and utilize for our in silico experiments was developed starting from the model in Occhipinti et al. (2007). The model contains the key metabolic pathways that need to be active whether the metabolic processes follow the classical or the ANLS hypothesis. Thus the preferred metabolic pathways to fuel the neuronal activity are determined only by the energetic needs of the two cell types and on substrate availability. The increased complexity of this model compared with others proposed in the literature (Aubert and Costalat 2005; Gruetter et al. 2001) requires the use of a robust and powerful methodology, described in the following text, to perform steady-state analysis. The main advantage of our approach to modeling cellular brain energy metabolism is that by performing BFBA, we can utilize a very detailed model while keeping the number of fixed input fluxes and transport rates to a bare minimum, letting the stoichiometry and the need of the systems decide what is a suitable distribution of the reactions fluxes and transport rates to attain steady state.
The brain is a strongly localized organ in the sense that the ratio of neurons and glial cells, their relative volumes as well as their morphology and functioning depend strongly on the regionality. Our in silico model is homogenized over a region of interest, so it is natural to assume that the domain consists of neurons and astrocytes that are morphologically and functionally of a given type. The model for an astrocyte-neuron system is a spatially homogenized multicompartment model that differentiates between blood and extracellular space (ECS) on one hand and two cell types, astrocytes and glutamatergic neurons. Furthermore, each cell type is subcompartmentalized into cytosol and mitochondria. Each compartment in turn is characterized by the biochemical reactions taking place in it (see appendix and Tables A1 and A2), and the various compartments are linked through the transport of some biochemical species between them.
TABLE A1.
Cytosol reactions and stoichiometry
| Cytosol Reactions | Stochiometry |
|---|---|
| 1. Hexokinase | GLC+ATP → G6P+ADP |
| 2. Glycogen synthesis | G6P+ATP → GLY+ADP+2Pi |
| 3. Glycogen phosphorylase | GLY+Pi → G6P |
| 4. Glucose 6-phosphate breakdown | G6P+ATP → 2GA3P+ADP |
| 5. Glyceraldehyde 3-phosphate dehydrogenase | GA3P+Pi+NAD+ → BPG+NADH |
| 6. Pyruvate production | BPG+2ADP → PYR+2ATP |
| 7. Lactate dehydrogenase | PYR+NADH ⇋ LAC+NAD+ |
| 8. Creatine kinase | PCR+ADP ⇋ CR+ATP |
| 9. ATP hydrolysis | ATP → ADP+Pi |
| 10. Malate dehydrogenase | OAA+NADH → MAL+NAD+ |
| 11. Aspartate aminotransferase | Asp+AKG → OAA+GLU |
| 12. Glutamine synthetase* | GLU+ATP → Gln+ADP+Pi |
| 13. Glutaminase | Gln → GLU |
Biochemical reactions in the cytosol of astrocyte and neuron.
, the reaction takes place only in the astrocytes.
TABLE A2.
Mitochondria reactions and stoichiometry
| Mitochondria Reactions | Stochiometry |
|---|---|
| 1. Aspartate aminotransferase | OAA+GLU → Asp+AKG |
| 2. Pyruvate dehydrogenase | PYR+CoA+NAD+ → ACoA+NADH + CO2 |
| 3. Pyruvate carboxylase* | PYR+ATP+CO2 → OAA+ADP+Pi |
| 4. Malic enzyme | PYR+CO2+NADH ⇋ MAL+NAD+ |
| 5. Citrate synthase | ACoA+OAA → CIT+CoA |
| 6. Isocitrate dehydrogenase | CIT+NAD+ → AKG+NADH+CO2 |
| 7. Alpha-ketoglutarate dehydrogenase | AKG+CoA+NAD+ → SCoA+NADH+CO2 |
| 8. Succinyl-CoA synthase | SCoA+ADP+Pi → SUC+CoA+ATP |
| 9. Succinate dehydrogenase | SUC+NAD+ → FUM+NADH |
| 10. Fumarase | FUM → MAL |
| 11. Malate dehydrogenase | MAL+NAD+ → OAA+NADH |
| 12. Glutamate dehydrogenase | AKG+NADH ⇋ GLU+NAD+ |
| 13. Glutamate decarboxylase† | GLU → GABA+CO2 |
| 14. GABA transaminase† | GABA+AKG → SSA+GLU |
| 15. Succinic semialdehyde dehydrogenase† | SSA+NAD+ → SUC+NADH |
| 16. Oxidative phosphorylation | O2+2NADH+6ADP+6Pi → 2H2O+2NAD++6ATP |
Biochemical reactions in the mitochondria of astrocyte and neuron.
, the reaction takes place only in the astrocytes;
, the corresponding reaction occurs only in neurons.
Biochemical pathways
In our computational model, based on the biochemical pathway chart shown in Fig. 1, both neurons and astrocytes are equipped with the glycolytic pathway in the cytosol domain and the TCA cycles and oxidative phosphorylation in the mitochondria. The TCA cycle reactions have a preferred direction that can be justified by thermodynamic considerations under baseline conditions. The virtual transport of reducing equivalents (NAD+/NADH) between cytosol and mitochondria goes via the malate-aspartate shuttle (Dennis and Clark 1978). Another shuttle mechanism, which is responsible for the transfer of reducing equivalents across the mitochondrial membrane, is the glycerol-3-phosphate shuttle that has not been included in the present model because it is considered of little importance in the brain (Nguyen et al. 2003; Siesjo 1978). Anaplerotic reactions, which serve to replenish pools of TCA cycle intermediates from precursors that are not TCA cycle intermediates, have been also included in the model. More specifically, pyruvate carboxylase, a mitochondrial reaction that occurs only in one direction leading to the formation of oxaloacetate from pyruvate and that has been detected only in astrocytes (Yu et al. 1983), has been included only in the mitochondria of the astrocyte. Both cytosolic and mitochondrial malic enzymes have been detected in astrocytes and neurons (Hassel 2001; Hassel and Brathe 2000) and for this reason have been modeled in the mitochondria of both cell types.
FIG. 1.

A schematic of the biochemical pathways in the 5 compartment model of energy metabolism for a cellular complex including astrocyte, glutamatergic neuron and a lumped blood and extracellular space (shaded area). The markers on the arrows indicate the malate/alpha-ketoglutarate and the glutamate/aspartate antiporter pairs of the malate-aspartate shuttle in each cell type.
Because our model is utilized to investigate brain glutamate metabolism, it includes the glutamate-glutamine cycle between astrocytes and glutamatergic neurons, referred to as V-cycle. Glutamate, which is the most abundant excitatory neurotransmitter in the brain (Fonnum 1984), is released by presynaptic glutamatergic neurons and rapidly removed by astrocytic glutamate transporters. Once inside the astrocytes, glutamate is metabolized into glutamine through the cytosolic astrocyte-specific enzyme glutamine synthetase (Martinez-Hernandez et al. 1976). Glutamine is further released from astrocytes and taken up by neurons that synthesize glutamate by phosphate-activated glutaminase (PAG), localized on the outer surface of the inner mitochondrial membrane (Kvamme et al. 2000). The biosynthesis of the neurotransmitter glutamate has been modeled following the model suggested by Palaiologos et al. (1988); this model explains the functional role of aspartate aminotransferase (AAT) in glutamatergic neurons. Because it has been shown that, in both cultured astrocytes and neurons, glutamate can be converted into α-ketoglutarate and enters the TCA cycle for further oxidation (Erecinska et al. 1988; Sonnenwald et al. 1997), the bidirectional biochemical reaction GLU⇋AKG has been included in the mitochondria of both cell types. In neurons, glutamate can be converted into GABA through the GABA shunt, comprising glutamate decarboxylase, GABA transaminase, and SSA dehydrogenase, that allows four of the five carbons lost from the TCA cycle in the form of AKG to reenter at the level of succinate (Siesjo 1978; Waagepetersen et al. 1999).
The transport rates of oxygen and glucose are modeled as influxes, whereas the carbon dioxide transports are effluxes. The lactate transports between the two cytosolic domains and the ECS are not forced in either direction, so our system is free to choose the preferred lactate efflux/influx pattern. This modeling strategy allows both cells to produce or utilize lactate in function of their metabolic needs without imposing the activation of either hypothesized metabolic channeling.
Multicompartment computational model
Our model can be used for both steady-state and dynamic studies of cellular brain energy metabolism. Although in this analysis, we focus on the steady-state case, hence we are not tracking metabolites and intermediates concentrations, which are assumed to remain constant in time, for sake of completeness, we present an overview of the computational model, which in the future will be employed for dynamic studies following the methodology proposed in (Calvetti and Somersalo 2006; Calvetti et al. 2006).
Metabolic substrates pass from the blood through the blood-brain barrier (BBB) and enter the extracellular space. Whereas in dynamic studies it is important to model the BBB and to account for a partitioning of ECS and blood, in our steady-state analysis, we combine them into a lumped perfectly mixed compartment, referred to as “ECS/blood,” where the substrate dynamics is described by convection and diffusion. The corresponding vector of substrate concentrations is denoted as Cb(t), where t is time. The system of differential equations describing the time evolution of the concentrations is of the form
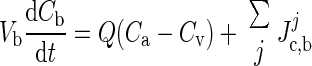 |
(1) |
where Vb is the volume of the ECS/blood compartment in the region of interest, Q = Q(t) is the blood flow, Ca − Cv is the vector of the differences between arterial and venous concentrations of the biochemical species (GLC, LAC, O2, CO2) tracked in the ECS/blood compartment, and the vector Jc,bj contains the net transport rates of the substrates between ECS/blood and the cytosol domains of the neurons (j = 1) and astrocytes (j = 2). The concentration vector Cb is a convex combination of venous and arterial concentration vectors, Cb = FCv + (1 − F)Ca, with a mixing ratio F, 0 < F < 1.
Consider the cytosol domain of one cell type j. We denote by Ccj the vector of cytosolic concentrations and write the mass balance equation as
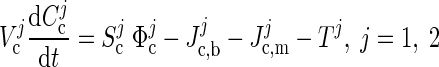 |
(2) |
where Vcj is the compartment domain volume, Φcj is the vector of reaction fluxes in the domain, and Scj is the corresponding stoichiometric matrix, expressing how many moles per unit time of each species is produced (positive entry) or consumed (negative entry) in each reaction. The transport rates Jc,bj and Jc,mj express the net substrate interchange between cytosol and ECS/blood and cytosol and mitochondria in the same cell. Observe that because not all metabolites are interchanged between the compartments, several components of the transport rates are known to vanish. The vector Tj, which denotes the neuronal activity vector, is present only in the cytosol of the neuron, and it is added to assign an energetic price for neuronal activity. In our model, the neuronal activity increases ATP hydrolysis, ATP →ADP + Pi, because it depletes ATP concentration (TATP1 < 0) and increases the ADP and phosphate concentrations (TADP1 = TPi1 = −TATP1 > 0), hence the T1 vector has only three nonzero elements.
Finally, we write the mass balance equation for the concentration vector in the jth mitochondria as
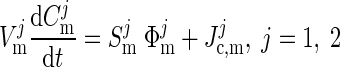 |
(3) |
where the notation is in agreement with the one in the preceding text.
We couple the neuronal activity with the transport of the neurotransmitter glutamate. Assuming no loss of neurotransmitter in the extracellular domain, we lump the transports of glutamate into a single transport between the cytosol compartments of neuron and astrocyte, JGLU,N→A, and write TATP1 = αGLUJGLU,N→A. The value of the activity coefficient αGLU is chosen on the basis of experimental information that 68% of the cellular metabolic rate is required for maintaining the entire activation machinery associated with the V-cycle (Hyder et al. 2006). In our model, the metabolic rate is identified with the total aerobic and anaerobic ATP production per each unit of glucose (Gjedde et al. 2002). Estimating the total ATP production as 2 + 36 = 38 units per unit of glucose, we set αGLU = 0.68 × 38 = 25.84. The activity level of the neuron is implemented by imposing a lower bound for the transport rate of glutamate as explained in the following text.
At steady state, the rates of change of the concentrations in Eqs. 1–3 are negligible, leading to a set of homogenous linear equations with respect to the reaction fluxes, transport rates, and concentration differences in the ECS/blood domain. Here we assume that we have an estimate for arterial and venous blood concentrations and consider r = Q(Ca − Cv) as data. Collecting the reaction fluxes and the nonvanishing components of the transport fluxes in the vector u, the steady-state equations can be written concisely as
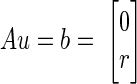 |
(4) |
that is, b is a vector of zeros (cytosol and mitochondria, Eqs. 2 and 3) and r as before. The system (Eq. 4) is underdetermined because the matrix A has a nontrivial null space. To properly handle the null space of A and to guarantee physiologically meaningful solutions, it is necessary to require that the unknowns satisfy some bound constraints, which include loose upper bounds for the absolute values of the unknowns and nonnegativity for reaction fluxes. Thermodynamics determines the preferred direction of reversible biochemical reactions and facilitated transport rates. Thermodynamic constraints cannot be implemented without information about the values of the concentrations of the substrates, but because at normal steady-state conditions the prevailing directions of many reversible reactions are known, we import this additional information by modeling them as unidirectional. Similarly, the preferred directions of some transport rates at baseline steady state are uncontroversial, thus we only model O2 and glucose as influx and CO2 as efflux. The information about the activity level of the astrocyte-neuron complex is encoded in the model by setting a lower bound for the glutamate efflux. In our simulations, we distinguish two activity levels: JGLU,N→A ≥ 0.8 mmol/min for high activity and JGLU,N→A ≥0.2 mmol/min for low activity, where we assume a reference volume for the brain of 1.5 l. These estimates for the lower bounds are based on the experimental results reported by Hyder et al. (2006) and Shulman et al. (2004).
The crucial bound constraint in our model is related to the regulation of glycolysis by oxidative phosphorylation. It is known that the glycolytic enzymes, HK and PFK, are inhibited by several factors, such as depressed pH and elevated levels of cytosolic ATP and activated by high ADP/AMP concentrations (Lowry and Passonneau 1964, 1966). The increased energetic need of a cell elevates ADP/AMP concentrations, thus activating the glycolytic enzymes. The concomitant production of pyruvate may result in an increased lactate production or in an increased oxidative phosphorylation, which efficiently restores the ATP/ADP equilibrium and redox states. Therefore in brain cells, where pyruvate is the main oxidative substrate, it is reasonable to posit that oxidative phosphorylation downregulates glycolysis.
Since the steady state computational model utilized for this study does not provide concentration information, the regulation of PFK by adenosine phosphates or pH is modeled in both cells using the oxidative phosphorylation reaction flux as a controller
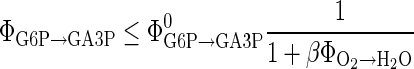 |
(5) |
where the indicated reaction fluxes are intended within the same cell type. The inequality (Eq. 5) conveys the information that an increase in the oxidative phosphorylation activity, ΦO2→H2O inhibits the PFK, which in our model regulates the G6P breakdown, ΦG6P→GA3P. While we do not exclude a prominent role of cytosolic redox in the regulation of glycolysis, because the redox states are affected by lactate dehydrogenase, malate aspartate shuttle, and glycolysis itself, it is not possible to test this alternative without concentration information.
In our computational experiments we set the value of the affinity parameter β to 1 and the saturation value ΦG6P→GA3P was found by a preliminary tuning run as will be explained later.
Bayesian flux balance analysis
The computational steady state model used for our in silico experiments is the stochastic extension of the linear system (Eq. 4), where the unknowns must satisfy the nonlinear inequality constraints Cu ≤ c(u), the source of nonlinearity in the bounds being the control constraints (Eq. 5). The Bayesian stochastic extension of the linear system (Eq. 4) is analyzed with sampling techniques as explained in Occhipinti et al. (2007), Calvetti et al. (2007), Heino et al. (2007), and Calvetti and Somersalo (2007). More specifically, we first replace the linear system (Eq. 4) by the stochastic model, b = Au + e, where the variables b, u, and e are random variables. The additive term e, or noise term, accounts for uncertainties in the measured rate r of transfer across the BBB as well as for the fact that the system may be close to, but not exactly at, a steady state. The stochastic reformulation of the problem is not only in better agreement with the stochastic nature of biological processes but also important from the mathematical and computational perspective. In fact, there is no guarantee that the system (Eq. 4) has a solution that satisfies the bound constraints. We assume that the noise term is a zero mean Gaussian random variable with covariance matrix
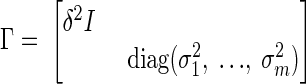 |
where δ2 is the variance expressing the uncertainty of the system being at steady state, and σj2 are the variances of the rates of transfer. The choices of these parameters are discussed in connection with the computed results. The likelihood probability density of b conditional on u is
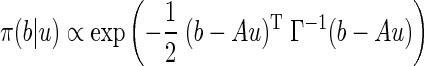 |
where “∝” means “proportional up to a normalizing factor.” The bound constraints are imported in the model via the prior density, which is defined as
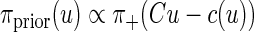 |
where π+ denotes a function that takes on the value 1 if all the components of its argument are positive and vanishes otherwise. By Bayes’ formula, the posterior probability density is πpost(u) = π(u|b) ∝ πprior(u)π(b|u). We explore the posterior probability density using a Markov Chain Monte Carlo (MCMC) scheme appropriate for the present problem. A large sample {u1, u2, …, uN} of vectors distributed according to the probability measure πpost is generated using a Markov transition rule that defines a move from the previous sample point uj−1 to the next one uj. The method of choice here is a hybrid of full scan Gibbs sampler (Geman and Geman 1984) and the hit-and-run algorithm (Calvetti et al. 2008; Smith 1984).
We express the vector u as the sum of two mutually orthogonal components, u = v + w, where w is in the null space of A, i.e., Aw = 0. Since the likelihood is independent of w, π(b|u) = π(b|v). Given the current sample point uj−1 = vj−1 + wj−1, we update first vj−1 → vj componentwise, drawing the kth new component value vkj from the Gaussian distribution with bound constraints
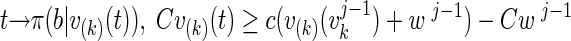 |
where we use the notation v(k)(t). = [v1j, …, vk−1j, t, vk+1j−1, …, vnj−1]T.
Conversely the null space component is independent of the likelihood, and the new update wj is drawn from the uniform distribution over the polyhedral domain
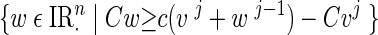 |
using the hit-and-run algorithm. The partition of the space into the null space of A and its orthogonal complement can be done in a straightforward manner using the singular value decomposition of the matrix A. Since the resulting update step uj → uj+1 is not reversible, we need to add an acceptance/rejection step as in the Metropolis-Hastings algorithm. It can be shown that with this modification the updating scheme becomes reversible.
This sampling algorithm is quite effective, with computing time in a standard personal 2GB computer using Matlab R2006b of the order of 25 min per 100,000 sample vectors.
RESULTS
In this section, we present the results of our computational experiments, and we explain how they fit into the ongoing debate about cellular brain energy metabolism. Before commenting on the distributions of the values of reaction fluxes and transport rates, we remark that the fluxes of the bidirectional biochemical reactions glycogen synthesis, glycogen phosphorylase, and creatine kinase were not included in the estimation problem because at steady state and under basal conditions, they are at equilibrium in the sense that their net flux is zero. Furthermore, to reduce the dimensionality of the null space of the matrix A, the bidirectional reactions lactate dehydrogenase, malic enzyme, glutamate dehydrogenase, and the bidirectional transport Jb⇋c,LACj, were estimated as net fluxes that could take on positive or negative values. Hence a negative value of ΦPYR⇋MAL means that the overall reaction is toward pyruvate formation.
The data, which are the input of our computational model, consist of the arterial-venous concentration differences in the ECS/blood domain. The values used for glucose and lactate are Ca,GLC − Cv,GLC = 0.54 mM and Ca,LAC − Cv,LAC = −0.18 mM (Gibbs et al. 1942) with SDs σj equal to 5% of the above mean values, however, not <0.01 mM, and δ = 0.001, whereas for oxygen and carbon dioxide, we used Ca,O2 − Cb,O2 = 2.99 mM and Ca,CO2 − Cb,CO2 = −2.99 mM (Gibbs et al. 1942) with SDs σj equal to 3. The value of Q is 1 l/min, and its possible variations between high and low activity are accounted for by a higher SD for r. The reaction fluxes and transport rates are expressed in μmol·gtissue−1·min−1.
Tuning runs and generation of the samples
Before generating the MCMC sample, tuning runs were performed to choose the saturation value of ΦG6P→GA3P0 with different activity levels, using a model without the PFK feedback control inequality (Eq. 5). The saturation value chosen, ΦG6P→GA3P0 = 0.8 mmol/min, is slightly higher than the upper bound for this flux suggested by the results of test runs with no control on PFK.
The MCMC sampling was preceded by a burn-in run of 600,000 sample points to make sure that all sample histories stabilize. Subsequently, a sample of 100,000 points was generated saving only every 10th vector from a Markov chain of size 1,000,000 to obtain a sample of more independent realizations. Two samples of size 100,000 were generated in this manner, corresponding to high and low neural activity, respectively. Diagnostics of the convergence of the samples, reported in detail in the supplemental materials,1 were performed by calculating the autocorrelation functions (ACF) of all components, providing an estimate of the correlation lengths of the chain (Calvetti and Somersalo 2007). More specifically, for each unknown, we plot the sample history, the ACF and the histogram as illustrated in Fig. 2. The width of the histogram gives an indication about how tightly the variable is estimated.
FIG. 2.
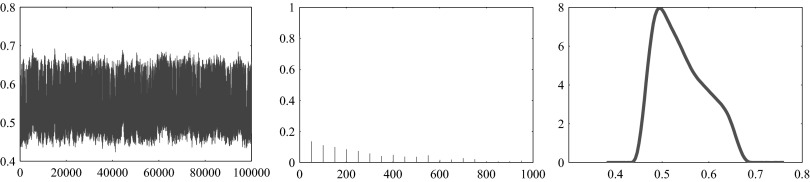
Example of diagnostics of the convergence of the sample. Sample history (left), autocorrelation function (center), and histogram (right) for the reaction flux ΦGLC→G6P are shown. The width of the histogram gives an indication about how tightly the variable is estimated.
For each activation level, we estimated the posterior mean and SD from the collected sample. Tables A3 and A4 of the appendix list the predicted mean values and SDs of all reaction fluxes and transport rates considered in the steady state model. A summary of the overall preferred biochemical pathways suggested by our computational model at the two different activation levels can be found in Fig. 3, where we also indicate the mean values of the key fluxes and transports rates.
TABLE A3.
Estimated reaction fluxes and transport rates
| Activity | Low |
High | ||
|---|---|---|---|---|
| Astrocyte | Neuron | Astrocyte | Neuron | |
| Jb→c,GLC | 0.22 ± 0.06 | 0.14 ± 0.06 | 0.36 ± 0.04 | 0.08 ± 0.03 |
| ΦBPG→PYR | 0.44 ± 0.12 | 0.28 ± 0.11 | 0.72 ± 0.07 | 0.16 ± 0.07 |
| ΦPYR⇋ LAC | 0.24 ± 0.19 | −0.12 ± 0.19 | 0.70 ± 0.07 | −0.58 ± 0.07 |
| ΦATP→ADP+Pi | 3.92 ± 1.68 |
2.13 ± 1.56 | 0.47 ± 0.21 | 0.15 ± 0.15 |
| Jb⇋c,LAC | −0.24 ± 0.19 |
0.12 ± 0.19 | −0.70 ± 0.07 | 0.59 ± 0.07 |
| ΦAap→OAA | 0.21 ± 0.10 | 0.40 ± 0.10 | 0.02 ± 0.01 | 0.74 ± 0.01 |
| ΦOAA→MAL | 0.21 ± 0.10 | 0.40 ± 0.10 | 0.02 ± 0.01 | 0.74 ± 0.01 |
| ΦOAA→Asp | 0.21 ± 0.10 | 0.40 ± 0.10 | 0.02 ± 0.01 | 0.74 ± 0.01 |
| Jc⇋m,MAL,AKG | 0.21 ± 0.10 | 0.40 ± 0.10 | 0.02 ± 0.01 | 0.74 ± 0.01 |
| Jc⇋m,Asp,GI,U | 0.21 ± 0.10 |
0.40 ± 0.10 | 0.02 ± 0.01 | 0.74 ± 0.01 |
| Jc→mPYR | 0.21 ± 0.10 | 0.40 ± 0.10 | 0.02 ± 0.01 | 0.74 ± 0.01 |
| Jc→m,ADP | 3.68 ± 1.72 | 7.40 ± 1.75 | 0.30 ± 0.19 | 13.93 ± 0.21 |
| Jc→m,Pi | 3.68 ± 1.72 | 7.40 ± 1.75 | 0.30 ± 0.19 | 13.93 ± 0.21 |
| Jm→c,ATP | 3.68 ± 1.72 |
7.40 ± 1.75 | 0.30 ± 0.19 | 13.93 ± 0.21 |
| ΦGLU→Gln | 0.22 ± 0.06 | 0.54 ± 0.01 | ||
| ΦGln→GLU | 0.22 ± 0.06 | 0.54 ± 0.01 | ||
| JN→A,GLU | 0.22 ± 0.06 | 0.54 ± 0.01 | ||
| JA→N,Gln | 0.22 ± 0.06 | 0.54 ± 0.01 | ||
Posterior mean values ± SD predicted by the model for the biochemical reactions and transport fluxes identifying the interaction between astrocyte and glutamatergic neuron at different neural activation levels (Low and High). The negative sign means that the overall reaction is toward the substrate or the compartment on the left side. The units are μmol·gtissue−1·min−1. The values of the means ± SD below a threshold of 10−3 are considered as zeros and denoted by ∼0.
TABLE A4.
More estimated reaction fluxes and transport rates
| Activity | Low |
High | ||
|---|---|---|---|---|
| Astrocyte | Neuron | Astrocyte | Neuron | |
| ΦPYR→ACoA | 0.21 ± 0.10 | 0.40 ± 0.10 | 0.02 ± 0.01 | 0.74 ± 0.01 |
| ΦPYR→OAA | 0.21 ± 0.16 | 0.02 ± 0.02 | ||
| ΦPYR⇋MAL | −0.21 ± 0.16 | ∼ 0 | 0.02 ± 0.02 | ∼ 0 |
| ΦACoA→CIT | 0.21 ± 0.10 | 0.40 ± 0.10 | 0.02 ± 0.01 | 0.74 ± 0.01 |
| ΦCIT→AKG | 0.21 ± 0.10 | 0.40 ± 0.10 | 0.02 ± 0.01 | 0.74 ± 0.01 |
| ΦAKG→SCoA | 0.21 ± 0.10 | 0.20 ± 0.13 | 0.02 ± 0.01 | 0.59 ± 0.13 |
| ΦSCoA→SUC | 0.21 ± 0.10 | 0.20 ± 0.13 | 0.02 ± 0.01 | 0.59 ± 0.13 |
| ΦSUC→FUM | 0.21 ± 0.10 | 0.40 ± 0.10 | 0.02 ± 0.01 | 0.74 ± 0.01 |
| ΦFUM→MAL | 0.21 ± 0.10 | 0.40 ± 0.10 | 0.02 ± 0.01 | 0.74 ± 0.01 |
| ΦMAL→GLU | 0.21 ± 0.16 | 0.80 ± 0.19 | 0.02 ± 0.02 | 1.48 ± 0.03 |
| ΦAKG⇋GLU | ∼ 0 |
∼ 0 | ∼ 0 | ∼ 0 |
| ΦGLU→GABA | 0.20 ± 0.13 | 0.15 ± 0.14 | ||
| ΦGABA→SSA | 0.20 ± 0.13 | 0.15 ± 0.14 | ||
| ΦSSA→SUC | 0.20 ± 0.13 |
0.15 ± 0.14 | ||
| ΦO2→H2O | 0.61 ± 0.29 | 1.20 ± 0.28 | 0.05 ± 0.03 | 2.22 ± 0.04 |
| Jb→c→m,O2 | 0.61 ± 0.29 | 1.20 ± 0.28 | 0.05 ± 0.03 | 2.22 ± 0.04 |
| Jm→c→b,CO2 | 0.61 ± 0.29 | 1.20 ± 0.28 | 0.05 ± 0.03 | 2.22 ± 0.04 |
Continuation of Table A3.
FIG. 3.

Schematics of preferred biochemical pathways in astrocyte and glutamatergic neuron during high (top) and low (bottom) neuronal activity. The values by the arrows are the mean values in μmol·gtissue−1·min−1 of the reaction and transport rates of the Markov Chain Monte Carlo (MCMC) sample generated from our computational model.
Interaction between astrocyte and glutamatergic neuron at high activity
At high activity level, brain energetics follows the original ANLS hypothesis (Pellerin and Magistretti 1994), with a glycolytic activity in astrocyte of 0.36 ± 0.04 μmol·g−1·min−1, about five times higher than in neuron. Almost all the end product of the astrocyte glycolytic process, pyruvate, is converted into lactate and released in the extracellular space from where it is taken up by the “hungry” neurons. The active glutamatergic neuron utilizes 0.59 ± 0.07 μmol·g−1·min−1 of the 0.70 ± 0.07 μmol·g−1·min−1 lactate released by the astrocyte, while the remaining is effluxed into the ECS. In neuron the oxidative phosphorylation flux is ∼45 times faster than in astrocyte, the malate-aspartate shuttle and the TCA cycle activity ∼37 times faster, and the exchange of ATP, ADP and Pi between cytosol and mitochondria ∼46 times faster. The GABA shunt in neuron is ∼0.15 ± 0.14 μmol·g−1·min−1, one-fifth of its TCA cycle flux.
Interaction between astrocyte and glutamatergic neuron at low activity
At low activity, the interaction between the two cell types follows the classical hypothesis with a glycolytic activity in neuron of 0.14 ± 0.06 μmol·g−1·min−1, only slightly lower than 0.22 ± 0.06 μmol·g−1·min−1 in astrocyte, and the oxidative activity of astrocytes, 0.61 ± 0.29 μmol·g−1·min−1 is half the one in neuron, 1.20 ± 0.28 μmol·g−1·min−1. In astrocyte, 0.21 ± 0.10 μmol·g−1·min−1 glucose equivalents enter the TCA cycle, the remaining 0.24 ± 0.19 μmol·g−1·min−1 being converted into lactate and effluxed into the ECS. Although the astrocyte-neuron lactate shuttle is active also during low neural activity, the influx of lactate from the ECS is lower, 0.12 ± 0.19 μmol·g−1·min−1, about one-fifth of the neuronal lactate uptake at high activity. Our results support the presence of pyruvate carboxylation and malic enzyme in astrocyte. The pyruvate carboxylation flux replenishes the loss of malate in the TCA cycle through the malic enzyme. The GABA shunt in neuron, at 0.20 ± 0.13 μmol·g−1·min−1, is about one-half of the TCA flux.
Validation of the in silico results
The values of the predicted cerebral glucose utilization, or cerebral metabolic rate (CMRGLC), total tricarboxylic acid cycle rate (VTCAtot), cerebral oxygen utilization (CMRO2), glutamate turnover (Vcycle), oxygen-glucose index (OGI) are presented in Table 1. VTCAtot is the sum of the fluxes of glucose equivalents entering the TCA cycles in astrocyte and neuron and the OGI is the ratio between the oxygen and glucose utilization. Our results, which are well in agreement with the values measured via 13C NMR spectroscopy reported in Gruetter et al. (2001) and Shen et al. (1999); support metabolic coupling between astrocytes and neurons during neurotransmission (Magistretti et al. 1993; Poitry et al. 2000). A comparison of our model predictions for the CMRGLC and the VTCAtot and the experimental data are presented in Fig. 4.
TABLE 1.
CMR of glucose, oxygen, OGI, Vcycle, and total glucose equivalents entering the TCA cycle in astrocyte and neuron at high and low activity
| Activity | Low | High |
|---|---|---|
| CMRGLC | 0.36 ± 0.02 | 0.44 ± 0.01 |
| CMRO2 | 1.81 ± 0.10 | 2.27 ± 0.05 |
| VTCAtot | 0.60 ± 0.03 | 0.76 ± 0.02 |
| Vcycle | 0.22 ± 0.06 | 0.54 ± 0.01 |
| OGI | 5.01 ± 0.08 | 5.22 ± 0.05 |
Estimated mean values ± SD in μmol·gtissue−1·min−1 for CMR of glucose, CMR of oxygen, total tricarboxylic acid cycle rate and Vcycle under low and high activities. We also report the mean value ± SD of the predicted oxygen-glucose index (OGI).
FIG. 4.

Comparison of our model predictions for CMRGLC (left) and VTCAtotal (right) at low (1st bar) and high (2nd bar) neuronal activity and of the experimental results reported by Shen et al. (1999) (3rd bar) and Gruetter et al. (2001) (4th bar).
Moreover, the values of 0.76 ± 0.02 and 0.60 ± 0.03 μmol·g−1·min−1 for the total TCA cycle fluxes predicted by our model for high and low neuronal activity, respectively, are in agreement with the values, in the range of 0.5–1 μmol·g−1·min−1, measured in awake human brain using 13C MRS summarized in the review of Hyder et al. (2006).
Table 1 indicates that our model predictions do not support the decrease in OGI ratio that has been observed during focal and global activation in normal normoxic human brain. This lack of decrease in OGI value is due to a 25% increase in oxygen utilization at high neuronal activity, a prediction that agrees with the observed average motor and secondary responses of oxidative metabolism to brain activation (Gjedde 2007; Vafaee et al. 1999). It has been suggested (Dienel and Cruz 2003) that the decoupling of glucose and oxygen utilization may depend on the experimental conditions and may not be a universal feature in neuronal activation. An explanation of the OGI drop observed during focal (Fox and Raichle 1986) or global (Madsen et al. 1995) stimulation may require modeling of the glycogen shunt dynamics in astrocyte (Dienel and Cruz 2003; Shulman et al. 2001). The results of our analysis suggest that at steady state, the ANLSH alone is not sufficient to explain the disproportionate increase in the rate of glucose utilization during activation.
Our model predicts that at low neuronal activity, astrocytes utilize ∼33% of the total oxygen consumption in agreement with the value of 30% reported by Hertz et al. (2007); hence supporting the emerging evidence that the energetic demands of activated astrocytes are higher and more complex than previously recognized (Dienel and Cruz 2004).
Finally the estimated neuronal lactate uptake from the ECS at high activity is about five times the corresponding lactate influx during low activity, suggesting a proportionality between neuronal lactate uptake and activity levels.
DISCUSSION
Our metabolic model, which identifies separate mitochondria and cytosol compartments and accounts for complete glycolytic pathways for both glutamatergic neuron and astrocyte, is a modification of the one proposed in Occhipinti et al. (2007). To avoid coercing the mathematical model of brain energetics toward either the classical or the ANLS hypothesis, we limit the information entered to a bare minimum, namely to the assumption that the system is at a near steady state and to the values of arterial-venous differences of glucose, lactate, oxygen, and carbon dioxide, without assuming a priori any imbalances of glucose uptake between the two cell types. We simulate two different levels of neuronal activity by setting the lower bound for the transport rate of glutamate from neuron to astrocyte at high activity four times higher than at low activity. We test whether the regulation of glycolysis by oxidative metabolism can explain the lactate trafficking from astrocyte to neuron by implementing symmetrically in both cells the simplified feedback control inequality (Eq. 5). Since our steady-state analysis does not provide estimates of the concentrations of metabolites and intermediates in the cellular compartments, we link the regulation of glycolysis to oxidative phosphorylation at the level of PFK. The distributions of all reaction fluxes and transport rates are determined by the system as needed to sustain a near steady state.
In addition to the hypothesis of glycolytic regulation, we suggest a mechanism that could be responsible for the control of the glycolysis, namely the allosteric downregulation of PFK and HK by the phosphorylation state. This can be seen as a follow up and extension to the attempts to reconciliate the ANLS hypothesis and the classical hypothesis already done in Hyder et al. (2006) and Aubert and Costalat (2005). It is known that the glycolytic enzymes are sensitive to other external conditions such as the cytosolic redox state via pH, and we do not exclude the possibility that the mechanism could be different from the proposed one. The present study is only concerned with steady-state analysis and does not provide any information about the concentrations of metabolites and intermediates. Therefore it is not possible to test if changes in the adenosine phosphate concentrations in cytosol are sufficient to explain the activation of the lactate shuttle, nor can we exclude the possibility that the control of glycolysis is exerted by the redox state. However, the statistical framework of our analysis makes it possible to consider the correlation between oxidative phosphorylation and the malate aspartate shuttle, on one hand, and lactate dehydrogenase on the other, which together with glycolysis itself are the main controllers of the redox state. From the corresponding scatters plots, shown in Figs. 5 and 6, it is clear that oxidative phosphorylation is highly correlated with the malate-aspartate shuttle at both activity levels and in both cell types, while its correlation with lactate dehydrogenase is much lower at high neuronal activity, not excluding the possibility that the primary glycolytic controller could indeed be the redox state or a more complex interplay between the redox and phosphorylation states. The observation that in some cellular systems a high glycolytic activity with lactate production can occur concurrently with high oxidative metabolism levels (Kemper et al. 2001) suggests that mechanisms behind the regulation of glycolysis can be more complex, in particular in the presence of alternative sources of oxidative substrates.
FIG. 5.

Scatter plots showing the correlation of the oxidative phosphorylation flux with the malate-aspartate shuttle rate (left) and the lactate dehydrogenase flux (right) at high neuronal activity for astrocyte (top) and for glutamatergic neuron (bottom). In both cell types, the oxidative phosphorylation reaction flux is high correlated with the malate-aspartate shuttle, therefore showing that the implementation of the phosphofructokinase (PFK) control on glycolysis through the oxidative phosphorylation flux is not in conflict with regulation mechanism based on redox states.
FIG. 6.

Scatter plots showing the correlation of the oxidative phosphorylation flux with the malate-aspartate shuttle rate (left) and the lactate dehydrogenase flux (right) at low neuronal activity for astrocyte (top) and for glutamatergic neuron (bottom). In both cell types, the oxidative phosphorylation reaction flux is high correlated with the malate-aspartate shuttle, therefore showing that the implementation of the PFK control on glycolysis through the oxidative phosphorylation flux is not in conflict with regulation mechanism based on redox states. In addition, there is a negative correlation between oxidative phosphorylation and the lactate dehydrogenase flux.
The inclusion of the main biochemical pathways that could be active under either hypothesis of brain energy metabolism makes our five compartment model substantially more complex than previously published ones (Aubert and Costalat 2005; Aubert et al. 2005, 2007; Gruetter et al. 2001). In this model, we also implement a physiological mechanism that regulates glycolysis and explains the lactate trafficking without assuming uneven partitioning of glucose uptake. In the debate on brain energy metabolism, the difference in availability of substrates to astrocyte and neuron has received a lot of attention without being definitely resolved. The closer proximity of astrocytes to the capillaries has been used to argue their easier—and more plentiful—access to glucose and hence their higher glycolytic activity. The presence of glucose transporters, mainly GLUT-1 in the membranes of astrocytes and of GLUT-3 in the membranes of neurons (Gjedde 2007), nonetheless undermines this argument. Recently it has been pointed out (Gjedde 2007) that “under normal circumstances, glucose transport is not rate limiting and glucose concentrations in the different cellular compartments of brain tissue are likely to be similar.” However, the experimental results reported by Loaiza et al. (2003) and Porras et al. (2004) indicate that increased extracellular glutamate concentration inhibits glucose uptake in neurons and activates it in astrocytes. The mechanism of this regulation and its connection to the ANLS is yet to be fully explained. The uneven partitioning of glucose uptake for the two cell types implemented in Aubert et al. (2005, 2007) and Occhipinti et al. (2007) could be therefore supported in the light of these findings. Our present model does not assume uneven partitioning of glucose uptake but it does not exclude it either. Thus it is the first model of the astrocyte-neuron cellular complex that permits both an even and an uneven partitioning of the transport of glucose as a function of neuronal activity without asymmetric regulation of glucose uptake and lactate trafficking. Our model predicts that at high activity the transport rate of glucose from ECS/blood to astrocyte is approximately five times higher than from ECS/blood to neuron, whereas it is only 1.5 times higher at low activity.
Our analysis suggests that indeed at high neuronal activity the glutamatergic neuron fuels its energetic needs by taking up astrocyte produced lactate. The shuttling of lactate from astrocyte to neuron is made possible by accounting for influx and efflux of lactate from both cell types into the ECS/blood compartment. In our model, both cells can efflux/influx lactate into/from the ECS/blood compartment, leaving it up to the system to decide the lactate trafficking needed to sustain a near steady state at a specified activity level. Our model suggests that at high neuronal activity lactate produced anaerobically by astrocyte is effluxed into the ECS/blood compartment, from where it is taken up by the neurons where it undergoes oxidation. While most of the astrocyte produced lactate is taken up by the neuron, a portion of it, independent of the activation level, leaks into the ECS/blood compartment, in agreement with published results. The mechanism whereby glucose transformed into lactate by astrocyte glycolysis undergoes oxidation in neuron, according to our model appears to be the way to bypass the impaired glycolysis in neuron.
The predictions of our model are qualitatively in agreement with the results reported in a recent review (Hyder et al. 2006). A direct quantitative comparison with the values reported there is not immediate because their model lumps glutamatergic and GABAergic neurons and different values for neuronal activity are used.
GRANTS
This work was partially supported by National Institute of General Medicine Grant GM-66309 to R. Occhipinti and D. Calvetti.
APPENDIX
In this appendix, we present the biochemical reactions included in the model and their stoichiometry, see Table A1 for cytosol and Table A2 for mitochondria. The posterior mean and SDs of the reaction fluxes and transport rates found by our Bayesian flux balance analysis at high and low neuronal activity are reported in Tables A3 and A4.
Footnotes
The online version of this article contains supplemental data.
REFERENCES
- Aubert and Costalat 2005.Aubert A, Costalat R. Interaction between astrocytes and neurons studied using a mathematical model of compartmentalized energy metabolism. J Cereb Blood Flow Metab 25: 1476–1490, 2005. [DOI] [PubMed] [Google Scholar]
- Aubert et al. 2005.Aubert A, Costalat R, Magistretti PJ, Pellerin L. Brain lactate kinetics: modeling evidence for neuronal lactate uptake upon activation. Proc Natl Acad Sci USA 102: 16448–16453, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubert et al. 2007.Aubert A, Pellerin L, Magistretti PJ, Costalat R. A coherent neurobiological framework for functional neuroimaging provided by a model integrating compartmentalized energy metabolism. Proc Natl Acad Sci USA 104: 4188–4193, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bak et al. 2006.Bak LK, Schousboe A, Sonnewald U, Waagepetersen HS. Glucose is necessary to mantain neurotransmitter homeostasis during synaptic activity in cultured glutamatergic neurons. J Cereb Blood Flow Metab 26: 1285–1297, 2006. [DOI] [PubMed] [Google Scholar]
- Calvetti et al. 2006.Calvetti D, Hageman R, Somersalo E. Large-scale Bayesian parameter estimation for a three compartment cardiac metabolism model during ischemia. Inverse Probl 22: 1797–1816, 2006. [Google Scholar]
- Calvetti et al. 2007.Calvetti D, Heino J, Somersalo E, Tunyan K. Bayesian stationary state flux balance analysis for a skeletal muscle metabolic model. Inverse Probl Imag 1: 247–263, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvetti et al. 2008.Calvetti D, Kuceyeski A, Somersalo E. Sampling-based analysis of a spatially distributed model for liver metabolism at steady state. Multiscale Model Simul 7: 407–431, 2008. [Google Scholar]
- Calvetti and Somersalo 2006.Calvetti D, Somersalo E. Large scale statistical parameter estimation in complex systems with an application to metabolic models. Multiscale Model Simul 5: 1333–1366, 2006. [Google Scholar]
- Calvetti and Somersalo 2007.Calvetti D, Somersalo E. Introduction to Bayesian Scientific Computing—Ten Lectures on Subjective Computing. Berlin: Springer Verlag, 2007.
- Chih et al. 2001.Chih C-P, Lipton P, Roberts EL Jr. Do active cerebral neurons really use lactate rather than glucose? Trends Neurosci 24: 573–578, 2001. [DOI] [PubMed] [Google Scholar]
- Chih and Roberts 2003.Chih C-P, Roberts EL Jr. Energy substrates for neurons during neural activity:a critical review of the astrocyte-neuron lactate shuttle hypothesis. J Cereb Blood Flow Metab 23: 1263–1281, 2003. [DOI] [PubMed] [Google Scholar]
- Dennis and Clark 1978.Dennis SC, Clark JB. The regulation of glutamate metabolism by tricarboxylic acid-cycle activity in rat brain mitochondria. Biochem J 172: 155–162, 1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dienel and Cruz 2003.Dienel GA, Cruz NF. Neighborly interactions of metabolically activated astrocytes in vivo. Neurochem Int 43: 339–354, 2003. [DOI] [PubMed] [Google Scholar]
- Dienel and Cruz 2004.Dienel GA, Cruz NF. Nutrition during brain activation: does cell to cell lactate shuttling contribute significantely to sweet and sour food for thought? Neurochem Int 45: 321–351, 2004. [DOI] [PubMed] [Google Scholar]
- Erecinska et al. 1988.Erecinska M, Zaleska MM, Nissim I, Nelson D, Dagani F, Yudkoff M. Glucose and synaptosomal glutamate metabolism: studies with [15N]glutamate. J Neurochem 51: 892–902, 1988. [DOI] [PubMed] [Google Scholar]
- Fonnum 1984.Fonnum F Glutamate: a neurotransmitter in mammalian brain. J Neurochem 42: 1–11, 1984. [DOI] [PubMed] [Google Scholar]
- Fox and Raichle 1986.Fox PT, Raichle ME. Focal physiological uncoupling of cerebral blood flow and oxidative metabolism during somatosensory stimulation of human subjects. Proc Natl Acad Sci USA 83: 1140–1144, 1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geman and Geman 1984.Geman S, Geman D. Stochastic relaxation, Gibbs distributions, and the Bayesian Restoration of images. IEEE Trans Pattern Anal Mach Intell 6: 721–741, 1984. [DOI] [PubMed] [Google Scholar]
- Gibbs et al. 1942.Gibbs EL, Lennox WG, Nims LF, Gibbs FA. Arterial and cerebral venous blood arterial-venous differences in man. J Biol Chem 144: 325–332, 1942. [Google Scholar]
- Gjedde 2007.Gjedde A Coupling of brain function to metabolism: evaluation of energy requirements. In: Handbook of Neurochemistry and Molecular Neurobiology Brain Energetics Integration of Molecular and Cellular Processes. Berlin: Springer Verlag, 2007, pt. IV, chapt. 5, p. 343–400.
- Gjedde et al. 2002.Gjedde A, Marrett S, Vafaee M. Oxidative and nonoxidative metabolism of excited neurons and astrocytes. J Cereb Blood Flow Metab 22: 1–14, 2002. [DOI] [PubMed] [Google Scholar]
- Gruetter et al. 2001.Gruetter R, Seaquist ER, Ugurbil K. A mathematical model of compartmentalized neurotransmitter metabolism in the human brain. Am J Physiol Endocrinol Metab 281: E100–E112, 2001. [DOI] [PubMed] [Google Scholar]
- Hassel and Brathe 2000.Hassel B, Brathe A. Neuronal pyruvate carboxylation supports formation of transmitter glutamate. J Neurosci 20: 1342–1347, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassel 2001.Hassel B Carboxylation and anaplerosis in neurons and glia. Mol Neurobiol 22: 21–40, 2001. [DOI] [PubMed] [Google Scholar]
- Heino et al. 2007.Heino J, Tunyan K, Calvetti D, Somersalo E. Bayesian flux balance analysis applied to a skeletal muscle metabolic model. J Theor Biol 248: 91–110, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertz et al. 2007.Hertz L, Peng L, Dieniel GA. Energy metabolism in astrocytes: high rate of oxidative metabolism and spatiotemporal dependence on glycolysis/glycogenolysis. J Cereb Blood Flow Metab 27: 219–249, 2007. [DOI] [PubMed] [Google Scholar]
- Hyder et al. 2006.Hyder F, Patel AB, Gjedde A, Rothman DL, Behar KL, Shulman RG. Neuronal-glial glucose oxidation and glutamatergic-GABAergic function. J Cereb Blood Flow Metab 26: 865–877, 2006. [DOI] [PubMed] [Google Scholar]
- Kasischke et al. 2004.Kasischke KA, Vishwasrao HD, Fisher PJ, Zipfel WR, Webb WW. Neural activity triggers neuronal oxidative metabolism followed by astrocytic glycolysis. Science 305: 99–103, 2004. [DOI] [PubMed] [Google Scholar]
- Kemper et al. 2001.Kemper WF, Lindstedt SL, Hartzler LK, Hicks JW, Conley KE. Shaking up glycolysis: sustained, high lactate flux during aerobic rattling. Proc Natl Acad Sci USA 98: 723–728, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvamme et al. 2000.Kvamme E, Roberg B, Torgner IA. Phosphate-activated glutaminase and mitochondrial glutamine transport in the brain. Neurochem Res 25: 1407–1419, 2000. [DOI] [PubMed] [Google Scholar]
- Leino et al. 1997.Leino RL, Gerhart DZ, van Bueren AM, McCall AL, Drewes LR. Ultrastructural localization of GLUT-1 and GLUT-3 glucose transporters in rat brain. J Neurosci Res 49: 617–626, 1997. [DOI] [PubMed] [Google Scholar]
- Loaiza et al. 2003.Loaiza A, Porras OH, Barros LF. Glutamate triggers rapid glucose transport stimulation in astrocytes as evidenced by real-time confocal microscopy. J Neurosci 23: 7337–7342, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry and Passonneau 1964.Lowry OH, Passonneau JV. The relationship between substrates and enzymes of glycolysis in brain. J Biol Chem 239: 31–42, 1964. [PubMed] [Google Scholar]
- Lowry and Passonneau 1966.Lowry OH, Passonneau JV. Kinetic evidence for multiple binding sites of phosphofructokinase. J Biol Chem 241: 2268–2279, 1966. [PubMed] [Google Scholar]
- Madsen et al. 1995.Madsen PL, Hasselbach SG, Hagemann LP, Olsen KS, Bulow J, Holm S, Wildschiodtz G, Paulson OB, Lassen NA. Persistent resetting of the cerebral oxygen/glucose uptake ratio by brain activation: evidence obtained with the Kety-Schmidt technique. J Cereb Blood Flow Metab 15: 485–491, 1995. [DOI] [PubMed] [Google Scholar]
- Magistretti et al. 1999.Magistretti PJ, Pellerin L, Rothman DL, Shulman RG. Energy on demand. Science 283: 496–497, 1999. [DOI] [PubMed] [Google Scholar]
- Magistretti et al. 1993.Magistretti PJ, Sorg O, Yu N, Martin JL, Pellerin L. Neurotransmitters regulate energy metabolism in astrocytes: implication for the metabolic trafficking between neural cells. Dev Neurosci 15: 306–312, 1993. [DOI] [PubMed] [Google Scholar]
- Martinez-Hernandez et al. 1976.Martinez-Hernandez A, Bell KP, Norenberg MD. Glutamine synthetase: glial localization in brain. Science 195: 1356–1358, 1976. [DOI] [PubMed] [Google Scholar]
- McKenna et al. 1993.McKenna MC, Tildon JT, Stevenson JH, Boatright R, Huang S. Regulation of energy metabolism in synaptic terminals and cultured rat brain astrocytes: differences revealed using aminooxyacetate. Dev Neurosci 15: 320–329, 1993. [DOI] [PubMed] [Google Scholar]
- Nguyen et al. 2003.Nguyen NH, Brathe A, Hassel B. Neuronal uptake and metabolism of glycerol and the neuronal expression of mitochondrial glycerol-3-phosphate dehydrogenase. J Neurochem 85: 831–842, 2003. [DOI] [PubMed] [Google Scholar]
- Occhipinti et al. 2007.Occhipinti R, Puchowicz MA, LaManna JC, Somersalo E, Calvetti D. Statistical analysis of metabolic pathways of brain metabolism at steady state. Ann Biomed Eng 6: 886–902, 2007. [DOI] [PubMed] [Google Scholar]
- Palaiologos et al. 1988.Palaiologos G, Hertz L, Schousboe A. Evidence that aspartate aminotransferase activity and ketodicarboxylate carrier function are essential for biosynthesis of transmitter glutamate. J Neurochem 51: 317–320, 1988. [DOI] [PubMed] [Google Scholar]
- Pellerin et al. 2007.Pellerin L, Bouzier-Sore AK, Aubert A, Serres S, Merle M, Costalat R, Magistretti PJ. Activity-dependent regulation of energy metabolism by astrocytes: an update. Glia 12: 1251–1262, 2007. [DOI] [PubMed] [Google Scholar]
- Pellerin and Magistretti 1994.Pellerin L, Magistretti PJ. Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc Natl Acad Sci USA 91: 10625–10629, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellerin and Magistretti 2004.Pellerin L, Magistretti PJ. Let there be (NADH) light. Science 305: 50–52, 2004. [DOI] [PubMed] [Google Scholar]
- Poitry et al. 2000.Poitry S, Poitry-Yamate C, Ueberfeld J, Macleish PR, Tsacopoulos M. Mechanisms of glutamate metabolic signaling in retinal glial (Muller) cells. J Neurosci 20: 1809–1821, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porras et al. 2004.Porras OH, Loaiza A, Barros LF. Glutamate mediates acute glucose transport inhibition in hippocampal neurons. J Neurosci 24: 9669–9673, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakrishna et al. 2001.Ramakrishna R, Edwards JS, McCulloch A, Palsson BO. Flux-balance analysis of mitochondrial energy metabolism: consequences of systemic stoichiometric constraints. Am J Physiol Regul Integr Comp Physiol 280: R695–704, 2001. [DOI] [PubMed] [Google Scholar]
- Riera et al. 2008.Riera JJ, Schousboe A, Waageptersen HS, Howarth C, Hyder F. The micro–architecture of the cerebral cortex: functional neuroimaging models and metabolism. NeuroImage 40: 1436–1459, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sappey-Marinier et al. 1992.Sappey-Marinier D, Calabrese G, Fein G, Hugg JW, Biggins C, Weiner MW. Effect of photic stimulation on human visual cortex lactate and phosphates using 1H and 31P magnetic resonance spectroscopy. J Cereb Blood Flow Metab 12: 584–592, 1992. [DOI] [PubMed] [Google Scholar]
- Shen et al. 1999.Shen J, Petersen KF, Behar KL, Brown P, Nixon TW, Mason GF, Petroff OA, Shulman GI, Shulman GR and Rothman DL. Determination of the rate of the glutamate/glutamine cycle in the human brain by in vivo13C NMR. Proc Natl Acad Sci USA 96: 8235–8240, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulman et al. 2001.Shulman RG, Hyder F, Rothman DL. Cerebral energetics and the glycogen shunt: neurochemical basis of functional imaging. Proc Natl Acad Sci USA 98: 6417–6422, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulman et al. 2004.Shulman RG, Rothman DL, Behar KL, Hyder F. Energetic basis of brain activity: implications for neuroimaging. Trends Neurosci 27: 489–495, 2004. [DOI] [PubMed] [Google Scholar]
- Siesjo 1978.Siesjo BK Brain Energy Metabolism. New York: Wiley, 1978.
- Smith 1984.Smith RL Efficient Monte Carlo procedures for generating points uniformly distributed over banded regions. Oper Res 32: 1296–1308, 1984. [Google Scholar]
- Sonnewald et al. 1997.Sonnewald U, Westergaard N, Schousboe A. Glutamate transport and metabolism in astrocytes. Glia 21: 56–63, 1997. [DOI] [PubMed] [Google Scholar]
- Vafaee et al. 1999.Vafaee MS, Meyer E, Marrett S, Paus T, Evans AC, Gjedde A. Frequency-dependent changes in cerebral metabolic rate of oxygen during activation of human visual cortex. J Cereb Blood Flow Metab 19: 272–277, 1999. [DOI] [PubMed] [Google Scholar]
- Vannucci et al. 1997.Vannucci SJ, Maher F, Simpson IA. Glucose transporter proteins in brain: delivery of glucose to neurons and glia. Glia 21: 2–21, 1997. [DOI] [PubMed] [Google Scholar]
- Waagepetersen et al. 1999.Waagepetersen HS, Sonnewald U, Schousboe A. The GABA paradox: multiple roles as metabolite, neurotransmitter, and neurodifferentiative agent. J Neurochem 73: 1335–1342, 1999. [DOI] [PubMed] [Google Scholar]
- Yu et al. 1983.Yu AC, Drejer J, Hertz L, Schousboe A. Pyruvate carboxylase activity in primary cultures of astrocytes and neurons. J Neurochem 41: 1484–1487, 1983. [DOI] [PubMed] [Google Scholar]
- Zielke et al. 2007.Zielke HR, Zielke CL, Baab PJ, Tildon JT. Effect of fluorocitrate on cerebral oxidation of lactate and glucose in freely moving rats. J Neurochem 101: 9–16, 2007. [DOI] [PubMed] [Google Scholar]