

Abstract
The cyclic AMP-dependent protein kinase (PKA) signaling pathway has been shown to be important in mechanisms of synaptic plasticity, although its direct and downstream signaling effects are not well understood. Using an in vitro model of eyeblink classical conditioning, we report that PKA has a critical role in initiating a signaling cascade that results in synaptic delivery of glutamate receptor 1 (GluR1)- and GluR4-containing α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs) in abducens motor neurons during conditioning. PKA and the Ca2+-calmodulin–dependent protein kinases (CaMKs) II and IV are activated early in conditioning and are required for acquisition and expression of conditioned responses (CRs). cAMP-response-element-binding protein (CREB) is also activated early in conditioning but is blocked by coapplication of inhibitors to PKA and the CaMKs, suggesting that CREB is downstream of those signaling cascades. Moreover, evidence suggests that PKA activates extracellular signal-regulated kinase, which is also required for conditioning. Imaging studies after conditioning further indicate that colocalization of GluR1 AMPAR subunits with the synaptic marker synaptophysin requires PKA, but is insensitive to the N-methyl-d-aspartate receptor (NMDAR) inhibitor d,l-AP5. PKA activation also leads to synaptic localization of GluR4 subunits that, unlike GluR1, is dependent on NMDARs and is mediated by CaMKII. Together with previous studies, our findings support a two-stage model of AMPAR synaptic delivery during acquisition of classical conditioning. The first stage involves synaptic incorporation of GluR1-containing AMPARs that serves to activate silent synapses. This allows a second stage of NMDAR- and protein kinase C–dependent delivery of GluR4 AMPAR subunits that supports the acquisition of CRs.
INTRODUCTION
The cyclic AMP–dependent protein kinase (PKA) signaling pathway has been shown to be pivotal for learning and synaptic plasticity (Nguyen and Woo 2003). However, the importance of PKA in different temporal phases of learning and plasticity has been a matter of considerable debate. In fear conditioning, for example, injection of a PKA inhibitor into the amygdala immediately after training impaired the consolidation of fear memory but left short-term memory largely intact (Schafe and LeDoux 2000). On the other hand, localized infusions of a PKA antagonist directly into the cerebellum just prior to delay eyeblink classical conditioning attenuated conditioned responses (CRs) during acquisition, but infusions after learning had been established failed to block CRs (Cartford et al. 2004). Genetically modified mice in which PKA anchoring could be conditionally inhibited also showed impaired acquisition of a spatial learning task (Nie et al. 2007). A number of studies have demonstrated a role for PKA signaling in late-phase long-term potentiation (L-LTP) in slices from the hippocampus (Frey et al. 1993; Huang et al. 1994; Nie et al. 2007; Weisskopf et al. 1994) and amygdala (Huang and Kandel 2007) and in transgenic mice in which PKA activity was reduced (Abel et al. 1997). A role for PKA in induction or early-phase LTP (E-LTP) is less clear. Inhibitors of adenylyl cyclase or PKA were found to diminish E-LTP in hippocampal slices but did not block it completely (Otmakhova et al. 2000). Moreover, transient N-methyl-d-aspartate receptor (NMDAR)–induced activation of PKA was increased after tetanic stimulation during the induction of LTP in hippocampal slices (Roberson and Sweatt 1996). These data also suggested that PKA does not have a direct role in expression of L-LTP, but rather has downstream effects on other effector proteins such as the cAMP-response-element-binding protein (CREB). Differences in learning paradigms and stimulation protocols have likely contributed to some of these conflicting results on the nature of PKA function in learning and plasticity.
The locus of PKA activity is also key to its function in plasticity mechanisms as indicated by evidence that PKA has both presynaptic and postsynaptic effects. Regulation of presynaptic release probability by PKA has been implicated in L-LTP (Bolshakov et al. 1997). The synaptic vesicle scaffolding protein Rab3-interacting molecule 1α (RIM1α) has been shown to be a PKA substrate critical for a presynaptic form of LTP (Powell 2006). For example, RIM1α is directly phosphorylated by PKA and induces presynaptic LTP in cultured cerebellar parallel fiber-Purkinje cell synapses (Lonart et al. 2003). Presynaptic action of PKA is also an important element in mechanisms of classical conditioning in Aplysia (Roberts and Glanzman 2003). Alternatively, postsynaptic trafficking of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs) and NMDARs has been demonstrated in response to PKA activation. Phosphorylation of glutamate receptor 1 (GluR1) and GluR4 AMPAR subunits by PKA mediates activity-dependent synaptic incorporation of receptors in hippocampal organotypic slices (Esteban et al. 2003). GluR1 subunits are phosphorylated at Ser831 and Ser845 by Ca2+-calmodulin–dependent protein kinase (CaMK) II and protein kinase C (PKC), and by PKA, respectively (Barria et al. 1997; Roche et al. 1996). Interestingly, the synaptic delivery of GluR1 driven by CaMKII requires the parallel activity of PKA (Esteban et al. 2003). Delivery of GluR1-containing AMPARs to perisynaptic sites prior to expression of LTP in CA1 hippocampal neurons has further been shown to require PKA signaling (Yang et al. 2008), supporting a model in which these receptors are rapidly mobilized to synaptic regions by PKA and then translocated into synapses by NMDAR-mediated Ca2+ influx to support LTP (Derkach et al. 2007). In addition to glutamate receptors, CREB is downstream of PKA and acts as a transcription factor to regulate gene expression. Many studies support the involvement of CREB not only in synaptic plasticity and learning, but also in neuropsychiatric disorders (Carlezon Jr et al. 2005). In addition to PKA, numerous intracellular signaling pathways including the CaMKs, PKC, and mitogen-activated protein kinase (MAPK) regulate the activation of CREB. Equally numerous are the target genes that are regulated by CREB. These include growth factors such as brain-derived neurotrophic factor (BDNF), known to be involved in plasticity mechanisms and synaptic delivery of AMPARs (Caldeira et al. 2007; Li and Keifer 2008, 2009).
Recently, we have made considerable progress in identifying some of the signaling pathways that generate CRs in an in vitro model of eyeblink classical conditioning. Evidence suggests that plasticity underlying conditioning occurs at the abducens motor neurons and is associated with synaptic incorporation of GluR1- and GluR4-containing AMPARs (Li and Keifer 2008, 2009; Mokin et al. 2007; Zheng and Keifer 2008). In this model, in place of tone and airpuff stimuli as used in behaving animals, weak electrical stimulation of the auditory nerve (the “tone” conditioned stimulus [CS]) is paired with strong stimulation of the trigeminal nerve (the “airpuff” unconditioned stimulus [US]) and results in a neural correlate of conditioned eyeblink responses recorded from the abducens nerve (see Keifer 2003 for a review). The synaptic delivery of GluR1 and GluR4 occurs sequentially during early stages of conditioning to promote CR acquisition. First, synaptic incorporation of GluR1 subunits precedes GluR4 to activate silent synapses (Mokin et al. 2007). This is followed by NMDAR-dependent synaptic incorporation of newly synthesized GluR4-containing AMPARs that are thought to support the acquisition and expression of CRs. Synaptic delivery of both GluR1 and GluR4 is regulated by MAPK family member extracellular signal-regulated kinase (ERK) signaling pathways (Keifer et al. 2007). Recent findings reveal that the coordinated activity of PKC and ERK controls the synaptic incorporation of GluR4-containing AMPARs, whereas delivery of GluR1 subunits is unaffected by inhibitors of PKC (Zheng and Keifer 2008). Here, we extend these findings on acquisition of conditioning to show that synaptic incorporation of AMPARs containing GluR1 subunits is dependent on PKA. PKA, the CaMKs (II and IV), and CREB are activated shortly after the onset of paired stimulation, followed by the activation of ERK. Furthermore, inhibition of NMDARs by d-2-amino-5-phosphonopentanoic acid (d,l-AP5) fails to block the synaptic insertion of GluR1-containing AMPARs. Taken together, these data support a two-stage model for the acquisition phase of in vitro classical conditioning in which PKA mediates the synaptic incorporation of GluR1-containing AMPARs followed by the NMDAR- and PKC-dependent delivery of GluR4 subunits that supports the acquisition of CRs.
METHODS
Conditioning procedures
Freshwater pond turtles Pseudemys scripta elegans, obtained from commercial suppliers, were anesthetized by hypothermia (by placing them in a freezer until torpid) and decapitated. Protocols involving the use of animals complied with the guidelines of the National Institutes of Health and the Institutional Animal Care and Use Committee. The brain stem was transected at the levels of the trochlear and glossopharyngeal nerves and the cerebellum was removed as described previously (Anderson and Keifer 1999). Therefore the preparation consisted of only the pons, with the cerebellar circuitry removed. The brain stem was continuously bathed in physiological saline (2–4 ml/min) containing (in mM): 100 NaCl, 6 KCl, 40 NaHCO3, 2.6 CaCl2, 1.6 MgCl2, and 20 glucose, which was oxygenated with 95% O2-5% CO2 and maintained at room temperature (22–24°C) at pH 7.6 (Anderson and Keifer 1999). Suction electrodes were used for stimulation and recording of cranial nerves. The US was a twofold threshold single-shock stimulus applied to the trigeminal nerve; the CS was a subthreshold 100 Hz, 1 s train stimulus applied to the ipsilateral posterior root of the eighth nerve that was below the threshold amplitude required to produce activity in the abducens nerve (Anderson and Keifer 1999; Keifer 2001; Keifer et al. 1995). The latter nerve will be referred to as the auditory nerve because it carries predominantly auditory fibers. Neural activity was recorded from the ipsilateral abducens nerve that normally projects to the extraocular muscles controlling movements of the eye, nictitating membrane, and eyelid. The CS–US interval was 20 ms, which is defined as the time between the offset of the CS and the onset of the US. This brief delay was found to be optimal for conditioning; however, conditioning is not supported using longer CS–US intervals (Keifer 2001). The intertrial interval between the paired stimuli was 30 s. A pairing session consisted of 50 CS–US presentations followed by a 30 min rest period in which there was no stimulation (Keifer et al. 1995). Conditioned responses were defined as abducens nerve activity that occurred during the CS and exceeded an amplitude of double the baseline recording level. Conditioned preparations were those that received paired CS–US stimulation, whereas pseudoconditioned control preparations received the same number of CS and US exposures that were explicitly unpaired using a CS–US interval randomly selected between 300 ms and 25 s.
Pharmacology
The following membrane-permeable compounds were dissolved in physiological saline and perfused through the bath to test for the function of PKA and the CaMKs in conditioning: the general CaMK inhibitor KN-62 (10 μM), the selective inhibitor of CaMKII autocamtide-2–related inhibitory peptide (AIP myristoylated, 5 μM; Calbiochem, San Diego, CA), the selective cAMP analog and competitive inhibitor of PKA activation Rp-cAMPs (50 μM), or its analog the phosphodiesterase-resistant PKA activator Sp-cAMPs (50 μM; Sigma, St. Louis, MO). Additionally, we tested the NMDAR antagonist d,l-2-amino-5-phosphonopentanoic acid (d,l-AP5, 100 μM; Tocris, Ellisville, MO) and the MEK–ERK inhibitor PD98059 (50 μM; Calbiochem). In some experiments, drug application was performed ≥30 min prior to beginning the first pairing session and continued throughout the conditioning procedure to test for the drug effects on acquisition of abducens CRs. In other experiments, preparations underwent the conditioning procedure in normal physiological saline and were tested for drug effects on expression of CRs. The drug was then washed from the bath and conditioning resumed in normal saline to test for CR recovery.
Western blot analysis
Turtle brain stems were pseudoconditioned, conditioned, or treated with drug and frozen in liquid nitrogen immediately after the physiological experiments and stored at −70°C. Tissue was homogenized in lysis buffer (20 mM Tris, pH 8.0; 1 mM EDTA; 1% NP40; 0.15 M NaCl; 10 mM Na4P2O7; and 5% glycine) with a protease (Roche, Mannheim, Germany) and phosphatase inhibitor cocktail (Sigma), rotated at 4°C for 2 h, centrifuged at 14,000 g for 20 min at 4°C, and the supernatants were aliquoted and stored at −70°C. Protein concentration was assessed using a BCA assay (Sigma) and protein sample concentrates were solubilized in 2× SDS/β-mercaptoethanol and boiled for 5 min before separation by 10% SDS–PAGE. After electrophoresis, membranes were blocked with 5% nonfat dry milk in Tris-buffered saline/0.1% Tween-20 for 1 h at room temperature. We used the phosphorylation site-directed antibodies against PKA Thr197 (p-PKA; Cell Signaling Technology, Danvers, MA), CaMKII Thr286 (p-CaMKII; Cell Signaling), CREB Ser133 (p-CREB; Cell Signaling), CaMKIV Thr196 (p-CaMKIV; Santa Cruz Biotechnology, Santa Cruz, CA), or ERK Thr183/Tyr185 (p-ERK; Promega, Madison, WI). For total protein we used PKA (t-PKA; Upstate, Lake Placid, NY), CaMKII (t-CaMKII; Chemicon), CREB (t-CREB; Cell Signaling), CaMKIV (t-CaMKIV; Abcam, Cambridge, MA), or ERK (t-ERK; Chemicon). Additionally, we used phosphorylation-specific antibodies to the GluR1 AMPAR subunit that recognizes Ser845 (Phosphosolutions, Aurora, CO) or Ser831 (Chemicon). Membranes were incubated with primary antibodies overnight at 4°C, washed, and incubated with horseradish peroxidase–conjugated (1:10,000) or fluorescently tagged secondary antibodies (1:5,000) for 1 h at room temperature. Loading controls were performed using primary antibodies to actin (1:1,000; Chemicon). Proteins were detected by the ECL-Plus chemiluminescence system (Amersham Pharmacia, Piscataway, NJ) or the Odyessy infrared imaging system (Li-Cor Biotechnology, Lincoln, NE). Immunoreactive signals were captured on Kodak X-omatic AR film and quantified by computer-assisted densitometry. Optical densities of the bands were determined relative to background levels. Quantifications of total and phosphoprotein were determined relative to actin. Ratios of phospho- to total protein were obtained for each experiment and averaged. Data are displayed as a percentage of normalized values from pseudoconditioned controls.
Glutamate receptor localization, confocal imaging, and data analysis
Immediately after the physiological experiments, brain stems were immersion fixed in cold 0.5% paraformaldehyde (Mokin and Keifer 2004). Tissue sections were cut at 30 μm and preincubated in primary antibody overnight at 4°C with gentle shaking. The primary antibodies used were a polyclonal antibody raised in goat that recognizes the GluR4 subunit of AMPA receptors (1:100, Santa Cruz 7614), a polyclonal raised in rabbit that recognizes GluR1 (1:200, Chemicon 1504), and a monoclonal antibody raised in mouse that recognizes synaptophysin (1:1,000, Sigma 5768). The specificity of the antibodies was confirmed by Western blot. After the primary antibodies, sections were rinsed and incubated with secondary antibodies for 2 h using a concentration of 1:100 for GluR4 and GluR1 or 1:200 for synaptophysin. The secondary antibodies were a Cy3-conjugated goat anti-rabbit immunoglobulin G (IgG) for GluR1, a Cy3-conjugated rabbit anti-goat IgG for GluR4, and a Cy2-conjugated goat anti-mouse IgG for synaptophysin (Jackson ImmunoResearch, West Grove, PA) that were used to visualize the primary antibodies. After incubation in the secondary antibodies, sections were rinsed, mounted on slides, and coverslipped. Images of labeled neurons in the principal or accessory abducens motor nuclei were obtained using an Olympus Fluoview 500 laser scanning confocal microscope. Tissue samples were scanned using a ×60 1.4 NA oil-immersion objective with dual excitation using a 488-nm argon laser and a 543-nm HeNe laser. Quantification of punctate staining of at least twofold greater intensity above background was performed independently by two investigators using stereological procedures (Mokin and Keifer 2006) with MetaMorph software (Universal Imaging, Downingtown, PA). Images of two consecutive optical sections were taken using confocal microscopy. Protein puncta were counted in one optical section (sample section) if they were not present in the optical section immediately below the sample section (look-up section) and if they were within the inclusion boundaries of the unbiased counting frame. Colocalized staining indicating the presence of glutamate receptor subunits at synaptic sites was determined when red and green puncta were immediately adjacent to one another or if they were overlapping. Data were analyzed using StatView software (SAS, Cary, NC) by ANOVA.
RESULTS
Inhibition of acquisition and expression of conditioning by selective PKA and CaMKII antagonists
The effects of selective PKA and CaMKII antagonists on the acquisition and expression of in vitro abducens classical conditioning were examined and these data are summarized in Figs. 1 and 2. Representative abducens nerve recordings from a preparation that exhibited conditioning and was treated with Rp-cAMPs, a selective cAMP analog and competitive inhibitor of PKA activation, are shown in Fig. 1A. The recordings show an abducens nerve conditioned response (CR, arrow) followed by the unconditioned response (UR) in the second pairing session prior to drug application (Normal). Application of 50 μM Rp-cAMPs blocked the expression of CRs but the UR was largely unaffected (Fig. 1A; Rp-cAMPs). Washout of the drug resulted in recovery of CRs (arrow; Wash). Acquisition curves of the mean percentage of abducens CRs under Rp-cAMPs treatment during the acquisition (left) and expression (right) phases of conditioning are shown in Fig. 1B. Application of Rp-cAMPs prior to conditioning resulted in no CR acquisition (Fig. 1B; n = 7). In cases examined for CR expression following conditioning, CRs were significantly attenuated to 9 ± 9% CRs (n = 6, P = 0.006) after two sessions of drug application. The CRs readily recovered after washout. Similar results were obtained with bath application of the selective CaMKII antagonist AIP (5 μM) as shown in Fig. 2. AIP also completely blocked acquisition of abducens CRs (Fig. 2B, left; n = 8) and significantly attenuated CR expression to 5 ± 3% CRs (Fig. 2B, right; n = 5; P = 0.0006), as can be seen in the physiological records (Fig. 2A). The CRs also readily recovered after washout of AIP. In other preparations (not shown), KN-62 (10 μM), a general CaMK inhibitor, also resulted in inhibition of both the acquisition (n = 6) and expression (n = 4, to 8 ± 2% CRs; P = 0.01) of abducens nerve CRs. These data provide strong physiological evidence supporting a role for PKA and the CaMKs in both the acquisition and expression phases of in vitro abducens classical conditioning.
FIG. 1.

Inhibition of cyclic AMP-dependent protein kinase (PKA) by Rp-adenosine-3′,5′-cyclic monophosphorothioate (Rp-cAMPs) suppresses acquisition and expression of classical conditioning. A: physiological records of abducens nerve recordings taken from an experiment in which the compound Rp-cAMPs blocked expression of conditioned responses (CRs). The traces show an abducens nerve CR (arrow) followed by the unconditioned response (UR) recorded in the 2nd pairing session prior to drug application (Normal). Record obtained during application of Rp-cAMPs in the 4th pairing session in which the expression of CRs was blocked but the UR was unaffected (Rp-cAMPs). Washout of drug in the 6th session resulted in reexpression of CRs (arrow, Wash). The conditioned stimulus (CS) and unconditioned stimulus (US) are indicated at the bottom. B: acquisition curves (means ± SD) of the percentage of abducens CRs following Rp-cAMPs treatment at the beginning of conditioning to test for acquisition (left) or after CRs had been obtained to test for CR expression (right). Application of Rp-cAMPs resulted in no acquisition and attenuated expression of CRs. *Indicates significant difference from session 2. P values are given in the text throughout.
FIG. 2.

The Ca2+-calmodulin–dependent protein kinase II (CaMKII) antagonist autocamtide-2–related inhibitory peptide (AIP) attenuates both the acquisition and expression of conditioning. A: physiological records of abducens nerve recordings taken from an experiment in which AIP reversibly blocked CR expression. B: acquisition curves of the percentage of abducens CRs show that AIP significantly attenuated both acquisition (left) and expression (right) of CRs. *Indicates significant difference from session 2.
Early onset of PKA, CaMK, and CREB protein phosphorylation during conditioning
The antibodies directed against total and phosphorylated PKA, CaMKII, CaMKIV, and CREB used in the present study were tested for their specificity by Western blot analysis of turtle and rat brain tissue (Fig. 3). The PKA antibody directed against total protein (t-PKA; Upstate, 06-903) identified a band in both species at about 42 kDa. A phosphorylation site-directed antibody that recognizes PKA when phosphorylated at Thr197 resulted in a single band in both rat and turtle at about 42 kDa (p-PKA; Cell Signaling, 4781). To examine CaMKII, an antibody to total protein (t-CaMKII; Chemicon, 8699) was used that recognizes a band in both rat and turtle at about 50 kDa. A phosphorylation site-directed antibody that recognizes CaMKII at Thr286 (p-CaMKII; Cell Signaling, 3361) also demonstrated one band in both species at about 50 kDa. An antibody that recognizes total CaMKIV (t-CaMKIV; Cell Signaling, 4032) and phosphorylated CaMKIV at Thr196 (p-CaMKIV; Santa Cruz, 28443R) demonstrated bands in both species at about 60 kDa. Finally, total CREB protein (t-CREB; Cell Signaling, 9197) and CREB phosphorylated at Ser133 (p-CREB; Cell Signaling, 9198) demonstrated one band in both species at about 43 kDa. These antibodies recognize similar molecular weight proteins in turtle brain compared with rat and were used for examination of in vitro classical conditioning.
FIG. 3.

Western blots demonstrating specificity of the antibodies used in the present study. Antibodies were tested on naive brain tissue from turtle (T) or rat (R). Bands appeared at the expected molecular weights in both species. See text for details.
The pattern of activation of PKA, CaMKII, CaMKIV, and CREB was examined during the time course of conditioning. Protein analysis was carried out after one session of pseudoconditioning (Ps1), 15 min of conditioning in the first pairing session (C15), one complete pairing session (or 25 min after conditioning stimulation onset, C1), two complete sessions (80 min, C2), or five pairing sessions (4 h, C5). Among these groups there was significant CR expression after only two (mean of 94% CRs) and five (100% CRs) pairing sessions. Blots for total and phosphoprotein are shown, whereas the ratio of protein is plotted in Fig. 4 (n = 4/group). The level of p-PKA was significantly increased after conditioning for 15 min and was maintained at elevated levels throughout the conditioning procedure, whereas there were no changes in total PKA (Fig. 4A; P < 0.0001, all groups vs. Ps1). Similarly, p-CaMKII was significantly increased after conditioning for 15 min and remained elevated, whereas no changes were detected in total protein (Fig. 4B; P = 0.0003, all groups vs. Ps1). For p-CaMKIV, protein was also significantly increased after conditioning for 15 min compared with pseudoconditioning and was elevated through the second pairing session and declined slightly by the fifth pairing session (Fig. 4C; P = 0.02, all groups vs. Ps1 except C5, which was P = 0.07). Finally, the level of p-CREB was significantly increased after 15 min of conditioning and again remained at elevated levels throughout (Fig. 4D; P < 0.0001, all groups vs. Ps1). These findings show that phosphorylation of all the proteins examined here—PKA, CaMKII, CaMKIV, and CREB—was significantly elevated after ≥15 min of paired stimulation and was maintained in that state throughout the conditioning procedures.
FIG. 4.

Characterization of the onset of protein phosphorylation during early conditioning as determined by phosphospecific antibodies and Western blotting. Blots for phospho- and total protein are shown, whereas quantitative data of the ratio of phospho- to total protein is plotted. Significant levels of phosphorylation for PKA (A), CaMKII (B), Ca2+-calmodulin–dependent protein kinase IV (CaMKIV, C), and cAMP-response-element-binding protein (CREB, D) occur within 15 min of application of the conditioning stimuli. Ps1, pseudoconditioning for one session; C15, conditioning for 15 min; C1, conditioning for one pairing session; C2, conditioning for 2 sessions; C5, conditioning for 5 sessions. *Indicates significant differences from Ps1.
Interaction of PKA, CaMK, and CREB in early conditioning
To make any firm conclusions on the function of PKA and CaMKII in conditioning, it was necessary to confirm the specificity of the pharmacological antagonists used in turtle brain. As shown in Fig. 5 (n = 4/group), Western blots revealed that preparations in which conditioning was blocked by the PKA inhibitor Rp-cAMPs (50 μM) for two pairing sessions significantly suppressed activation of p-PKA (Fig. 5A; P = 0.0002, Rp2 vs. C2), but not p-CaMKII (Fig. 5B; P = 0.45, Rp2 vs. C2), compared with the conditioned group. On the other hand, preparations in which CRs were blocked by treatment with the CaMKII antagonist AIP (5 μM) showed inhibition of p-CaMKII (Fig. 5B; P = 0.003, AIP2 vs. C2), whereas p-PKA was unaffected (Fig. 5A; P = 0.25, AIP2 vs. C2) compared with conditioning. Therefore these compounds are effective in selectively blocking activation of either the PKA or CaMKII signaling pathways in turtle. Using similar treatment groups, we also determined whether phosphorylation of CREB was affected by inhibitors of PKA or the CaMKs during conditioning. Although levels of p-CREB were substantially elevated after conditioning compared with pseudoconditioned controls, application of Rp-cAMPs during the conditioning procedure significantly reduced activation of CREB (Fig. 5C; P = 0.02, Rp2 vs. C2), although these levels remained relatively high compared with pseudoconditioning. Preparations treated by coapplication of both Rp-cAMPs and the general CaMK antagonist KN-62 resulted in complete suppression of CREB activation during conditioning (P = 0.77, Rp2 + KN62 vs. Ps2). Therefore since this antibody recognizes CREB phosphorylated at Ser133, the PKA, CaMKIV, and ribosomal S6 kinases (RSKs) binding site, these data indicate that CREB is activated at least by both PKA and CaMKIV during conditioning. Consistent with these findings, the PKA activator Sp-cAMPs (50 μM) significantly increased phosphorylation of CREB by 336% after only 15 min of bath application (data not shown; P < 0.0001 above pseudoconditioned levels), consistent with the conclusion that PKA activates CREB early in conditioning.
FIG. 5.

Selectivity of PKA and CaMKII antagonists and effects on p-CREB. A: bath application of Rp-cAMPs during 2 sessions of paired stimulation significantly reduced the conditioning-related increase in PKA phosphorylation to pseudoconditioned levels, whereas AIP had no significant effect. B: application of AIP for 2 sessions suppressed phosphorylation of CaMKII, whereas Rp-cAMPs failed to have a significant effect. C: application of Rp-cAMPs for 2 pairing sessions significantly attenuated phosphorylation of CREB compared with conditioning. Coapplication of Rp-cAMPs with the general CaMKII antagonist KN-62 for 2 sessions completely inhibited CREB phosphorylation to levels similar to pseudoconditioning. *Indicates significant differences from Ps2; # indicates significant differences from C2.
PKA activates ERK in early conditioning
Our previous studies showed that the ERK signaling pathway was central to the synaptic localization of GluR1- and GluR4-containing AMPARs during conditioning (Keifer et al. 2007; Zheng and Keifer 2008). Evidence here further suggests that PKA regulates ERK early in conditioning and these data are shown in Fig. 6 (n = 4/group). Western blots show that treatment with the PKA inhibitor Rp-cAMPs for two pairing sessions suppressed p-ERK to levels similar to pseudoconditioning (Fig. 6A; P = 0.28, Rp2 vs. Ps2), whereas the CaMKII antagonist AIP did not significantly affect the elevated levels of p-ERK (P < 0.0001, AIP vs. Ps2). Alternatively, as shown in Fig. 6B, application of the PKA activator Sp-cAMPs for two sessions resulted in significantly enhanced p-ERK to similar levels obtained after conditioning (P = 0.004, Sp2 vs. Ps2). When incubated in the selective MEK–ERK inhibitor PD98059 (50 μM) for two pairing sessions, preparations showed little change in the elevated state of PKA phosphorylation observed after conditioning (Fig. 6C; P < 0.0001, PD2 vs. Ps2). These data suggest that PKA is upstream from and activates ERK in the early stages of conditioning.
FIG. 6.

Evidence that PKA is downstream from and activates extracellular signal-regulated kinase (ERK). A: bath application of Rp-cAMPs for 2 pairing sessions inhibited the conditioning-related activation of ERK to pseudoconditioned levels. Application of AIP for 2 sessions was ineffective in inhibiting ERK. B: application of the PKA activator Sp-cAMPs alone for the equivalent time period of 2 pairing sessions (with no electrical stimulation) induced phosphorylation of ERK to levels similar to those obtained after conditioning. C: application of the selective MEK–ERK antagonist PD98059 for 2 sessions failed to affect the conditioning-induced activation of PKA, suggesting that ERK does not feed back onto PKA signaling pathways. *Indicates significant differences from Ps2.
Phosphorylation of PKA and CaMKII is not affected by NMDAR antagonist d,l-AP5
A previous study had shown that acquisition of CRs was NMDAR dependent (Keifer 2001) and therefore we were interested in whether the early activation of PKA and CaMKII during conditioning was similarly sensitive to the NMDAR antagonist d,l-AP5. To examine this, preparations were treated with d,l-AP5 during paired stimulation for 15 min or one complete pairing session (C1, 25 min) and analyzed by Western blot. These data are summarized in Fig. 7 (n = 3/group). Compared with conditioning for one session, neither p-PKA (P < 0.0001, all groups vs. Ps1) nor p-CaMKII (P = 0.01, C1 and C15 + d,l-AP5 vs. Ps1; P = 0.03, C1 + d,l-AP5 vs. Ps1) demonstrated any sensitivity to blockade of NMDARs during the earliest phases of the conditioning procedure.
FIG. 7.

The N-methyl-d,l-aspartate receptor (NMDAR) antagonist d,l-2-amino-5-phosphonopentanoic acid (d,l-AP5) does not affect phosphorylation of either PKA or CaMKII in early conditioning. A: application of d,l-AP5 during conditioning for 15 min (C15) or one pairing session (C1) failed to attenuate phosphorylated levels of PKA, which were similar to values obtained after conditioning in normal saline. B: application of d,l-AP5 also did not affect phosphorylation of CaMKII in early conditioning. *Indicates significant differences from Ps1.
Effects of PKA and CaMKII inhibition on synaptic delivery of AMPARs
Immunocytochemistry and confocal imaging were performed to assess the synaptic localization of AMPARs in abducens motor neurons in conditions in which levels of PKA or CaMKII were manipulated during the conditioning procedures. These data are summarized in Fig. 8 (n = 3/group). First, preparations were treated with the PKA antagonist Rp-cAMPs during conditioning for two sessions and immunocytochemical results were compared with conditioning and pseudoconditioning for two sessions in normal saline. As reported previously (Li and Keifer 2008, 2009; Mokin et al. 2007; Zheng and Keifer 2008), normal conditioning resulted in significantly enhanced punctate staining for synaptophysin (P < 0.0001) and colocalization of GluR1- (P < 0.0001) and GluR4-containing (P < 0.0001) AMPARs with synaptophysin compared with pseudoconditioning. These findings are illustrated in the confocal images shown in Fig. 8A (compare C2 with Ps2) and can be seen in the histograms (Fig. 8B). Data suggest that the increased punctate staining for synaptophysin is due to the enhanced size of presynaptic boutons apposed to abducens motor neurons following conditioning (Li and Keifer, unpublished data). Bath application of Rp-cAMPs during conditioning not only resulted in complete blockade of the acquisition of CRs but also dramatically suppressed the levels of synaptophysin (P < 0.0001) and the synaptic localization of both GluR1 and GluR4 AMPARs (P < 0.0001 and P < 0.0001, respectively) compared with the conditioned group (Fig. 8, A and B). We next determined whether inhibition of CaMKII by application of AIP for two sessions would have similar effects on AMPAR synaptic incorporation as Rp-cAMPs. In contrast to PKA inhibition, AIP failed to alter staining for synaptophysin (P = 0.08) as well as synaptic delivery of GluR1 compared with conditioning (P = 0.28). However, incorporation of GluR4 subunits was significantly suppressed (P < 0.0001). These changes in synaptic AMPAR localization cannot be attributed to alterations in synaptophysin alone because there were no significant differences in the ratio of GluR1 or GluR4 with synaptophysin among the treatment groups (P = 0.33). Therefore inhibition of PKA resulted in attenuation of the synaptic insertion of both GluR1- and GluR4-containing AMPARs, whereas inhibition of CaMKII reduced only GluR4 delivery.
FIG. 8.

Synaptic incorporation of glutamate receptor 1 (GluR1)- and GluR4-containing α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs) is dependent on PKA, whereas GluR4 incorporation also requires CaMKII. A: confocal images of selected abducens motor neurons from the different experimental groups show punctate staining for synaptophysin (Syn) and colocalization of GluR1 (GluR1 + Syn) and GluR4 AMPAR subunits (GluR4 + Syn) with synaptophysin. Colocalization of AMPARs (red) with Syn (green) is defined by overlapping (yellow) or adjacent puncta and are indicated by the arrowheads. B: quantitative analysis of Syn, GluR1 + Syn, and GluR4 + Syn punctate staining for the different experimental groups is plotted. Preparations conditioned for 2 pairing sessions (C2) showed significantly greater punctate staining for Syn, GluR1 + Syn, and GluR4 + Syn compared with the pseudoconditioned group (Ps2). Treatment with the PKA inhibitor Rp-cAMPs for 2 sessions (Rp2) significantly reduced the conditioning-related increase in synaptophysin and the colocalization of GluR1 and GluR4 with synaptophysin. Application of the CaMKII blocker AIP for 2 sessions (AIP2) did not affect elevated levels of Syn or GluR1 + Syn, but significantly attenuated GluR4 + Syn. Application of the PKA activator Sp-cAMPs (Sp2) increased punctate staining for synaptophysin and induced synaptic incorporation of GluR1 and GluR4 subunits. Coapplication of Sp-cAMPs with d,l-AP5 (Sp2 + d,l-AP5) for 2 sessions had no effect on GluR1 synaptic insertion but significantly attenuated GluR4. *Indicates significant differences from Ps2. Scale bar = 2 μm.
PKA activation results in synaptic incorporation of GluR1 and GluR4 subunits, but only GluR4 was d,l-AP5 sensitive
The question then arose as to whether activation of PKA alone would induce synaptic delivery of GluR1 and GluR4 subunits. To test this, the PKA activator Sp-cAMPs was bath applied for the equivalent time period of two pairing sessions. As can be seen from the data in Fig. 8, Sp-cAMPs resulted in enhanced levels of synaptophysin (P = 0.0001) and synaptic delivery of both GluR1 and GluR4 AMPAR subunits (P < 0.0001 and P < 0.0001, respectively, compared with Ps2) to levels similar to those observed after conditioning. These data strongly support a role for PKA in initiating a signal transduction cascade resulting in synaptic incorporation of GluR1- and GluR4-containing AMPARs. In a previous study (Li and Keifer 2009) we found that synaptic incorporation of GluR4 subunits after conditioning was NMDAR dependent, whereas GluR1 was not. Therefore whether Sp-cAMPs–induced AMPAR delivery was also similarly dependent on NMDARs was addressed. To examine this question, preparations were incubated in Sp-cAMPs and the NMDAR antagonist d,l-AP5 for the equivalent of two sessions. These cases showed significant synaptic delivery of GluR1-containing AMPARs compared with pseudoconditioning (Fig. 8; P < 0.0001). In contrast, delivery of GluR4-containing AMPARs was severely attenuated by d,l-AP5 (P < 0.0001, Sp2 + d,l-AP5 vs. C2). From these data it can be concluded that, like conditioning, only GluR4-containing AMPARs require NMDAR activation during PKA-induced synaptic delivery, whereas GluR1-containing AMPARs do not.
GluR1 is phosphorylated at Ser845 and Ser831 during conditioning
The immunocytochemical data strongly indicated that PKA activation resulted in synaptic incorporation of GluR1 subunits. Since phosphorylation of GluR1 subunits at Ser845 and Ser831 is implicated in synaptic delivery (Esteban et al. 2003), we assessed whether this was the case during early conditioning using phosphorylation site-directed antibodies. These data are shown in Fig. 9 and indicate that GluR1 undergoes significant phosphorylation at both the PKA binding site Ser845 (P = 0.01) and the CaMKII/PKC binding site Ser831 (P < 0.0001) after 25 min of conditioning (C1).
FIG. 9.
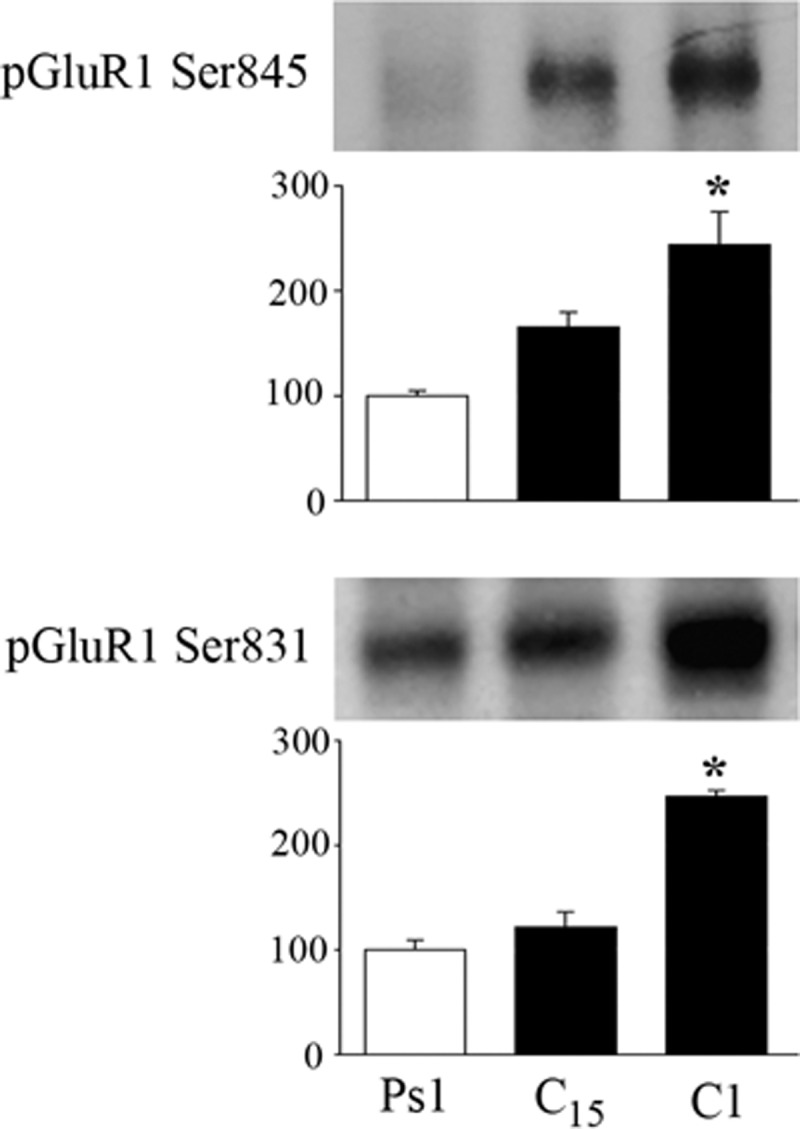
GluR1 AMPAR subunits are phosphorylated in early stages of conditioning. Western blots using phosphorylation site-selective antibodies indicate that GluR1 subunits are phophorylated at both Ser845 and Ser831. Quantitative analysis shows that the average levels of phosphorylation are significant after one pairing session (C1) or after 25 min of paired stimulation. *Indicates significant differences from Ps1.
DISCUSSION
Temporally paired, but not unpaired, stimulation of auditory and trigeminal synaptic inputs results in a neural correlate of a learned behavioral response in this in vitro analog of eyeblink classical conditioning. This model system allows us to tease apart molecular mechanisms that underlie a simple form of associative learning and reveals a complex interplay of intracellular responses that form a behaviorally relevant contingency between two otherwise unrelated inputs. The present study presents several new findings that are key to understanding conditioning in this model system. First, PKA and the CaMKs (II and IV) are activated early in conditioning, within 15 min of paired stimulation, and are required for acquisition and expression of CRs. Second, CREB is also activated early in conditioning but is blocked by coapplication of inhibitors to PKA and the CaMKs, suggesting that CREB is downstream of those signaling cascades. Third, PKA activates ERK, which has been previously shown to undergo phosphorylation during conditioning with a time course that occurs after PKA (Zheng and Keifer 2008). Finally, PKA is required for synaptic delivery of GluR1-containing AMPARs in abducens motor neurons that is NMDAR independent and leads to delivery of GluR4 subunits that is sensitive to d,l-AP5. Together with previous studies, our findings support a two-stage model of AMPAR trafficking during classical conditioning that is the result of multiple signal transduction elements shown in Fig. 10. The first stage occurs during the first pairing session, but before CRs can be recorded, and involves synaptic incorporation of GluR1-containing AMPARs that act to unsilence auditory nerve synapses (Mokin et al. 2007). GluR1 insertion allows enough postsynaptic depolarization from the CS that NMDARs become activated, allowing Ca2+ entry to initiate a second stage of synthesis and incorporation of GluR4 AMPAR subunits that are hypothesized to generate the CRs.
FIG. 10.

Two-stage model of the acquisition phase of in vitro classical conditioning. The first stage of acquisition occurs during the 1st pairing session but prior to when CRs are recorded (top line) and involves synaptic insertion of GluR1-containing AMPARs to activate silent synapses. This process does not involve protein synthesis of GluR1 but the translocation of existing receptors. Shortly after the onset of paired CS–US stimulation PKA, CaMKII, and CaMKIV are phosphorylated. Activation of CREB is depicted downstream of PKA and the CaMKs because inhibitors of these block phosphorylation of CREB. Western analysis further shows that brain-derived neurotrophic factor (BDNF), a known target for CREB, is synthesized after phosphorylation of PKA and the CaMKs (at 25 min) and that ERK is activated shortly after that, which leads to synaptic incorporation of GluR1-containing AMPARs. Data also suggest that GluR1 subunits are directly phosphorylated at Ser845 and Ser831, PKA, and CaMKII sites, which could provide an additional or parallel route for GluR1 modification and trafficking. The 2nd stage of acquisition occurs during the 2nd pairing session when CRs are recorded (bottom line) and involves NMDAR-dependent synthesis and synaptic insertion of GluR4-containing AMPARs that are thought to replace GluR1. The synaptic incorporation of GluR4 subunits is associated with the acquisition of CRs. This stage requires protein kinase C (PKC) and is also ERK dependent.
During the first stage of conditioning in our model, data indicate that PKA and the CaMKs (II and IV) are activated shortly after the onset of paired CS–US stimulation. PKA and the CaMKs (probably CaMKIV) are thought to activate CREB through known interactions at Ser133 (Carlezon Jr et al. 2005), which is suggested by the finding that inhibitors of both PKA and the CaMKs suppress CREB phosphorylation at this site. There are undoubtedly multiple target genes for CREB during conditioning. In this system, one of them is likely to be BDNF (Fig. 10). Our previous work has demonstrated that BDNF is required for conditioning (Li and Keifer 2008) and that protein levels are significantly increased after 25 but not 15 min (or one pairing session) of conditioning, after activation of PKA, CaMK, and CREB has already occurred (Li and Keifer, unpublished data). Furthermore, ERK has been shown to be activated slightly later in conditioning and is required for synaptic incorporation of both GluR1- and GluR4-containing AMPARs (Keifer et al. 2007; Zheng and Keifer 2008). Our current findings show that the PKA inhibitor Rp-cAMPs suppressed ERK expression, whereas the PKA activator Sp-cAMPs stimulated ERK. Similarly, Rp-cAMPs inhibited synaptic incorporation of GluR1 and GluR4 subunits, whereas Sp-cAMPs resulted in insertion. Moreover, application of the ERK antagonist PD98059 during paired stimulation had no effect on the conditioning-related increase in p-PKA, suggesting that ERK does not affect activation of PKA. Taking these data into account, ERK is thought to be downstream of PKA (Fig. 10) and leads to synaptic delivery of GluR1 (as well as GluR4, discussed in the following text). Delivery of GluR1 subunits is thought to be rapid and does not involve protein synthesis of GluR1 but instead uses existing receptor proteins (Mokin et al. 2007). In hippocampal pyramidal neurons synaptic delivery of GluR1 is achieved through interactions of PKA with CaMKII and Ras-ERK signaling (Esteban et al. 2003) and evidence suggests this may be the case here. Incorporation of GluR1 could also be achieved directly by phosphorylation of Ser845 and Ser831 by PKA and CaMKII, respectively. However, in this model of classical conditioning synaptic insertion of GluR1 also requires signaling pathways using BDNF. BDNF is a necessary and sufficient element for GluR1 insertion because not only does application of BDNF alone induce activation of ERK and synaptic incorporation of GluR1-containing AMPARs (Li and Keifer 2008, 2009), but treatment of preparations with Sp-cAMPs plus BDNF antibodies also suppresses the synaptic incorporation of GluR1 that can be induced by Sp-cAMPs alone (this study; Zheng and Keifer, unpublished data). Similar to the present findings, a BDNF-like growth factor is required for induction of ERK and enhancement of a serotonin-dependent form of long-term facilitation in Aplysia (Purcell et al. 2003). Application of serotonin results in a persistent activation of PKA that is thought to be upstream of BDNF (or related molecule) elevation. A similar situation in which cAMP drives BDNF release and activation of ERK is also visualized in the CA1 region of the hippocampus following induction of late-phase LTP (Patterson et al. 2001).
The second stage of synaptic AMPAR delivery during conditioning involves replacement of GluR1 with GluR4 subunits (Mokin et al. 2007) and appears to be dependent on completion of the first phase of GluR1 synaptic incorporation. This step occurs during the second pairing session and involves the NMDAR-dependent synthesis and synaptic insertion of GluR4-containing AMPARs that are hypothesized to support the generation of CRs. Our previous imaging studies using the NMDAR antagonist d,l-AP5 showed that synaptic insertion of GluR4 AMPAR subunits during conditioning, but not GluR1, was NMDAR dependent and resulted in blockade of CR acquisition (Li and Keifer 2009; Mokin and Keifer 2004; Mokin et al. 2006). We have also found that BDNF-induced synaptic insertion of GluR4 is sensitive to d,l-AP5, whereas GluR1 is not (Li and Keifer 2009). These findings are extended here by data showing that, although the PKA activator Sp-cAMP–induced synaptic incorporation of both GluR1 and GluR4, only GluR4 was suppressed by coapplication with d,l-AP5. The NMDAR dependence of Sp-cAMP–induced synaptic delivery of GluR4 AMPARs is likely to be mediated by the enhanced expression of BDNF (data not shown) and direct phosphorylation of NMDARs shown to increase channel activity (Xu et al. 2006). Therefore synaptic insertion of GluR4 subunits is an NMDAR-dependent process, whereas GluR1 is not (Fig. 10). Further work suggests that synaptic incorporation of GluR4 requires CaMKII because it is blocked by AIP (this study) and Ca2+-dependent and -independent isoforms of PKC since it is suppressed by the PKC inhibitors chelerythrine chloride or ζ-pseudosubstrate inhibitory peptide (termed ZIP) (Zheng and Keifer 2008). The coordinated actions of these kinases with ERK result in GluR4 insertion and expression of CRs (Keifer et al. 2007). The exact roles of CaMKII and PKC in this model of conditioning are currently poorly understood. Although CaMKII undergoes phosphorylation early in conditioning, we have been unable to pinpoint a specific function for it. The GluR4 subunit has been reported to be phosphorylated at Ser842 by PKA, PKC, and CaMKII in vitro (Carvalho et al. 1999), although the potential interactions of these kinases at this site and effect on trafficking are unknown. Likewise, PKCζ is strongly activated within 40 min of conditioning onset; however, its expression is not sufficient for conditioning because it may be phosphorylated in conditions under which CRs are not expressed during pharmacological treatment (Zheng and Keifer 2008).
Both GluR1 and GluR4 AMPAR subunits appear to require ERK activation for synaptic delivery during conditioning (Keifer et al. 2007; Zheng and Keifer 2008). The question arises as to how ERK signaling specificity is achieved for PKA-dependent GluR1 and PKC-dependent GluR4 delivery. One possibility is that selective actions of these kinases are maintained by intracellular compartmentalization. Intriguing findings by Shalin et al. (2006) suggest this possibility because PKC-coupled activation of a membrane fraction of ERK was found to occur independently of PKA-coupled ERK activity. Data suggest this is accomplished through kinase suppressor of Ras (KSR1) that scaffolds ERK and selective downstream effectors into functionally related pools of proteins. Spatially localized kinase signaling may also be achieved by forming macromolecular complexes such as PKA with AMPARs through A-kinase anchoring proteins (AKAPs; Tavalin et al. 2002). Supporting these observations, genetic disruption of mechanisms that promote compartmentalized signaling have been shown to impair not only LTP but also associative learning (Nie et al. 2007; Shalin et al. 2006).
Multistep models of AMPAR trafficking during LTP have been proposed and implicate PKA in initiating early stages of signal transduction cascades responsible for synaptic plasticity (Derkach et al. 2007; Oh et al. 2006; Yang et al. 2008; see also, Sun et al. 2005). For LTP, PKA signaling is thought to phosphorylate and mobilize GluR1-containing AMPARs to the surface membrane of extrasynaptic sites. This is followed by NMDAR-dependent Ca2+ influx that induces activation of signaling pathways including CaMKII and PKC, resulting in translocation of receptors by lateral mobility into synapses. Trafficking of GluR1-containing AMPARs to the surface membrane is correlated with phosphorylation at Ser845 and occurs in the presence of d,l-AP5, suggesting that NMDARs are not required (Oh et al. 2006), similar to our current findings on classical conditioning. In the classical conditioning paradigm used here, pairing of the CS and US leads to rapid phosphorylation of PKA, the CaMKs, and CREB, whereas the same stimuli that are unpaired during pseudoconditioning do not. Phosphorylation of PKA and CaMKII in the early stages of conditioning is not d,l-AP5 sensitive. NMDARs have been viewed to be coincidence detectors because they require pre- and postsynaptic activation to function. However, the initial cellular responses to paired stimulation here are not NMDAR mediated. How are coincident stimuli detected if not by NMDARs? Some insight has been gained on this point recently by studies of a cellular analog of operant conditioning in neuron B51 of Aplysia (Lorenzetti et al. 2008). Conditioning was found to be dependent on the synergistic action of PKC and dopamine, resulting in increased adenylyl cyclase that in turn increased cAMP/PKA to generate cellular changes in B51. These authors concluded that adenylyl cyclase itself may serve as an early coincidence detector for inputs controlling behavior and the reinforcement. In their model, contingent activation of two pathways, but not either pathway alone, results in increased levels of adenylyl cyclase, leading to synthesis of cAMP to a threshold value required to produce cellular changes. This idea is attractive because this mechanism can occur rapidly in a few steps and without invoking NMDAR-mediated mechanisms that may occur later. To determine whether such a scheme is possible in our in vitro model of classical conditioning, it will be important to show that stimulation of the trigeminal and auditory nerves alone results in increased adenylyl cyclase and cAMP/PKA production for adenylyl cyclase to be a point of CS–US convergence and that paired stimulation enhances this effect. Although many questions remain with regard to our specific model of classical conditioning, growing evidence indicates that mechanisms underlying activity-dependent synaptic plasticity and learning are conserved and some general principles are beginning to emerge. Among these is the complex interplay of signal transduction elements required for multistep trafficking of AMPARs to synapses.
GRANTS
This work was supported by National Institutes of Health Grants NS-051187 and P20 RR-015567, designated as a Center of Biomedical Research Excellence to J. Keifer.
Acknowledgments
We thank Dr. Frances Day for assistance with the confocal microscopy.
REFERENCES
- Abel et al. 1997.Abel T, Nguyen PV, Barad M, Deuel TAS, Kandel ER, Bourtchuladze R. Genetic demonstration of a role for PKA in the late phase of LTP and in hippocampus-based long-term memory. Cell 88: 615–626, 1997. [DOI] [PubMed] [Google Scholar]
- Anderson and Keifer 1999.Anderson CW, Keifer J. Properties of conditioned abducens nerve responses in a highly reduced in vitro brain stem preparation from the turtle. J Neurophysiol 81: 1242–1250, 1999. [DOI] [PubMed] [Google Scholar]
- Barria et al. 1997.Barria A, Derkach V, Soderling T. Identification of the Ca2+/calmodulin-dependent protein kinase II regulatory phosphorylation site in the alpha-amino-3-hydroxyl-5-methyl-4-isoxazole-proprionate-type glutamate receptor. J Biol Chem 272: 32727–32730, 1997. [DOI] [PubMed] [Google Scholar]
- Bolshakov et al. 1997.Bolshakov VY, Golan H, Kandel ER, Siegelbaum SA. Recruitment of new sites of synaptic transmission during the cAMP-dependent late phase of LTP at CA3–CA1 synapses in the hippocampus. Neuron 19: 635–651, 1997. [DOI] [PubMed] [Google Scholar]
- Caldeira et al. 2007.Caldeira MV, Melo CV, Pereira DB, Carvalho R, Correia SS, Backos DS, Carvalho AL, Esteban JA, Duarte CB. Brain-derived neurotrophic factor regulates the expression and synaptic delivery of alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor subunits in hippocampal neurons. J Biol Chem 282: 12619–12628, 2007. [DOI] [PubMed] [Google Scholar]
- Carlezon et al. 2005.Carlezon WA Jr, Duman RS, Nestler EJ. The many faces of CREB. Trends Neurosci 28: 436–445, 2005. [DOI] [PubMed] [Google Scholar]
- Cartford et al. 2004.Cartford MC, Samec A, Fister M, Bickford PC. Cerebellar norepinephrine modulates learning of delay classical eyeblink conditioning: evidence for post-synaptic signaling via PKA. Learn Mem 11: 732–737, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho et al. 1999.Carvalho AL, Kameyama K, Huganir RL. Characterization of phosphorylation sites on the glutamate receptor 4 subunit of the AMPA receptors. J Neurosci 19: 4748–4754, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derkach et al. 2007.Derkach VA, Oh MC, Guire ES, Soderling TR. Regulatory mechanisms of AMPA receptors in synaptic plasticity. Nat Rev Neurosci 8: 101–113, 2007. [DOI] [PubMed] [Google Scholar]
- Esteban et al. 2003.Esteban JA, Shi S-H, Wilson C, Nuriya M, Huganir RL, Malinow R. PKA phosphorylation of AMPA receptor subunits controls synaptic trafficking underlying plasticity. Nat Neurosci 6: 136–143, 2003. [DOI] [PubMed] [Google Scholar]
- Frey et al. 1993.Frey U, Huang YY, Kandel ER. Effects of cAMP simulate a late stage of LTP in hippocampal CA1 neurons. Science 260: 1661–1664, 1993. [DOI] [PubMed] [Google Scholar]
- Huang and Kandel 2007.Huang YY, Kandel ER. Low-frequency stimulation induces a pathway-specific late phase of LTP in the amygdala that is mediated by PKA and dependent on protein synthesis. Learn Mem 14: 497–503, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang et al. 1994.Huang YY, Li XC, Kandel ER. cAMP contributes to mossy fibre LTP by initiating both a covalently mediated early phase and macromolecular synthesis-dependent late phase. Cell 79: 69–79, 1994. [DOI] [PubMed] [Google Scholar]
- Keifer 2001.Keifer J In vitro eye-blink classical conditioning is NMDA receptor-dependent and involves redistribution of AMPA receptor subunit GluR4. J Neurosci 21: 2434–2441, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keifer 2003.Keifer J In vitro classical conditioning of the turtle eyeblink reflex: approaching cellular mechanisms of acquisition. Cerebellum 2: 55–61, 2003. [DOI] [PubMed] [Google Scholar]
- Keifer et al. 1995.Keifer J, Armstrong KE, Houk JC. In vitro classical conditioning of abducens nerve discharge. J Neurosci 15: 5036–5048, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keifer et al. 2007.Keifer J, Zheng Z, Zhu D. MAPK signaling pathways mediate AMPA receptor trafficking in an in vitro model of classical conditioning. J Neurophyiol 97: 2067–2074, 2007. [DOI] [PubMed] [Google Scholar]
- Li and Keifer 2008.Li W, Keifer J. Coordinate action of pre- and postsynaptic brain-derived neurotrophic factor is required for AMPAR trafficking and acquisition of in vitro classical conditioning. Neurosci 155: 686–697, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li and Keifer 2009.Li W, Keifer J. BDNF-induced synaptic delivery of AMPAR subunits is differentially dependent on NMDA receptors and requires ERK. Neurobiol Learn Mem 91: 243–249, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonart et al. 2003.Lonart G, Schoch S, Kaeser PS, Larkin CJ, Sudof TC, Linden DJ. Phosphorylation of RIM1α by PKA triggers presynaptic long-term potentiation at cerebellar parallel fiber synapses. Cell 115: 49–60, 2003. [DOI] [PubMed] [Google Scholar]
- Lorenzetti et al. 2008.Lorenzetti FD, Baxter DA, Byrne JH. Molecular mechanisms underlying a cellular analog of operant reward learning. Neuron 59: 815–828, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mokin and Keifer 2004.Mokin M, Keifer J. Targeting of GluR4-containing AMPA receptors to synaptic sites during in vitro classical conditioning. Neuroscience 128: 219–228, 2004. [DOI] [PubMed] [Google Scholar]
- Mokin and Keifer 2006.Mokin M, Keifer J. Quantitative analysis of immunofluorescent punctate staining of synaptically localized proteins using confocal microscopy and stereology. J Neurosci Methods 157: 218–224, 2006. [DOI] [PubMed] [Google Scholar]
- Mokin et al. 2006.Mokin M, Lindahl JS, Keifer J. Immediate-early gene-encoded protein Arc is associated with synaptic delivery of GluR4-containing AMPA receptors during in vitro classical conditioning. J Neurophysiol 95: 215–224, 2006. [DOI] [PubMed] [Google Scholar]
- Mokin et al. 2007.Mokin M, Zheng Z, Keifer J. Conversion of silent synapses into the active pool by selective GluR1–3 and GluR4 AMPAR trafficking during in vitro classical conditioning. J Neurophysiol 98: 1278–1286, 2007. [DOI] [PubMed] [Google Scholar]
- Nguyen and Woo 2003.Nguyen PV, Woo NH. Regulation of hippocampal synaptic plasticity by cyclic AMP-dependent protein kinases. Prog Neurobiol 71: 401–437, 2003. [DOI] [PubMed] [Google Scholar]
- Nie et al. 2007.Nie T, McDonough CB, Huang T, Nguyen PV, Abel T. Genetic disruption of protein kinase A anchoring reveals a role for compartmentalized kinase signaling in theta-burst long-term potentiation and spatial memory. J Neurosci 27: 10278–10288, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh et al. 2006.Oh MC, Derkach VA, Guire ES, Soderling TR. Extrasynaptic membrane trafficking regulated by GluR1 serine 845 phosphorylation primes AMPA receptors for long-term potentiation. J Biol Chem 281: 752–758, 2006. [DOI] [PubMed] [Google Scholar]
- Otmakhova et al. 2000.Otmakhova NA, Otmakhov N, Mortenson LH, Lisman JE. Inhibition of the cAMP pathway decreases early long-term potentiation at CA1 hippocampal synapses. J Neurosci 20: 4446–4451, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson et al. 2001.Patterson SL, Pittenger C, Morozov A, Martin KC, Scanlin H, Drake C, Kandel ER. Some forms of cAMP-mediated long-lasting potentiation are associated with release of BDNF and nuclear translocation of phospho-MAP kinase. Neuron 32: 123–140, 2001. [DOI] [PubMed] [Google Scholar]
- Powell 2006.Powell CM Gene targeting of presynaptic proteins in synaptic plasticity and memory: across the great divide. Neurobiol Learn Mem 85: 2–15, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell et al. 2003.Purcell AL, Sharma SK, Bagnall MW, Sutton MA, Carew TJ. Activation of a tyrosine kinase-MAPK cascade enhances the induction of long-term synaptic facilitation and long-term memory in Aplysia. Neuron 37: 473–484, 2003. [DOI] [PubMed] [Google Scholar]
- Roberson and Sweatt 1996.Roberson ED, Sweatt JD. Transient activation of cyclic AMP-dependent protein kinase during hippocampal long-term potentiation. J Biol Chem 271: 30436–30441, 1996. [DOI] [PubMed] [Google Scholar]
- Roberts and Glanzman 2003.Roberts AD, Glanzman DL. Learning in Aplysia: looking at synaptic plasticity from both sides. Trends Neurosci 26: 662–670, 2003. [DOI] [PubMed] [Google Scholar]
- Roche et al. 1996.Roche KW, O'Brien RJ, Mammen AL, Bernhardt J, Huganir RL. Characterization of multiple phosphorylation sites on the AMPA receptor GluR1 subunit. Neuron 16: 1179–1188, 1996. [DOI] [PubMed] [Google Scholar]
- Schafe and LeDoux 2000.Schafe GE, LeDoux JE. Memory consolidation of auditory Pavlovian fear conditioning requires protein synthesis and protein kinase A in the amygdala. J Neurosci 20: RC96(1–5), 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalin et al. 2006.Shalin SC, Hernandez CM, Dougherty MK, Morrison DK, Sweatt JD. Kinase suppressor of Ras1 compartmentalizes hippocampal signal transduction and subserves synaptic plasticity and memory formation. Neuron 50: 765–779, 2006. [DOI] [PubMed] [Google Scholar]
- Sun et al. 2005.Sun X, Zhao Y, Wolf ME. Dopamine receptor stimulation modulates AMPA receptor synaptic insertion in prefrontal cortex neurons. J Neurosci 25: 7342–7351, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavalin et al. 2002.Tavalin SJ, Colledge M, Hell JW, Langeberg LK, Huganir RL, Scott JD. Regulation of GluR1 by the A-kinase anchoring protein 79 (AKAP79) signaling complex shares properties with long-term depression. J Neurosci 22: 3044–3051, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisskopf et al. 1994.Weisskopf MG, Castillo PE, Zalutsky RA, Nicoll RA. Mediation of hippocampal mossy fibre long-term potentiation by cyclic AMP. Science 265: 1878–1882, 1994. [DOI] [PubMed] [Google Scholar]
- Xu et al. 2006.Xu F, Plummer MR, Len G-W, Nakazawa T, Tamamoto T, Black IB, Wu K. Brain-derived neurotrophic factor rapidly increases NMDA receptor channel activity through Fyn-mediated phosphorylation. Brain Res 1121: 22–34, 2006. [DOI] [PubMed] [Google Scholar]
- Yang et al. 2008.Yang Y, Wang X, Frerking M, Zhou Q. Delivery of AMPA receptors to perisynaptic sites precedes the full expression of long-term potentiation. Proc Natl Acad Sci USA 105: 11388–11393, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng and Keifer 2008.Zheng Z, Keifer J. Protein kinase C-dependent and -independent signaling pathways regulate synaptic GluR1 and GluR4 AMPAR subunits during in vitro classical conditioning. Neuroscience 156: 872–884, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]