

Abstract
Trypanosomiasis remains a significant disease across the sub-Saharan African continent, with 50,000 to 70,000 individuals infected. The utility of current therapies is limited by issues of toxicity and the need to administer compounds intravenously. We have begun a program to pursue lead optimization around MDL 73811, an irreversible inhibitor of S-adenosylmethionine decarboxylase (AdoMetDC). This compound is potent but in previous studies cleared rapidly from the blood of rats (T. L. Byers, T. L. Bush, P. P. McCann, and A. J. Bitonti, Biochem. J. 274:527-533). One of the analogs synthesized (Genz-644131) was shown to be highly active against Trypanosoma brucei rhodesiense in vitro (50% inhibitory concentration, 400 pg/ml). Enzyme kinetic studies showed Genz-644131 to be approximately fivefold more potent than MDL 73811 against the T. brucei brucei AdoMetDC-prozyme complex. This compound was stable in vitro in rat and human liver microsomal and hepatocyte assays, was stable in rat whole-blood assays, did not significantly inhibit human cytochrome P450 enzymes, had no measurable efflux in CaCo-2 cells, and was only 41% bound by serum proteins. Pharmacokinetic studies of mice following intraperitoneal dosing showed that the half-life of Genz-644131 was threefold greater than that of MDL 73811 (7.4 h versus 2.5 h). Furthermore, brain penetration of Genz-644131 was 4.3-fold higher than that of MDL 73811. Finally, in vivo efficacy studies of T. b. brucei strain STIB 795-infected mice showed that Genz-644131 significantly extended survival (from 6.75 days for controls to >30 days for treated animals) and cured animals infected with T. b. brucei strain LAB 110 EATRO. Taken together, the data strengthen validation of AdoMetDC as an important parasite target, and these studies have shown that analogs of MDL 73811 can be synthesized with improved potency and brain penetration.
Sleeping sickness, or human African trypanosomiasis (HAT), afflicts 50,000 to 70,000 people across sub-Saharan Africa, with 17,000 new cases reported in the year 2004 (9) and 10,769 reported in 2007 (31). Untreated, the disease is inevitably fatal. Current treatments include drugs first developed over 50 years ago, and while not without efficacy, some have high toxicity and generally need to be administered by intravenous (i.v.) infusion—hardly a practical solution in locations where this disease is prevalent (17). Despite the obvious need for new, easily administered therapies, the rate of development of new drugs for HAT by the pharmaceutical industry has been negligible. Recognizing this, the Drugs for Neglected Diseases Initiative (DNDi) was formed in 2003 to facilitate the formation of partnerships among industry, academia, and public-sector organizations to develop affordable solutions for this urgent unmet medical need (www.dndi.org). Studies presented here were conducted under one such partnership.
Polyamines are small-molecule cationic structures that are critical to the survival of eukaryotic cells, including trypanosomes (2, 4, 19). Difluoromethyl ornithine (DFMO) is an inhibitor of ornithine decarboxylase, a key enzyme in the polyamine biosynthetic pathway. DFMO is an effective and relatively well tolerated agent for the treatment of the central nervous system (CNS) second stage of HAT caused by Trypanosoma brucei gambiense. However, DFMO must be given as an i.v. infusion of up to 30 g per day four times per day for 14 days. The amount required and frequency of dosing demonstrate the limitations of this drug in terms of potency, cost, and logistics of administration in the field (10, 16). Nevertheless, the extraordinary efficacy of DFMO serves to validate the polyamine pathway as an attractive target for new drug discovery for trypanosomiasis. A potent, safe, orally bioavailable inhibitor of polyamine biosynthesis would represent a major breakthrough for the treatment of this disease.
A second key enzyme in the polyamine biosynthetic pathway is S-adenosylmethionine decarboxylase (AdoMetDC). AdoMetDC is allosterically activated by a novel mechanism unique to trypanosomatid parasites; the functional form of the enzyme is a heterodimer between the active subunit and a paralog (termed prozyme) that arose through gene duplication (32). Gene knockout and small interfering RNA experiments have indicated that suppression of this enzyme by knockdown of either AdoMetDC or its regulatory subunit prozyme is lethal to the parasite (33). Inhibitors of this enzyme have been shown to be highly efficacious in killing trypanosomes in vitro (3, 14) and in curing T. brucei-infected mice (6, 8). However, these agents lack both the potency and, especially, the pharmacokinetic (PK) and tissue (CNS) distribution characteristics (11) that are essential to meet the improved target product profile for a new antitrypanosomal drug.
MDL 73811 is an irreversible inhibitor of AdoMetDC and is believed to form a Schiff base with a pyruvate group within the active site of the enzyme (13). The compound displays a high level of selectivity (>100-fold) for killing parasites compared with its toxicity for mammalian cells (24). Reasons for this selectivity are not clear, but it has been postulated that the compound is more readily taken up by the parasites (12). Alternatively, selectivity may result from differences in enzyme turnover (33). The mechanism of killing is believed to be associated with the buildup of S-adenosylmethionine (11). Recent studies utilizing RNA interference silencing, however, suggest that a major method by which AdoMetDC inhibition kills trypanosomes is by depleting reserves of trypanothione (33). Finally, MDL 73811 induces increased expression of the prozyme in T. brucei bloodstream form parasites, providing strong evidence that the primary target of MDL 73811 responsible for parasite death is AdoMetDC inhibition (33). MDL 73811 has been demonstrated to reduce parasitemia within 5 h and to effect cures of acute infections in T. brucei brucei- and T. brucei rhodesiense-infected mice when administered at a dose of 20 mg/kg of body weight twice a day (BID) for 4 days (11, 24). This compound was originally developed in the 1980s by Merrell-Dow as an anticancer therapy.
Despite its trypanocidal activity, MDL 73811 is not itself an attractive drug candidate for a number of reasons. This compound is not effective as monotherapy against the CNS stage of infection, although it is curative when given in combination with DFMO (6). It has been hypothesized that DFMO may in some way temporarily alter the blood-brain barrier (BBB) to effect this; however, data to support this contention are currently lacking. Recent studies (28) showed that DFMO by itself penetrated the BBB poorly and that addition of 250 μM DFMO did not improve the uptake of other solutes, although a statistically significant increase in BBB penetration was seen at day 28 and later after the trypanosome infection. Finally, the drug has poor oral bioavailability and was thought to have limited metabolic stability (P. Casara, personal communication). We therefore initiated a program to synthesize analogs of MDL 73811 and determine whether they could overcome these issues. Studies reported here describe initial progress.
MATERIALS AND METHODS
Chemicals.
MDL 73811 and analogs were synthesized as described in the supplemental material.
Trypanosomes in vitro.
In vitro trypanosome killing assays were performed as previously described using Trypanosoma brucei rhodesiense strain STIB 900, a clone which is known to be susceptible to all currently used drugs. Briefly, serial dilutions of drugs (90 to 0.123 μg/ml) in supplemented minimal essential medium (GIBCO-BRL catalog no. 072-1100) were inoculated with 104 bloodstream trypomastigotes and incubated 72 h, and then viability was determined using Alamar Blue (27).
Trypanosomes in vivo.
Collaborators utilized two different protocols. In vivo studies at the Swiss Tropical Institute (STI) were performed as previously described (30) under a protocol reviewed and approved by the local veterinary authorities of the Canton Basel-Stadt. Mice were infected with 1 × 104 trypanosomes (T. b. brucei strain STIB 795) on day 0 and then treated once/day (QD) intraperitoneally (i.p.) with 50 mg/kg test compound for 4 days starting on day 3. Animals were assessed by microscopic examination of blood smears twice/week through day 30. Untreated animals generally were moribund and were euthanized by days 7 to 9. Studies conducted at Pace University (under a protocol approved by the university's Institutional Animal Care and Use Committee) utilized the LAB 110 EATRO strain of T. b. brucei as previously described (8). Briefly, groups of five animals were infected i.p. on day 0 with 2.5 × 105 parasites, and dosing was initiated on day 1. Genz-644131 and MDL 73811 were dosed at 50 mg/kg/day i.p. either QD for 7 days or split and administered BID for 4 days. Treatment controls received 2 mg/kg pentamidine QD i.p. for 4 days. Animals were assessed by microscopic examination of at least 20 fields of wet blood smears twice/week through day 40 before being considered cured. In this model, untreated animals generally were moribund and were euthanized by days 3 to 4.
Enzyme kinetics.
T. brucei AdoMetDC-prozyme complex and human AdoMetDC were expressed in Escherichia coli and copurified by Ni2+-agarose and anion-exchange chromatography as previously described (7, 22, 32). Previous studies indicated that the activity of the enzyme complex consisting of recombinant His-tagged AdoMetDC enzyme and Flag-tagged recombinant prozyme was equivalent to that of the native complex in lysates (32). AdoMetDC activity was determined by trapping of labeled 14CO2, also as previously described (7, 22, 32). Kinetic analysis of the described inhibitors was conducted by the methods of Kitz and Wilson, which describes analysis of time-dependent irreversible inhibition (23). Enzyme (0.1 μM) was preincubated with inhibitor at various concentrations (0.1, 0.3 0.6, and 1.0 μM) in buffer (100 mM HEPES [pH 8.0], 50 mM NaCl,1 mM dithiothreitol) at 37°C over a time range of 1 to 22 min. Aliquots were removed at various time points and diluted 10-fold into assay mix containing 1 mM AdoMetDC, and the activity remaining (vi) relative to that of the no-inhibitor control at the same time point (vo) was determined in a 10-min assay. Data were fitted to equation 1 to determine the observed rate constant (kobs). The first-order rate constant of inactivation (kinact) and the apparent Ki (Kiapp) were determined by fitting kobs to equation 2. Data are reported as kinact/Kiapp ratios, which represent the apparent second-order rate constants defining the efficiency of enzyme inactivation by the inhibitor.
 |
(1) |
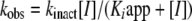 |
(2) |
where 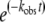 is the negative logarithm of the apparent first-order rate constant (kobs) at time t and [I] is the inhibitor concentration.
is the negative logarithm of the apparent first-order rate constant (kobs) at time t and [I] is the inhibitor concentration.
In vitro pharmaceutical properties.
Solubility was determined using a kinetic method. Briefly, the compound was dissolved in dimethyl sulfoxide, was diluted to 0.5% in phosphate-buffered saline, pH 7.4 (PBS), was allowed to equilibrate for 16 to 24 h, and then was filtered using a 1.2-μm filter, and the concentration was measured using a 96-well plate UV spectrophotometer.
Passive permeability was measured using the parallel artificial membrane permeation assay (PAMPA). Compound was added to the donor well (pH 6.5 in PBS), which was separated from the acceptor well (pH 7.4 in PBS) by a phospholipid membrane in dodecane. Samples were incubated 4 h, and then the concentrations were measured in donor and acceptor wells to calculate permeability (1, 21).
Metabolic stability was determined using both rat and human liver microsomes. Compound was incubated with rat or human liver microsomes at 0.5 mg/ml protein and NADPH cofactors. Postincubation at 0, 10, 20, 30, and 45 min, samples were withdrawn for liquid chromatography-tandem mass spectrometry (LC-MS-MS) analysis. Half-life (t1/2) was determined by plotting ln(peak area ratio) versus time (min). The intrinsic clearance was calculated based on the well-stirred model (26).
The logarithm of the concentration ratio was calculated by measuring the partitioning of compound between octanol and PBS (pH 7.4) after shaking and phase separation.
Plasma protein binding was calculated by diluting a 25 mM stock solution in DMSO to 5 μM in plasma, dialyzing for 4 h against PBS (pH 7.4) at 37°C, and determining the concentration remaining in each compartment.
Cytochrome P450 (CYP) inhibition was determined for human CYP isozymes 1A2, 2D6, 3A4, 2C9, and 2C19 by measuring the inhibition of each isozyme's ability to process its specific fluorescein-labeled substrate. This assay tested inhibition at a single concentration (5 μM) (15, 29).
In vivo PKs.
Groups of six animals (split into two subgroups to minimize the number/volume of blood collections) were administered doses either i.v. or by mouth (p.o.) ranging from 10 mg/kg to 50 mg/kg. Blood samples were drawn at 15 min, 30 min, 2 h, 4 h, 8 h, and 24 h and were subjected to LC-MS-MS to determine drug levels.
Brain penetration was determined by harvesting brains at selected time points and by weighing and extracting total tissue homogenate for LC-MS-MS analysis. The amount of test compound associated with the brain was calculated by subtracting an amount assumed to be attributable to blood vessels (3%, times the blood exposure levels).
RESULTS
MDL 73811 and Genz-644131 are highly active against T. b. rhodesiense and purified T. b. brucei AdoMetDC-prozyme enzyme complex in vitro.
MDL 73811 and its analogs were tested for their ability to kill parasites in vitro and for activity against the purified AdoMetDC prozyme-enzyme complex. Results (Table 1) show that MDL 73811 is highly active in vitro against the parasite (50% inhibitory concentration [IC50], 0.004 μg/ml; 0.0014 μM) and also against the T. brucei AdoMetDC heterodimer (kinact/Kiapp = 1.5 μM−1 min−1). Some analogs (Genz-644390, Genz-644043, Genz-644053) showed a >100-fold loss of activity against the parasite. However, one analog (Genz-644131) showed 10-fold-greater potency against the parasite (IC50 = 400 pg/ml; 0.0001 μM) and ∼5-fold activity against the purified T. brucei AdoMetDC heterodimer AdoMetDC (kinact/Kiapp = 7.8 M−1 min−1). Similar kinetic constants of inactivation were obtained for the human enzyme (Table 1). The efficiency of inactivation measured for Genz-644043 is likely to be an underestimate, because the enzyme activity was nearly completely depleted by the first measurable time point (2.5 min) for all concentrations of inhibitor above 0.1 μM. Additionally, the results were affected by stoichiometric inhibition, because the enzyme concentration ([E]) required to obtain reliable data was similar to the inhibitor concentration ([E] = 0.1 μM). It was therefore not possible to test lower inhibitor concentrations.
TABLE 1.
Activities against parasites and purified AdoMetDC
 |
ND, not determined.
ADME profiles of MDL 73811 and its analogs are generally favorable.
Results from in vitro absorption, distribution, metabolism, and excretion (ADME) profiling studies are shown in Table 2. The aqueous solubility of all the analogs was >47 μg/ml. Compound stability was examined in a number of assays because previous work suggested rapid clearance of MDL 73811 from the blood of rodents (11). Rates of clearance by microsomes and hepatocytes were low, and CYP inhibition was minimal. Plasma protein binding (41%) by the most active analog, Genz-644131, was approximately half that of MDL 73811 in either human (72.9%) or mouse (69.8%) plasma. The only poor in vitro ADME characteristic of these compounds was their membrane permeability, which was low.
TABLE 2.
In vitro ADME profiling
| Compound | Solubility (μg/ml) | Permeability (10−6 cm/s) | Stability clearance (ml/min/kg)
|
IC50 (μM) for human CYP isozyme:
|
% of protein bound in plasma
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Microsomes
|
Hepatocytes
|
||||||||||||
| Rat | Human | Rat | Human | 3A4 | 2D6 | 2C9 | 2C19 | 1A2 | Human | Mouse | |||
| MDL 73811 | >47 | 0.13 | <11 | <4.2 | <11 | <4.2 | >5 | >5 | >5 | >5 | >5 | 72.9 | 69.8 |
| Genz-643990 | >47 | 0.00 | <11 | <4.2 | <11 | <4.2 | >5 | >5 | >5 | >5 | >5 | 23.3 | 47.7 |
| Genz-644043 | >47 | 0.00 | <11 | <4.2 | <11 | <4.2 | >5 | >5 | >5 | >5 | >5 | 32.2 | 37.2 |
| Genz-644053 | >51 | 0.36 | <11 | <4.2 | <11 | <4.2 | >5 | >5 | >5 | >5 | >5 | 39.0 | 42.2 |
| Genz-644131 | >49 | 0.22 | <11 | <4.2 | <11 | <4.2 | >5 | >5 | >5 | >5 | >5 | 41.2 | 35.7 |
Additional in vitro studies were done to characterize MDL 73811 and to create a benchmark against which to compare future analogs. MDL 73811 was highly stable in rat and human plasma, with no degradation over a 4-h period (100% remained). Similarly, MDL 73811 was highly stable in rat whole blood (100% remaining) and showed low permeability, no evidence of efflux in CaCo-2 cells ([3.25 ± 0.2] × 10−6 and [2.7 ± 0.2] × 10−6 cm/s for apical (Ap) to basolateral surface (BL) and for BL to Ap, respectively), and modest partitioning into red blood cells (partitioning coefficient, 0.28). Because these results strongly suggested that neither blood stability nor partitioning into red blood cells was responsible for clearance from plasma, Genz-644131 was not tested for these parameters.
PK parameters of Genz-644131 in rats.
Two studies were performed assessing PK parameters in rats (Table 3). Oral bioavailability was 2%; the t1/2 ranged from 3.2 to 7.0 h between the two studies. Only 4% was cleared in urine by 8 h postadministration; by 24 h, 21% was cleared in urine and 0.2% in bile.
TABLE 3.
PK parameters of Genz-644131 in rats
| Study | Route of administration of Genz-644131 | Mean value (SD)a
|
Elimination route | % of dose (collection interval, h) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Dose level (mg/kg) | Tmax (h) | Cmax (ng/ml) | AUC (ng·h/ml) | t1/2 (h) | Vss (liter/kg) | CL (ml/min/kg) | F (%) | ||||
| 1 | i.v. | 3 | 681 (290) | 3.17 (0.02) | 853 (632) | 81 (35) | Urine | 4.02 (0-8) | |||
| p.o. | 10 | 0.25 (0) | 39 (5) | 49 (13) | 1.53 (1.11) | 2.1 (0.60) | Bile | 0.20 (0-24) | |||
| 2 | i.v. | 3 | 1,550 (696) | 7.03 (1.01) | 2,830 (2,120) | 38 (19) | Urine | 21.1 (0-24) | |||
Tmax, time to maximum concentration of drug in serum; Vss, volume of distribution at steady state; CL, clearance; F, percent oral bioavailability.
Genz-644131 shows a longer t1/2 and brain penetration in vivo in mice than MDL 73811.
Selected PK parameters were examined following single intraperitoneal administration at 50 mg/kg in mice. Results (Table 4) show that the t1/2 of Genz-644131 is approximately threefold longer than that of MDL 73811 (7.43 h versus 2.48 h). Similarly, brain penetration is also ∼4.3-fold higher for Genz-644131. This is noteworthy because of the importance of brain penetration for treating stage 2 disease.
TABLE 4.
Selected systemic PK parameters and brain penetration following single-dose i.p administration in mice
| Compound | AUC in blood (μg·h/ml) | t1/2 (h) | AUC in brain (μg·h/ml) | AUC in brain/AUC in blood (%) | Cmax in blood (ng/ml) | Cmax in brain (ng/g) |
|---|---|---|---|---|---|---|
| MDL 73811 | 20.69 | 2.48 | 0.36 | 1.70 | 44,800 | 80 |
| Genz-643990 | 16.32 | 1.93 | 0.14 | 0.86 | 33,000 | 30 |
| Genz-644053 | 22.08 | 1.66 | 0.40 | 1.79 | 38,950 | 110 |
| Genz-644131 | 17.92 | 7.43 | 1.30 | 7.28 | 36,900 | 660 |
Genz-644131 shows a longer t1/2 and higher IC50 levels in the blood of mice over time than does MDL 73811.
PK studies were conducted with mice to establish the benchmark values for MDL 73811. Animals were injected with either 50 mg/kg or 20 mg/kg MDL 73811, and levels in blood were measured over 8 h. Results show that by 8 h, remaining drug following a 50-mg/kg dose was still ∼10-fold above the IC50 after correction for protein binding (Fig. 1A). At 8 h, following a single dose of 20-mg/kg MDL 73811, levels had fallen to approximately the IC50. Based upon these studies, two situations were modeled. The first follows the hypothetical clearance of the single original 50-mg/kg dose from 8 h through 24 h and predicts that levels of MDL 73811 will fall below the IC50 by 12 h (Fig. 1A). The second situation assumes that animals are dosed two more times (at 8 and 16 h) with 20-mg/kg doses. Under this scenario, blood levels are expected to remain ∼10-fold above the IC50 throughout the 24-h period.
FIG. 1.

Measured and predicted levels of MDL 73811 and Genz-644131 in the blood of mice. (A) Mice were injected i.p. with MDL 73811 either at 50 mg/kg (open circles) or 20 mg/kg (open triangles); samples were collected and analyzed over an 8-h period. Based upon those values, the predicted exposure level was modeled. Filled circles indicated predicted levels following a single injection of 50 mg/kg, plotted from 8 to 24 h. Filled triangles indicated predicted blood levels modeled on thrice daily injections over a 24-h period. The dashed line indicates the calculated IC50 level, corrected for protein binding. (B) Mice were injected once with 50 mg/kg Genz-644131, and blood levels were sampled over 8 h (open circles). Predicted values extending clearance of the 50-mg/kg dose through 24 h are modeled (open triangles). Filled triangles indicate predicted values for a hypothetical dose of 5 mg/kg, based upon results with 50 mg/kg. The dashed line indicates the calculated IC50 level, corrected for protein binding.
A similar, single-dose study was done with Genz-644131 administered at 50 mg/kg (Fig. 1B). Because of the increased potency, lower protein binding and longer t1/2, plasma levels of Genz-644131 were expected to remain ∼3 orders higher than the IC50 corrected for plasma protein binding of the compound. Based on these observations, a model showing the elimination profile of a 5-mg/kg dose was developed (Fig. 1B). Even at this dose, blood levels of Genz-644131 were expected to remain >10-fold above the IC50 for 24 h.
Both MDL 73811 and Genz-644131 significantly increase the survival of T. b. brucei-infected mice.
Mice were infected on day 0 with T. b. brucei strain STIB 795, and treatment was administered to groups of four animals at 50 mg/kg once per day for 4 days starting on day 3. Animals were then monitored for survival (daily) and for parasitemia twice/week through day 30. Results (Table 5) show that the mean survival time for untreated control animals was 6.75 days but that treated animals survived though day 30 (when the study was terminated and animals were euthanized). By day 26, one animal in the MDL 73811-treated group was deemed cured, and no parasites were found upon examination of blood smears. This animal remained free from parasitemia through day 30. In the Genz-644131-treated group, there was no detectible parasitemia in any of the animals through day 20, though by day 30, parasites were again found in three out of four animals.
TABLE 5.
Antiparasitic activity in T. b. brucei strain STIB 795-infected mice
| Treatment groupa | No. cured/no. infected on dayb:
|
Survival (days) | ||
|---|---|---|---|---|
| 20 | 26 | 30 | ||
| Control | 0/4 | 6.75 | ||
| MDL 73811 | ND | 1/4 | 1/4 | >30 |
| Genz-644131 | 4/4 | ND | 1/4 | >30 |
The dose of both MDL 73811 and Genz-644131 was 50 mg/kg i.p. on days 3 to 6.
ND, not done.
Additionally, we studied the effect of increased dose frequency and duration. Results (Table 6) show that treatment with 50 mg/kg/day either BID for 4 days or QD for 7 days resulted in sterile cure (no parasites through day 40) of the T. b. brucei LAB 110 EATRO strain.
TABLE 6.
Antiparasitic activity in T. b. brucei strain LAB 110 EATRO-infected mice
| Treatment group | Dose (days) | No. cured/no. infected | Survival (days) |
|---|---|---|---|
| Untreated control | 0/5 | 5 | |
| Pentamidine | 2 mg/kg QD (1-4) | 5/5 | >40 |
| MDL 73811 | 50 mg/kg/day BID (1-4) | 5/5 | >40 |
| Genz-644131 | 50 mg/kg/day BID (1-7) | 5/5 | >40 |
| MDL 73811 | 50 mg/kg/day QD (1-7) | 5/5 | >40 |
| Genz-644131 | 50 mg/kg/day QD (1-7) | 5/5 | >40 |
DISCUSSION
Despite the obvious unmet medical need, progress in developing new drugs for HAT has been very slow. Studies initiated in the late 1970s and early 1980s demonstrated that the polyamine biosynthetic pathway was essential in trypanosomes and that inhibitors could be potential drug candidates (2, 5, 14). DFMO, which inhibits ornithine decarboxylase, has become widely used in treating HAT yet has issues of administration requiring large amounts of drug to be infused i.v. over a 2-week period (10, 20).
Irreversible inhibitors of AdoMetDC, another enzyme in the polyamine biosynthetic pathway, have also shown considerable promise (6, 8, 11). However, drug characteristics were not optimal; MDL 73811 is not curative against the CNS stage of infection when used as monotherapy (6), and studies examining levels in the blood of rats appeared to indicate that the compound is cleared with a t1/2 of 10 to 20 min (11). We therefore set out to characterize the ADME properties of MDL 73811 to provide a benchmark against which to compare subsequent analogs that hopefully will begin to address some of these issues.
MDL 73811 (32) and four analogs were synthesized. Compounds were tested for efficacy against trypanosomes in vitro, and two were tested against the purified T. brucei AdoMetDC-prozyme heterodimer (Table 1). All halogenated compounds were ≥100-fold less active than MDL 73811 against trypanosomes. The activities of two of these analogs against the purified enzyme were also lower than that of MDL 73811, though it was not reduced by the same proportion. While the reasons for this are uncertain, the uptake of MDL 73811 by trypanosomes is mediated by purine transporters (12, 18), and the halogenated compounds may disrupt the electron distribution required by the transporter. In contrast, the activity of Genz-644131 against both T. brucei blood form parasites in whole-cell assays and the purified AdoMetDC prozyme-enzyme heterodimer was increased (10- and 5-fold, respectively) compared with that of MDL 73811.
In vitro, these compounds were found to be stable in rat and human liver microsomes and hepatocyte stability tests, did not inhibit human CYP enzymes, and had relatively low plasma protein binding (Table 2). Further studies examined compound stability in rat and human plasma as well as whole blood from rats (see above). Over a 4-h period, there was no apparent change in compound structure, nor was there significant uptake into red blood cells. Additional studies examined whether lung endothelial cells (human microvascular lung endothelial cells) or kidney cells (renal proximal tubule epithelial cells) altered the stability of these AdoMetDC inhibitors. Again, there was no apparent change in the chemical structure of the compound after a 4-h exposure (data not shown). In vivo PK studies showed that by 24 h, ∼21% of Genz-644131 was cleared in urine while only 0.2% was cleared in bile (Table 3). Because MDL 73811 and its analogs appear stable in these in vitro assays and the proportion excreted is low, the clearance mechanism(s) remains unknown.
PK studies of mice examined the clearance of compound from plasma and penetration into the brain (Table 4). While differences in areas under the concentration-time curve (AUC) and maximum concentrations of the drug in serum (Cmax) were not great, the t1/2 for Genz-644131 was threefold higher than that of MDL 73811 in plasma, and PK modeling studies suggest that this difference could be sufficient to maintain plasma levels above the IC50 over a 24-h period with a single dose of Genz-644131 as low as 5 mg/kg (Fig. 1B).
It is noteworthy that the t1/2 values for MDL 73811 in plasma in rats were much higher in our studies than in the earlier studies of Byers and coworkers (11) (3 to 7 h here versus ∼20 min in the Byers et al. study). While some animal-to-animal variation is expected in studies of this sort using small numbers of animals, it is likely that this difference also reflects the increased sensitivity of compound detection methods utilized in the present study.
Our studies demonstrate that the Cmax in brain for Genz-644131 is 3.6-fold higher than that of MDL 73811 (Table 4). Since the efficacy of Genz-644131 is currently being tested in a mouse model for stage 2 disease, the effect of this increased penetration is not yet known. However, previous work showed that MDL 73811 was not sufficient as monotherapy to cure CNS stage disease, although there was an additive effect when it was used with DFMO (6). Nonetheless, studies here showed that through chemical modification, this property of MDL 73811 can be improved.
Initial in vivo efficacy studies showed that both MDL 73811 and Genz-644131 were effective in clearing parasites from the blood following infection and significantly prolonging survival time. For MDL 73811, these results were similar to those reported previously (6, 8). Those studies also demonstrated that to achieve a cure, animals required multiple daily dosing (i.p. injection three times/day for 7 days) or continuous delivery via an osmotic pump. In the case of Genz-644131, 4 days of treatment with a single daily i.p. injection reduced parasitemia below the level of detection through day 20 postinfection. Parasites again became detectable by day 30, indicating that 4 days of treatment was not sufficient to achieve cure against T. b. brucei strain STIB 795. In studies against T. b. brucei LAB 110 EATRO, BID treatment or QD treatment with 50 mg/kg/day was curative through day 40 postinfection. Achieving sterile cure in one study but not the other could be due to several factors, including parasite strain-specific differences, differences resulting from initiating dosing on day 3 (STI) versus on day 1 (Pace University), or differences in polyamine concentrations resulting from diet, which have been shown to affect parasite proliferation rates in vivo (25).
Studies presented here confirm the earlier work demonstrating that AdoMetDC is a valid target for drugs against HAT. Preliminary efforts examining analogs of MDL 73811 demonstrate that structural modifications can alter fundamental properties associated with potency and drug disposition (e.g., t1/2 in plasma and brain penetration). Ongoing studies are examining the mechanism of clearance in hopes of improving plasma levels and brain penetration of future analogs.
Supplementary Material
Acknowledgments
We thank Patrick Casara, Simon Croft, Reto Brun, and Robert Don for helpful discussions throughout these studies.
This work was supported in part by funding from DNDi, resources provided by Genzyme, the National Institutes of Health (grant R01 AI34432 to M.A.P.), a predoctoral fellowship (GM007062 to E.K.W.), and the Welch Foundation (grant I-1257 to M.A.P.).
Footnotes
Published ahead of print on 16 March 2009.
Supplemental material for this article may be found at http://aac.asm.org/.
REFERENCES
- 1.Avdeef, A., P. E. Nielsen, and O. Tsinman. 2004. PAMPA: a drug absorption in vitro model. 11. Matching the in vivo unstirred water layer thickness by individual-well stirring in microtitre plates. Eur. J. Pharm. Sci. 22:365-374. [DOI] [PubMed] [Google Scholar]
- 2.Bacchi, C. J. 1981. Content, synthesis, and function of polyamines in trypanosomatids: relationship to chemotherapy. J. Protozool. 28:20-27. [DOI] [PubMed] [Google Scholar]
- 3.Bacchi, C. J., R. Brun, S. L. Croft, K. Alicea, and Y. Buhler. 1996. In vivo trypanocidal activities of new S-adenosylmethionine decarboxylase inhibitors. Antimicrob. Agents Chemother. 40:1448-1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bacchi, C. J., G. Y. Lipschik, and H. C. Nathan. 1977. Polyamines in trypanosomatids. J. Bacteriol. 131:657-661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bacchi, C. J., H. C. Nathan, S. H. Hutner, P. P. McCann, and A. Sjoerdsma. 1980. Polyamine metabolism: a potential therapeutic target in trypanosomes. Science 210:332-334. [DOI] [PubMed] [Google Scholar]
- 6.Bacchi, C. J., H. C. Nathan, N. Yarlett, B. Goldberg, P. P. McCann, A. J. Bitonti, and A. Sjoerdsma. 1992. Cure of murine Trypanosoma brucei rhodesiense infections with an S-adenosylmethionine decarboxylase inhibitor. Antimicrob. Agents Chemother. 36:2736-2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beswick, T. C., E. K. Willert, and M. A. Phillips. 2006. Mechanisms of allosteric regulation of Trypanosoma cruzi S-adenosylmethionine decarboxylase. Biochemistry 45:7797-7807. [DOI] [PubMed] [Google Scholar]
- 8.Bitonti, A. J., T. L. Byers, T. L. Bush, P. J. Casara, C. J. Bacchi, A. B. Clarkson, Jr., P. P. McCann, and A. Sjoerdsma. 1990. Cure of Trypanosoma brucei brucei and Trypanosoma brucei rhodesiense infections in mice with an irreversible inhibitor of S-adenosylmethionine decarboxylase. Antimicrob. Agents Chemother. 34:1485-1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blum, J. A., C. Burri, C. Hatz, L. Kazumba, P. Mangoni, and M. J. Zellweger. 2007. Sleeping hearts: the role of the heart in sleeping sickness (human African trypanosomiasis). Trop. Med. Int. Health 12:1422-1432. [DOI] [PubMed] [Google Scholar]
- 10.Burri, C., and R. Brun. 2003. Eflornithine for the treatment of human African trypanosomiasis. Parasitol. Res. 90(Suppl. 1):S49-S52. [DOI] [PubMed] [Google Scholar]
- 11.Byers, T. L., T. L. Bush, P. P. McCann, and A. J. Bitonti. 1991. Antitrypanosomal effects of polyamine biosynthesis inhibitors correlate with increases in Trypanosoma brucei brucei S-adenosyl-l-methionine. Biochem. J. 274:527-533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Byers, T. L., P. Casara, and A. J. Bitonti. 1992. Uptake of the antitrypanosomal drug 5′-([(Z)-4-amino-2-butenyl]methylamino)-5′-deoxyadenosine (MDL 73811) by the purine transport system of Trypanosoma brucei brucei. Biochem. J. 283:755-758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Casara, P., P. Marchal, J. Wagner, and C. Danzin. 1989. 5′-{[(Z)-4-Amino-2-butenyl]methylamino}-5′-deoxyadenosine: a potent enzyme-activated irreversible inhibitor of S-adenosyl-l-methionine decarboxylase from Escherichia coli. J. Am. Chem. Soc. 111:9111-9113. [Google Scholar]
- 14.Chang, K. P., R. F. Steiger, C. Dave, and Y. C. Cheng. 1978. Effects of methylglyoxal bis(ganylhydrazone) on trypanosomatid flagellates: inhibition of growth and nucleoside incorporation in Trypanosoma brucei. J. Protozool. 25:145-149. [DOI] [PubMed] [Google Scholar]
- 15.Crespi, C. L., V. P. Miller, and B. W. Penman. 1997. Microtiter plate assays for inhibition of human, drug-metabolizing cytochromes P450. Anal. Biochem. 248:188-190. [DOI] [PubMed] [Google Scholar]
- 16.Docampo, R., and S. N. Moreno. 2003. Current chemotherapy of human African trypanosomiasis. Parasitol. Res. 90(Suppl. 1):S10-S13. [DOI] [PubMed] [Google Scholar]
- 17.Fairlamb, A. H. 2003. Chemotherapy of human African trypanosomiasis: current and future prospects. Trends Parasitol. 19:488-494. [DOI] [PubMed] [Google Scholar]
- 18.Goldberg, B., D. Rattendi, D. Lloyd, J. R. Sufrin, and C. J. Bacchi. 2001. In situ kinetic characterization of methylthioadenosine transport by the adenosine transporter (P2) of the African Trypanosoma brucei brucei and Trypanosoma brucei rhodesiense. Biochem. Pharmacol. 61:449-457. [DOI] [PubMed] [Google Scholar]
- 19.Heby, O., S. C. Roberts, and B. Ullman. 2003. Polyamine biosynthetic enzymes as drug targets in parasitic protozoa. Biochem. Soc. Trans. 31:415-419. [DOI] [PubMed] [Google Scholar]
- 20.Jannin, J., and P. Cattand. 2004. Treatment and control of human African trypanosomiasis. Curr. Opin. Infect. Dis. 17:565-571. [DOI] [PubMed] [Google Scholar]
- 21.Kansy, M., F. Senner, and K. Gubernator. 1998. Physicochemical high throughput screening: parallel artificial membrane permeation assay in the description of passive absorption processes. J. Med. Chem. 41:1007-1010. [DOI] [PubMed] [Google Scholar]
- 22.Kinch, L. N., J. R. Scott, B. Ullman, and M. A. Phillips. 1999. Cloning and kinetic characterization of the Trypanosoma cruzi S-adenosylmethionine decarboxylase. Mol. Biochem. Parasitol. 101:1-11. [DOI] [PubMed] [Google Scholar]
- 23.Kitz, R., and I. B. Wilson. 1962. Esters of methanesulfonic acid as irreversible inhibitors of acetylcholinesterase. J. Biol. Chem. 237:3245-3249. [PubMed] [Google Scholar]
- 24.Marasco, C. J., Jr., D. L. Kramer, J. Miller, C. W. Porter, C. J. Bacchi, D. Rattendi, L. Kucera, N. Iyer, R. Bernacki, P. Pera, and J. R. Sufrin. 2002. Synthesis and evaluation of analogues of 5′-([(Z)-4-amino-2-butenyl]methylamino)-5′-deoxyadenosine as inhibitors of tumor cell growth, trypanosomal growth, and HIV-1 infectivity. J. Med. Chem. 45:5112-5122. [DOI] [PubMed] [Google Scholar]
- 25.Nishimura, K., T. Yanase, N. Araki, Y. Ohnishi, S. Kozaki, K. Shima, M. Asakura, W. Samosomsuk, and S. Yamasaki. 2006. Effects of polyamines on two strains of Trypanosoma brucei in infected rats and in vitro culture. J. Parasitol. 92:211-217. [DOI] [PubMed] [Google Scholar]
- 26.Obach, R. S., J. G. Baxter, T. E. Liston, B. M. Silber, B. C. Jones, F. MacIntyre, D. J. Rance, and P. Wastall. 1997. The prediction of human pharmacokinetic parameters from preclinical and in vitro metabolism data. J. Pharmacol. Exp. Ther. 283:46-58. [PubMed] [Google Scholar]
- 27.Raz, B., M. Iten, Y. Grether-Buhler, R. Kaminsky, and R. Brun. 1997. The Alamar Blue assay to determine drug sensitivity of African trypanosomes (T.b. rhodesiense and T.b. gambiense) in vitro. Acta Trop. 68:139-147. [DOI] [PubMed] [Google Scholar]
- 28.Sanderson, L., M. Dogruel, J. Rodgers, B. Bradley, and S. A. Thomas. 2008. The blood-brain barrier significantly limits eflornithine entry into Trypanosoma brucei brucei infected mouse brain. J. Neurochem. 107:1136-1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stresser, D. M., A. P. Blanchard, S. D. Turner, J. C. Erve, A. A. Dandeneau, V. P. Miller, and C. L. Crespi. 2000. Substrate-dependent modulation of CYP3A4 catalytic activity: analysis of 27 test compounds with four fluorometric substrates. Drug Metab. Dispos. 28:1440-1448. [PubMed] [Google Scholar]
- 30.Thuita, J. K., S. M. Karanja, T. Wenzler, R. E. Mdachi, J. M. Ngotho, J. M. Kagira, R. Tidwell, and R. Brun. 2008. Efficacy of the diamidine DB75 and its prodrug DB289, against murine models of human African trypanosomiasis. Acta Trop. 108:6-10. [DOI] [PubMed] [Google Scholar]
- 31.W. H. O. 2008, posting date. Human African trypanosomiasis. World Health Organization, Geneva, Switzerland. http://www.who.int/trypanosomiasis_african/disease/en/index.html.
- 32.Willert, E. K., R. Fitzpatrick, and M. A. Phillips. 2007. Allosteric regulation of an essential trypanosome polyamine biosynthetic enzyme by a catalytically dead homolog. Proc. Natl. Acad. Sci. USA 104:8275-8280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Willert, E. K., and M. A. Phillips. 2008. Regulated expression of an essential allosteric activator of polyamine biosynthesis in African trypanosomes. PLoS Pathog. 4:e1000183. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.