

Abstract
Antibiotic treatment of Staphylococcus aureus infections is often problematic due to the slow response to therapy and the high frequency of infection recurrence. The intracellular persistence of staphylococci has been recognized and could offer a good explanation for these treatment difficulties. Knowledge of the interplay between intracellular antibiotic activity and the overall outcome of infection is therefore important. Several intracellular in vitro models have been developed, but few experimental animal models have been published. The mouse peritonitis/sepsis model was used as the basic in vivo model exploring a quantitative ex vivo extra- and intracellular differentiation assay. The intracellular presence of S. aureus was documented by electron microscopy. Five antibiotics, dicloxacillin, cefuroxime, gentamicin, azithromycin, and rifampin (rifampicin), were tested in the new in vivo model; and the model was able to distinguish between their extra- and intracellular effects. The intracellular effects of the five antibiotics could be ranked as follows as the mean change in the log10 number of CFU/ml (Δlog10 CFU/ml) between treated and untreated mice after 4 h of treatment: dicloxacillin (3.70 Δlog10 CFU/ml) > cefuroxime (3.56 Δlog10 CFU/ml) > rifampin (1.86 Δlog10 CFU/ml) > gentamicin (0.61 Δlog10 CFU/ml) > azithromycin (0.21 Δlog10 CFU/ml). We could also show that the important factors during testing of intracellular activity in vivo are the size, number, and frequency of doses; the time of exposure; and the timing between the start of infection and treatment. A poor correlation between the intracellular accumulation of the antibiotics and the actual intracellular effect was found. This stresses the importance of performing experimental studies, like those with the new in vivo model described here, to measure actual intracellular activity instead of making predictions based on cellular pharmacokinetic and MICs.
Staphylococcus aureus is a major human pathogen that causes both community- and hospital-acquired infections (35). It causes a diverse array of infections ranging from relatively minor skin and wound infections to more serious and life-threatening diseases such as pneumonia (20, 46), endocarditis (48), osteomyelitis (17, 29), arthritis (1), and meningitis (40). Some of these types of S. aureus infections, e.g., endocarditis, are associated with high rates of mortality (25 to 50%), despite antimicrobial treatment (48, 49, 57). Furthermore, S. aureus infections are often persistent and are associated with treatment difficulties, such as a slow response to antibiotic treatment and recurrences, that lead to an extended duration of antimicrobial therapy (11, 13, 31). The antimicrobial treatment of S. aureus infections has also become more difficult due to the emergence of multidrug-resistant strains (3, 4).
Several factors may help explain the capacity of staphylococci to avoid the actions of antibiotics. Biofilm formation might be the main reason for a deficient antibiotic effect when foreign bodies are involved in the staphylococci infections (12, 15, 53). Otherwise, the intracellular presence of the bacteria could offer a good explanation for the slow response to antibiotics, since bacteria located intracellularly might be protected from the effects of antibiotics (55).
S. aureus has classically been classified as an extracellular pathogen (21). Conversely, several reports have established that S. aureus internalizes and survives within professional and even nonprofessional mammalian phagocytes (7, 19, 24, 25, 26, 27). The attitude is therefore changing toward classifying S. aureus as a facultative/opportunistic intracellular pathogen (13, 36, 41, 42, 55).
Having an intracellular target for antimicrobial therapy is more complex than having an extracellular target, because intracellular antimicrobial activity further depends on the penetration into and accumulation in the cell, cellular metabolism, the subcellular disposition, and the bioavailability of the drug. The bacterial responsiveness to antibiotics can also change intracellularly (54, 55). Antimicrobial activity is therefore often impaired intracellularly (6, 56).
To date, this knowledge of the intracellular presence of S. aureus has not influenced the choice of antibiotic to be used for the treatment for S. aureus infections. Penicillinase-stable penicillins, for instance, are considered the mainstay of treatment for methicillin-susceptible S. aureus infections (5, 23, 35), even though penicillins are usually considered not to penetrate cells (8, 30, 50).
Recurrent S. aureus infections may also, at least partly, be explained by the intracellular presence of the bacteria. Gresham et al. demonstrated that polymorphonuclear neutrophils with intracellular S. aureus isolated from the peritoneums of infected mice could cause a new infection by intraperitoneal injection of these cells into healthy mice (24). They also demonstrated that intracellular survival was linked to the global regulator sar, which regulates multiple virulence factors in S. aureus. These two observations could together indicate that intracellular survival is a part of the pathogenesis of S. aureus.
Appropriate models for the testing of the intracellular activities of antimicrobials against S. aureus are needed. Several in vitro models that use various cells and cell lines are available for the study of intracellular S. aureus (6, 10, 18, 24, 44, 51), but only a few in vivo models have been developed.
Here we present an in vivo model that can be used to study the intracellular activities of antimicrobials against S. aureus.
(Part of this study was presented at the 16th European Congress of Clinical Microbiology and Infectious Diseases, Nice, France.)
MATERIALS AND METHODS
Bacterial strains and growth conditions.
A clinical strain of S. aureus from a patient with bacteremia, strain E19977 (Statens Serum Institut, Copenhagen, Denmark), was used throughout the study. The strain is penicillin resistant and methicillin susceptible. The organism was grown and quantified on 5% blood agar plates. Inocula were prepared by measurement of the optical density at 546 nm. The accurate bacterial count (CFU/ml) was quantified by the use of 10-fold dilutions obtained by spotting each dilution (20 μl) in duplicate on agar plates. The detection limit was 25 CFU/ml. Saline with 0.1% (vol/vol) Triton X-100 (T-8787; Sigma-Aldrich Inc., St. Louis, MO) was used in the dilutions to prevent the clumping of the bacteria during whirl mixing, as increased whirl mixing compounds the effect of bacterial clumping. Dilution and spotting were performed in one procedure, and dilutions containing both bacteria and Triton X-100 were never stored for more than 1 min before they were spotted on agar plates. Bacterial cell lysis did not occur due to the short time of exposure to Triton X-100. Mucin (M-2378; Sigma-Aldrich Inc.) was used as an adjuvant for the inoculation of mice. The mucin stock solution was prepared in saline, sterilized, and adjusted to physiological pH before use.
Antimicrobial agents.
When possible, antibiotics were procured as the commercial product registered in Denmark for parenteral use; dicloxacillin (DCX) as Diclocil (Bristol-Myers Squibb Company, New York, NY) cefuroxime (CXM) as Zinacef (GlaxoSmithKline, Brentford, Middlesex, United Kingdom), azithromycin (AZM) as Zitromax (Pfizer Inc., New York, NY), and gentamicin (GEN) as Garamycin (Schering-Plough Corporation, Kenilworth, NJ). Rifampin (RIF; rifampicin) was procured from Sigma-Aldrich Inc. RIF stock solutions were made in dimethyl sulfoxide (D-8418; Sigma-Aldrich Inc.). The final concentration of dimethyl sulfoxide in the solutions used for the animal studies never exceeded 10%.
In vitro studies.
The MICs were determined by the Etest, according to the manufacturer's instructions (AB Biodisk, Solna, Sweden).
A previously described (34) in vitro kinetic model was used for the in vitro kinetic studies. The model simulates antibiotic elimination in a bacterial culture without a change to the inoculum.
Bacteria in exponential phase of growth were applied to the model at a final density of 106.3 CFU/ml. Clinical pharmacokinetic conditions were simulated (Table 1). Growth controls were included in each test run. Samples for quantitative cultures were withdrawn after 0, 2, 4, 6, 8, 12, and 24 h.
TABLE 1.
MICs, antibiotic parameters in humans, and experimental dosages used for single-dose in vivo and in vitro studies of activities of antibiotics against S. aureus E19977
| Antibiotic | MIC (mg/liter) | Clinical parameters
|
Experimental dosage
|
Reference(s) | ||||
|---|---|---|---|---|---|---|---|---|
| Human dosage and routea | Human Cmaxb (mg/liter) (total drug) | Protein binding in human serum (%) | Half-life in humans (h) | In vivo (mg/kg) | In vitroc (mg/liter) | |||
| DCX | 0.5d | 1,000 mg p.o. | 30 | 95 | 1.0 | 200e | 1.5 | 37, 38, 43, 47 |
| CXM | 2.0 | 1,000 mg i.m. | 26-40 | 30 | 1.5 | 200e | 20.0 | 37, 38, 43, 47 |
| AZM | 0.5 | 1,000 mg p.o.f | 1f | 50 | 10.0g | 1 | 0.5 | 16 |
| GEN | 0.38 | 240 mg i.m. | 10 | 10 | 2.0 | 10 | 9.0 | 16, 39 |
| RIF | <0.016 | 600 mg p.o. | 10 | 80 | 3.0 | 10 | 2.0 | 16, 28 |
Conventional dosages and route of administration to humans (p.o., per oral administration; i.m., intramuscular administration).
Commonly observed Cmax in serum after administration of the drug at the dosages and by the route stated for humans.
The initial concentration added to the in vitro kinetic model corresponding to the free peak concentration of the antibiotic in humans after administration of a single dose.
The MIC for DCX was estimated by using an oxacillin Etest. The MICs for DCX and oxacillin are identical, according to the Clinical and Laboratory Standards Institute (14).
Murine dosages expected to yield simulated Cmaxs in mice that are the same as those in humans. For these drugs, however, it was not possible to find appropriate data from the literature, which is why these dosages were estimated on the basis of calculation of the surface area (22).
The most conventional dosage used is 500 mg given orally, but due to the accumulation of the drug, a dosage of 1,000 mg and the corresponding Cmax were used to obtain more actual conditions for the drug.
Half-life corresponding to the initial half-life of azithromycin.
Mouse peritonitis/sepsis model.
The mouse peritonitis/sepsis model has been described previously (22, 32). In brief, outbred female NMRI mice (HsdWin:NMRI; weight, 25 to 30 g; Harlan Netherlands, Horst, The Netherlands) were used throughout the study. The mice had free access to chow and water. The mice were inoculated by intraperitoneal (i.p.) injection of 0.5 ml bacterial suspension containing 107.7 CFU/ml and 5% (wt/vol) mucin. To achieve a sterile immune response, 0.5 ml of a 2.0 mg/ml zymosan A suspension (Z-4250; Sigma-Aldrich Inc.) was injected i.p., as described previously (33).
Mice were euthanized before sample collection. A peritoneal wash was performed by injecting 2.0 ml sterile saline i.p. The resulting fluid, which contained both murine cells and bacteria, was collected with a pipette after the peritoneum was opened. Antibiotic treatment (0.25 ml) was administered subcutaneously. Humane end points consisted of clinical signs of irreversible sickness. If the mouse had these signs, the mouse was immediately euthanized. All animal experiments were approved by the Danish Animal Experimentation Inspectorate (license no. 2004/561-835).
WBC count and differentiation in peritoneal wash.
The white blood cell (WBC) count was determined with an automatic hematology analyzer (Medonic CA620 VET; Boule Medical AB, Stockholm, Sweden). WBC differentials were performed with the analyzer; and the WBCs were discriminated into granulocytes, midsize cells, and lymphocytes.
Light microscopy.
The sample preparations were fixed with a flame, stained with methylene blue (1%; SSI Diagnostics, Hillerød, Denmark), and studied by light microscopy at ×1,000 magnification.
Electron microscopy.
The suspension of cells and bacteria was fixed by adding glutaraldehyde in 0.1 M cacodylate buffer (pH 7.2) containing 0.01 M CaCl2 to a final concentration of 3% (vol/vol). After 60 min of fixation at 4°C, the samples were centrifuged at 300 × g for 15 min. To increase the cell concentration, three-fourths of the supernatant was removed. Procedures for further preparation of the samples were as described previously (2).
Separation of intracellular and extracellular S. aureus in the peritoneal wash.
Samples from two mice were pooled (1:1) to ensure a minimum sample volume of 2.0 ml. The total amount of bacteria in the pooled sample was quantified before any other procedure was performed. Then, 1.5 ml of the pooled samples was transferred to micro-test tubes (Eppendorf AG, Hamburg, Germany) and the tubes were centrifuged at 300 × g and 25°C for 10 min. The extracellular amount of bacteria in the supernatant was quantified.
The pellet was resuspended in Hanks balanced salt solution (H-9269; Sigma-Aldrich Inc.) with 100 μg/ml of lysostaphin (L-7386; Sigma-Aldrich Inc.). The suspension was incubated at room temperature for 7 min to kill the remaining extracellular bacteria. A cell-free bacterial suspension was run in parallel as a control for extracellular killing, and a 6-log10-unit decrease in colony counts was recorded for these controls. The supernatant from the lysostaphin-treated samples was also cultured as a control for extracellular killing. The lysostaphin was removed by washing the samples four times with 2.0 ml fresh Hanks balanced salt solution. The pellet was finally resuspended in 1.5 ml of cold sterile water for cell lysis, and the intracellular bacterial count was quantified.
WBC counts were obtained and light microscopy was performed during critical steps in the separation procedure.
Statistical methods.
Normality tests (D'Agostino and Pearson omnibus normality test and probability plots) were performed with all data sets. Some data were log transformed to obtain a normal distribution. In general, the data sets were log10 normally distributed. Parametric tests were performed with the log10-transformed data.
The mean colony counts among the treatment groups were compared by one-way analysis of variance, followed by the Tukey-Kramer multiple-comparison test (mutual comparison of treatment groups) or Dunnett's multiple-comparison test (comparison of results for the treatment groups to those for the control group). The paired t test was used to compare the means of matched groups. A P value of <0.05 was considered significant. For analysis of dose-effect relationships, the Hill equation (variable slope) was employed. The Prism program (version 5; GraphPad Software Inc., San Diego, CA) was used for illustration and statistical analysis.
RESULTS
MIC determination and time-kill curves in vitro.
The MICs for DCX, CXM, AZM, GEN, and RIF against S. aureus E19977 are shown in Table 1. The strain was susceptible to all five antibiotics, according to the guidelines of the Clinical and Laboratory Standards Institute (14). The in vitro time-kill profiles of the five antibiotics were tested by use of the in vitro kinetic model. Peak (maximum) concentrations (Cmaxs) corresponding to the free drug (fCmax) (Table 1) were applied, and the killing was recorded for 24 h (Fig. 1). GEN, DCX, CXM, and RIF all showed bactericidal effects (>2-log10-unit decreases). GEN was the most effective drug at these concentrations, with a 4-log10-decrease achieved within the first 4 h. RIF had a slower but more persistent bactericidal effect than DCX and CXM (a 2-log10-unit decrease within the first 12 h), which could be due to the longer half-life applied for this drug in the model (Table 1). AZM showed a bacteriostatic effect for the first 8 h, but in line with the antibiotic washout, growth occurred in parallel with the control growth. For all five antibiotics tested, regrowth of the bacteria occurred (Fig. 1).
FIG. 1.

Results of time-kill curve studies performed by use of the in vitro kinetic model by applying both concentrations corresponding to the fCmax measured in humans after administration of a single dose of the antibiotic and simulated human serum elimination half-lives for the five antibiotics studied. The graph shows the change in CFU/ml in the culture flask over a 24-h period (mean and range, n = 2) for RIF, AZM, CXM, DCX, and GEN. The left ordinate displays the actual numbers of CFU/ml, and the right ordinate displays the changes in the numbers of log10 CFU/ml compared to the amount of bacteria present before the antibiotic was applied. The gray area displays decreased colony counts compared to the starting bacterial count, and the white area displays increased colony counts compared to the starting bacterial count.
In vivo model.
Intracellular infection by S. aureus E19977 was documented by light and electron microscopy. On the basis of the findings of light microscopy, the proportion of macrophage-like cells found to be infected was estimated to be >60% after 6 h of infection.
The cells harvested from the peritoneums of the mice were fragile. They were sensitive to, e.g., agitation, turbulence, and fast temperature drops.
Figure 2 displays electron micrographs of the peritoneal wash after 6 h of staphylococcal infection and shows bacteria both intra- and extracellularly. The majority of the intracellular bacteria seemed to be located in vacuoles (Fig. 2A to D). Some apparently spacious vacuoles are seen in Fig. 2A and B, and some more closely spaced vacuoles are observed in Fig. 2C and D. As displayed in Fig. 2B and D, it seems plausible that S. aureus can divide intracellularly.
FIG. 2.

Electron microscopy of peritoneal wash after staphylococcal infection showing both intracellular and extracellular bacteria. Panel B is an enlargement of panel A, and panel D is an enlargement of panel C.
The number of granulocytes increased over time in the peritoneum after inoculation of S. aureus E19977. The numbers of granulocytes/ml (mean ± standard deviation or range) in the peritoneal wash after inoculation were 6.13 × 105 (range, 3.0 × 105 to 1.2 × 106 [n = 4]) at 0 h, 1.07 × 106 ± 3.47 × 105 (n = 20) at 2 h, and 2.49 × 106 ± 5.88 × 105 (n = 27) at 6 h. The increased numbers of granulocytes from 2 h to 6 h was significant (P < 0.0001).
The median total bacterial counts were 105.5 CFU/ml (interquartile [IQ] range, 105.4 to 105.8 CFU/ml; n = 20) 2 h after inoculation and 107.4 CFU/ml [IQ range, 107.1 to 107.5 CFU/ml; n = 28) 6 h after inoculation if the mice were untreated. The inocula injected ranged from 107.7 to 108.1 CFU/ml.
The cell-free bacterial suspension used as a control for extracellular killing by lysostaphin revealed that sedimentation of the bacteria also occurred during centrifugation. After centrifugation, a significant decrease in the bacterial count of up to 1 log10 unit was observed (mean log10 difference, 0.85; 95% confidence interval, 0.74 to 0.95; P < 0.0001; n = 16).
The rate of appearance of false-positive intracellular bacteria during the separation assay was shown to be low; uninfected WBCs were harvested from the mouse peritoneum after a sterile immune response. Concentrations of 107, 106, and 105 WBCs/ml were all spiked with 107, 106, 105, and 104 extracellular CFU/ml; and each WBC-CFU combination was tested by the intra- and extracellular separation assay.
False-positive intracellular bacteria occurred, and this occurrence was most dependent on the amount of spiked bacteria. The occurrence of false-positive findings was also, to some extent, dependent on the amount of cells. For samples with 107 WBCs/ml, 1.620% ± 0.007% of the spiked bacteria were counted as intracellular during the separation assay. For samples containing 106 WBCs/ml, 0.518% ± 0.002% were counted as intracellular, and for samples containing 105 WBCs/ml, 0.080% ± 0.001% were counted as intracellular. Microscopy of the cells after the separation procedure revealed that the false-positive “intracellular” bacteria were almost all extracellular bacteria trapped between the cell surfaces and were not extracellular bacteria forced into the cells. This trapping apparently protected the bacteria from the lysostaphin treatment and from washout, and they were thereby counted as part of the intracellular pool.
Tests with five different antibiotics.
To verify the new in vivo setup, five antibiotics with theoretical differences in intracellular penetration and antibacterial properties were tested in the model. The antibiotics tested were DCX, CXM, AZM, GEN, and RIF. Mice were treated 2 h after bacterial challenge with single doses expected to yield simulated human Cmaxs in mice (Table 1) (group size, n = 16). The mice were sampled 4 h after treatment onset. Untreated control groups were included and were sampled 2 and 6 h after bacterial challenge.
The changes in the numbers of CFU/ml after 4 h of treatment i.p. (total, extracellular, and intracellular) are shown in Fig. 3A to I.
FIG. 3.

Bacterial counts in peritoneums of mice after 4 h of treatment (from 2 to 6 h of infection) with a single dose (Table 1) of AZM, GEN, RIF, CXM, and DCX 2 h after bacterial challenge. The graph shows the change in CFU/ml (median ± IQ range; n = 8). Data for the control groups at 2 h and 6 h are the medians ± IQ ranges for all studies (n = 11). (A to C) Changes in the total, intracellular, and extracellular numbers of CFU/ml, respectively, for the β-lactams DCX and CXM; (D to F) changes in the total, intracellular, and extracellular numbers of CFU/ml, respectively, for GEN and AZM; (G to I) changes in the total, intracellular and extracellular numbers of CFU/ml, respectively, for RIF. The left ordinate displays the actual numbers of CFU/ml, and the right ordinate displays the changes in the log10 numbers of CFU/ml compared to the amount of bacteria before the start of treatment. The gray areas display decreased colony counts compared to the starting bacterial count, and the white areas display increased colony counts compared to the starting bacterial count. Note that the scales for each compartment (total, extracellular, and intracellular) are different.
The bacterial counts in the peritoneums of untreated mice increased 1.94 log10 units in total, 3.65 log10 units intracellularly, and 1.88 log10 units extracellularly.
All antibiotics except AZM produced significant decreases in colony counts in total, extracellularly, and intracellularly compared to the colony counts in the untreated control group by the Dunnett's multiple-comparison test.
The effects of the five antibiotics were compared by Tukey's multiple-comparison test. The effects of the antibiotics on the total colony counts, the extracellular colony counts, and the intracellular colony counts were compared. The levels of significance between groups are marked by asterisks: *, 0.05 > P > 0.01; **, 0.01 > P > 0.001; ***, P < 0.001; and NS, nonsignificant.
The effects of the five antibiotics fell into two significantly different groups when the total effects were compared, as follows:
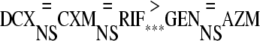 |
DCX, CXM, and RIF showed the best total effect, and their effects did not differ significantly from each other. GEN and AZM showed the poorest effect.
The five antibiotics fell into four significantly different groups when the extracellular effects were compared, as follows:
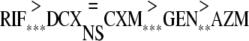 |
RIF showed the best effect extracellularly, while DCX, CXM, and GEN showed intermediate effects. AZM showed the poorest effect.
Finally, when the intracellular effects were compared, the antibiotics fell into three groups, as follows:
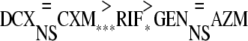 |
DCX and CXM showed the best intracellular effects, while RIF showed an intermediate effect and GEN and AZM showed the poorest intracellular effects.
The effect of GEN in vivo was very poor compared to the effect of GEN seen in vitro (Fig. 1). The effect of RIF intracellularly differed remarkably from its effect extracellularly (Fig. 3G to I).
Timing of treatment onset and bacterial challenge.
The influence of different timing intervals between inoculation and treatment onset was explored. Mice were treated 1, 2, or 3 h after inoculation with a single dose of DCX (60 mg/kg of body weight) or RIF (10 mg/kg) (four mice per group). All mice were sampled after 4 h of treatment; but the duration of infection was 5, 6, or 7 h, respectively. Vehicle-treated control groups were included; and they were also sampled 5, 6, or 7 h after inoculation.
The change in log10 CFU/ml (Δlog10 CFU/ml) between the treated and the untreated mice after 4 h of treatment according to the time of treatment after inoculation is shown in Fig. 4. The values of Δlog10 CFU/ml between the treated and the untreated mice were calculated by using untreated mice with the same times of infection (i.e., 5, 6, or 7 h). The effects of the two drugs in all three fractions (total, intracellular, and extracellular) were influenced by the time of treatment; i.e., the effect decreased with an increasing time between inoculation and treatment onset. For DCX the difference was most noticeable for the intracellular count, and for RIF the difference was most noticeable for the extracellular count.
FIG. 4.

Difference in colony counts (Δlog10 CFU/ml) between the treated and untreated mice in relation to the time of treatment after inoculation (1, 2, and 3 h). The mice treated with DCX received 60 mg/kg, and the mice treated with RIF received 10 mg/kg. The Δlog10 CFU/ml was based on the numbers of CFU/ml in the untreated mice infected for the same duration (5, 6, and 7 h, respectively). All treated mice received treatment for 4 h.
Dose-response relationship extra- and intracellularly.
Mice were treated with five different doses of DCX (200, 60, 30, 20, or 10 mg/kg; 4 to 16 mice per group) 2 h after inoculation. The mice were sampled after 4 h of treatment. Vehicle-treated control groups were included.
The Δlog10 CFU/ml correlated to the dose given (log10 mg/kg) is shown in Fig. 5. Significant dose-response correlations were recorded both for the total count and when the counts were separated into the extra- and intracellular compartments. The untreated control group formed a part of the dose-response curve by including the colony count for the control group as a very small concentration (0.3 mg/kg). The static dose for each compartment was calculated by interpolation in GraphPad Prism software. The static doses were 47.2 mg/kg for the intracellular compartment (Fig. 5B), 18.2 mg/kg for the extracellular compartment (Fig. 5C), and 59.8 mg/kg for the effect in total (Fig. 5A).
FIG. 5.

Dose-response curves for DCX showing the change in the numbers of log10 CFU/ml in the peritoneal wash in mice after 4 h of treatment in relation to dose. (A) Change in the total count; (B) change in extracellular count; (C) change in intracellular count. The left ordinate displays the actual numbers of log10 CFU/ml, and the right ordinate displays the changes in the numbers of log10 compared to the amount of bacteria before the start of treatment. The dark gray areas display decreases in colony counts compared to the starting level of bacteria, and the light gray areas display increases in colony counts compared to the starting level of bacteria. The median for the untreated control group (n = 12) forms a part of the dose-response curve, displayed as a small concentration (0.3 mg/kg).
Effect of one dose versus effect of three doses on extra- and intracellular S. aureus at 24 h.
Mice were treated with one or three doses of DCX (200 mg/kg) or RIF (60 mg/kg) and received the first dose 2 h after inoculation (four mice per group). The three-dose regimen was administered every 8 h. The mice were sampled 19 h after treatment onset. Untreated control groups were included, but only for 6 h of infection. At this point, untreated mice met the clinical signs of irreversible sickness and were euthanized.
Time-kill curves displaying the changes in the colony counts (CFU/ml) in the peritoneums of the mice over time after treatment with both RIF and DCX in relation to the number of doses given are shown in Fig. 6. RIF did not show a dose-dependent effect on the total, extracellular, or intracellular colony counts. On the contrary, the infection outcome was highly affected by the number of doses given in the mice treated with DCX.
FIG. 6.

In vivo time-kill curves showing the change in the numbers of CFU/ml in the peritoneums of mice (mean ± range, n = 2) compared to the number of doses of DCX (200 mg/kg) and RIF (10 mg/kg). (A) Change in the total count; (B) change in extracellular count; (C) change in intracellular count. The left ordinate displays the actual numbers of CFU/ml, and the right axis displays the changes in the numbers of log10 CFU/ml compared to the amount of bacteria before the start of treatment. The gray areas display decreased colony counts compared to the starting bacterial count, and the white areas display increased colony counts compared to the starting bacterial count. The arrows at the abscissa indicate the times of dosing the second and third doses of DCX and RIF.
In total, a decrease in the colony counts of approximately 2 log10 units during the 19 h of treatment was estimated for the mice receiving one and three doses of RIF and mice receiving three doses of DCX. For the mice receiving only one dose of DCX, however, regrowth appeared at between 4 and 19 h of treatment. Therefore, compared to the bacterial level before treatment, a decrease of less than 1 log10 unit after 19 h of treatment was estimated for these mice.
In the extracellular compartment, a reduction of approximately 2.5 log10 units appeared within the first 4 h of treatment for mice treated with RIF, and no regrowth appeared for the following 15 h, irrespective of the number of doses. For mice receiving three doses of DCX, a reduction of 2 log10 units appeared during the 19 h of treatment. For the mice receiving only one dose, a reduction of less than 1 log10 unit occurred.
None of the dosing regimens were able to reduce the colony counts below the bacterial level intracellularly at the start of treatment. The result obtained with the three-dose regimen with DCX, however, was static compared to the starting bacterial level, while the one-dose regimen with DCX resulted in an increase of the colony count of 1 log10 unit. Treatment with RIF resulted in an increase in the colony count of more than 1 log10 compared to the bacterial level at the start of treatment for the first 4 h; this was followed by a decrease of 1 log10 unit for the following 15 h of treatment, irrespective of the number of doses given.
No change in MIC between that for the original bacteria injected and that for the bacteria retrieved from the mice after 19 h of treatment with RIF was observed.
DISCUSSION
An in vivo model was developed to study the relationship between the intracellular presence of S. aureus and antimicrobial effects of five antibiotics.
As exemplified by the test with the five different antibiotics (DCX, CXM, AZM, GEN, and RIF), the model allowed a distinction between antistaphylococcal effects extracellularly and intracellularly (Fig. 3).
When we compared the present results with the results of in vitro studies with various cell lines, we found that they were similar; Barcia-Macay et al. studied the intra- and extracellular antistaphylococcal effects of 16 different antibiotics in vitro (6) and reported an impaired intracellular antibacterial effect compared to the extracellular effect after 24 h of exposure to drugs at concentrations corresponding to the MIC, 10× MIC, and Cmax (human doses). Similar to our findings for AZM, they reported decreases in counts of less than 1 log10 unit both intra- and extracellularly, irrespective of the antibiotic concentration. For oxacillin, they showed, as we did for DCX in vivo, a good intracellular effect, but only at high concentrations. Finally, when they tested RIF and GEN, they showed an intracellular effect markedly lower than the extracellular effect (6).
When data from the new in vivo model are processed, some methodological pitfalls should be considered. (i) When the extracellular and intracellular counts for one sample were added, the sum was always less than the total count for the same sample, indicating a loss of bacteria during the ex vivo separation assay. During centrifugation of the cell suspension in order to isolate the cells and the intracellular bacteria from the extracellular bacteria, sedimentation of the extracellular bacteria from the supernatant also occurred. This resulted in an underestimation of the extracellular count and could at least partly explain the loss of bacteria during the separation assay. Cell lysis might also occur during the separation assay, which could confuse the separation of intra- and extracellular bacteria, and it could also contribute to the loss of bacteria during the lysostaphin washout step. Cell lysis, however, was very dependent on the sample processing procedure and was restrained by careful sample handling during the whole separation assay. Thus, only the results for samples that have undergone the exact same isolation procedure should be compared. Furthermore, it is important that the counts for each fraction are evaluated separately, whether it is the total, extracellular, or intracellular bacterial counts.
(ii) The separation assay induced false-positive intracellular bacteria that consisted of up to 1.6% of the extracellular bacteria. This bias, however, would influence the conclusions only in situations with a high extracellular bacterial load and a low intracellular bacterial load.
(iii) After 2 h of infection, the variation in the intracellular bacterial counts (IQ range, 102.4 to 104.1 CFU/ml; n = 16) was larger than the variation in the total and the extracellular counts (Fig. 3). Furthermore, the intracellular count increased more dramatically (3.65 log10 units) in the control group than the total and the extracellular counts did (1.94 and 1.88 log10 units, respectively). This indicates that the number of intracellular bacteria is not static; i.e., it encompasses both potential intracellular bacterial growth (Fig. 2B and D) and extracellular bacteria that are internalized because of phagocytosis. Such a dynamic nature of infection must, however, be expected throughout the entire infectious process, since phagocytosis and, to some extent, cell lysis continue. Furthermore, the migration of new cells to the infection site also continues. Owing to this increased initial variation, standardization of the intracellular inoculum before antibiotic treatment is difficult.
In general, as shown by us and others, it is difficult to predict the intracellular effects of antibiotics. The finding of a poor intracellular effect for AZM was surprising, since AZM is known to accumulate to a great extent inside cells and to thus have a potentially good intracellular effect (6, 9, 54). On the contrary, the β-lactams showed a good intracellular effect, even though they do not accumulate in cells (9, 45, 55). This indicates that intracellular accumulation alone is not an indicator of intracellular activity, as was previously assumed. According to Van Bambeke et al. (55), the intracellular activities of antibiotics depend on a wide range of other factors besides intracellular presence, which explains the impaired intracellular activities of antibiotics that are often recorded. The impaired effect could be caused by (i) different subcellular locations of the antibiotic and the bacteria or (ii) increased MICs intracellularly due to the impaired expression of the antibacterial activity of the antibiotic (e.g., drug metabolism, changed local pH, or protein binding) or altered bacterial responsiveness (e.g., a changed bacterial metabolism or growth rate). All these parameters make prediction of the actual intracellular effect difficult, which increases the relevance of experimental models for measurement of intracellular activity.
The results obtained for RIF and DCX in this study evidently show how the antibiotic exposure time can change the intracellular activity profile of an antibiotic; the results presented in Fig. 6 display very different activity-time profiles for the two antibiotics. The interpretation of the effect obtained for the animals that received a single dose depends on whether the colony counts were obtained after 4 or 21 h. With the short-term results alone, we would conclude that DCX is superior to RIF intracellularly. However, with the longer-term results, we would conclude that RIF is superior to DCX intracellularly after the administration of a single dose. Since the onset of the extracellular killing activity of RIF is very rapid, RIF must reach the infection site quickly. Intracellular accumulation of RIF is expected, but the time required for RIF to reach intracellular equilibrium and its subcellular location could be slow (the equilibrium time is unknown), which would explain the slow intracellular response (6, 9). The results for DCX show another pattern (Fig. 6) and indicate a lack of accumulation of DCX, since there was a reasonable short-term effect but no long-term effect after the administration of a single dose. Furthermore, the rapid onset of the intracellular effect of DCX could reflect rapid penetration and the rapid achievement of subcellular equilibrium.
Knowledge of the dose-response relationship is also crucial when antibiotic activity extra- and intracellularly is explored, as shown by the results of the dose-response study with DCX (Fig. 5). The dose-response curves clearly display the need for the use of higher doses to obtain an effect in the intracellular compartment compared to the doses required to achieve an effect in the extracellular compartment.
Finally, our results revealed that the time between inoculation and treatment onset in vivo were also critical to the final outcome of the infection (Fig. 4). Changes in the subcellular placement of intracellular S. aureus during different infection stages have been reported by several study groups (7, 24, 52). These subcellular changes would theoretically affect the factors mentioned by Van Bambeke et al. (55) and would result in a change in the antimicrobial effect over time. Therefore, the timing of treatment onset should also be considered when intracellular activity studies are planned and when results are compared.
Both the in vivo and the in vitro models used to assess intracellular antibacterial effect have some advantages, disadvantages, and limitations. The in vivo model has the advantage of allowing the study of concepts in a whole-body system; for example, it includes a fully functional immune system, whole-body drug kinetics are occurring, and the nature of the intracellular infection is dynamic. However, the infection course in vivo is more difficult to standardize, as mentioned above, and it can be difficult to differentiate between an actual antibiotic effect and other in vivo influences. Furthermore, one can only speculate how the response in the mouse model resembles the response that would take place in the actual clinical situation, since immune defenses are highly species specific. The advantage of using cell lines instead of an in vivo model is that the intracellular effect can be explored in different cell types of different host origins, including those of human origin. A cell line model also allows the exploration of isolated mechanisms and effects. However, in vitro models cannot easily simulate drug kinetics as they exist in animals and cannot evaluate the interplay between a fully functional immune system and antibiotic treatment. So far the in vivo animal model and in vitro cell line models complement each other. Continued research may show that the in vitro models alone may sufficiently predict the intracellular effect.
In conclusion, a new in vivo model was developed to explore the extra- and intracellular activities of antimicrobials against S. aureus. The new model complements existing in vitro models well by providing the opportunity to perform more complex studies in a whole-organism system. Studies of particular interest that could be performed with the new model include dose-response studies, drug development and screening assays, pharmacokinetic and pharmacodynamic studies, combination treatments, staphylococcal virulence studies, relapse studies, and others. Studies with this model could be complemented by in vitro studies with cell line models to emphasize the conclusions. When studies with the new in vivo model are planned, the study design should be considered carefully, since exposure time, dose selection and frequency, and the time between inoculation and treatment onset are highly critical to the final conclusions.
Acknowledgments
Jytte Mark Andersen, Leila Borggild, Dorte Truelsen, and Marie Aster Knudsen are thanked for technical assistance during the animal studies; and Elizabeth Engels is thanked for technical assistance performing the electron microscopy.
This work was supported by the M. L. Joergensen and Gunnar Hansens Foundation.
Footnotes
Published ahead of print on 17 February 2009.
REFERENCES
- 1.Al Nammari, S. S., P. Bobak, and R. Venkatesh. 2007. Methicillin resistant Staphylococcus aureus versus methicillin sensitive Staphylococcus aureus adult haematogenous septic arthritis. Arch. Orthop. Trauma Surg. 127:537-542. [DOI] [PubMed] [Google Scholar]
- 2.Andersen, L. P., J. Blom, and H. Nielsen. 1993. Survival and ultrastructural changes of Helicobacter pylori after phagocytosis by human polymorphonuclear leukocytes and monocytes. APMIS 101:61-72. [PubMed] [Google Scholar]
- 3.Appelbaum, P. C. 2006. MRSA—the tip of the iceberg. Clin. Microbiol. Infect. 12(Suppl. 2):3-10. [DOI] [PubMed] [Google Scholar]
- 4.Appelbaum, P. C. 2006. The emergence of vancomycin-intermediate and vancomycin-resistant Staphylococcus aureus. Clin. Microbiol. Infect. 12(Suppl. 1):16-23. [DOI] [PubMed] [Google Scholar]
- 5.Bamberger, D. M., and S. E. Boyd. 2005. Management of Staphylococcus aureus infections. Am. Fam. Physician 72:2474-2481. [PubMed] [Google Scholar]
- 6.Barcia-Macay, M., C. Seral, M. P. Mingeot-Leclercq, P. M. Tulkens, and F. Van Bambeke. 2006. Pharmacodynamic evaluation of the intracellular activities of antibiotics against Staphylococcus aureus in a model of THP-1 macrophages. Antimicrob. Agents Chemother. 50:841-851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bayles, K. W., C. A. Wesson, L. E. Liou, L. K. Fox, G. A. Bohach, and W. R. Trumble. 1998. Intracellular Staphylococcus aureus escapes the endosome and induces apoptosis in epithelial cells. Infect. Immun. 66:336-342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brown, K. N., and A. Percival. 1978. Penetration of antimicrobials into tissue culture cells and leucocytes. Scand. J. Infect. Dis. Suppl., p. 251-260. [PubMed]
- 9.Carryn, S., H. Chanteux, C. Seral, M. P. Mingeot-Leclercq, F. Van Bambeke, and P. M. Tulkens. 2003. Intracellular pharmacodynamics of antibiotics. Infect. Dis. Clin. N. Am. 17:615-634. [DOI] [PubMed] [Google Scholar]
- 10.Carryn, S., F. Van Bambeke, M. P. Mingeot-Leclercq, and P. M. Tulkens. 2002. Comparative intracellular (THP-1 macrophage) and extracellular activities of beta-lactams, azithromycin, gentamicin, and fluoroquinolones against Listeria monocytogenes at clinically relevant concentrations. Antimicrob. Agents Chemother. 46:2095-2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chang, F. Y., J. E. Peacock, Jr., D. M. Musher, P. Triplett, B. B. MacDonald, J. M. Mylotte, A. O'Donnell, M. M. Wagener, and V. L. Yu. 2003. Staphylococcus aureus bacteremia: recurrence and the impact of antibiotic treatment in a prospective multicenter study. Medicine (Baltimore) 82:333-339. [DOI] [PubMed] [Google Scholar]
- 12.Chuard, C., J. C. Lucet, P. Rohner, M. Herrmann, R. Auckenthaler, F. A. Waldvogel, and D. P. Lew. 1991. Resistance of Staphylococcus aureus recovered from infected foreign body in vivo to killing by antimicrobials. J. Infect. Dis. 163:1369-1373. [PubMed] [Google Scholar]
- 13.Ciampolini, J., and K. G. Harding. 2000. Pathophysiology of chronic bacterial osteomyelitis. Why do antibiotics fail so often? Postgrad. Med. J. 76:479-483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clinical and Laboratory Standards Institute. 2008. Performance standards for antimicrobial susceptibility Testing; 15th informational supplement (M100-S15). Clinical and Laboratory Standards Institute, Wayne, PA.
- 15.Costerton, J. W., P. S. Stewart, and E. P. Greenberg. 1999. Bacterial biofilms: a common cause of persistent infections. Science 284:1318-1322. [DOI] [PubMed] [Google Scholar]
- 16.den Hollander, J. G., J. D. Knudsen, J. W. Mouton, K. Fuursted, N. Frimodt-Moller, H. A. Verbrugh, and F. Espersen. 1998. Comparison of pharmacodynamics of azithromycin and erythromycin in vitro and in vivo. Antimicrob. Agents Chemother. 42:377-382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ellington, J. K., M. Harris, L. Webb, B. Smith, T. Smith, K. Tan, and M. Hudson. 2003. Intracellular Staphylococcus aureus. A mechanism for the indolence of osteomyelitis. J. Bone Joint Surg. Br. 85:918-921. [PubMed] [Google Scholar]
- 18.Elliott, G. R., P. K. Peterson, H. A. Verbrugh, M. R. Freiberg, J. R. Hoidal, and P. G. Quie. 1982. Influence of subinhibitory concentrations of penicillin, cephalothin, and clindamycin on Staphylococcus aureus growth in human phagocytic cells. Antimicrob. Agents Chemother. 22:781-784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Esen, M., B. Schreiner, V. Jendrossek, F. Lang, K. Fassbender, H. Grassme, and E. Gulbins. 2001. Mechanisms of Staphylococcus aureus induced apoptosis of human endothelial cells. Apoptosis 6:431-439. [DOI] [PubMed] [Google Scholar]
- 20.Ferrara, A. M. 2007. Treatment of hospital-acquired pneumonia caused by methicillin-resistant Staphylococcus aureus. Int. J. Antimicrob. Agents 30:19-24. [DOI] [PubMed] [Google Scholar]
- 21.Finlay, B. B., and P. Cossart. 1997. Exploitation of mammalian host cell functions by bacterial pathogens. Science 276:718-725. [DOI] [PubMed] [Google Scholar]
- 22.Frimodt-Moller, N., J. D. Knudsen, and F. Espersen. 1999. The mouse peritonitis/sepsis model, p. 127-136. In O. Zak and M. A. Sande (ed.), Handbook of animal models of infection. Academic Press, London, United Kingdom.
- 23.Grayson, M. L. 2006. The treatment triangle for staphylococcal infections. N. Engl. J. Med. 355:724-727. [DOI] [PubMed] [Google Scholar]
- 24.Gresham, H. D., J. H. Lowrance, T. E. Caver, B. S. Wilson, A. L. Cheung, and F. P. Lindberg. 2000. Survival of Staphylococcus aureus inside neutrophils contributes to infection. J. Immunol. 164:3713-3722. [DOI] [PubMed] [Google Scholar]
- 25.Hamill, R. J., J. M. Vann, and R. A. Proctor. 1986. Phagocytosis of Staphylococcus aureus by cultured bovine aortic endothelial cells: model for postadherence events in endovascular infections. Infect. Immun. 54:833-836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hess, D. J., M. J. Henry-Stanley, E. A. Erickson, and C. L. Wells. 2003. Intracellular survival of Staphylococcus aureus within cultured enterocytes. J. Surg. Res. 114:42-49. [DOI] [PubMed] [Google Scholar]
- 27.Hudson, M. C., W. K. Ramp, N. C. Nicholson, A. S. Williams, and M. T. Nousiainen. 1995. Internalization of Staphylococcus aureus by cultured osteoblasts. Microb. Pathog. 19:409-419. [DOI] [PubMed] [Google Scholar]
- 28.Jayaram, R., S. Gaonkar, P. Kaur, B. L. Suresh, B. N. Mahesh, R. Jayashree, V. Nandi, S. Bharat, R. K. Shandil, E. Kantharaj, and V. Balasubramanian. 2003. Pharmacokinetics-pharmacodynamics of rifampin in an aerosol infection model of tuberculosis. Antimicrob. Agents Chemother. 47:2118-2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jensen, A. G., F. Espersen, P. Skinhoj, and N. Frimodt-Moller. 1998. Bacteremic Staphylococcus aureus spondylitis. Arch. Intern. Med. 158:509-517. [DOI] [PubMed] [Google Scholar]
- 30.Johnson, J. D., W. L. Hand, J. B. Francis, N. King-Thompson, and R. W. Corwin. 1980. Antibiotic uptake by alveolar macrophages. J. Lab. Clin. Med. 95:429-439. [PubMed] [Google Scholar]
- 31.Johnson, L. B., M. O. Almoujahed, K. Ilg, L. Maolood, and R. Khatib. 2003. Staphylococcus aureus bacteremia: compliance with standard treatment, long-term outcome and predictors of relapse. Scand. J. Infect. Dis. 35:782-789. [DOI] [PubMed] [Google Scholar]
- 32.Knudsen, J. D., N. Frimodt-Moller, and F. Espersen. 1995. Experimental Streptococcus pneumoniae infection in mice for studying correlation of in vitro and in vivo activities of penicillin against pneumococci with various susceptibilities to penicillin. Antimicrob. Agents Chemother. 39:1253-1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kolaczkowska, E., B. Arnold, and G. Opdenakker. 2008. Gelatinase B/MMP-9 as an inflammatory marker enzyme in mouse zymosan peritonitis: comparison of phase-specific and cell-specific production by mast cells, macrophages and neutrophils. Immunobiology 213:109-124. [DOI] [PubMed] [Google Scholar]
- 34.Lowdin, E., I. Odenholt, S. Bengtsson, and O. Cars. 1996. Pharmacodynamic effects of sub-MICs of benzylpenicillin against Streptococcus pyogenes in a newly developed in vitro kinetic model. Antimicrob. Agents Chemother. 40:2478-2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lowy, F. D. 1998. Staphylococcus aureus infections. N. Engl. J. Med. 339:520-532. [DOI] [PubMed] [Google Scholar]
- 36.Lowy, F. D. 2000. Is Staphylococcus aureus an intracellular pathogen? Trends Microbiol. 8:341-343. [DOI] [PubMed] [Google Scholar]
- 37.Mandell, G. L., J. E. Bennett, and R. Dolin. 2005. Principles and practice of infectious diseases, vol. 2. Churchill Livingstone, New York, NY.
- 38.O'Grady, F., H. P. Lambert, R. G. Finch, and D. Greenwood. 1996. Antibiotic and chemotherapy. Churchill Livingstone, New York, NY.
- 39.Onyeji, C. O., D. P. Nicolau, C. H. Nightingale, and L. Bow. 2000. Modulation of efficacies and pharmacokinetics of antibiotics by granulocyte colony-stimulating factor in neutropenic mice with multidrug-resistant Enterococcus faecalis infection. J. Antimicrob. Chemother. 46:429-436. [DOI] [PubMed] [Google Scholar]
- 40.Pedersen, M., T. L. Benfield, P. Skinhoej, and A. G. Jensen. 2006. Haematogenous Staphylococcus aureus meningitis. A 10-year nationwide study of 96 consecutive cases. BMC Infect. Dis. 6:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Proctor, R. A., P. van Langevelde, M. Kristjansson, J. N. Maslow, and R. D. Arbeit. 1995. Persistent and relapsing infections associated with small-colony variants of Staphylococcus aureus. Clin. Infect. Dis. 20:95-102. [DOI] [PubMed] [Google Scholar]
- 42.Proctor, R. A., C. von Eiff, B. C. Kahl, K. Becker, P. McNamara, M. Herrmann, and G. Peters. 2006. Small colony variants: a pathogenic form of bacteria that facilitates persistent and recurrent infections. Nat. Rev. Microbiol. 4:295-305. [DOI] [PubMed] [Google Scholar]
- 43.Putnam, W. S., J. M. Woo, Y. Huang, and L. Z. Benet. 2005. Effect of the MDR1 C3435T variant and P-glycoprotein induction on dicloxacillin pharmacokinetics. J. Clin. Pharmacol. 45:411-421. [DOI] [PubMed] [Google Scholar]
- 44.Qazi, S. N., E. Counil, J. Morrissey, C. E. Rees, A. Cockayne, K. Winzer, W. C. Chan, P. Williams, and P. J. Hill. 2001. agr expression precedes escape of internalized Staphylococcus aureus from the host endosome. Infect. Immun. 69:7074-7082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Renard, C., H. J. Vanderhaeghe, P. J. Claes, A. Zenebergh, and P. M. Tulkens. 1987. Influence of conversion of penicillin G into a basic derivative on its accumulation and subcellular localization in cultured macrophages. Antimicrob. Agents Chemother. 31:410-416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Risson, D. C., E. D. O'Connor, R. W. Guard, J. M. Schooneveldt, and G. R. Nimmo. 2007. A fatal case of necrotising pneumonia due to community-associated methicillin-resistant Staphylococcus aureus. Med. J. Aust. 186:479-480. [DOI] [PubMed] [Google Scholar]
- 47.Roder, B. L., N. Frimodt-Moller, F. Espersen, and S. N. Rasmussen. 1995. Dicloxacillin and flucloxacillin: pharmacokinetics, protein binding and serum bactericidal titers in healthy subjects after oral administration. Infection 23:107-112. [DOI] [PubMed] [Google Scholar]
- 48.Roder, B. L., D. A. Wandall, N. Frimodt-Moller, F. Espersen, P. Skinhoj, and V. T. Rosdahl. 1999. Clinical features of Staphylococcus aureus endocarditis: a 10-year experience in Denmark. Arch. Intern. Med. 159:462-469. [DOI] [PubMed] [Google Scholar]
- 49.Sanabria, T. J., J. S. Alpert, R. Goldberg, L. A. Pape, and S. H. Cheeseman. 1990. Increasing frequency of staphylococcal infective endocarditis. Experience at a university hospital, 1981 through 1988. Arch. Intern. Med. 150:1305-1309. [PubMed] [Google Scholar]
- 50.Sanchez, M. S., C. W. Ford, and R. J. Yancey, Jr. 1988. Evaluation of antibiotic effectiveness against Staphylococcus aureus surviving within the bovine mammary gland macrophage. J. Antimicrob. Chemother. 21:773-786. [DOI] [PubMed] [Google Scholar]
- 51.Seral, C., J. M. Michot, H. Chanteux, M. P. Mingeot-Leclercq, P. M. Tulkens, and F. Van Bambeke. 2003. Influence of P-glycoprotein inhibitors on accumulation of macrolides in J774 murine macrophages. Antimicrob. Agents Chemother. 47:1047-1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shompole, S., K. T. Henon, L. E. Liou, K. Dziewanowska, G. A. Bohach, and K. W. Bayles. 2003. Biphasic intracellular expression of Staphylococcus aureus virulence factors and evidence for Agr-mediated diffusion sensing. Mol. Microbiol. 49:919-927. [DOI] [PubMed] [Google Scholar]
- 53.Stewart, P. S., and J. W. Costerton. 2001. Antibiotic resistance of bacteria in biofilms. Lancet 358:135-138. [DOI] [PubMed] [Google Scholar]
- 54.Tulkens, P. M. 1991. Intracellular distribution and activity of antibiotics. Eur. J. Clin. Microbiol. Infect. Dis. 10:100-106. [DOI] [PubMed] [Google Scholar]
- 55.Van Bambeke, F., M. Barcia-Macay, S. Lemaire, and P. M. Tulkens. 2006. Cellular pharmacodynamics and pharmacokinetics of antibiotics: current views and perspectives. Curr. Opin. Drug Discov. Dev. 9:218-230. [PubMed] [Google Scholar]
- 56.Van Bambeke, F., S. Carryn, C. Seral, H. Chanteux, D. Tyteca, M. P. Mingeot-Leclercq, and P. M. Tulkens. 2004. Cellular pharmacokinetics and pharmacodynamics of the glycopeptide antibiotic oritavancin (LY333328) in a model of J774 mouse macrophages. Antimicrob. Agents Chemother. 48:2853-2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Watanakunakorn, C. 1994. Staphylococcus aureus endocarditis at a community teaching hospital, 1980 to 1991. An analysis of 106 cases. Arch. Intern. Med. 154:2330-2335. [PubMed] [Google Scholar]