

Abstract
Histone deacetylases (HDAC) are potential targets for the development of new antimalarial drugs. The growth of Plasmodium falciparum and other apicomplexans can be suppressed in the presence of potent HDAC inhibitors in vitro and in vivo; however, in vivo parasite suppression is generally incomplete or reversible after the discontinuation of drug treatment. Furthermore, most established HDAC inhibitors concurrently show broad toxicities against parasites and human cells and high drug concentrations are required for effective antimalarial activity. Here, we report on HDAC inhibitors that are potent against P. falciparum at subnanomolar concentrations and that have high selectivities; the lead compounds have mean 50% inhibitory concentrations for the killing of the malaria parasite up to 950 times lower than those for the killing of mammalian cells. These potential drugs improved survival and completely and irreversibly suppressed parasitemia in P. berghei-infected mice.
Malaria parasites infect hundreds of millions of people and lead to more than 1 million deaths annually, predominantly in sub-Saharan Africa and Asia. The principal malarial parasite in humans responsible for malaria-associated mortality is Plasmodium falciparum. Recently, a number of drug-resistant strains of P. falciparum have evolved, and these strains are resistant to the majority of the existing antimalarial drugs (4, 19). Even though antimalarials like quinine, artemisinin, and their derivatives offer effective treatment, the need to develop new drugs for the treatment of infections caused by resistant parasite strains has become a clear, current priority. Initiatives to identify new molecular targets, to structurally modify existing antimalarials, and to identify agents that can be used in combination to overcome parasite drug resistance are in progress (4, 19).
Histone acetylation is a reversible process and is controlled by histone acetyltransferases (HATs) and histone deacetylases (HDACs). HATs are divided into three families on the basis of the number of conserved motifs and include the GNAT, MYST, and p300/CBP families. Of these, homologues of the first two families (P. falciparum GCN5 [PfGCN5] and P. falciparum MYST [PfMYST]) have been described in P. falciparum (11) and may play critical roles in parasite development and virulence. Recently, the inhibition of PfGCN5 has been shown to alter the expression of approximately 5% of plasmodial genes and blocked the growth of chloroquine-sensitive and -resistant P. falciparum strains (7).
A genomic search revealed five putative deacetylase-encoding genes, including the partially characterized P. falciparum HDAC1 (PfHDAC1) gene (13) and the P. falciparum Sir2 gene (23). Sequence analyses of PfHDAC1 showed that it has a high degree of homology among apicomplexans, raising the possibility that inhibitors with the potential for broad clinical application against apicomplexan diseases may be developed. The HDACs offer attractive molecular targets since they are involved in the regulation of the chromatin structure and transcription.
It is believed that acetylation on histone lysine residues changes the conformation of chromatin from a condensed, closed form to an open, transcriptionally active form. HDAC inhibitors affect the balance of reversible acetylation to modify the expression of genes involved in cell growth, cell cycle progression, and apoptosis. Human cells contain at least 18 HDACs with functional redundancy capabilities (5, 15). The HDACs are distributed among four classes, with HDACs 1, 2, 3, and 8 belonging to class I; HDACs 4, 5, 6, 7, 9, and 10 belonging to class II, sirtuins (NAD+-dependent deacetylases) 1 to 7 belonging to class III; and HDAC 11 belonging to class IV. The presence of only five putative HDACs in P. falciparum potentially offers fewer redundant targets for inhibition than is the case for human cells.
HDACs have been targets for antimalarial drug discovery for the past 10 years, and some inhibitors have shown broad antiprotozoal growth-inhibitory activities (8, 20). Generally, high concentrations of drugs have been required to inhibit parasite growth. Furthermore, inhibitors show little selectivity for Plasmodium cells compared with their selectivity for human cells and incompletely and/or reversibly suppress parasite growth in vivo after drug treatment. For example, treatment of P. berghei-infected mice with apicidin, a broad-spectrum HDAC inhibitor, resulted in a significant reduction in the level of parasitemia but no cures (8).
The compounds evaluated in the present study were developed for isoform-specific HDAC inhibition (6, 14). Here, we report on novel potent HDAC inhibitors that suppress the growth of P. falciparum with 50% inhibitory concentration (IC50s) that are up to 950 times lower than the concentration required for inhibition of the growth of mammalian cells. In vivo, the lead compound YC-II-88 cured mice infected with P. berghei and showed no clinically apparent toxicity to the mice.
MATERIALS AND METHODS
Materials.
Type O-positive human blood and heat-inactivated serum were obtained from BiocheMed (Winchester, VA), Sybr green was obtained from Invitrogen (Carlsbad, CA), an HDAC fluorescent activity assay kit was obtained from BioMol (San Diego, CA), and all other reagents were from Sigma (St. Louis, MO). ICR mice were purchased from Charles River Laboratories (Wilmington, MA).
Culture and synchronization of P. falciparum.
Geographically representative strains of P. falciparum banked in the Departments of Biochemistry and Molecular Biology, Georgetown University Medical Center, or obtained from the MR4 malaria parasite depository resource (ATCC, Manassas, VA) or Carole Long (NIAID, NIH, Rockville, MD) were used in these assays. All parasite strains were maintained as described previously (1, 25). In brief, parasites were maintained in complete medium (RPMI 1640 medium supplemented with 25 mM HEPES, 29 mM sodium bicarbonate, 0.005% hypoxanthine, p-aminobenzoic acid [2 mg/liter], gentamicin sulfate [50 mg/liter], 10% type O-positive human serum with O-positive human red blood cells [RBCs] at a 2% hematocrit). The cultures were maintained at 37°C in an atmosphere of 90% N2, 5% O2, and 5% CO2. The culture medium was exchanged every 2 days.
The cultures were synchronized by using a modified Percoll-sorbitol method (18). Briefly, infected RBCs with mature schizonts were separated on a 55% Percoll solution, washed twice with serum-free medium, and mixed with 20 volumes of noninfected RBCs (NIRBCs) and maintained in culture. Thin-film slides of the culture were made every 30 min to observe the first signs of schizont rupture, the presence of free merozoites, or newly invaded rings. Two hours later (the time for merozoite release and the invasion of new RBCs), the cells were harvested by centrifugation, diluted with 5 volumes of prewarmed 5% sorbitol, and incubated at 37°C for 5 min. The cultures were washed with serum-free medium and maintained as described above. The cultures were synchronized every 2 to 3 weeks.
Inhibition of Plasmodium HDAC activity.
The compounds were provided by Alan Kozikowski (Department of Medicinal Chemistry and Pharmacognosy, University of Illinois at Chicago). The activities of the compounds against P. falciparum HDAC (PfHDAC) were assessed with an HDAC fluorescent activity assay kit, according to the manufacturer's protocol (BioMol) (14). Trophozoite or schizont crude whole-cell extracts were used in the assays for PfHDAC-inhibitory activity. Trophozoites and schizonts were separated on a 55% Percoll solution, washed several times, and diluted in a reaction solution (0.1 M KCl, 20 mM HEPES, pH 7.9, 20% glycerol, 0.2 mM EDTA, 0.5 mM dithiothreitol, 0.5 mM phenylmethylsulfonyl fluoride). The cells were lysed by freezing-thawing and were stored at −80°C until use. To verify the anti-PfHDAC activities, various amounts of crude parasite extracts were mixed with Fluor de Lys substrate and 1 μM trichostatin A or 0.1% dimethyl sulfoxide (Me2SO), and the mixtures were incubated for 15 min at 37°C in HDAC buffer. The reactions were stopped by the addition of Fluor de Lys developer, and fluorescence measurements were made at an excitation wavelength of 360 nm and an emission wavelength of 460 nm. All samples were assayed in triplicate and included a blank (an RBC extract with 0.1% Me2SO). After comparison of the fluorescence intensities for samples with different amounts of parasite extracts, 1 × 106 infected RBCs were determined to be optimal for this assay. To assess the PfHDAC-inhibitory activities of the compounds, we repeated the experiments with the indicated concentrations of selected compounds (K.2, I.2, W.2, YC-II-84, YC-II-88, YC-II-90, AG-THIA-01, AG-b, and suberoylanilide hydroxamic acid). Fluorescent readings were normalized and were used to calculate the percent anti-PfHDAC activities of the compounds by comparing the readings to those for the controls (RBC extracts). Dose-response curves of the anti-PfHDAC activities versus the concentrations were used to determine the IC50s. These values were calculated by using the nonlinear regression curve-fitting option within Prism (version 3.0) software (GraphPad Software Inc., San Diego, CA).
Screening of drugs for antimalarial activities.
A total of 22 compounds were screened for their antimalarial growth-inhibitory activities with two strains of P. falciparum, 3D7 (chloroquine sensitive) and FCB1 (chloroquine resistant), by a standard growth inhibition assay (GIA) (24). In brief, parasite cultures at the ring stage were incubated at 1% parasitemia with 500 nM of the indicated compounds (see Fig. 2) in a 96-well plate at 37°C for 48 h in an atmosphere of 90% N2, 5% CO2, and 5% O2. Complete medium containing 0.1% Me2SO was used as the negative control; at this concentration, the Me2SO did not affect parasite growth in vitro. Wells containing NIRBCs in complete medium were used to obtain background readings. We included the results only for assays in which the levels of parasitemia of the Me2SO controls increased at least threefold from the initial level of parasitemia. The levels of parasitemia were determined by fluorescent-activated cell sorting (FACS) by counting 100,000 Sybr green-stained cells with a FACScan instrument. The percent parasite growth inhibition was determined by using the following formula: [(N − B) − (S − B)]/(N − B), where N is the level of parasitemia in the Me2SO control well, B is the readout determined from the wells containing NIRBCs, and S is the level of parasitemia of the sample. The assays were run in triplicate and were repeated at least twice.
FIG. 2.
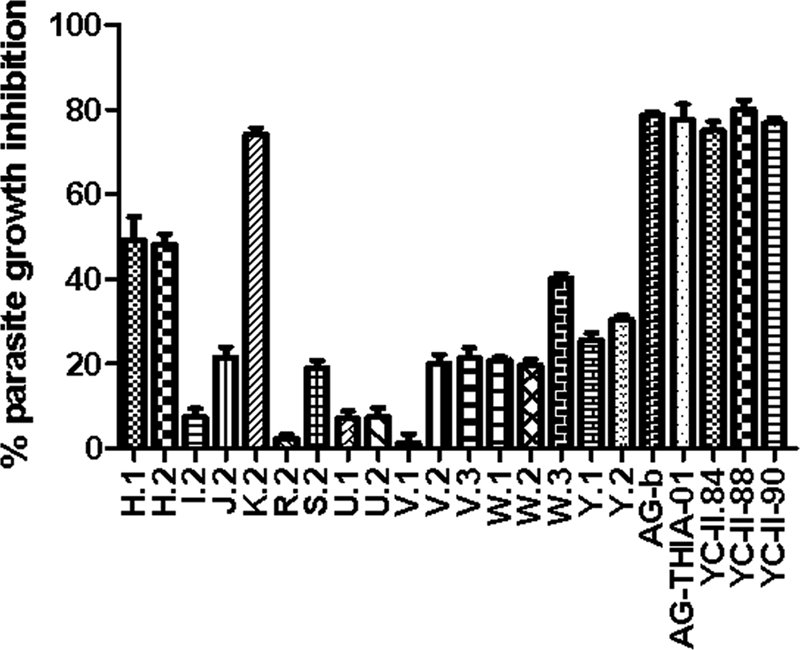
Screening of HDAC inhibitors for their antimalarial activities. Antimalarial activities were screened by a GIA (24), with modifications, by using P. falciparum strain 3D7. The levels of parasitemia were determined by FACS and were used to calculate the percent parasite growth inhibition. Assays were performed twice in triplicate, and the averages and the standard errors of the means are presented. Similar results were obtained with strain FCB1 (data not shown).
We treated several geographically representative P. falciparum strains at the ring stage with graded concentrations of each compound and determined the percent parasite growth inhibition. A plot of the percent parasite growth inhibition versus drug concentration was made, and IC50s were calculated by using a nonlinear regression curve fit. The specificity indices (SIs) were calculated as the IC50 of the drug for the mammalian cell/IC50 of the drug for P. falciparum. The antiproliferative activities of these compounds in mammalian cells have been reported previously (14).
Quality control for GIA.
Experiments were repeated by an independent scientist by using the same assay to provide a minimum of six data points for each drug concentration. The average and standard errors are reported for each drug concentration. For selected compounds, the IC50s were also validated by independent laboratories at Georgetown University and NIAID, NIH.
Assessment of reversibility of growth inhibition by HDAC inhibitor.
To determine if parasite growth resumes after HDAC inhibitor withdrawal, we treated a P. falciparum 3D7 culture with YC-II-88 at 2.5 nM (the minimum concentration required to achieve complete parasite suppression in a 48-h GIA) for various intervals. The drug (YC-II-88) was removed, and the levels of parasitemia were measured every 12 h for 4 days to determine the percent parasite growth inhibition.
Assessment of stage-specific effects of HDAC inhibitor.
The erythrocytic cycle of P. falciparum involves three stages of development: the ring, trophozoite, and schizont stages. Various degrees of cell maturation and cell division occur at each stage. Since HDAC inhibitors alter growth primarily by affecting these processes, we investigated the differential toxicities of the drugs to the various parasite stages (the ring, trophozoite, and schizont stages). Parasite rings (which appear from 1 to 16 h postinvasion), trophozoites (which appear from 16 to 32 h after merozoite invasion), and schizonts (which appear 32 to 48 h postinvasion) were exposed to 1.25 nM YC-II-88 for 14 h. The levels of parasitemia were determined at the end of drug treatment and 16 and 32 h later. All assays included positive and negative controls (continuous exposure from 0 to 48 h with 1.25 nM YC-II-88 and 0.1% Me2SO, respectively). The levels of parasitemia were compared to those for the controls to assess the percent growth inhibition and growth recovery. The assays were performed twice in triplicate.
In vivo toxicity and efficacy studies with mice.
The animal protocols used here were approved by the Animal Care and Use Committee of Georgetown University Medical Center. To determine the dose for use in vivo, we first determined the minimum toxic dose of YC-II-88, the lead compound. Groups of nonimmune ICR mice (five mice per group; Charles River Laboratories) were injected with 0, 0.5, 5, 50, or 100 mg/kg of body weight/day of YC-II- 88 divided into two doses for 5 days and 14 days. The mice were observed for signs of distress (ruffled hair, abdominal distention, porphyria, movement, and feeding). The mice were killed on day 5 or 14, and the internal organs (brain, lungs, heart, spleen, liver, kidney, adrenal, and bone marrow) were harvested for pathological examinations (gross and microscopic). The only pathological change was an increase in the erythroid mass, which was noted in a subset of drug-treated mice. This was scored by the pathologist as mild, moderate, or severe. The data established a maximum dose of YC-II-88 of 50 mg/kg/day for use in the efficacy studies.
In vivo efficacy studies were performed with cryopreserved P. berghei NK56 parasites injected intraperitoneally into ICR mice. The levels of parasitemia were monitored every 2 days until they reached 10% or more. The blood was diluted with phosphate-buffered saline, and 106 parasites were injected intraperitoneally into five groups of ICR mice (five mice per group). In our hands, 100% of the mice infected in this way developed parasitemia by day 4 or earlier and died 8 to 14 days postinoculation. Each group of mice was simultaneously injected (intraperitoneally) with a graded concentration (0, 0.05, 0.5, 5, or 50 mg/kg/day) of YC-II-88 divided into two doses for 4 days.
The mice were monitored daily for clinical signs of distress. On day 4, blood was collected from a tail snip for two thin blood smears. The smears were stained with Giemsa, and the levels of parasitemia were determined manually by counting 1,500 cells from 10 random fields. The numbers of parasites on each slide were counted by two experienced microscopists, providing a minimum of four readings per time point per mouse. A reading from an independent, third microscopist was obtained when the first two microscopists reported levels of parasitemia that differed by 25% or greater. Slides with no observed parasites were confirmed by retesting by staining with Sybr green. Sybr green is more sensitive than Giemsa strain and detects very early rings that could otherwise be missed.
We monitored the mice for a total of 6 weeks or until they died. During this time, parasitemia and hematocrit levels were measured every 4 days. The rates of survival were measured from the time of the initiation of treatment. Differences in hematocrit levels, survival rates, levels of parasitemia, and other parameters were compared by a paired Student t test with the assumption of equal variance.
RESULTS
Effects of HDAC inhibitors on PfHDAC activities.
To determine the inhibitory activities of the HDAC inhibitors, P. falciparum crude extracts were assayed and were found to be inhibited by known hydroxamic acid HDAC inhibitors (TSA and SAHA) in a concentration-dependent manner. The HDAC inhibitors showed no activity against NIRBC extracts (data not shown). Among the compounds tested, YC-II-88 exhibited the highest level of inhibitory activity and had an IC50 of 0.23 nM, approximately 550-fold lower than the IC50 of SAHA (127 nM) (Fig. 1 and Table 1). YC-II-84, YC-II-90, AG-THIA-01, AG-b, and K.2 also demonstrated PfHDAC-inhibitory activities at lower levels. Furthermore, the mercaptoacetamide W.2 (6) demonstrated low levels of PfHDAC-inhibitory activity (IC50, 4,555 nM) (Table 1). Although YC-II-88 also inhibited human HDACs in HeLa cells at the highest level, its IC50 for human HDACs (3.9 nM) was still 17 times greater than that for the PfHDAC (0.23 nM) (Table 1).
FIG. 1.
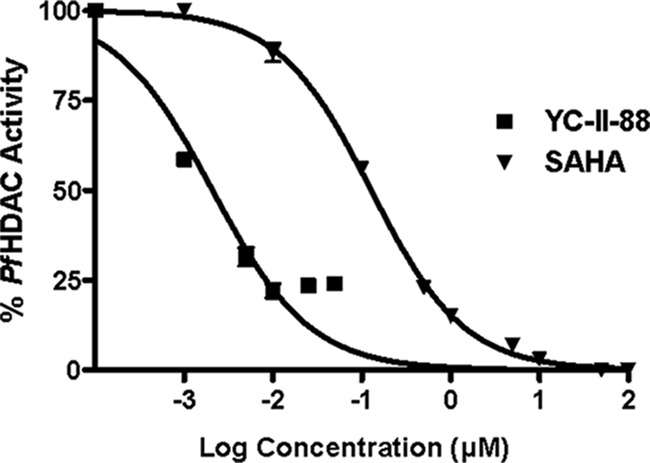
Inhibition of PfHDAC activity. Trophozoite- and schizont-containing crude cell lysates were the sources of PfHDAC activity. Parasite lysates corresponding to 106 parasites were mixed with the indicated concentrations of YC-II-88 or SAHA and used to measure the anti-PfHDAC activity by using a fluorimetric HDAC activity assay kit from BioMol, and the manufacturer's protocol was used, with slight modifications (14). TSA (1 μM) and Me2SO were included as negative and positive controls, respectively. Assays were performed twice in triplicate. The standard errors of the means are presented as bars.
TABLE 1.
Effects of HDAC inhibitors on HDAC activities in P. falciparum and HeLa cellsa
 |
HADC inhibitors were mixed at the concentrations indicated in the text with trophozoite or schizont crude cell lysates or HeLa cell nuclear extracts (HeLa is a human cervical cancer cell line) to determine the inhibition of Plasmodium and human HDAC activities. All experiments were performed in triplicate. The IC50 indicates the concentration of the HDAC inhibitor that conferred 50% HDAC enzyme activity inhibition.
Specificity index (SI) = IC50 HeLa cell extract/IC50 Pf3D7.
HDAC inhibitors selectively inhibit P. falciparum growth and not human cell lines.
Of the 22 different compounds tested, 6 (YC-II-84, YC-II-88, YC-II-90, AG-THIA-01, AG-b, and K.2) completely inhibited parasite growth at 500 nM (Fig. 2). These results were similar for the chloroquine-sensitive strain (P. falciparum 3D7) (Fig. 2) and the multidrug-resistant strain (P. falciparum FCB1) (data not shown). These six compounds and SAHA, W.2, and I.2 were selected for determination of their IC50s. Consistent with its HDAC-inhibitory activity, YC-II-88 showed the greatest inhibition of parasite growth and had an IC50 of 1.25 nM for P. falciparum 3D7 (Table 2). Other compounds (YC-II-90, AG-THIA-01, AG-b, YC-II-84, and K.2) also had IC50s in the low nanomolar range (Table 2). SAHA and YC-II-88 had IC50s for P. falciparum 3D7of 1.25 nM and 900 nM, respectively, which indicates that YC-II-88 had an estimated 600- to 2,350-fold greater potency than SAHA for the various parasite strains tested. The antiplasmodial growth inhibition correlated with the inhibition of PfHDAC activity with a correlation coefficient of 0.9.
TABLE 2.
HDAC inhibitors inhibit the growth of P. falciparum strainsa
| Compound | IC50 (nM) for the following P. falciparum strain (source):
|
||||||
|---|---|---|---|---|---|---|---|
| 7G8 (BRA) | Dd2 (INDO) | FCR3 (GAM) | 3D7 (UNK) | HB3 (HON) | V1/S (VIE) | FCB1 (BRA) | |
| SAHA | NDc | 750.00 | ND | 900.00 | 950.00 | 725.00 | 820.00 |
| K.2b | 250.00 | 185.00 | 175.00 | 500.00 | 250.00 | 250.00 | 125.00 |
| AG-b | 160.00 | 170.00 | 150.00 | 220.00 | 180.00 | 175.00 | 95.00 |
| AG-THIA-01 | 8.00 | 6.00 | 12.00 | 15.00 | 7.00 | 6.00 | 6.00 |
| YC-II-84 | 100.00 | 85.00 | 115.00 | 129.00 | 110.00 | 115.00 | 80.00 |
| YC-II-88 | 0.40 | 0.60 | 0.60 | 1.25 | 0.90 | 1.00 | 0.60 |
| YC-II-90 | 10.00 | 12.00 | 22.00 | 30.00 | 20.00 | 23.00 | 15.00 |
| I.2 | 10,010.00 | ND | 9,575.00 | 10,950.00 | 9,550.00 | 8,115.00 | 6,550.00 |
| W.2 | 5,550.00 | 7,550.00 | 6,750.00 | 9,750.00 | 8,990.00 | 9,910.00 | 6,780.00 |
Geographically representative strains of P. falciparum were incubated with the indicated HDAC inhibitors, and the levels of parasitemia were determined by FACS to determine IC50s. Abbreviations: BRA, Brazil; INDO, Indochina; GAM, The Gambia; UNK, unknown; HON, Honduras; VIE, Vietnam.
The structure of K.2 has been published elsewhere (14).
ND, not determined.
The level of inhibition of the growth of the other P. falciparum strains tested was similar to that observed for P. falciparum 3D7 (Table 2). YC-II-88 consistently had greater growth-inhibitory activity than any of the other compounds tested, with IC50s ranging from 0.4 to 1.25 nM. AG-THIA-01 and YC-II-90 were also active, but their IC50s were about 1 log unit higher than the IC50 of YC-II-88. K.2, YC-II-84, and AG-b inhibited all strains tested, and their IC50s were about 2 log units higher than the IC50 of YC-II-88. Similar potencies against chloroquine-sensitive strains (strains 3D7 and HB3) and chloroquine-resistant strains (strains 7G8, Dd2, V1/S, FCB1, and FCR3) of P. falciparum were also observed.
The HDAC inhibitors also inhibited the growth of several human cell lines, but at concentrations much higher than those required to inhibit P. falciparum strains. The preferential inhibition of P. falciparum was determined by calculating the SI for each compound. The lead compound, YC-II-88, inhibited the P. falciparum strains with SIs of 160 to 950 (Table 3).
TABLE 3.
YC-II-88 selectively inhibits P. falciparum growtha
| Strain or cell line | YC-II-88 IC50 (nM) |
|---|---|
| P. falciparum | |
| 7G8 | 0.4 |
| 3D7 | 1.25 |
| Human cell linesb | |
| SQ20B | 320 |
| PC3 | 210 |
| MCF7 | 380 |
| HeLa | 200 |
The IC50s for the human cell lines were compared to the IC50s for the P. falciparum strains to calculate the SIs. The SIs ranged from 160 to 950.
The toxicity data for human cell lines have been published previously (14).
We also evaluated the reversibility of the antiplasmodial activities of these compounds. Exposure to YC-II-88 for 48 h resulted in the completely irreversible suppression of the parasites after YC-II-88 was withdrawn (data not shown). Shorter treatments also resulted in irreversible parasite suppression if the trophozoite window was included. The parasite levels did not recover by 3 days after drug withdrawal (Fig. 3a). A similar result was observed with compound K.2 (Fig. 3a).
FIG. 3.

HDAC inhibitor-induced parasite inhibition is irreversible and is most significant in mature parasite stages. (a) A synchronized P. falciparum 3D7 culture was treated with the minimum concentrations of YC-II-88 (2.5 nM) and K.2 (1,000 nM) found to completely inhibit P. falciparum 3D7 continuously over 4 days (solid lines) or from 10 to 34 h postinvasion (dashed line). Parasite growth was assessed every 12 h for 4 days by FACS, and the degree of growth inhibition was compared to that for the controls (parasites treated with Me2SO). The data are representative of those from three separate experiments. (b) Rings (1 to 16 h postinvasion), trophozoites (16 to 30 h), and schizonts (32 to 48 h) were treated with 1.25 nM YC-II-88 (the IC50 for P. falciparum 3D7) or Me2SO for 14 h. The assay was performed twice in triplicate. The standard errors of the means are presented as bars.
The erythrocytic cycle of the malaria parasite requires approximately 48 h for development and maturation to transform rings through the trophozoite and schizont stages to the infective merozoites stage, which is capable of reinvading RBCs to restart the cycle. Since various developmental processes (cell division, stage-specific gene expression, DNA synthesis) occur at different stages of the cycle, we investigated the parasite stages (rings [0 to 16 h], trophozoites [16 to 32 h], and schizonts [32 to 48 h]) for their sensitivities to HDAC inhibitor-induced parasite death. We treated the various parasite stages with 1.25 nM YC-II-88 for 14 h to determine parasite growth inhibition (Fig. 3b). Analyses of the data indicate that trophozoites (16 to 32 h) were the most sensitive to HDAC inhibitor-induced parasite death (25% and 35% growth inhibition at the end of treatment and 16 after treatment, respectively) compared to the sensitivities of the schizonts (20% and 25% growth inhibition at the end of treatment and 16 after treatment, respectively) and rings (<5% and 15% at 16 and 32 h, respectively).
Toxicity of YC-II-88 in vivo.
Both short (5-day) and long (14-day) exposures to YC-II-88 produced no clinical signs of distress or mortality in mice after the injection of 100 mg/kg/day (Table 4). However, histological examinations of the harvested organs (lung, brain, adrenal, spleen, liver, bone marrow, and kidney) revealed an increased erythroid mass in the spleen, liver, and bone marrow, which was scored as mild, moderate, or severe. Twenty percent (one of five) of the mice injected with 100 mg/kg/day showed a mild increase in erythroid mass. However, 5-day exposures to doses of 0.5, 5, and 50 mg/kg/day of YC-II-88 resulted in no increase in erythroid mass. Longer exposures (14 days) to YC-II-88 increased the erythroid mass only with the two highest doses. It should be noted that the erythroid cells from the drug-exposed and the control mice showed no morphological differences.
TABLE 4.
Results of in vivo toxicity studiesa
| Exposure time and treatment dose (mg/kg/day) | No. of mice that:
|
No. of mice with the following increase in erythroid mass/total no. of mice:
|
|||
|---|---|---|---|---|---|
| Died | Had clinical signs of distress | Mild | Moderate | Severe | |
| 5 days | |||||
| 100 | 0/5 | 0/5 | 1/5 | 0/5 | 0/5 |
| 50 | 0/5 | 0/5 | 0/5 | 0/5 | 0/5 |
| 5 | 0/5 | 0/5 | 0/5 | 0/5 | 0/5 |
| 0.5 | 0/5 | 0/5 | 0/5 | 0/5 | 0/5 |
| Control | 0/5 | 0/5 | 0/5 | 0/5 | 0/5 |
| 14 days | |||||
| 100 | 0/5 | 0/5 | 2/5 | 1/5 | 0/5 |
| 50 | 0/5 | 0/5 | 2/5 | 0/5 | 0/5 |
| 5 | 0/5 | 0/5 | 0/5 | 0/5 | 0/5 |
| Control | 0/5 | 0/5 | 0/5 | 0/5 | 0/5 |
Groups of ICR mice (five mice per group) were injected with YC-II-88 at the indicated daily doses for 5 or 14 days. Control mice were injected with saline containing 0.1% Me2SO. The mice were observed for death and clinical signs of distress. The increase in erythroid mass (in the bone marrow, liver, and spleen) was scored as mild, moderate, or severe. In case two organs from the same mouse showed different scores, the higher score was assigned.
HDAC inhibitors completely inhibit the growth of P. berghei in mice and improve survival.
Five groups of mice (five mice per group) were injected with YC-II-88 intraperitoneally and were simultaneously infected with P. berghei. The levels of parasitemia were determined microscopically by the use of thin blood smears stained with Giemsa or Sybr green on day 4. None of the mice treated with 50 mg/kg/day of YC-II-88 showed detectable parasites or developed clinical signs of distress. Mice injected with split doses, 5 mg/kg/day and 0.5 mg/kg/day, had detectable parasites on day 4, but the levels of parasitemia were lower than those for the control mice (P < 0.001 and P = 0.02, respectively). Mice treated with 0.05 mg/kg/day had levels of parasitemia comparable to those for the control mice (P = 0.08). The IC50 of YC-II-88 for the control P. berghei isolate was about 0.5 mg/kg/day (Fig. 4a).
FIG. 4.

HDAC inhibitors irreversibly inhibit Plasmodium in vivo and increase the rate of survival. Five groups of mice (five mice per group) were infected with 106 P. berghei cells intraperitoneally and were treated simultaneously with the indicated doses of YC-II-88 divided into two daily doses for 4 days. The percentage of parasite growth inhibition was plotted against the drug concentration (a). The mice were monitored for a total of 6 weeks or until they died; and the levels of parasitemia (b), hematocrit levels (c), and rates of survival (d) are shown. The standard errors of the means are presented as bars in panels a, b, and c. CTRL, control.
Drug treatments were discontinued on day 4, and the levels of parasitemia (Fig. 4b) and hematocrit (Fig. 4c) were assessed for 6 weeks or until the mice died. On day 4 (the last day of treatment), the mice in the control group and the group treated with 0.05 mg/kg/day had comparable levels of parasitemia. By day 8, the levels of parasitemia in the group treated with 0.5 mg/kg/day were comparable to those in the control group. The levels of parasitemia in the group treated with 5 mg/kg/day reached the levels in the control group on day 20 of the experiment. None of the mice in the group treated with 50 mg/kg/day developed parasitemia during the 6-week duration of the experiment (Fig. 4b).
The hematocrit levels declined in the mice in all treatment groups except the group receiving 50 mg/kg/day (Fig. 4c). The hematocrit levels of the control mice dropped the fastest and reached an average of about 18% on day 10 after infection. The rate of decrease in the hematocrit level in the group treated with 50 mg/kg/day were comparable to those observed in the group treated with 0.05 mg/kg/day and the control group. Mice treated with 0.5 and 5 mg/kg/day of YC-II-88 had slower rates of decline in hematocrit levels. The hematocrits of the mice receiving 50 mg/kg/day remained stable for the 6 weeks of the experiment (Fig. 4c).
Even though the mice in the control group and the group treated with 0.5 mg/kg/day had comparable levels of parasitemia starting on day 8 of the experiment, the control mice lived an average of 11.7 days, while the mice in the group treated with 0.5 mg/kg/day lived an average of 23.6 days (P < 0.001) (Fig. 4d). The control mice also died as their levels of parasitemia reached 50%, whereas the mice in the groups treated with 0.5 and 5 mg/kg/day lived an average of about 10 days with levels of parasitemia of 50% or more before they died. The mice in the group treated with 0.05 mg/kg/day group lived an average of 3 days longer than the mice in the Me2SO-treated control group (15.3 days), but this difference was not significant (Fig. 4d).
DISCUSSION
HDACs are potential targets for the treatment of various human diseases (10, 12, 21) and for antimalarial drug development (2, 3, 8, 9, 20). These are particularly attractive targets for antimalarial drug therapy because, unlike mammalian cells, HDACs are more limited and potentially less redundant in Plasmodium species. The P. falciparum genome reveals five putative HDACs, two of which have been characterized (13, 23), and sequence analysis of PfHDAC1 (accession no. PFI1260c) reveals 65% sequence homology with human class 1 HDAC but up to 96% sequence homology with HDAC1 of other Plasmodium species.
Several HDAC inhibitors with antiprotozoal activity in vitro and in vivo have been reported (2, 8, 20). For example, apicidin, a fungal metabolite, inhibits plasmodial HDAC activity and parasite growth with mean inhibitory concentrations in the low-nanomolar range. However, apicidin failed to completely suppress parasitemia in P. berghei-infected mice (8). Suberic acid bisdimethylamide completely suppressed parasitemia in P. berghei-infected mice, but the parasitemia recurred 8 to 12 days posttreatment (3). In this investigation, we screened HDAC inhibitors to find compounds that had high potencies and specificities for P. falciparum in vitro and in vivo and that could irreversibly suppress parasites in vivo.
We evaluated these HDAC inhibitors showing high potencies against a variety of geographically representative drug-sensitive and multidrug-resistant strains of P. falciparum (Table 2). In vitro, these drugs had mean inhibitory concentrations as low as 0.4 nM for P. falciparum, a value that is several orders of magnitude lower than the values for previously described HDAC inhibitors. The growth-inhibitory activities were directly related to the enzyme-inhibitory activities. In our experiments, the lead compound, YC-II-88, completely and irreversibly suppressed parasitemia in vitro (Fig. 3) and in vivo (Fig. 4a). It is of note that increased concentrations of this drug could not suppress HDAC activity in vitro below 25%. This may be attributable to selective HDAC inhibition but is not fully understood. The strain of P. berghei used in the in vivo efficacy studies (strain NK56) is chloroquine sensitive; however, effects similar to those obtained in vitro are anticipated with chloroquine-resistant strains (Fig. 2 and Table 2). Recently, Dow and colleagues (9) reported the results of a challenge experiment with P. berghei in mice in which they failed to effect a cure. The oral administration of YC-II-88 resulted in no cures in P. berghei-infected mice at doses up to 640 mg/kg/day for 3 days. They speculate that the failure of monotherapy in mice may be due to the metabolic stability of YC-II-88, which exhibits a short half-life after the administration of a single oral dose of 50 mg/kg. However, we observed cures in P. berghei-infected mice treated with YC-II-88 intraperitoneally at doses of 50 mg/kg/day divided into two daily doses for 4 days. We reason that the different routes of delivery may contribute to the outcomes, since the solubility and stability of hydroxamates are adversely affected by the low pH observed in the stomach (16). Another possibility is related to the model system used, since ours was a prophylactic model, whereas Dow et al. used a curative mode (9).
HDACs regulate various cellular processes, including DNA synthesis, cell division and differentiation, apoptosis, and others. Inhibitors of HDACs arrest cell division and stimulate apoptosis-related processes (22). For this reason, HDAC inhibitors show selective toxicity against growing malignant cells and not against healthy cells. As predicted, among the three stages of the erythrocytic cycle, trophozoites (in which DNA and most macromolecular synthesis occur) and schizonts (in which schizogony occurs) were more susceptible to HDAC inhibitor-induced cell death than rings (Fig. 3).
Since HDACs show a high degree of sequence homology among various species, HDAC inhibitors (like apicidin and SAHA) show little selectivity among species (8). The lack of selectivity raises the possibility of side effects, should these drugs be used to treat human parasitic diseases. We observed a relatively high degree of selective toxicity against P. falciparum compared to the toxicity observed against mammalian cells (Table 3). The selectivity was greater for six of the compounds (K.2, YC-II-84, YC-II-88, YC-II-90, AG-THIA-01, and AG-b) than for SAHA. The lead compound, YC-II-88, was up to 950 times more toxic to P. falciparum than to human cells in vitro (Tables 2 and 3). Parasitemia was completely suppressed in P. berghei-infected mice (Fig. 4a) treated with YC-II-88 at 50 mg/kg/day, and there were no observable clinical side effects (Table 4). We did not investigate higher infective doses of P. berghei or mice with already patent parasitemia.
The results of the toxicity studies indicated that mice injected with 50 mg/kg/day of YC-II-88 for 4 days would not show no any histological changes (Table 4), but longer exposures or a higher dose (100 mg/kg/day) resulted in increases in erythroid mass (Table 4). The mechanism underlying the HDAC inhibitor-induced erythroid mass increase is not understood; however, it has been reported that HDAC inhibitors promote the differentiation of erythroid cells (17). Hematocrit levels were comparable in control and drug-exposed mice in the toxicity studies (data not shown). No histologic changes were observed in the other organs examined (heart, kidney, lungs, brains, and adrenals).
Acknowledgments
We thank Alan Kozikowski for providing the compounds; Carole Long for providing P. falciparum 3D7 and performing growth inhibition assays; Paul Roepe (Georgetown University, Washington, DC) for providing P. falciparum strains FCB1, Dd2, and HB3; Simon Metenou (NIAID, NIH, Bethesda, MD) for reading the slides; the MR4 malaria parasite depository resource for providing several P. falciparum and P. berghei strains; and Manny Subramanian (Best Medical International, Springfield, VA) and Geoffrey Dow (WRAIR, Silver Spring, MD) for helpful discussions.
This work was supported by NIH/NCI grant P01CA074175 and a fellowship from Krishnan Suthanthiran of Best Medical International, Springfield, VA.
Footnotes
Published ahead of print on 17 February 2009.
REFERENCES
- 1.Agbor-Enoh, S. T., R. N. Achur, M. Valiyaveettil, R. Leke, D. W. Taylor, and D. C. Gowda. 2003. Chondroitin sulfate proteoglycan expression and binding of Plasmodium falciparum-infected erythrocytes in the human placenta during pregnancy. Infect. Immun. 71:2455-2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andrews, K. T., T. N. Tran, A. J. Lucke, P. Kahnberg, G. T. Le, G. M. Boyle, D. L. Gardiner, T. S. Skinner-Adams, and D. P. Fairlie. 2008. Potent antimalarial activity of histone deacetylase inhibitor analogues. Antimicrob. Agents Chemother. 52:1454-1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andrews, K. T., A. Walduck, M. J. Kelso, D. P. Fairlie, A. Saul, and P. G. Parsons. 2000. Anti-malarial effect of histone deacetylation inhibitors and mammalian tumor cytodifferentiating agents. Int. J. Parasitol. 30:761-768. [DOI] [PubMed] [Google Scholar]
- 4.Bosman, A., and K. N. Mendis. 2007. A major transition in malaria treatment: the adoption and deployment of artemisinin-based combination therapies. Am. J. Trop. Med. Hyg. 77:193-197. [PubMed] [Google Scholar]
- 5.Butler, K. V., and A. P. Kozikowski. 2008. Chemical origins of isoform selectivity in histone deacetylase inhibitors. Curr. Pharm. Des. 14:505-528. [DOI] [PubMed] [Google Scholar]
- 6.Chen, B., P. A. Petukhov, M. Jung, E. Eliseeva, A. Velena, A. Dritschilo, and A. P. Kozikowski. 2005. Chemistry and biology of mercaptoacetamindes as novel histone deacetylase inhibitors. Bioorg. Med. Chem. Lett. 15:1389-1392. [DOI] [PubMed] [Google Scholar]
- 7.Cui, L., J. Miao, T. Furuya, Q. Fan, L. Xinyi, P. K. Rathod, X. Su, and L. Cui. 2008. Histone acetyltransferase inhibitor anacardic acid causes changes in global gene expression during in vitro Plasmodium falciparum development. Eukaryot. Cell 7:1200-1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Darkin-Rattray, S. J., A. M. Gurnett, R. W. Myers, P. M. Dulski, T. M. Crumley, J. J. Allocco, C. Cannova, P. T. Meinke, S. L. Colletti, M. A. Bednarek, S. B. Singh, M. A. Goetz, A. W. Dombrowski, J. D. Polishook, and D. M. Schmatz. 1996. Apicidin: a novel antiprotozoal agent that inhibits parasite histone deacetylase. Proc. Natl. Acad. Sci. USA 12:13143-13147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dow, D. S., Y. Chen, K. T. Andrews, D. Caridha1, L. Gerena1, M. Gettayacamin, J. Johnson, Q. Li, V. Melendez, N. Obaldia, T. N. Tran, and A. P. Kozikowski. 2008. Antimalarial activity of phenylthiazolyl-bearing hydroxamate-based histone deacetylase inhibitors. Antimicrob. Agents Chemother. 52:3467-3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Elaut, G., V. Rogiers, and T. Vanhaecke. 2007. The pharmaceutical potential of histone deacetylase inhibitors. Curr. Pharm. Des. 13:2584-2620. [DOI] [PubMed] [Google Scholar]
- 11.Fan, Q., L. An, and L. Cui. 2004. PfADA2, a Plasmodium falciparum homologue of the transcriptional coactivator ADA2 and its in vivo association with the histone acetyltransferase PfGCN5. Gene 336:251-261. [DOI] [PubMed] [Google Scholar]
- 12.Griffiths, E. A., and S. D. Gore. 2008. DNA methyltransferase and histone deacetylase inhibitors in the treatment of myelodysplastic syndromes. Semin. Hematol. 45:23-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Joshi, M. B., D. T. Lin, P. H. Chiang, N. D. Goldman, H. Fujioka, M. Aikawa, and C. Syin. 1999. Molecular cloning and nuclear localization of a histone deacetylase homologue in Plasmodium falciparum. Mol. Biochem. Parasitol. 99:11-19. [DOI] [PubMed] [Google Scholar]
- 14.Jung, M., A. Velena, B. Chen, P. A. Petukhov, A. P. Kozikowski, and A. Dritschilo. 2005. Novel HDAC inhibitors with radiosensitizing properties. Radiat. Res. 163:488-493. [DOI] [PubMed] [Google Scholar]
- 15.Khan, N., M. Jeffers, S. Kumar, C. Hackett, F. Boldog, N. Khramtsov, X. Qian, E. Mills, S. C. Berghs, N. Carey, P. Finn, L. S. Collins, A. Tumber, J. W. Ritchie, P. B. Jensen, H. S. Lichenstein, and M. Sehested. 2000. Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem. J. 409:581-589. [DOI] [PubMed] [Google Scholar]
- 16.Konsoula, R., and M. Jung 2008. In vitro plasma stability, permeability and solubility of mercaptoacetamide histone deacetylase inhibitors. Int. J. Pharm. 361:19-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kosugi, H., M. Towatari, S. Hatano, K. Kitamura, H. Kiyoi, T. Kinoshita, M. Tanimoto, T. Murate, K. Kawashima, H. Saito, and T. Naoe. 1999. Histone deacetylase inhibitors are the potent inducer/enhancer of differentiation in acute myeloid leukemia: a new approach to anti-leukemia therapy. Leukemia 13:1316-1324. [DOI] [PubMed] [Google Scholar]
- 18.Kutner, S., W. V. Breuer, H. Ginsburg, S. Aley, and Z. I. Cabantchik. 1985. Characterization of permeation pathways in the plasma membrane of human erythrocytes infected with early stages of Plasmodium falciparum: association with parasite development. J. Cell. Physiol. 125:521-527. [DOI] [PubMed] [Google Scholar]
- 19.Laufer, M. K., A. A. Djimdé, and C. V. Plowe. 2007. Monitoring and deterring drug-resistant malaria in the era of combination therapy. Am. J. Trop. Med. Hyg. 77(6 Suppl.):160-169. [PubMed] [Google Scholar]
- 20.Mai, A., I. Cerbara, S. Valente, S. Massa, L. A. Walker, and B. L. Tekwani. 2004. Antimalarial and antileishmanial activities of aroyl-pyrrolyl- hydroxyamides, a new class of histone deacetylase inhibitors. Antimicrob. Agents Chemother. 48:1435-1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marks, P. A., and M. Dokmanovic. 2005. Histone deacetylase inhibitors: discovery and development as anticancer agents. Expert Opin. Investig. Drugs 14:1497-1511. [DOI] [PubMed] [Google Scholar]
- 22.Mehnert, J. M., and W. K. Kelly. 2007. Histone deacetylase inhibitors: biology and mechanism of action. Cancer J. 13:23-29. [DOI] [PubMed] [Google Scholar]
- 23.Merrick, C. J., and M. T. Duraisingh. 2007. Plasmodium falciparum Sir2: an unusual sirtuin with dual histone deacetylase and ADP-ribosyltransferase activity. Eukaryot. Cell 6:2081-2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nikodem, D., and E. Davidson. 2000. Identification of a novel antigenic domain of Plasmodium falciparum merozoite surface protein-1 that specifically binds to human erythrocytes and inhibits parasite invasion, in vitro. Mol. Biochem. Parasitol. 30:79-91. [DOI] [PubMed] [Google Scholar]
- 25.Trager, W. 1971. A new method for intraerythrocytic cultivation of malaria parasites (P. coatneyi and P. falciparum). J. Protozool. 18:239-242. [DOI] [PubMed] [Google Scholar]