

Abstract
The bifunctional Saccharomyces cerevisiae Skn7 transcription factor regulates osmotic stress response genes as well as oxidative stress response genes; however, the mechanisms involved in these two types of regulation differ. Skn7 osmotic stress activity depends on the phosphorylation of the receiver domain aspartate, D427, by the Sln1 histidine kinase. In contrast, D427 and the SLN1-SKN7 phosphorelay are dispensable for the oxidative stress response, although the receiver domain is required. The majority of oxidative stress response genes regulated by Skn7 also are regulated by the redox-responsive transcription factor Yap1. It is therefore possible that the nuclearly localized Skn7 does not itself respond to the oxidant but simply cooperates with Yap1 when it translocates to the nucleus. We report here that oxidative stress leads to a phosphatase-sensitive, slow-mobility Skn7 variant. This suggests that Skn7 undergoes a posttranslational modification by phosphorylation following exposure to oxidant. Oxidant-dependent Skn7 phosphorylation was eliminated in strains lacking the Yap1 transcription factor. This suggests that the phosphorylation of Skn7 is regulated by Yap1. Mutations in the receiver domain of Skn7 were identified that affect its oxidative stress function. These mutations were found to compromise the association of Yap1 and Skn7 at oxidative stress response gene promoters. A working model is proposed in which the association of Yap1 with Skn7 in the nucleus is a prerequisite for Skn7 phosphorylation and the activation of oxidative stress response genes.
Skn7 is a response regulator in the Saccharomyces cerevisiae SLN1 two-component osmotic stress signal transduction pathway. In this pathway, the phosphorylation of the Sln1 sensor kinase controls the phosphorylation of the phosphorelay intermediate, Ypd1, and two downstream response regulators, Ssk1, which controls the HOG1 osmotic stress mitogen-activated protein kinase (MAPK) cascade, and Skn7, which directly affects the transcription of SLN1-SKN7-responsive genes such as the mannosyltransferase gene OCH1 (21) (Fig. 1A).
FIG. 1.

Skn7 function, structure, and sequence. (A) The role of the Skn7 transcription factor in two different signal transduction pathways. SLN1-P, YDP1-P, SSK1-P, and SKN7-D427-P are the phosphorylated forms of each protein. Skn7 is phosphorylated on the D427 residue in response to the activation of the SLN1 pathway (left), and Skn7 works with a second transcription factor, Yap1, to activate OXR genes (right). The OXR function of Skn7 is independent of SLN1 and the D427 residue. “ox” refers to unknown modifications to the Yap1 transcription factor in response to oxidative stress. (B) Domain structure of Skn7. HSF, the HSF-like N-terminal DBD of Skn7; CC, coiled-coil domain involved in some protein interactions; Receiver, domain that undergoes aspartyl phosphorylation in the response regulators of two-component-type phosphorelay pathways; Q-rich, C-terminal domain rich in glutamine. The amino acid residues comprising each domain are given below the cartoon. (C) ClustalW-based (38) sequence alignment between the receiver domain of four SKN7 homologs and the prokaryotic response regulator CheY. Ec_, Escherichia coli; Sc_, Saccharomyces cerevisiae; Sp_, Schizosaccharomyces pombe; Ca_, Candida albicans; and Cn_, Cryptococcus neoformans. The alignment was adjusted manually using the alignment editor GeneDoc (26). Columns consisting of identical or highly conserved amino acids are shown in gray. Residues with known importance in the response regulator family are marked with an asterisk. Conserved Ser/Thr residues are shown in white type on a black background. Dashed lines represent gaps in the alignment. Residues comprising a putative tripartite MAPK docking motif are boxed and labeled “Basic,” “LXL” (leucine, any amino acid residue, leucine), and “φ” (hydrophobic).
The N terminus of Skn7 is similar to the conserved DNA binding domain (DBD) of heat shock factor (Hsf1). The Skn7 DBD is followed by coiled-coil, receiver, and glutamine-rich domains (4, 5) (Fig. 1B). Aspartate 427 (D427) in the Skn7 receiver domain is the phospho-accepting residue in the SLN1 osmotic stress pathway. The mutation of D427 to asparagine (D427N) eliminates SLN1-activated Skn7 activity (4, 25).
SKN7 also plays an important role in oxidative stress resistance (16, 27, 33, 40). The role of Skn7 in oxidative stress is not dependent on SLN1. Although D427 in the Skn7 receiver domain is not required for the oxidative stress response (OXR) (24), the receiver domain itself is necessary and may be sufficient for this activity (6). Skn7 cooperates with the Yap1 transcription factor in activating many of the key OXR genes (e.g., TRX2, TRR1, TSA1, GPX2, AHP1, CCP1, and CTT1) (11, 24, 39). Skn7 and Yap1 each are capable of directly binding to oxidative stress-responsive Skn7 and Yap1 response elements in the promoters of OXR genes. However, the presence of a robust Skn7 and Yap1 codependent complex at the promoters of several OXR genes examined using electrophoretic mobility gel shifts (11, 24) also suggests the possibility of direct interaction between the two proteins. This Skn7- and Yap1-dependent complex is necessary for the normal induction of OXR genes; however, a clear demonstration of direct interaction between the Skn7 and Yap1 proteins has not yet been reported (16, 24).
The activation of Yap1 upon oxidative stress involves the C-terminal cysteine-rich domain, which contains a nuclear export signal embedded within three repeated cysteine-containing motifs (8, 9, 19). Yap1 is oxidized by Gpx3, which leads to the formation of a Cys303-Cys598 intramolecular disulfide bond, thus causing a conformational change that masks the Yap1 nuclear export signal and allows the protein to accumulate in the nucleus, where it helps to activate OXR genes (9, 19). Cysteine-based redox sensing also is important in the oxidative stress activation of the Escherichia coli OxyR and SoxR transcription factors (43) and mammalian NF-κB and c-jun (14, 28, 43).
In addition to the aspartyl phosphorylation required in the context of the SLN1-SKN7 osmotic stress pathway (4, 21, 25), Skn7 has been reported to undergo serine/threonine phosphorylation (4), although the function of this type of phosphorylation has not been investigated. In this study, we examine the requirements for the Skn7 response to an oxidant. We find an oxidant-dependent reduction in the mobility of the Skn7 protein that is sensitive to phosphatase. The oxidant-dependent phosphorylation of Skn7 is eliminated in strains lacking Yap1. Receiver domain mutations are identified that affect the oxidative phenotype by reducing the Skn7 association with Yap1 and, in one case, reduce oxidant-dependent phosphorylation. Oxidative stress resistance can be restored in these mutants by the overexpression of YAP1, but the presence of excess Yap1 did not restore phosphorylation. These results indicate that Skn7 phosphorylation is neither sufficient nor absolutely necessary for the OXR. We conclude that Skn7 phosphorylation is not involved in recruiting Yap1; rather, it plays a role in stabilizing the Skn7-Yap1 complex.
MATERIALS AND METHODS
Strains and yeast techniques.
Yeast strains used in this work include the SKN7+ strain JF1565 (MATα his3Δ200 leu2Δ1 ura3-52 trp1Δ63 lys2Δ202 canR cyhR) and its isogenic derivatives, JF1904 (skn7Δ::TRP1), JF2272 (yap1Δ), JF2312 (yap1Δ skn7Δ::TRP1), and JF2052 (skn7Δ::TRP1 sln1-21). One-hybrid analysis was performed in the yeast strain PJ69-4A (MATatrp1-901 leu2-3,112 ura3-52 his3-200 gal4Δ gal80Δ LYS2::GAL1-HIS3 GAL2-ADE2 met2::GAL7-lacZ) (12). JF2413 was obtained by the disruption of YAP1 with the Kanr selectable marker gene in PJ69-4A. Media were prepared as described by Sherman et al. (31a). Yeast cells were grown on standard yeast extract-peptone-dextrose (YPD) or synthetic complete medium lacking the specified amino acid (SC-aa). Yeast transformation was performed with the Frozen-EZ yeast transformation II kit (Zymo Research).
Plasmids.
The plasmids used in this study are listed in Table 1. The SKN7 open reading frame (ORF) plus 1,293 bp upstream and 304 bp downstream was introduced into the pBluescript II SK(+/−) cloning vector (Stratagene) as a 4,042-bp NotI-XhoI fragment. A 2× MYC tag sequence with HindIII and SpeI sites was fused to the 3′ end (nucleotide SKN7+1866) of the SKN7 ORF. Full-length wild-type and mutant SKN7 derivatives were introduced into plasmid pRS315 (CEN; LEU2) or pRS425 (2μm; LEU2) (32). SKN7-green fluorescent protein fusion (SKN7-GFP) plasmids were constructed by cloning the GFP sequence (GFP mut3; S65G S72A; GenBank accession number U73901) into pRS315-SKN7 plasmids at the 3′end (SKN7+1866) of the SKN7 ORF at HindIII and SpeI sites. One-hybrid plasmids (pXH1956 to pXH1970) were constructed by the PCR amplification of SKN7 positions +1051 to +1866, including the 2× MYC tag from the pXH1853 to pXH1856 and pXH1858 templates, and the ligation of the products into the BamHI and SalI sites downstream of GAL4-DBD in the plasmid pGBD-C2 (12).
TABLE 1.
Plasmids used in this study
| Plasmid | Descriptiona | Vector | Reference |
|---|---|---|---|
| PXH1641 | CCP1 (−309/+14)-lacZ | pSEYC102 | 11 |
| PXH1801 | SKN7 (WT) | pRS315 | 32 |
| PXH1833 | skn7-C160A | pRS315 | |
| PXH1834 | skn7-C300A | pRS315 | |
| PXH1835 | skn7-C392A | pRS315 | |
| PXH1836 | skn7-C401A | pRS315 | |
| PXH1837 | skn7-C499A | pRS315 | |
| PXH1853 | SKN7 (WT) (myc2x)b | pRS315 | |
| PXH1854 | skn7-T437A (myc2x) | pRS315 | |
| PXH1855 | skn7-T455A (myc2x) | pRS315 | |
| PXH1856 | skn7-T449A (myc2x) | pRS315 | |
| PXH1857 | skn7-T437S (myc2x) | pRS315 | |
| PXH1858 | skn7-D427N (myc2x) | pRS315 | |
| PXH1946 | skn7-T437E (myc2x) | pRS315 | |
| PXH1859 | SKN7 (WT) (GFP) | pRS315 | |
| PXH1860 | skn7-T437A (GFP) | pRS315 | |
| PXH1861 | skn7-T455A (GFP) | pRS315 | |
| PXH1862 | skn7-T449A (GFP) | pRS315 | |
| PXH1863 | skn7-T437S (GFP) | pRS315 | |
| PXH1864 | skn7-D427N (GFP) | pRS315 | |
| PXH1938 | SKN7 (WT) (myc2x) (high copy) | pRS425 | |
| PXH1939 | skn7-T449A (myc2x) (high copy) | pRS425 | |
| PXH1941 | skn7-dock2 (myc2x) (high copy) | pRS425 | |
| PXH1956 | GAL4 DBD-SKN7 (+1051/+1866) (WT) (myc2x) | pBD-C(2) | 12 |
| PXH1966 | GAL4 DBD-skn7 (+1051/+1866)-T437A (myc2x) | pBD-C(2) | |
| PXH1967 | GAL4 DBD-skn7 (+1051/+1866)-T455A (myc2x) | pBD-C(2) | |
| PXH1968 | GAL4 DBD-skn7 (+1051/+1866)-T449A (myc2x) | pBD-C(2) | |
| PXH1970 | GAL4 DBD-skn7 (+1051/+1866)-D427N (myc2x) | pBD-C(2) | |
| pSEY18-YAP1 | YAP1 (high copy) | pSEY18 |
WT, wild type.
myc2x, two copies of the myc epitope tag.
Site-directed mutagenesis of DNA sequences.
Desired point mutations were made using the QuikChange site-directed mutagenesis kit (Stratagene). DNA fragments containing the region to be mutagenized first were cloned into the plasmid pBluescript II SK(+/−) to create the template. Pairs of mutagenic primers complementary to each strand were extended using Pfu Turbo DNA polymerase (Stratagene). The methylated, nonmutated parental DNA template was digested with DpnI, and the nicked circular double-stranded DNA product was transformed into Escherichia coli DH5α supercompetent cells (Zymo Research). Mutated plasmids were recovered from bacteria, confirmed by sequence analysis, and recloned into the desired yeast expression vector.
β-Galactosidase assays.
Overnight cultures for β-galactosidase assays were subcultured in fresh SC-Trp medium, incubated with aeration at 30°C, and harvested in log phase (∼ 2 × 107 cells/ml) by centrifugation. Where indicated, cultures were incubated for 30 min in the presence of 0.5 mM tert-butyl hydroperoxide (t-BOOH; Sigma) prior to harvesting. Crude extracts were prepared by glass bead lysis and cleared by centrifugation. Extract concentrations were determined using the Bio-Rad protein assay. β-Galactosidase activity was calculated in Miller units normalized to the protein concentration (23). Final activities are expressed as the averages from three independent transformants.
Stress assays.
Log-phase yeast cultures were diluted to 2 × 106 cells/ml. A series of 10-fold dilutions were spotted on YPD plates or YPD plates containing the indicated concentrations of oxidants. Plates were incubated at 30°C for 2 to 3 days.
Northern analysis.
Total RNA was isolated from log-phase cultures using the hot acidic phenol method (2) and separated on formaldehyde gels (1.2% agarose). Following transfer to nylon membranes (Whatman), RNA blots were hybridized with appropriate [α-32P]dATP-labeled probes in PerfectHyb solution (Sigma) at 68°C overnight and washed twice in 2× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate), 0.1% sodium dodecyl sulfate (SDS) and twice in 0.5× SSC, 0.1% SDS. Quantification was performed by phosphorimager analysis (Quantity One; Bio-Rad).
Western analysis.
Glass bead extracts were prepared by vortexing log-phase culture pellets in protein extraction buffer (20 mM Tris-HCl [pH 8.0], 150 mM NaCl, 1 mM EDTA, 10 mM MgCl2, 5% glycerol, 1× protein inhibitor cocktail [Roche], and 1 mM phenylmethylsulfonyl fluoride [PMSF]) in the presence of glass beads. Lysates were cleared by centrifugation for 15 min at 4°C. For phosphorylation experiments, trichloroacetic acid (TCA) extracts were prepared from cultures harvested at log phase and treated, where indicated, with t-BOOH for 20 min (oxidative stress), 0.4 M NaCl for 5 min (osmotic stress), or 1 U/ml zymolyase for 30 min (wall stress) or were shifted to 39°C for 20 min (heat stress). Cell pellets were frozen and then disrupted by being vortexed in water in the presence of glass beads. TCA was added to 20% and alternately (10 times) vortexed for 30 s and incubated on ice for 1 min. Following the addition of 1 ml ice-cold 5% TCA, the contents were centrifuged on high for 15 min in the cold. The protein pellet was washed twice with cold acetone and allowed to dry at room temperature for 5 min. Protein pellets were resuspended in protein buffer (20 mM HEPES, pH 8.0, 5 mM EDTA, 20% glycerol, 7 mM β-mercaptoethanol, and protease inhibitors [1 mM PMSF and 1× protein inhibitor cocktail; Roche]). Lambda phosphatase (New England Biolabs) was added to 100 U for 30 min at 30°C. Phosphatase inhibitors included 5 mM EDTA, 1 mM sodium orthovanadate, and 10 mM sodium pyrophosphate (final concentrations). Samples were subjected to SDS-polyacrylamide gel electrophoresis (PAGE) using 10% (29:1) gels. Electrophoresis was performed for various times at 200 V. Separated proteins were transferred onto Protran nitrocellulose membranes (Whatman). The expression of the Skn7-Myc fusion protein was examined by immunoblot analysis with anti-Myc antibody (c-Myc <A-14>; 1:1,000; Santa Cruz Biotechnology) followed by goat anti-mouse secondary antibody (1:3,000; Santa Cruz Biotechnology). Immune complexes were visualized by chemiluminescence (Pierce Supersignal West Pico chemiluminescent substrate).
Electrophoretic mobility shift analysis.
The preparation of yeast protein extracts, protein-DNA binding reaction mixtures, and the electrophoretic fractionation of complexes were performed as described previously (41), with minor modifications. DNA probes were generated by PCR amplification or directly synthesized and end labeled with T4 polynucleotide kinase and [γ-32P]dATP. Labeled DNA probes were precipitated in 2.0 M NH4− acetate (NH4−OAc) and then in 0.3 M NaOAc and three volumes of ethanol and resuspended in Tris-EDTA buffer. Binding reaction mixtures (20 μl) included 5 to 15 μg yeast protein extract, 0.5 μg poly(dI-dC), and 0.5 to 1 ng (5 to 20 counts per s) DNA in EMSA buffer (25 mM Tris-HCl, pH 7.5, 50 mM NaCl, 2 mM EDTA, 7 mM MgCl2, 10% glycerol). Reaction mixtures were incubated at 37°C for 15 min. Following preelectrophoresis at 100 V for 1 h, the reaction mixtures were loaded on 4% nondenaturing polyacrylamide (29:1 acrylamide:bisacrylamide) gels in 0.5× TBE (89 mM Tris, 89 mM borate, 2.4 mM EDTA, pH 8.0), and complexes were separated at 200 V for 2 h. Vacuum-dried gels were exposed and subjected to phosphorimager analysis (Bio-Rad).
RESULTS
The Skn7 receiver domain is phosphorylated in an oxidant-dependent manner.
To determine if phosphorylation is important for the SKN7-mediated OXR, the mobility of Skn7 was evaluated after SDS-PAGE. Skn7 from wild-type cells reproducibly exhibited a slower mobility in extracts prepared from oxidant-treated cultures than in extracts prepared from untreated cultures (Fig. 2A). The alteration in mobility was eliminated upon treatment with phosphatase prior to electrophoresis and was restored when phosphatase inhibitors were added to the reaction mixture (Fig. 2A). These results indicate that Skn7 is modified by phosphorylation upon oxidative stress. The oxidant-dependent shift in mobility is not expected to be due to aspartyl phosphorylation, since acyl-phosphorylated amino acids such as aspartate are labile in acid as well as base (10) conditions and will not survive the TCA extraction procedure. We nonetheless confirmed that the oxidant-dependent shift did not depend on D427, the phospho-accepting residue in the SLN1-SKN7 osmotic stress pathway; the oxidant-dependent alteration in mobility was unaffected in a D427N mutant (Fig. 2C). Furthermore, no change in the mobility of Skn7 was observed in extracts subjected to osmotic, heat, or wall stress (Fig. 2D). These results indicate that the change in mobility of Skn7 is a specific response to oxidant exposure.
FIG. 2.

Slow-migrating Skn7 variant seen in oxidative stress-treated cultures is eliminated by phosphatase treatment and in a yap1Δ strain. TCA extracts prepared from JF1904 (skn7Δ) or JF2312 (skn7Δ yap1Δ) cultures carrying SKN7 or skn7-D427N expression plasmids and left unexposed (−) or exposed (+) to an oxidant (ox) or other (heat shock, wall, and osmotic) stress treatments were subjected to SDS-10% PAGE for 2.5 h at 200 V. Lambda phosphatase (Ph) was added where indicated to 100 U for 30 min at 30°C according to the manufacturer's recommendations. Representative experiments are shown. (A) Extracts were prepared from oxidant-treated and untreated cultures of the skn7Δ strain JF1904, carrying the SKN7 plasmid pXH1853. Phosphatase inhibitors (In) were added to all lanes except the lane labeled Ph. A dashed white line is added to the figure based on the migration of two untreated wild-type samples run as controls on the outside edges of the gel. (B) Extracts were prepared from oxidant-treated and untreated cultures of the skn7Δ strain JF1904 (left two lanes) and the skn7Δ yap1Δ strain JF2312 (right two lanes), each carrying the SKN7 plasmid pXH1853. (C) Extracts were prepared from oxidant-treated and untreated cultures of the skn7Δ strain JF1904, carrying the SKN7 plasmid pXH1853 (left two lanes) or the skn7-D427N plasmid pXH1858 (right two lanes). (D) Extracts were prepared from treated and untreated cultures of the skn7Δ strain JF1904, carrying the SKN7 plasmid pXH1853. The two lanes on the left are without or with oxidant treatment, while the two lanes on the right are without or with the indicated alternative stress treatments, including osmotic (0.4 M NaCl for 5 min), heat shock (39°C for 20 min), and wall stress (1 U/ml zymolyase for 30 min).
To determine whether the oxidant-specific transcription factor Yap1 is required for Skn7 phosphorylation, the same assay was conducted in a strain lacking Yap1. The oxidant-dependent phosphorylation of Skn7 (Fig. 2B, lane 2) was found to be at least partially dependent on Yap1. The levels of the slow-mobility Skn7 variant were detectably reduced in extracts prepared from oxidant-treated yap1Δ cells compared to levels of YAP1 cells (Fig. 2B, compare lane 4 to 2).
Identification of Skn7 receiver domain residues involved in the OXR.
Since the phosphorylation of Skn7 was both oxidant dependent and dependent on the oxidative stress transcription factor Yap1, we investigated whether the phosphorylation of Skn7 is important in the regulation of OXR genes. The alignment of the receiver domain of Saccharomyces cerevisiae Skn7 (ScSkn7) and its orthologs in Schizosaccharomyces pombe (SpPrr1), Candida albicans (CaSkn7), and Cryptococcus neoformans (CnSkn7), which were selected based on their involvement in OXRs, with the Escherichia coli response regulator CheY revealed two classes of conserved serine and threonine residues, those conserved in all response regulators and those conserved only among SKN7 orthologs (Fig. 1C). Residues conserved in both SKN7 orthologs and in CheY (Fig. 1B), including Asp12, Asp13, Asp56, Thr87, and Lys109 of CheY (Glu383, Asp384, Asp427, Thr455, and Lys477 in Skn7), are important for the phosphorylation of the receiver domain aspartate or subsequent changes in transcription factor activity due to aspartyl phosphorylation (1, 7, 20, 22, 29, 30, 35). We speculated that serine and threonine residues conserved in SKN7 orthologs but not in bacterial CheY, which does not have an OXR function, may be important in the oxidative stress function of the receiver domain. To test this, we selected six threonine residues that are conserved among at least three SKN7 orthologs (residues 437, 439, 449, 455, 465, and 480) (Fig. 1C) for further study. Each was individually mutated to alanine and introduced into an skn7Δ mutant to assess the complementation of the oxidative stress sensitivity phenotype. Like the D427N osmotic response mutation used here as a negative control, the T439A, T455A, T465A, and T480A mutants fully complemented the oxidative stress-sensitive phenotype. T455A is shown as an example. The skn7-T437A and skn7-T449A mutants exhibited modest and severe sensitivity to t-BOOH, respectively (Fig. 3A).
FIG. 3.

Oxidant sensitivity of skn7 mutants. Yeast strain JF1904 (skn7Δ) was transformed with low-copy (A) or high-copy (B) expression plasmids carrying wild-type or mutated SKN7 genes fused to sequences encoding the Myc epitope tag. Tenfold serial dilutions starting at 106 cells/ml were spotted, and the plates were incubated at 30°C for 2 to 3 days. (C and D) α-myc Western analysis showing normal protein levels in the skn7 mutant strains. Ponceau S staining (C) or anti-Hog1 Western analysis (D) was used to confirm the equal loading of samples. Low-copy plasmids were the following: SKN7+, pXH1853; skn7Δ, pRS315; skn7-T437A, pXH1854; skn7-T455A, pXH1855; skn7-T449A, pXH1856; and skn7-D427N, pXH1858. High-copy plasmids were the following: SKN7+, pXH1938; skn7Δ, pRS425; and skn7-T449A, pXH1939.
Western analysis of Myc-tagged fusions confirmed that Skn7-T437A protein levels were similar to that of the wild-type Skn7 protein (Fig. 3C), whereas Skn7-T449A protein levels appeared to be slightly reduced. To test whether the skn7-T449A phenotype was due to reduced protein levels, experiments were conducted with high-copy expression plasmids. We found that strains carrying a high-copy skn7-T449A expression plasmid had at least normal levels of the mutant Skn7 protein but nonetheless remained very sensitive to oxidant (Fig. 3B and D). These results indicate that the oxidative stress-sensitive phenotype of skn7-T449A is attributable to the loss of an Skn7 activity and not to a reduction in the abundance of the Skn7 protein. GFP-tagged derivatives of Skn7-T437A and Skn7-T449A proteins showed correct nuclear localization (data not shown), thus ruling out mislocalization as an explanation for the oxidative stress phenotype.
Thr437 and Thr449 affect Skn7 receiver domain activation by oxidants.
To determine the effects of T and A mutations on Skn7 activation, the C terminus of Skn7 (amino acid [aa] 351 to 622), including the Skn7 receiver domain, was fused to the Gal4 DBD to create a hybrid that could bind to DNA at Gal4 upstream activating sequence (UASG) sites. The activity of the Gal4 DBD-Skn7 hybrid protein was induced 1.6-fold upon treatment with t-BOOH, showing, as previously reported (13, 16), that the Skn7 receiver domain is capable of mediating an OXR (Fig. 4). As expected, there was no effect of the D427N osmotic response mutation on activation by oxidative stress. The oxidant-dependent induction of the UASG-lacZ reporter was eliminated in the absence of YAP1 (Fig. 4), indicating that the oxidative stress activation of the Skn7 receiver domain is dependent on Yap1. Since there are no Yap1 binding sites in the reporter construct, the requirement for Yap1 is consistent with the possibility of Yap1 function via a protein-protein interaction with Skn7.
FIG. 4.
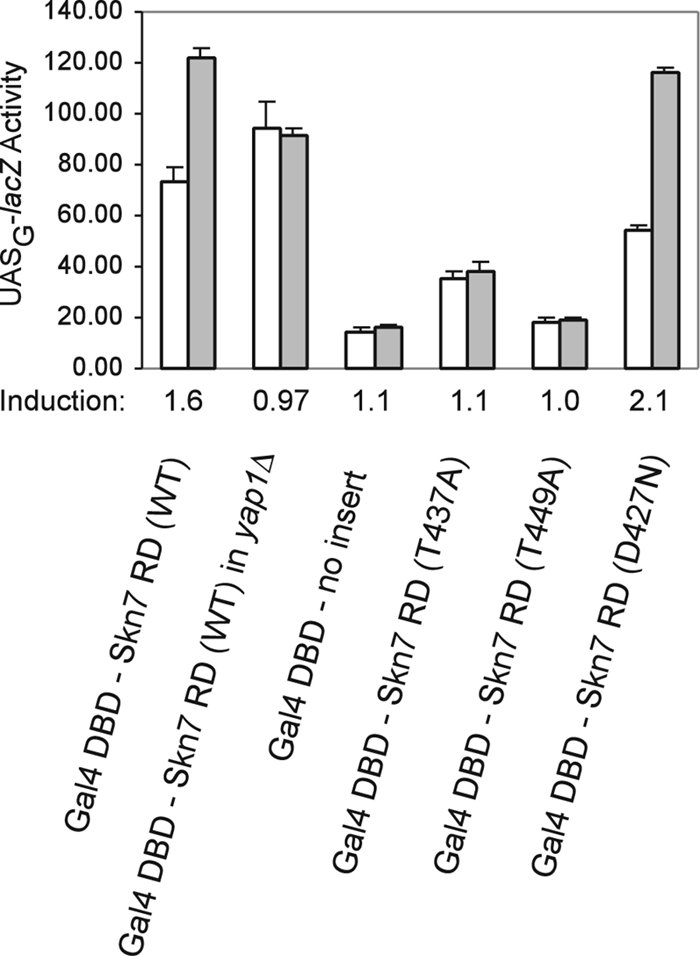
skn7 receiver domain (RD) mutations affect Skn7 transcriptional activation in response to oxidant. Fusions of the Skn7 C-terminal sequence (aa 351 to 622; wild-type [WT] and mutant derivatives as indicated) to the DBD of Gal4 were introduced into wild-type yeast strain PJ69-4A (12), which carries the UASG-lacZ reporter. The Gal4 DBD plasmid (pGBD-C2) also was introduced as a negative control. The yap1Δ strain was generated from PJ69-4A (yap1Δ::KAN). Log-phase cultures were untreated or were treated with 0.5 mM t-BOOH for 30 min (white bars, untreated; filled bars, treated). β- Galactosidase activities are given in Miller units (23) normalized to the protein concentration and are the average activities of three or four independent transformants. Error bars are the standard deviations from the means. Below the plot is the induction (n-fold) due to oxidant treatment.
The T449A and T437A threonine mutations were introduced into the Gal4 DBD-Skn7 hybrid, and the activities of the mutant hybrid fusion proteins were determined. The T449A and T437A fusions decreased basal activity (Fig. 4) relative to that of the wild type. However, the key result is that each mutant also eliminated the transcriptional activation of the hybrid protein by oxidant (Fig. 4). This was expected from the previously observed oxidative stress phenotypes of these mutants (Fig. 3). Taken together, these results suggest that the T449 and T437 residues play a role in the oxidant-dependent modulation of Skn7 receiver domain activity.
Thr437 is specific for the OXR.
To determine if SKN7 mutants with oxidant-sensitive phenotypes are defective specifically in oxidative stress, skn7 T/A alleles were tested for the activation of OCH1 and NCA3, genes that are induced by the SLN1-SKN7 osmotic stress signaling pathway (21 and J. S. Fassler, K. E. Mulford, and X.-J. He, unpublished data). Controls showing the expected increase in the expression of NCA3 and OCH1 in an activated sln1 mutant (sln1*) and the reduction of sln1* activation in the skn7Δ and skn7-D427N mutants are shown in Fig. 5. The skn7-T437A mutant exhibited almost normal SLN1-SKN7 signaling, 1.8-fold increased expression of NCA3 by the mutant compared to 1.9-fold increased expression by normal SLN1-SKN7 and 1.6-fold increased expression of OCH1 by the mutant compared to 1.8-fold increased expression by normal SLN1-SKN7, indicating that the mutation causes a specific defect in the OXR. In contrast, the skn7-T449A mutation eliminated the sln1* activation of OCH1 and NCA3 (Fig. 5), indicating that the T449 residue plays a role in osmotic stress activation and oxidative stress activation.
FIG. 5.
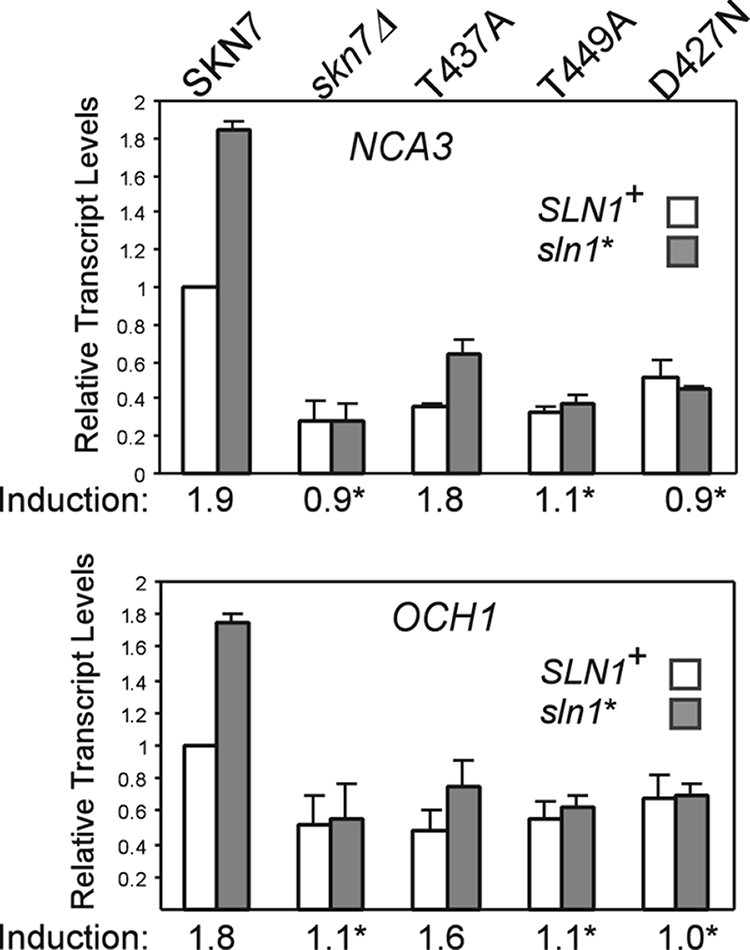
Effect of skn7 mutations on the SLN1-SKN7 target genes. Total RNA (15 μg) was isolated from the log-phase cultures of the skn7Δ yeast strains (SLN1+ [JF1904] and sln1* [JF2052]) carrying the indicated SKN7 expression plasmids or empty vector (skn7Δ) and subjected to formaldehyde agarose gel electrophoresis. The expression of the OCH1 and NCA3 genes was determined by hybridization to the appropriate probes and to a PGK1 probe for normalization. Transcript levels were determined using Quantity One software (Bio-Rad). The expression of the untreated SKN7+ culture was set to 1.0. Induction values (averages from at least three experiments) are plotted and provided below the x axis for each pair of bars. Asterisks indicate induction values that differ significantly (P < 0.05) from that of SKN7+. Plasmids carrying SKN7 alleles were the following: SKN7+, pXH1853; skn7-T437A, pXH1854; skn7-T449A, pXH1856; and skn7-D427N, pXH1858.
skn7-T449A is defective in binding to DNA.
The effects of the skn7-T437A and skn7-T449A mutations on DNA binding and on the formation of complexes involving both Skn7 and Yap1 were evaluated using electrophoretic mobility shift assays. Skn7 binding to DNA was monitored using a fragment of the OCH1 promoter (−355/−154) that includes Skn7 binding sites but no Yap1 binding sites (21). The skn7-T437A mutation did not appreciably affect binding, while the skn7-T449A mutation eliminated binding to the OCH1 promoter fragment (Fig. 6). This explains why the defect in the skn7-T449A mutant is not limited to the oxidative stress pathway; the skn7-T449A mutation affects all promoters by disrupting the ability of the Skn7 protein to bind to DNA. Skn7 binding also was evaluated in a yap1Δ strain. The Skn7 complexes observed in extracts lacking the Yap1 protein mirrored what was seen in YAP1+ extracts, confirming that binding to the OCH1 promoter is independent of Yap1.
FIG. 6.
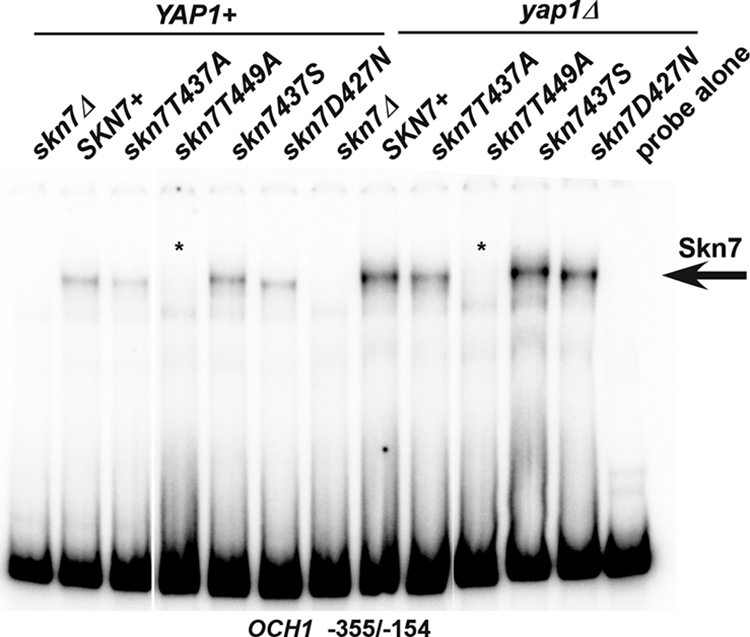
Effect of receiver domain mutations on Skn7 DNA binding. Shown are electrophoretic mobility shift assays in which protein extracts were prepared from YAP1 skn7Δ (JF1904) or yap1Δ skn7Δ (JF2312) yeast strains carrying wild-type or mutant derivatives of SKN7 and incubated with a labeled DNA probe corresponding to positions −355 to −154 of the OCH1 promoter. This fragment of the OCH1 promoter contains Skn7 but no Yap1 binding sites. Complexes were separated by nondenaturing polyacrylamide gel electrophoresis at 200 V for 2 h. A single SKN7-dependent complex is labeled with an arrow. Plasmids carrying SKN7 alleles were the following: SKN7+, pXH1853; skn7-T437A, pXH1854; skn7-T449A, pXH1856; skn7-T437S, pXH1857; and skn7-D427N, pXH1858.
skn7-T437A affects formation of a Skn7-Yap1 complex at oxidative stress gene promoters.
A protein complex including both Skn7 and Yap1 is necessary for the induction of several OXR genes, including TRX2, CCP1, and AHP1 (11, 24). In view of the importance and specificity of T437 in the OXR, we examined its role in the formation of the SKN7- and YAP1-dependent protein complexes using TRX2 (−210/−142) and CCP1 (−322/−245) promoter sequences, which include both Skn7 and Yap1 binding sites. Extracts prepared from SKN7+ strains form two major complexes with either the TRX2 or the CCP1 promoter fragment (11) (Fig. 7). The faster of the two major bands consists of DNA and either Skn7 or Yap1 (11); a band remains visible at that position in both skn7Δ (lane 1) and the yap1Δ (lane 8) extracts. A second major band, migrating more slowly, is evident only in extracts prepared from strains having both wild-type Skn7 and Yap1 (11) and thus is dependent on both proteins. The absence of this complex in the skn7-T437A mutant extracts reflects an inability to form a Skn7-Yap1 complex on the DNA. In contrast, the control skn7-D427N mutation did not affect DNA binding or complex formation with Yap1. As expected, the Skn7-T449A protein could not bind to these promoters, either in the presence or absence of Yap1 (Fig. 7).
FIG. 7.
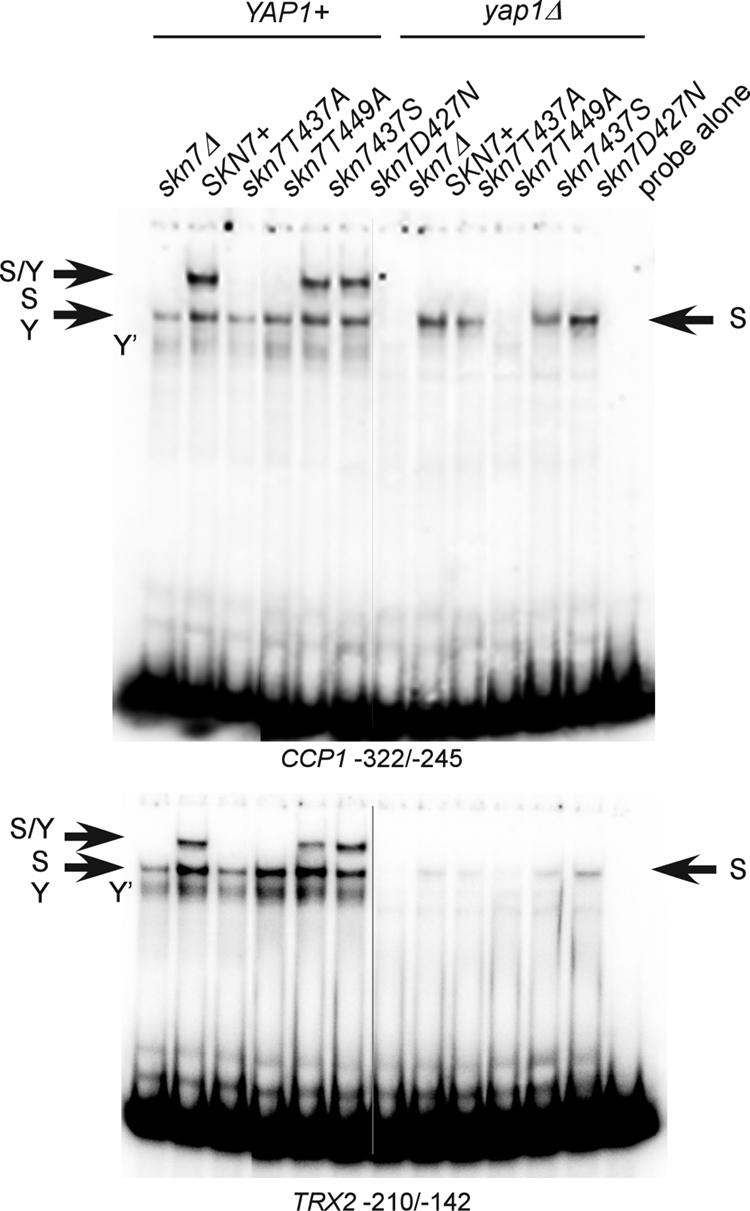
Effect of SKN7 receiver domain mutations on Skn7-Yap1 complex formation. Shown are electrophoretic mobility shift assays in which protein extracts were prepared from a YAP1 skn7Δ (JF1904) or yap1Δ skn7Δ (JF2312) yeast strain carrying wild-type or mutant derivatives of SKN7 and incubated with labeled DNA probes as indicated. Complexes were separated by nondenaturing polyacrylamide gel electrophoresis at 200 V. The CCP1 and TRX2 promoter fragments (−322 to −245 and −210 to −142, respectively) used as probes each contain both Skn7 and Yap1 binding sites. S/Y, complex dependent on both Skn7 and Yap1; S, complex dependent on Skn7; Y, complex dependent on Yap1; Y′, minor complex dependent on Yap1. The S and Y complexes from YAP1+ extracts tend to comigrate. Plasmids carrying SKN7 alleles were the following: SKN7+, pXH1853; skn7-T437A, pXH1854; skn7-T455A, pXH1855; skn7-T449A, pXH1856; skn7-T437S, pXH1857; and skn7-D427N, pXH1858.
Mutations in a nearby MAPK dock-like sequence are oxidant sensitive and affect Skn7-Yap1 complex formation.
The T437 residue is located just downstream of the aspartate (D427) that is phosphorylated during SLN1-SKN7 osmotic stress signaling but that is not required for the OXR. We noted that the sequences in the vicinity of D427 (KYRYDLVLMD427IVM) (underlined residues correspond to the three motifs boxed in Fig. 1C) bear a resemblance to a tripartite D domain, which serves as a dock for the association of MAPKs. The dock, found in some transcription factors known to interact with MAPKs (31), consists of up to three motifs (a basic patch consisting of between one and three basic residues, a φXφ motif, and/or a downstream hydrophobic motif). To test the role of the putative dock in oxidative stress, a 2-bp mutation was introduced in the hydrophobic motif. The skn7-dock2 mutant (I428A, V429A) had normal protein levels but exhibited an oxidant-sensitive phenotype (Fig. 8A and B). In addition, the skn7-dock2 mutation eliminated the formation of the Skn7-Yap1 complex but did not prevent Skn7 association with DNA (Fig. 8C).
FIG. 8.
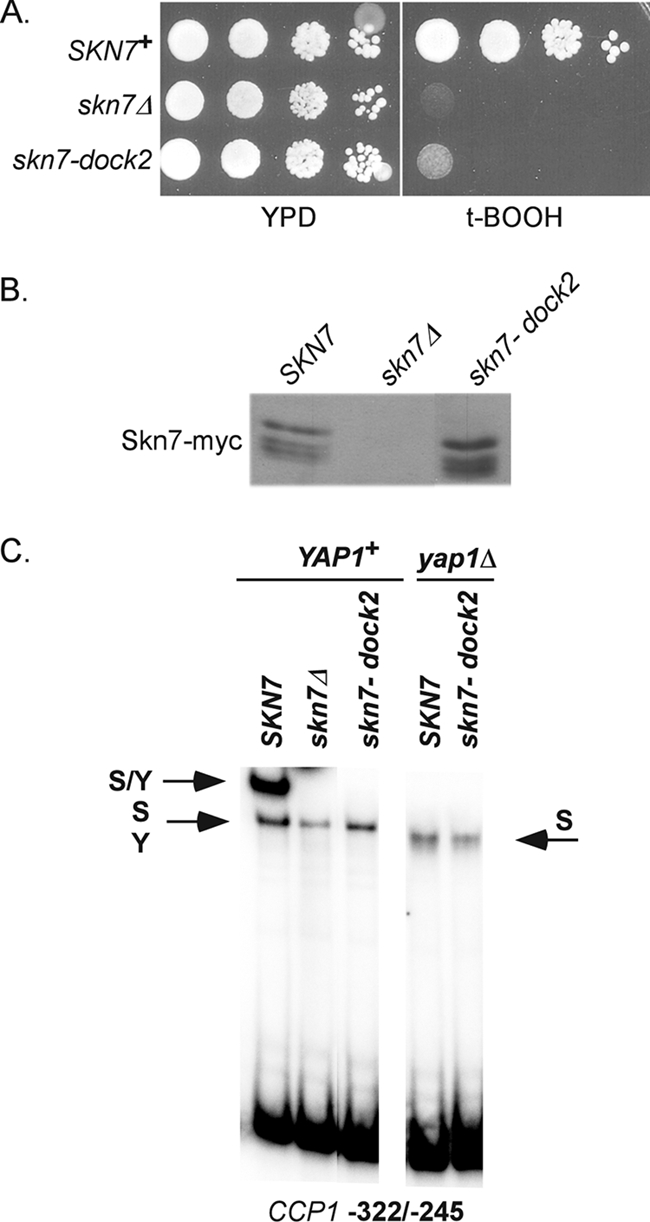
Dock mutant is oxidative stress sensitive and defective in Skn7-Yap1 complex formation. (A) Tenfold serial dilutions starting at 106 cells/ml were spotted on media with and without oxidant, and the plates incubated at 30°C for 2 to 3 days. The skn7Δ strain, JF1904, was transformed with the following high-copy SKN7 expression plasmids: SKN7+, pXH1938; skn7Δ, pRS425; and skn7-dock2, pXH1941. (B) α-myc Western analysis showing normal protein levels in the skn7-dock2 mutant. (C) Gel shift analysis of the skn7-dock2 mutant with CCP1 promoter sequences containing both Skn7 and Yap1 binding sites. S/Y, complex dependent on both Skn7 and Yap1; S, complex dependent on Skn7; Y, complex dependent on Yap1. The S and Y complexes from YAP1+ extracts comigrate.
Oxidative stress-dependent phosphorylation of Skn7 in skn7 mutants.
The oxidative stress-sensitive mutations skn7-T437A and skn7-dock2 each reduced the formation of the Skn7-Yap1 complex in gel shift experiments. To determine the relationship between Yap1-Skn7 complex formation and Skn7 phosphorylation, the migration of the Skn7-T437A and the Skn7-dock2 mutant proteins isolated from untreated and oxidant-treated cells was examined. Skn7 isolated from the skn7-dock2 mutant did not exhibit altered migration following oxidant exposure (Fig. 9B), indicating the importance of the I428 and V429 residues in the oxidative dependent phosphorylation of Skn7. In contrast, the Skn7-T437A protein migrated more slowly after oxidant treatment, indicating that the T437A residue is not absolutely required for the phosphorylation of the Skn7 protein.
FIG. 9.
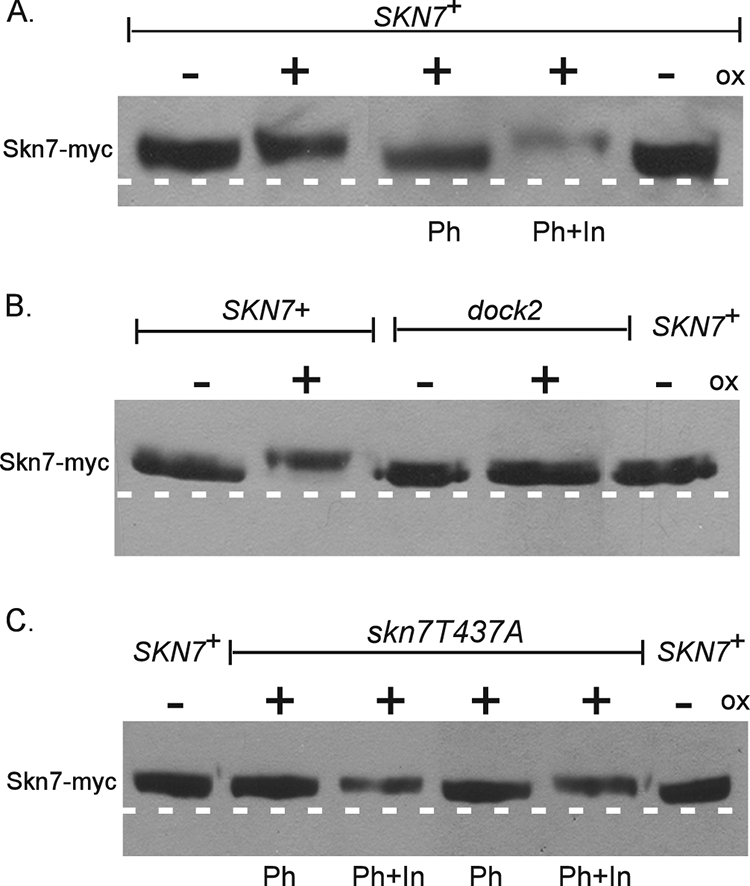
Effects of skn7-dock2 and skn7-T437A mutations on the oxidant (ox)-dependent phosphorylation of Skn7. Shown is the oxidant-dependent phosphorylation of (A) wild-type SKN7 (as seen in Fig. 2), (B) skn7-dock2, and (C) skn7-T437A. TCA extracts prepared from JF1904 (skn7Δ) cultures carrying SKN7 expression plasmids (SKN7+, pXH1853; skn7-dock2, pXH1941; and skn7-T437A, pXH1854) and left untreated (−) or treated (+) with oxidant (2 mM H2O2 for 20 min) were subjected to SDS-10% PAGE for 2.5 h at 200 V. Lambda phosphatase (Ph) was added where indicated to 40 U (C, lanes 2 and 3) or 100 U (A and C, lanes 4 and 5) for 30 min at 30°C according to the manufacturer's recommendations. (A and C) Phosphatase inhibitors (In) were added to all lanes except for those labeled Ph. A dashed white line is added to the figure based on the migration of untreated wild-type control samples on the outside edges of each gel.
Characterization of skn7-T437 substitution mutants.
The role of the T437 residue in oxidative stress was further examined by evaluating the functionality of T437 substitutions. Aspartic acid and glutamic acid phosphomimetic substitutions were constructed to further assess the potential for T437 phosphorylation. Both skn7-T437E and skn7-T437D (data not shown) mutants were more sensitive to t-BOOH than the moderately sensitive skn7-T437A (Fig. 10A), although the protein levels were equivalent (Fig. 10B). A skn7-T437S mutant, in contrast, exhibited wild-type resistance to t-BOOH (Fig. 10A). Northern analysis showed that the induction of the oxidative response gene CCP1 was eliminated in skn7-T437E, while induction in skn7-T437S was similar to that of the wild type (Fig. 10C). The formation of an Skn7-Yap1 complex on OXR gene promoters also was normal in the skn7-T437S mutant (Fig. 7).
FIG. 10.
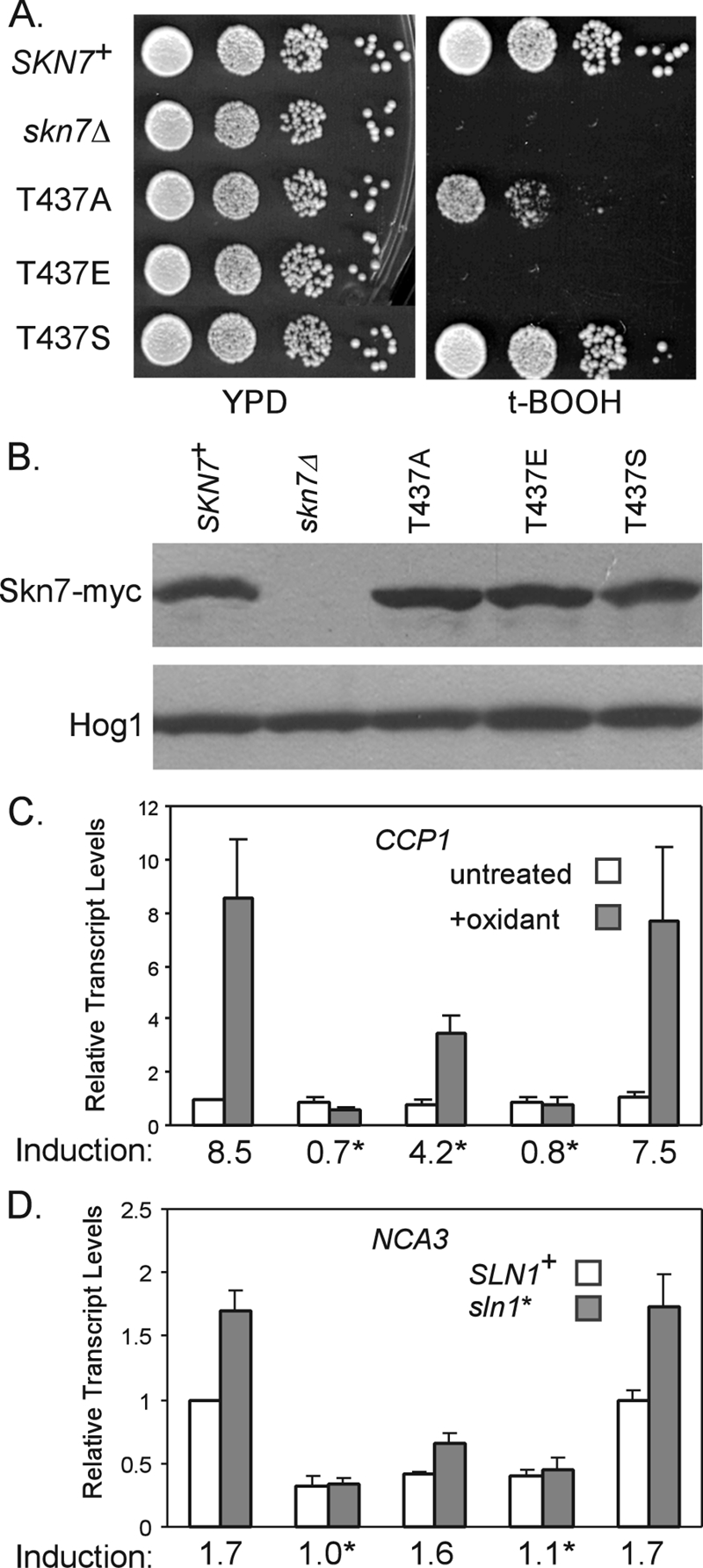
Characterization of the skn7-T437 substitution mutants. CEN expression plasmids carrying Myc-tagged wild-type or T437 substitution derivatives of SKN7 were introduced into the skn7Δ strains (wild type, JF1904; sln1*, JF2052). (A) Tenfold serial dilutions of log-phase cultures starting at 106/ml were spotted onto the indicated plates and were incubated at 30°C for 2 to 3 days. (B) α-myc Western analysis was used to evaluate Skn7-myc expression. Equal loading was confirmed using anti-Hog1 antibody. (C) Effect of skn7-T437 substitutions on the expression of the OXR gene CCP1 and (D) the SLN1-SKN7 target gene NCA3. Transcript levels were normalized to values for PGK1. Induction values (averages from at least three experiments) are plotted and provided below the x axis for each pair of bars. Asterisks indicate values that differ significantly (P < 0.05) from those for SKN7+. Plasmids carrying SKN7 alleles included the following: SKN7+, pXH1938; skn7-T437A, pXH1854; skn7-T437E, pXH1946; and skn7-T437S, pXH1857.
NCA3 was used to evaluate the effect of the skn7-T437 substitution mutants on the SLN1-SKN7 osmotic response pathway activity. The sln1* activation of NCA3 expression seen in SKN7+ strains was retained in the skn7-T437S mutant (Fig. 10D) but was substantially attenuated in the skn7-T437E mutant. In summary, a serine at T437 sustains both the osmotic and oxidative stress pathway functions of Skn7; an alanine at T437 specifically reduces the oxidative stress function of Skn7 (Fig. 3, 4), and an aspartic acid or glutamic acid at T437 seriously impairs both pathways. The functionality of skn7-T437S is consistent with the possibility of the phosphorylation of a hydroxyl group at this residue. However, the complete loss of SKN7 function in the phosphomimetic substitution mutants indicates that the constitutive presence of a negative charge at this position is detrimental to the overall function of the Skn7 protein.
Yap1 overexpression suppresses the oxidant sensitivity of mutations in the receiver domain.
Gel shift analysis showed defects in Skn7 complex formation with Yap1 in the skn7-T437A and skn7-dock2 mutants. To further investigate the potential for the direct association of the Skn7 and Yap1 proteins, the effect of YAP1 overexpression on the oxidant-sensitive phenotypes of these mutants was tested. The growth of both mutants on oxidant plates was improved by YAP1 overexpression. In contrast, YAP1 overexpression did not improve the oxidant-sensitive phenotypes of the skn7-T449A or skn7Δ mutant (Fig. 11A). These results are consistent with the hypothesis that the T437 residue and the dock-like region are important for the Skn7-Yap1 interaction at oxidative stress gene promoters. To determine if phosphorylation also is restored by YAP1 overexpression, the migration of Skn7 was examined in extracts prepared from an untreated or an oxidant-treated skn7-dock2 mutant strain containing a high-copy YAP1 expression plasmid. The migration of the Skn7-dock2 protein was not affected by the addition of oxidant, even in the presence of levels of YAP1 sufficient to restore the strain to oxidative stress resistance (Fig. 11B). These results suggest that the recruitment of Yap1 is important for a normal OXR and that the phosphorylation of Skn7 is dispensable when Yap1 protein levels are elevated.
FIG. 11.
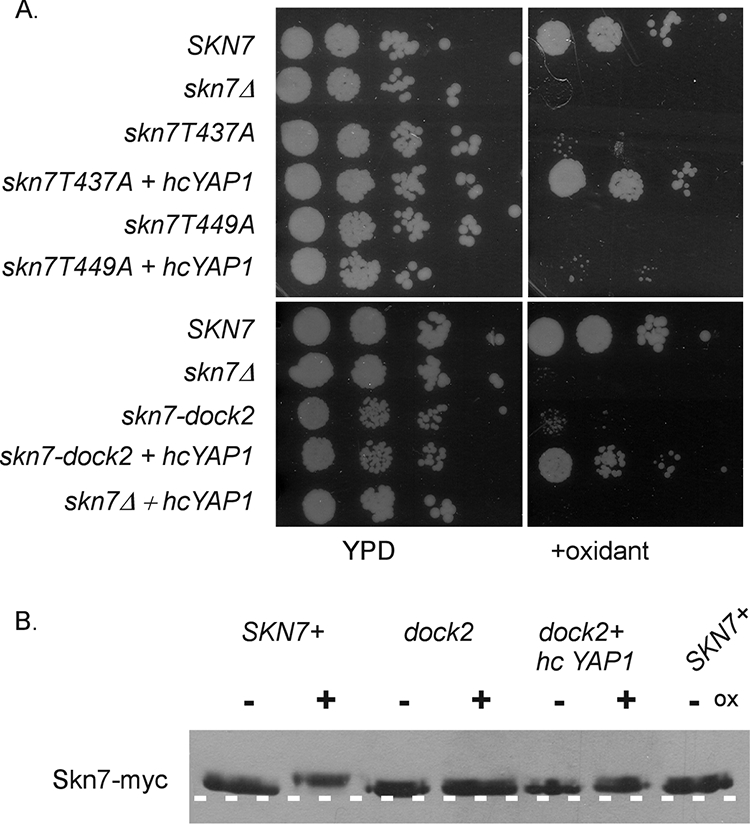
Effect of YAP1 overexpression on oxidant (ox) sensitivity and the phosphorylation of oxidative stress-defective skn7 mutants. (A) Cultures of skn7 mutants with and without a high-copy YAP1 expression plasmid were spotted (10-fold dilutions starting at 106cells/ml) on plates lacking or containing oxidant. Yeast strains included JF1904 (skn7Δ) carrying the following SKN7 expression plasmids: SKN7+, pXH1853; skn7-T437A, pXH1854; skn7-T449A, pXH1939; skn7-dock2, pXH1941; and the high-copy (hc) YAP1 expression plasmid (pSEY18-YAP1). (B) Effect of the high-copy YAP1 expression plasmid on the migration of the skn7-dock2 mutant protein. TCA extracts were prepared from JF1904 (skn7Δ) carrying the skn7-dock2 expression plasmid (pXH1941) and the high-copy YAP1 expression plasmid (pSEY18-YAP1) or vector. Extracts were prepared from cultures treated with 2 mM H2O2 (20 min) (+) or left untreated (−) and were subjected to SDS-10% PAGE for 2.5 h at 200 V. A dashed white line is added to the figure based on the migration of two untreated wild-type samples run as controls on the outside edges of the gel.
DISCUSSION
The oxidation of critical cysteine residues affords a direct mechanism for sensing the cellular oxidation state employed by a variety of transcription factors, including yeast Yap1 (9, 17), mammalian c-jun and NF-κB (14, 28), and the bacterial OXR transcription factor OxyR (42). To determine whether the Skn7 transcription factor also is a redox sensor, all five cysteine residues in Skn7 (including three in the receiver domain and two upstream of it) were individually mutated to alanine (X.-J. He and J. S. Fassler, unpublished data). Mutant derivatives of SKN7 were introduced into an skn7Δ strain, and the extent of complementation was evaluated. Each skn7 C/A mutant was indistinguishable from the wild-type strain, and a triple mutant including all three receiver domain mutations exhibited only minor defects in growth on oxidative stress plates. Quantitative measurements using an oxidative stress-responsive reporter plasmid, pXH1641 containing CCP1 (−309/∼+14)-lacZ (11), confirmed that the cysteines in Skn7 are not critical for the oxidant-dependent induction of the CCP1 reporter gene (He and Fassler, unpublished). The lack of effect in the C/A mutants indicates that neither individual cysteines nor any pair of cysteines in the receiver domain are required for the Skn7 OXR. Based on these results, Skn7 appears unlikely to be a cysteine-based redox sensor.
We therefore turned our attention to previous reports of the serine/threonine phosphorylation of Skn7 (4), asking whether Skn7 undergoes phosphorylation in response to an oxidant. Our results show that the migration of Skn7 in denaturing polyacrylamide gels is slower after oxidant treatment, and that this change in mobility is sensitive to phosphatase. On the basis of these data, we conclude that the phosphorylation of SKN7 is one aspect of the OXR. An important question is whether this phosphorylation is important for the normal OXR conferred by Skn7. The oxidative stress sensitivity of a mutation in receiver domain sequences that resemble MAPK docking sites found in other transcription and MAPK-interacting factors (36, 37) is consistent with the hypothesis that Skn7 phosphorylation is important for an Skn7-mediated OXR. The sensitivity and absence of oxidant-dependent phosphorylation in this mutant raised the possibility that one of the six MAPKs in S. cerevisiae is involved in the oxidative stress-dependent phosphorylation of Skn7. In previous reports, a deletion of the SLT2 MAPK gene caused modest sensitivity on minimal medium plates containing 0.2 mM hydrogen peroxide (15), and the growth of a strain with a deletion of the PBS2 gene, a MAPK kinase in the HOG1 MAPK pathway, was sensitive to 0.5 mM t-BOOH in liquid YPD cultures (3). We therefore investigated the potential contribution of each of the six S. cerevisiae MAPKs in the OXR (data not shown). Two experimental tests were used. In the first test, all six MAPK mutants were found to be relatively oxidative stress resistant on YPD plates containing t-BOOH at levels causing the complete absence of growth of an skn7 deletion mutant (data not shown). The second test used the Gal4 DBD-Skn7 receiver domain fusion in strains lacking individual MAPKs. Strains lacking the Hog1 kinase exhibited a very modest reduction in the oxidative stress activation of the Gal4 DBD-Skn7 hybrid to 70% of wild-type levels (P = 0.004). None of the other mutants showed any effect at all. Consistent with the modest effect of the hog1Δ mutation, we observed no effect on the oxidative stress-dependent phosphorylation of Skn7 in extracts prepared from strains lacking Hog1 (data not shown). We conclude that neither Hog1 nor any of the other five MAPKs makes a significant contribution to the OXR or are responsible for Skn7 phosphorylation in response to oxidative stress.
MAPK phosphorylation sites typically take the form (S/T)P (34), and the sole example of this motif in the Skn7 receiver domain is T449. The skn7-T449A mutant was sensitive to oxidative stress (Fig. 3) and defective in the oxidant-dependent phosphorylation of Skn7 (data not shown). However, we found that several Skn7-T449 substitutions mutants (Skn7-T449A, Skn7-T449E, and Skn7-T449D) were defective in binding to DNA in gel shift experiments (Fig. 6) and inactive when fused to the Gal4 DBD (Fig. 4), indicating that the T449A protein is nonspecifically defective in all Skn7 activities. Hence, the T449 residue is not specifically involved in the OXR but rather in DNA binding. Taken together, our data suggest that neither the putative MAPK dock nor the putative MAPK phosphorylation site in the Skn7 receiver domain is important for the oxidative stress-dependent phosphorylation of Skn7. With no strong evidence for the MAPK-mediated phosphorylation of Skn7 in response to an oxidant, we presume that the oxidant-dependent phosphorylation of Skn7 can be attributed to another class of kinase that has yet to be identified.
Our analysis of conserved threonine residues in the Skn7 receiver domain identified T437 as being required for the normal function of Skn7 in the OXR. The alanine substitution of T437 caused a mild oxidative stress-sensitive phenotype but did not abrogate the oxidant-dependent phosphorylation of Skn7. Additional substitutions at this position were generated to further investigate the likelihood of phosphorylation at this site. The fact that neither skn7-T437D nor skn7-T437E mutants complemented the oxidative stress sensitivity of an skn7Δ mutant does not rule out phosphorylation at T437, but neither does it provide support for the idea. The observation that the T437S mutant was fully functional is consistent with the requirement for a hydroxyl at this position but does not rule out alternatives to phosphorylation.
Both the T437A and the dock2 I428A V429A mutants were defective in forming Skn7-Yap1-dependent complexes at the promoters of OXR genes. Many OXR genes are activated by the joint action of the Skn7 and Yap1 transcription factors. The activation of Yap1 upon oxidative stress has been studied extensively (8, 9, 18, 19), but the precise mechanism by which these two factors coactivate OXR genes is not yet clear. The effect of Yap1 (as well as Skn7) on the GAL1-lacZ reporter, despite the absence of the Yap1 binding sites in the UASG, suggests that Yap1 can mediate its effect indirectly, presumably via an association with Skn7. The identification of skn7 mutations defective in OXR and in the formation of Skn7-Yap1-DNA complexes on OXR gene promoters suggests that this type of interaction is an important part of the Skn7 response to oxidant. The failure of the skn7-T437A mutant to form normal levels of the Skn7-Yap1 complex despite the continued phosphorylation of the Skn7 protein, and the suppression of this phenotype by the overexpression of YAP1, indicates that the activation of Skn7 in the OXR is a matter of recruiting the Yap1 transcription factor and not simply a matter of phosphorylating Skn7.
The oxidative stress phenotype of the I428A V429A (dock2) mutant likewise is suppressed by YAP1 overexpression. The suppression is not accompanied by the restoration of oxidant-dependent phosphorylation; this supports the argument that the oxidative-dependent phosphorylation of Skn7 is not required for Skn7 to activate OXR gene expression. Furthermore, it shows that the need for the phosphorylation of Skn7 can be bypassed by increasing the concentration of Yap1. This observation results in an apparent paradox, because it suggests that the purpose of Skn7 phosphorylation is the enhanced recruitment of Yap1, a conclusion that is in apparent contradiction to our observation that oxidant-dependent phosphorylation is eliminated in the yap1Δ mutant (Fig. 2B). If phosphorylation is needed for Skn7 to recruit Yap1, then how can the absence of Yap1 prevent phosphorylation? Several possible explanations come to mind. First, Yap1 might weakly bind unphosphorylated Skn7, and once bound, it might stimulate phosphorylation that then strengthens the interaction. Second, the Yap1 protein might protect phosphorylated Skn7 from phosphatase; in the absence of Yap1, Skn7 would appear unphosphorylated. Alternatively, the expression of the Skn7 kinase might be stimulated by Yap1. Based on our analysis of oxidative stress-sensitive mutants, we favor the first idea and propose the following two-stage working model for an Skn7-Yap1 interaction in the OXR (Fig. 12). Upon the nuclear localization of Yap1 in response to an oxidizing environment, Yap1 binds weakly to unphosphorylated DNA-bound Skn7. A Skn7 kinase is then recruited to the Skn7-Yap1 complex, leading to the phosphorylation of Skn7, which in turn stabilizes the complex. The Skn7-Yap1 activation complex then recruits transcription machinery to mount a full-fledged OXR. In this model, the I428 and V429 residues of the Skn7 receiver domain are important for the initial weak interaction with Yap1 (Fig. 12). Since the mutant cannot attract Yap1, it cannot recruit the Skn7 kinase. This explains why both the yap1Δ mutant and the skn7-I428A,V429A (dock2) mutant are defective in phosphorylation. In contrast, we propose that the T437 residue, the mutation of which does not eliminate phosphorylation but does affect Skn7-Yap1 complex formation, plays a role in a secondary interaction (Fig. 12) between phospho-Skn7 and Yap1. One interesting aspect of this model is the frequent occurrence of tandem Skn7 and Yap1 binding sites in OXR genes. Our data suggest that each binding site is dispensable for an OXR, since the proteins can interact with one another. However, promoters of osmotic response genes such as OCH1 bind Skn7 but not Yap1 and do not exhibit oxidative stress responsiveness. Thus, in genes like OCH1, something must prevent the recruitment of Yap1 to Skn7-DNA complexes. The basis for the differences between OXR and osmotic stress promoters remains an open question.
FIG. 12.
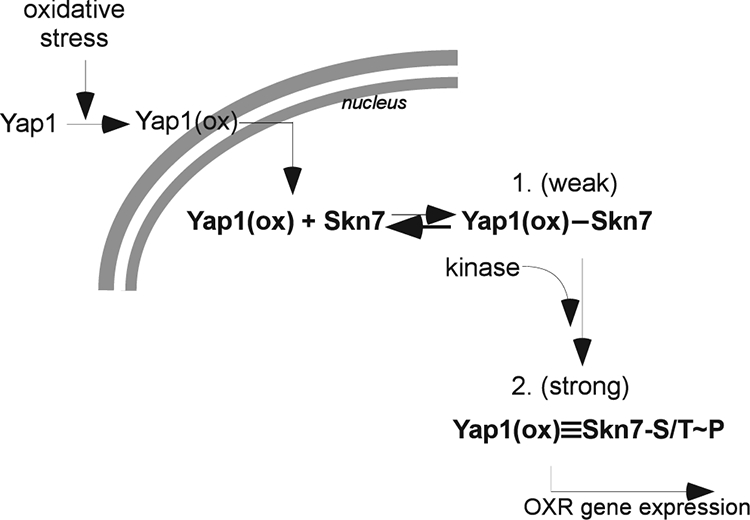
Working model for Skn7 participation in the activation of OXR genes. The nuclear localization of Yap1 in an oxidizing environment [Yap1(ox)] leads to a weak interaction with DNA-associated Skn7 via the I428 and V429 residues in the Skn7 receiver domain. The Skn7-Yap1 (DNA) complex recruits a kinase that reinforces the Skn7-Yap1 interaction via serine/threonine phosphorylation of Skn7 (Skn7-S/T∼P). The strong association between Skn7 and Yap1 that is a result of Skn7 phosphorylation is required for the efficient transcriptional activation of OXR genes.
Acknowledgments
We are grateful to Greg Gingerich for providing technical assistance with electrophoretic mobility shift assays. We thank Scott Moye-Rowley for Yap1 reagents and Bob Malone and Dan Weeks for productive discussions and constructive comments on the manuscript.
Funding for this research was provided by GM56719 from the National Institutes of Health. K.E.M. was supported in part by a fellowship from the Center for Biocatalysis and Bioprocessing (University of Iowa).
Footnotes
Published ahead of print on 20 March 2009.
REFERENCES
- 1.Appleby, J. L., and R. B. Bourret. 1999. Activation of CheY mutant D57N by phosphorylation at an alternative site, Ser-56. Mol. Microbiol. 34915-925. [DOI] [PubMed] [Google Scholar]
- 2.Ausubel, F. M., R. Brent, R. E. Kingston, D. E. Moore, J. G. Seidman, J. A. Smith, and K. Struhl. 1989. Current protocols in molecular biology. John Wiley and Sons, New York, NY.
- 3.Bilsland, E., C. Molin, S. Swaminathan, A. Ramne, and P. Sunnerhagen. 2004. Rck1 and Rck2 MAPKAP kinases and the HOG pathway are required for oxidative stress resistance. Mol. Microbiol. 531743-1756. [DOI] [PubMed] [Google Scholar]
- 4.Brown, J. L., H. Bussey, and R. C. Stewart. 1994. Yeast Skn7 functions in a eukaryotic two-component regulatory pathway. EMBO J. 135186-5194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brown, J. L., S. North, and H. Bussey. 1993. SKN7, a yeast multicopy suppressor of a mutation affecting cell wall beta-glucan assembly, encodes a product with domains homologous to prokaryotic two-component regulators and to heat shock transcription factors. J. Bacteriol. 1756908-6915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Charizanis, C., H. Juhnke, B. Krems, and K. D. Entian. 1999. The oxidative stress response mediated via Pos9/Skn7 is negatively regulated by the Ras/PKA pathway in Saccharomyces cerevisiae. Mol. Gen. Genet. 261740-752. [DOI] [PubMed] [Google Scholar]
- 7.Cho, H. S., S. Y. Lee, D. Yan, X. Pan, J. S. Parkinson, S. Kustu, D. E. Wemmer, and J. G. Pelton. 2000. NMR structure of activated CheY. J. Mol. Biol. 297543-551. [DOI] [PubMed] [Google Scholar]
- 8.Delaunay, A., A. D. Isnard, and M. B. Toledano. 2000. H2O2 sensing through oxidation of the Yap1 transcription factor. EMBO J. 195157-5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Delaunay, A., D. Pflieger, M. B. Barrault, J. Vinh, and M. B. Toledano. 2002. A thiol peroxidase is an H2O2 receptor and redox-transducer in gene activation. Cell 111471-481. [DOI] [PubMed] [Google Scholar]
- 10.Duclos, B., S. Marcandier, and A. Cozzone. 1991. Chemical properties and separation of phosphoamino acids by thin-layer chromatography and/or electrophoresis. Methods Enzymol. 20110-21. [DOI] [PubMed] [Google Scholar]
- 11.He, X. J., and J. S. Fassler. 2005. Identification of novel Yap1 and Skn7 binding sites involved in the oxidative stress response of Saccharomyces cerevisiae. Mol. Microbiol. 581454-1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.James, P., J. Halladay, and E. A. Craig. 1996. Genomic libraries and a host strain designed for highly efficient two-hybrid selection in yeast. Genetics 1441425-1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Juhnke, H., C. Charizanis, F. Latifi, B. Krems, and K. D. Entian. 2000. The essential protein fap7 is involved in the oxidative stress response of Saccharomyces cerevisiae. Mol. Microbiol. 35936-948. [DOI] [PubMed] [Google Scholar]
- 14.Klatt, P., E. P. Molina, M. G. De Lacoba, C. A. Padilla, E. Martinez-Galesteo, J. A. Barcena, and S. Lamas. 1999. Redox regulation of c-Jun DNA binding by reversible S-glutathiolation. FASEB J. 131481-1490. [DOI] [PubMed] [Google Scholar]
- 15.Krasley, E., K. F. Cooper, M. J. Mallory, R. Dunbrack, and R. Strich. 2006. Regulation of the oxidative stress response through Slt2p-dependent destruction of cyclin C in Saccharomyces cerevisiae. Genetics 1721477-1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krems, B., C. Charizanis, and K. D. Entian. 1996. The response regulator-like protein Pos9/Skn7 of Saccharomyces cerevisiae is involved in oxidative stress resistance. Curr. Genet. 29327-334. [DOI] [PubMed] [Google Scholar]
- 17.Kuge, S., M. Arita, A. Murayama, K. Maeta, S. Izawa, Y. Inoue, and A. Nomoto. 2001. Regulation of the yeast Yap1 nuclear export signal is mediated by redox signal-induced reversible disulfide bond formation. Mol. Cell. Biol. 216139-6150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuge, S., and N. Jones. 1994. YAP1 dependent activation of TRX2 is essential for the response of Saccharomyces cerevisiae to oxidative stress by hydroperoxides. EMBO J. 13655-664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuge, S., N. Jones, and A. Nomoto. 1997. Regulation of yAP-1 nuclear localization in response to oxidative stress. EMBO J. 161710-1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee, S. Y., H. S. Cho, J. G. Pelton, D. Yan, E. A. Berry, and D. E. Wemmer. 2001. Crystal structure of activated CheY. Comparison with other activated receiver domains. J. Biol. Chem. 27616425-16431. [DOI] [PubMed] [Google Scholar]
- 21.Li, S., S. Dean, Z. Li, J. Horecka, R. J. Deschenes, and J. S. Fassler. 2002. The eukaryotic two-component histidine kinase Sln1 regulates OCH1 via the transcription factor, Skn7. Mol. Biol. Cell 13412-424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lukat, G. S., A. M. Stock, and J. B. Stock. 1990. Divalent metal ion binding to the CheY protein and its significance to phosphotransfer in bacterial chemotaxis. Biochemistry 295436-5442. [DOI] [PubMed] [Google Scholar]
- 23.Miller, J. H. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY.
- 24.Morgan, B. A., G. R. Banks, W. M. Toone, D. Raitt, S. Kuge, and L. H. Johnston. 1997. The Skn7 response regulator controls gene expression in the oxidative stress response of the budding yeast Saccharomyces cerevisiae. EMBO J. 161035-1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morgan, B. A., N. Bouquin, G. F. Merrill, and L. H. Johnston. 1995. A yeast transcription factor bypassing the requirement for SBF and DSC1/MBF in budding yeast has homology to bacterial signal transduction proteins. EMBO J. 145679-5689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nicholas, K. B., H. B. Nicholas, Jr., and D. W. Deerfield II. 1997. GeneDoc: analysis and visualization of genetic variation. http://www.psc.edu/biomed/genedoc.
- 27.Ohmiya, R., C. Kato, H. Yamada, H. Aiba, and T. Mizuno. 1999. A fission yeast gene (prr1+) that encodes a response regulator implicated in oxidative stress response. J. Biochem. (Tokyo) 1251061-1066. [DOI] [PubMed] [Google Scholar]
- 28.Pineda-Molina, E., P. Klatt, J. Vazquez, A. Marina, M. Garcia de Lacoba, D. Perez-Sala, and S. Lamas. 2001. Glutathionylation of the p50 subunit of NF-kappaB: a mechanism for redox-induced inhibition of DNA binding. Biochemistry 4014134-14142. [DOI] [PubMed] [Google Scholar]
- 29.Sanders, D. A., B. L. Gillece-Castro, A. M. Stock, A. L. Burlingame, and D. E. Koshland, Jr. 1989. Identification of the site of phosphorylation of the chemotaxis response regulator protein, CheY. J. Biol. Chem. 264:21770-21778. [PubMed] [Google Scholar]
- 30.Schuster, M., R. E. Silversmith, and R. B. Bourret. 2001. Conformational coupling in the chemotaxis response regulator CheY. Proc. Natl. Acad. Sci. USA 986003-6008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sharrocks, A. D., S. H. Yang, and A. Galanis. 2000. Docking domains and substrate-specificity determination for MAP kinases. Trends Biochem. Sci. 25448-453. [DOI] [PubMed] [Google Scholar]
- 31a.Sherman, F., G. R. Fink, and J. B. Hicks. 1986. Methods in yeast genetics, p. 163-167. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY.
- 32.Sikorski, R. S., and P. Hieter. 1989. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 12219-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Singh, P., N. Chauhan, A. Ghosh, F. Dixon, and R. Calderone. 2004. SKN7 of Candida albicans: mutant construction and phenotype analysis. Infect. Immun. 722390-2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Songyang, Z., K. P. Lu, Y. T. Kwon, L. H. Tsai, O. Filhol, C. Cochet, D. A. Brickey, T. R. Soderling, C. Bartleson, D. J. Graves, A. J. DeMaggio, M. F. Hoekstra, J. Blenis, T. Hunter, and L. C. Cantley. 1996. A structural basis for substrate specificities of protein Ser/Thr kinases: primary sequence preference of casein kinases I and II, NIMA, phosphorylase kinase, calmodulin-dependent kinase II, CDK5, and Erk1. Mol. Cell. Biol. 166486-6493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stock, A. M., E. Martinez-Hackert, B. F. Rasmussen, A. H. West, J. B. Stock, D. Ringe, and G. A. Petsko. 1993. Structure of the Mg2+-bound form of CheY and mechanism of phosphoryl transfer in bacterial chemotaxis. Biochemistry 3213375-13380. [DOI] [PubMed] [Google Scholar]
- 36.Tanoue, T., M. Adachi, T. Moriguchi, and E. Nishida. 2000. A conserved docking motif in MAP kinases common to substrates, activators and regulators. Nat. Cell Biol. 2110-116. [DOI] [PubMed] [Google Scholar]
- 37.Tanoue, T., and E. Nishida. 2003. Molecular recognitions in the MAP kinase cascades. Cell Signal. 15455-462. [DOI] [PubMed] [Google Scholar]
- 38.Thompson, J. D., D. G. Higgins, and T. J. Gibson. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 224673-4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tsuzi, D., K. Maeta, Y. Takatsume, S. Izawa, and Y. Inoue. 2004. Distinct regulatory mechanism of yeast GPX2 encoding phospholipid hydroperoxide glutathione peroxidase by oxidative stress and a calcineurin/Crz1-mediated Ca2+ signaling pathway. FEBS Lett. 569301-306. [DOI] [PubMed] [Google Scholar]
- 40.Wormley, F. L., Jr., G. Heinrich, J. L. Miller, J. R. Perfect, and G. M. Cox. 2005. Identification and characterization of an SKN7 homologue in Cryptococcus neoformans. Infect. Immun. 735022-5030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu, G., and J. S. Fassler. 1993. SPT13/GAL11 of Saccharomyces cerevisiae negatively regulates activity of the MCM1 transcription factor in Ty1 elements. Mol. Cell. Biol. 1363-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zheng, M., F. Aslund, and G. Storz. 1998. Activation of the OxyR transcription factor by reversible disulfide bond formation. Science 2791718-1721. [DOI] [PubMed] [Google Scholar]
- 43.Zheng, M., and G. Storz. 2000. Redox sensing by prokaryotic transcription factors. Biochem. Pharmacol. 591-6. [DOI] [PubMed] [Google Scholar]