

Abstract
Full term pregnancy early in life is the most effective natural protection against breast cancer in women. Rats treated with chemical carcinogen are similarly protected by a previous pregnancy from mammary carcinogenesis. Proliferation and differentiation of the mammary gland does not explain this phenomenon, as shown by the relative ineffectiveness of perphenazine, a potent mitogenic and differentiating agent. Here, we show that short term treatment of nulliparous rats with pregnancy levels of estradiol 17β and progesterone has high efficacy in protecting them from chemical carcinogen induced mammary cancers. Because the mammary gland is exposed to the highest physiological concentrations of estradiol and progesterone during full term pregnancy, it is these elevated levels of hormones that likely induce protection from mammary cancer. Thus, it appears possible to mimic the protective effects of pregnancy against breast cancer in nulliparous rats by short term specific hormonal intervention.
Women who have undergone a full term pregnancy before 20 years of age have one-half the risk of developing breast cancer compared with nulliparous women (1). This protective effect of early pregnancy is universal, occurring among women of all ethnic groups worldwide, and is clearly of major consideration in devising strategies for the prevention of breast cancer. Rats and mice that undergo a full term pregnancy also have a greatly reduced susceptibility to chemically induced mammary carcinogenesis as compared with nulliparous animals and are used as experimental models (2–5). During pregnancy, many hormones act to cause proliferation and differentiation of the mammary gland in preparation for lactation. The mammary gland is exposed to high levels of ovarian (estrogens and progestins), pituitary (prolactin and growth hormone), and placental (placental lactogens) hormones (4, 6). After pregnancy, under the influence of lactogenic hormones, the mammary gland becomes fully lactational. After the weaning of the offspring, involution of the differentiated lobuloalveolar structures occurs, characterized by apoptotic events owing to the reduction in lactogenic hormones. The protective effects of pregnancy have been reported in rats undergoing pregnancy before or subsequent to exposure to chemical carcinogens (2–5).
The mechanisms for the protective effect of pregnancy have not been defined. The most widely accepted explanation is the one put forth by Russo and Russo (4) that the protective effect is attributable to the pregnancy-induced differentiation of the target structures for carcinogenesis, terminal end buds and terminal ducts. In their view, these structures show high proliferation and hence are more susceptible than are lobuloalveolar structures that have a low level of proliferation (4, 7). In essence, pregnancy may cause the removal or modification of a population of cells that are highly susceptible to the development of breast cancer. There are, however, other alterations that also might explain refractoriness to mammary carcinogenesis. After pregnancy, the mammary cells have an enhanced capability for DNA repair (4). Pregnancy also has been found to cause a subsequent reduction of blood levels of mammogenic hormones in women and in rats and a decrease in the receptors for epidermal growth factor and estrogen in the mammary glands of parous rats (8).
Hormonal prevention strategies have used exogenous hormonal treatment to induce lobuloalveolar differentiation of terminal end buds to protect against mammary carcinogenesis in rats. Treatments with human chorionic gonadotropins or combinations of estradiol and progesterone, either before or after treatment with chemical carcinogens, both induce lobuloalveolar differentiation and are protective (7, 9–13). The most accepted explanation for this induced refractoriness to mammary carcinogenesis is that either the mammary target cells for cancer induction are altered by prior hormonal treatment to a nonsusceptible state (4, 13) or, in the case of hormonal treatment after carcinogen administration, the preneoplastic cells induced by the carcinogen treatment differentiate (11, 12) or undergo apoptosis (14).
Our objective was to examine whether a variety of different agents that could induce a near lactational differentiation in the rat mammary gland also would provide protection from chemical carcinogen-induced mammary carcinogenesis. We chose the rat mammary carcinogenesis experimental system as a model for human breast cancer because mammary cancers can be reproducibly induced at a high frequency, the cancers are adenocarcinomas and are predominantly ductal in origin (similar to many human breast cancers), a full term pregnancy in the rat also induces refractoriness to mammary cancer development, and the rat has been used as a model system for the development of tamoxifen and retinoid adjuvant therapies now being used in humans (2, 3, 15, 16).
Our question in this study regarded whether the artificial induction by any agent of lobuloalveolar development and lactogenesis with subsequent involution would consistently make nulliparous rats refractory to mammary carcinogenesis. We hypothesized that, by short term administration of exogenous agents, it might be possible to induce a pregnancy-like morphogenesis of lobuloalveolar structures as well as lactogenesis in the mammary glands of young nulliparous rats, which are highly susceptible to mammary carcinogenesis. The differentiation thus induced before or after carcinogen exposure, and the involution that inevitably follows after cessation of treatment, might result in refractoriness mimicking that of pregnancy.
METHODS AND MATERIALS
Animals.
Virgin Lewis rats (Harlan–Sprague–Dawley) were fed food (Teklad, Madison, WI) and water ad libitum and were kept on a 12-hour light and 12-hour dark cycle.
Peripheral Blood Serum Levels of Estradiol 17β (E) and Progesterone (P).
Five 9-week-old virgin rats (Harlan–Sprague–Dawley) per group were placed in the following groups: untreated controls, those injected in the dorsal cervical s.c. area with 5 mg/kg perphenazine (PPZ) (Sigma) dissolved in 0.03 M HCl (5 mg/ml) five times per week for 3 weeks, or those implanted with one silastic capsule (Baxter Healthcare; size 0.078 inch i.d. × 0.125 inch o.d., 2 cm in length) containing 30 mg of estradiol (Sigma) plus one silastic capsule containing 30 mg of progesterone (Sigma). Trunk blood was collected from the vena cava at termination, 3 weeks after the start of treatment. Estradiol and progesterone were measured by radioimmunoassay kit from Diagnostic Products (Los Angeles) (8).
Effects of N-Methyl-N-nitrosourea (MNU) Exposure, Followed by Treatment with Differentiating Agents, on Prevention of MNU-Induced Mammary Carcinogenesis.
Effect of PPZ plus or minus E plus P. At 7 weeks of age, the rats were treated with a single i.p. injection of MNU (Ashe Stevens Detroit) at a dose of 50 mg/kg (17). At 9 weeks of age, the rats were divided into four groups: (i) control, (ii) those receiving five daily treatments of 5 mg/kg PPZ for 3 weeks; (iii) those receiving five daily treatments of PPZ for 3 weeks plus 30 mg of E and 30 mg of P administered by s.c. implantation of silastic capsules for 3 weeks; and (iv) those receiving 30 mg of E and 30 mg of P in silastic capsules for 3 weeks. The silastic capsules were removed after 3 weeks of treatment.
Effect of E plus P treatment on MNU-induced mammary carcinogenesis.
Female Lewis rats were given a single injection i.p. of 50 mg/kg MNU at 7 weeks of age (17). Two or four weeks later, the rats were implanted s.c. with a silastic capsule containing 30 mg or 200 μg of E and a silastic capsule containing 30 mg of P. The silastic capsules were removed after 3 weeks of treatment.
Effect of the dose of E on mammary carcinogenesis.
Rats were treated with 50 mg/kg of MNU at 7 weeks of age. At 9 weeks of age, the rats were implanted with silastic capsules containing either 2 mg, 200 μg, or 20 μg of E plus 30 mg of P. The silastic capsules were removed 3 weeks later.
Effect of estradiol and/or progesterone on MNU-induced mammary carcinogenesis.
Rats were treated with 50 mg/kg of MNU at 7 weeks of age. Silastic capsules containing 30 mg of E and/or 30 mg of P were implanted s.c. at 9 weeks of age. The silastic capsules were removed 3 weeks later.
Effect of the duration of E plus P treatment on mammary carcinogenesis.
Rats were treated with 50 mg/kg of MNU at 7 weeks of age. At 9 weeks of age, the rats were implanted with silastic capsules containing 30 mg of E plus 60 mg of P or 30 mg of E plus 90 mg of P. The silastic capsules were removed 1 week later (30 mg of E plus 60 mg of P, 30 mg of E plus 90 mg of P) or 2 weeks later (30 mg of E plus 60 mg of P).
Effect of E plus P treatment before MNU exposure.
Rats were implanted with silastic capsules containing 30 mg of E plus 30 mg of P, 60 mg of E plus 30 mg of P, or 200 μg of E plus 30 mg of P at 7 weeks of age. The silastic capsules were removed 2 weeks later. At 14 weeks of age, the rats were treated with 50 mg/kg of MNU.
Mammary Carcinogenesis.
Rats were palpated weekly for 9 months to monitor for mammary cancers. The carcinomatous nature of palpated tumors was confirmed by histopathological examination.
Statistics.
Significance of differences compared with controls was calculated by using the χ2 statistic for 2 × 2 contingency tables.
RESULTS
We chose perphenazine, a dopamine receptor inhibitor, and the natural steroid hormones estradiol 17β and progesterone as the agents to be tested. Perphenazine is a compound that has been used to cause the acute release of prolactin from the anterior pituitary of rats by blocking the inhibitory influence of dopamine from the hypothalamus (18). This release of prolactin causes the proliferation and differentiation of the mammary cells to a near lactational state after a short period of treatment. Estradiol and progesterone are mammogenic steroids that are present at high levels during pregnancy. The effects of perphenazine or estradiol plus progesterone treatment first were tested on 9-week-old virgin rats to determine the extent of proliferation and differentiation induced by the treatment and to determine the resultant blood levels of estradiol and progesterone. Three weeks of treatment with either perphenazine or estradiol and progesterone induced a high level of proliferation and lobuloalveolar morphogenesis in the mammary glands (Fig. 1). Secretion was visible in the mammary glands. Peripheral blood levels of estradiol in perphenazine-treated rats were comparable to those in nonpregnant controls, and estradiol plus progesterone-treated rats had an elevated level of estradiol compared with nonpregnant controls but below the peak concentrations found in pregnancy. Progesterone levels in perphenazine-treated rats were elevated and comparable to peak levels in pregnant rats. Progesterone levels in estradiol plus progesterone-treated rats were approximately double the levels of nonpregnant controls, below the level reported in pregnant rats (6) (Table 1).
Figure 1.

(A) Histological section of the mammary gland of an untreated 12-week-old virgin Lewis rat. Note the presence of empty ducts with small alveolar buds. (B) Histological section of the differentiated mammary gland of a virgin rat treated for 3 weeks with 30 mg of estradiol and 30 mg of progesterone in silastic capsules. Note the presence of a dilated duct and alveoli filled with secretion and fat droplets. (C) Histological section of the differentiated mammary gland of a virgin rat treated for 3 weeks with 5 mg/kg daily perphenazine. Note the secretion filled mammary ducts and alveoli. (×200.)
Table 1.
Peripheral blood serum levels of estradiol and progesterone
| Treatment | Estradiol, pg/ml | Progesterone, ng/ml |
|---|---|---|
| Virgin control | 18.3 ± 8.8 | 12.7 ± 2.1 |
| Pregnant* | 55–630 | 45–130 |
| Perphenazine | 16.6 ± 7.8 | 101.5 ± 10.3 |
| Estradiol plus progesterone | 168.8 ± 22.1 | 25.8 ± 6.6 |
Shown are serum levels of estradiol and progesterone in virgin, pregnant, and rats treated with 5 mg/kg perphenazine or 30 mg of E plus 30 mg of P.
Pregnancy levels are derived from Numan (6) and represent the range during pregnancy.
The effect of perphenazine and/or estradiol plus progesterone treatment on mammary carcinogenesis then was tested. Seven-week-old virgin Lewis rats were treated with a single i.p. injection of MNU (17). We chose to treat with carcinogen before hormonal treatment because, although the timing of the carcinogenic insult in women is unknown, epidemiological studies suggest that young women are highly susceptible to the initiation of breast cancer (19, 20). Two weeks after carcinogen treatment, the rats were treated with perphenazine for three weeks and/or with estradiol plus progesterone (Fig. 2). Mammary cancer incidence in these rats was recorded for a period of 9 months after MNU treatment. Ninety percent of control rats (2.3 cancers/rat) treated with MNU and seventy-three percent of the rats (1.3 cancers/rat) treated with perphenazine developed mammary cancers. Thus, there were slight decreases in incidence and multiplicity of cancers in perphenazine-treated rats as compared with controls. Treatment with either a combination of perphenazine and estradiol plus progesterone or estradiol plus progesterone without perphenazine greatly protected the rats from mammary carcinogenesis. Of the rats treated with perphenazine and estradiol and progesterone, 16% (0.16 cancers/rat) developed mammary cancer. Mammary cancers developed in 9% (0.1 cancers/rat) of the rats treated with estradiol plus progesterone during the 9-month-period of observation after MNU treatment. Rats treated with estradiol plus progesterone alone had up to a 96% reduction in mammary cancers compared with controls.
Figure 2.
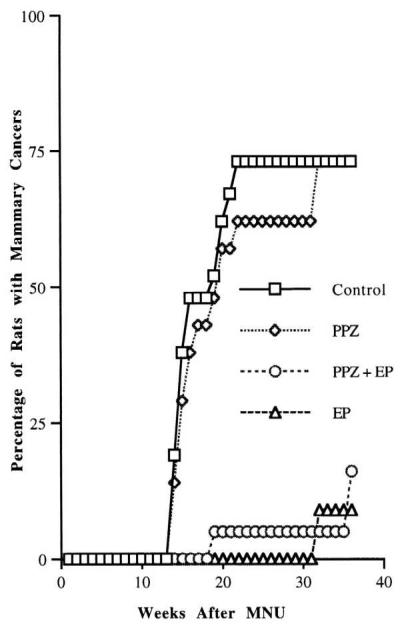
Effect of perphenazine or estradiol plus progesterone treatment on mammary carcinogenesis in MNU-exposed rats. Rats treated with PPZ plus E plus P or E plus P had a significant reduction in mammary cancer incidence compared with controls or PPZ-treated rats during the 9 months of observation. P < 0.005.
The effect of estradiol plus progesterone treatment on the reduction of mammary cancers in rats was repeated in four different experiments (Table 2). The protective effect of estradiol plus progesterone treatment was highly reproducible and statistically significant (P ≤ 0.05–0.001). In three of five experiments, the cancer incidence was 0% at 6 months after carcinogen treatment. Experiments were extended to 9 months to determine whether there was an increase in cancer incidence. Rats treated with estradiol plus progesterone for a 3-week period after MNU treatment developed up to 93% fewer mammary carcinomas during the 9-month observation period compared with controls not receiving hormonal treatment.
Table 2.
Effect of estradiol (E) plus progesterone (P) treatment on MNU-induced mammary carcinogenesis
| Treatment | Mammary cancer incidence | Percent of rats with mammary cancer | Average number of cancers per rat |
|---|---|---|---|
| Control | 18/20 | 90 | 2.3 ± 0.4 |
| 30 mg of E plus 30 mg of P* | 1/11§ | 9 | 0.1 ± 0.1§ |
| Control | 9/9 | 100 | 2.8 ± 0.1 |
| 30 mg of E plus 30 mg of P* | 5/10‡ | 50 | 0.6 ± 0.2§ |
| Control | 9/9 | 100 | 2.8 ± 0.5 |
| 30 mg of E plus 30 mg of P* | 0/8 | 0 | 0§ |
| Control | 10/10 | 100 | 2.8 ± 0.6 |
| 30 mg of E plus 30 mg of P† | 1/9 | 11 | 0.1 ± 0.1§ |
| Control | 9/9 | 100 | 2.7 ± 0.5 |
| 200 μg of E plus 30 mg of P* | 1/10§ | 10 | 0.1 ± 0.1§ |
Rats were treated with MNU at 7 weeks of age.
Rats treated 2 weeks later with E plus P for 3 weeks.
Rats treated 4 weeks later with E plus P for 3 weeks.
P = 0.05 compared to control.
P < 0.005 compared to control.
A dose of 200 μg of E was as effective as 2 mg of E in inducing protection from mammary carcinogenesis (Table 3). The mammary cancer incidence of rats treated with 20 μg of E (67%) was not statistically different from that of controls (100%), but there was a significant decrease in the number of mammary cancers per rat: 0.8 vs. 3.6, respectively.
Table 3.
Effect of the dose of estradiol on mammary carcinogenesis
| Treatment | Mammary cancer incidence | Percent of rats with mammary cancer | Average number cancers per rat |
|---|---|---|---|
| Control | 7/7 | 100 | 3.6 ± 1.2 |
| 2 mg of E plus 30 mg of P | 2/9 | 22* | 0.2 ± 0.1‡ |
| 200 μg of E plus 30 mg of P | 3/11 | 27* | 0.4 ± 0.2‡ |
| 20 μg of E plus 30 mg of P | 7/11 | 67† | 0.8 ± 0.2‡ |
Rats were treated with MNU at 7 weeks of age. At 9 weeks of age, the rats were treated with various dosages of E plus 30 mg of P for 3 weeks.
P < 0.0025 compared to control.
P < 0.07 compared to control.
P < 0.05 compared to control.
The effect of either estradiol or progesterone given singly or together next was tested to determine the ability to induce protection from mammary carcinogenesis (Table 4). Of the control rats, 100% developed mammary cancer by 9 months (2.8 cancers/rat). Treatment with estradiol alone provided protection from mammary carcinogenesis. Of the treated rats, 38% developed mammary cancer (0.5 cancers/rat). In contrast, treatment with progesterone alone actually enhanced mammary carcinogenesis in terms of cancer load compared with controls. Of the rats treated with progesterone, 100% developed mammary cancers (3.5 cancers/rat). Thus, compared with controls, there was a >83% reduction in cancer number in rats treated with estradiol and up to a 93% reduction in rats treated with estradiol plus progesterone. Progesterone-treated rats had an increase of 126% in the number of cancers compared with controls.
Table 4.
Effect of estradiol (E) or progesterone (P) given singly on MNU-induced mammary carcinogenesis
| Treatment | Mammary cancer incidence | Percent of rats with mammary cancer | Average number of cancers per rat |
|---|---|---|---|
| Control | 8/8 | 100 | 2.8 ± 0.5 |
| 30 mg of E plus 30 mg of P | 1/9 | 11* | 0.1 ± 0.1‡ |
| 30 mg of E | 3/8 | 38† | 0.5 ± 0.2‡ |
| 30 mg of P | 9/9 | 100 | 3.5 ± 0.7 |
Rats were treated with MNU at 7 weeks of age. At 9 weeks of age, they were treated with E or P for 3 weeks.
P = 0.005 compared to control.
P = 0.025 compared to control.
P < 0.0003 compared to control.
The data in Table 5 indicate that treatments with 30 mg of E plus 60 mg of P for a 1- or 2-week period were equally effective in providing protection. Treatment with 30 mg of E plus 90 mg of P for 1 week was also highly effective. Short term treatments for one-third the length of pregnancy in the rat were as effective as 3 weeks of treatment in other experiments.
Table 5.
Effect of the duration of estradiol plus progesterone treatment on mammary carcinogenesis
| Treatment | Mammary cancer incidence | Percent of rats with mammary cancer | Average number cancers per rat |
|---|---|---|---|
| Control | 8/9 | 88 | 1.6 ± 0.2 |
| 30 mg of E plus 60 mg of P, 1 week | 1/6 | 16* | 0.2 ± 0.2† |
| 30 mg of E plus 60 mg of P, 2 weeks | 0/7 | 0* | 0† |
| 30 mg of E plus 90 mg of P, 1 week | 0/8 | 0* | 0† |
Rats were treated with MNU at 7 weeks of age. At 9 weeks of age, the rats were treated with E plus P for 1 or 2 weeks.
P < 0.005 compared to control.
P < 0.001 compared to control.
Treatment of rats with E plus P before MNU administration was also highly effective in protecting from mammary carcinogenesis (Table 6). The mammary cancer incidence was suppressed greatly in rats treated with either 30 mg of E plus 30 mg of P (0%), 60 mg of E plus 30 mg of P (17%), or 200 μg of E plus 30 mg of P (7%) compared with controls (55%).
Table 6.
Effect of estradiol plus progesterone treatment before MNU exposure
| Treatment | Mammary cancer incidence | Percent of rats with mammary cancer | Average number cancers per rat |
|---|---|---|---|
| Control | 6/11 | 55 | 0.7 ± 0.2 |
| 30 mg of E plus 30 mg of P | 0/10 | 0* | 0† |
| 60 mg of E plus 30 mg of P | 2/12 | 17* | 0.2 ± 0.2‡ |
| 200 μg of E plus 30 mg of P | 1/15 | 7* | 0.1 ± 0.1§ |
At 7 weeks of age, rats were treated with E plus P for 2 weeks. At 14 weeks of age, they were treated with MNU.
P < 0.007 compared to control.
P < 0.001 compared to control.
P < 0.03 compared to control.
P < 0.005 compared to control.
DISCUSSION
Both full term pregnancy and estradiol plus progesterone treatment shortly before or after carcinogen exposure result in a significant long term protection from mammary carcinogenesis. Similar to pregnancy, both perphenazine treatment and estradiol plus progesterone treatment induce growth and differentiation; involution in the mammary gland occurs after cessation of treatment. Assays of blood levels of estradiol and progesterone indicate that, with perphenazine treatment, only progesterone levels are increased to a pregnancy level. There was no increase in estradiol levels compared with untreated virgin rats. Perphenazine treatment did not cause a drastic reduction in mammary cancer incidence.
Treatment with estradiol plus progesterone results in an ≈10-fold increase in blood levels of estradiol and a 2-fold increase in progesterone, respectively, compared with controls. These values are lower than peak values seen in pregnancy, yet these levels are highly protective against mammary carcinogenesis (6). The experiments in which estradiol or progesterone were administered singly also suggest that it is the pregnancy levels of estradiol that are the most significant in protection from mammary carcinogenesis. However, the addition of some progesterone enhances the effect of estradiol.
Pregnancy is highly protective against mammary carcinogenesis. Our results show that such protection can be achieved by treatment with physiological (pregnancy) levels of estradiol plus progesterone. Our results show that even lower levels of estradiol are effective because silastic capsules containing only 200 micrograms of estradiol were also highly effective in preventing mammary carcinogenesis. Our data also suggest that the duration of treatment may be shortened to 1 week, one-third the pregnancy period in rats.
Because treatment with perphenazine or with estradiol and progesterone both result in pregnancy-like growth and differentiation of the mammary gland, these findings suggest that simple lobuloalveolar differentiation and involution are insufficient to induce pregnancy-like refractoriness to carcinogenesis. Recently, Medina’s laboratory found that full lobuloalveolar differentiation is not necessary to induce protection with exogenous estradiol plus progesterone treatment (13).
Thus, estradiol and progesterone treatment must have a specific effect on a transformable subpopulation of mammary epithelial cells in the mammary gland. It appears that treatment with sustained pregnancy levels of estradiol and progesterone for a short period of time results in the modification of this neoplastically transformed population. It is known that full term pregnancy offers the maximum physiological protection from mammary carcinogenesis whereas the interruption of pregnancy reduces the protective effect (4, 5).
Based on our findings, a possible explanation for this phenomenon is that, during a full term pregnancy, the mammary gland is exposed to the highest physiological concentrations of estradiol and progesterone. These modify the early transformed mammary cells while causing extensive proliferation of normal mammary cells. Interruption of pregnancy decreases the exposure to high levels of estradiol and progesterone and thus also decreases protection.
Huggins et al. originally reported that high levels of estradiol and progesterone given for 30 days, beginning 15 days after carcinogen administration, inhibited the appearance of mammary cancers in rats treated with the polycyclic hydrocarbon carcinogen 7,12-dimethylbenz[a]anthracene (9). Huggins suggested that treatment with high levels of ovarian hormones killed the potential cancer cells because of a differential sensitivity to hormones (21). Normal cells proliferate and differentiate in the presence of high levels of hormones, but cancer cells are killed. Grubbs demonstrated that treatment with high levels of estradiol and progesterone after MNU treatment was as effective as ovariectomy in preventing mammary carcinogenesis (11). He suggested that the primary action of the hormones is to cause differentiation of the preneoplastic cells.
The experiments of Russo and Russo (4), Grubbs et al. (12), and Medina and colleagues (13) showing that treatment with hormones before the initiation of cancer cells with a carcinogen is highly protective suggest that some target cells involved in mammary carcinogenesis must be removed or altered (4, 12). Russo and Russo and Grubbs et al. (4, 7, 11, 12) suggested that the primary action of the hormones is to cause normal differentiation of potential target cells. The interpretation of Medina and colleagues is that hormone treatment results in persistent alterations in the intracellular pathways governing proliferation responses to carcinogens (13).
An enigma in mammary cancer prevention studies in rats has been the fact that certain hormonal treatments provide a high level of protection (e.g., human chorionic gonadotropin) whereas others (e.g., placental lactogens) do not, although differentiation is induced in both cases (4, 7). A possible explanation may lie in the ratio of estradiol to progesterone resulting from the different treatments, as in our comparison of the effects of perphenazine with those of estradiol plus progesterone. The effect of estradiol and progesterone treatment also appears to be different from that of antiestrogens because only a brief treatment with estradiol and progesterone is sufficient to afford protection whereas antiestrogens must be administered continuously (15, 16). Treatment with estradiol plus progesterone provides prolonged protection from mammary carcinogenesis, in that the effect persists over a long period of time and the treatment need not be administered continuously.
In contrast to previous findings on the induction of protection to mammary carcinogenesis by administering exogenous hormones (7, 9–13), we have been able to determine that the protective effect is associated with the sustained release of pregnancy levels of estradiol along with some progesterone. This effect is achieved in a period that is only one-third that of the entire pregnancy.
Although all of these experimental findings could be explained on the basis of the death or modification of the target cells for mammary cancer or preneoplastic cells, other explanations are also possible. Previous studies in parous rats have shown an altered hormonal environment, altered sensitivity of mammary epithelial cells to hormones, and alterations in receptor levels, any of which might explain these results based on an inadequate hormonal environment to support mammary carcinogenesis (3, 8). We now are testing which of these alternatives are involved in the protection against mammary carcinogenesis in parous and in E-plus-P-treated rats. Whether the protective effect is a direct effect of estradiol or results from the formation of downstream estrogen metabolites that may have their own unique receptors or effectors remains to be determined (22).
The results convincingly show that short term treatment with appropriate levels of estradiol plus progesterone is highly effective in preventing mammary carcinogenesis. This provides a simple method, using natural hormones at physiological levels over a short period of time, to modify mammary epithelial cell populations and/or the host hormonal environment associated with the genesis of overt mammary cancers.
Acknowledgments
We thank Dr. Jean Nandi for critically reviewing and editing the manuscript. We thank Kathleen Buckley, Joanne Leung, and Nate Rogers for excellent technical assistance and Carol Slatten and Judith Yee for administrative assistance. This work was supported by National Cancer Institute Grants CA 05388, CA 63369, and CA 62598.
ABBREVIATIONS
- E
estradiol 17β
- P
progesterone
- PPZ
perphenazine
- MNU
N-methyl-N-nitrosourea
References
- 1.MacMahon B, Cole P, Lin M, Lowe C R, Mirra A P, Ravinihar B, Salber E J, Valaoras V G, Yuasa S. Bull W H O. 1970;43:209–221. [PMC free article] [PubMed] [Google Scholar]
- 2.Welsch C W. Cancer Res. 1985;45:3415–3443. [PubMed] [Google Scholar]
- 3.Moon R C. In: Breast Cancer: Advances in Biology and Therapeutics. Calvo F, Crepin M, Magdelenat H, editors. Paris: John Libbey Eurotext; 1996. pp. 19–22. [Google Scholar]
- 4.Russo I, Russo J. Environ Health Perspect. 1996;104:938–967. doi: 10.1289/ehp.96104938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sinha D K, Pazik J E, Dao T L. Br J Cancer. 1988;57:390–394. doi: 10.1038/bjc.1988.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Numan M. In: The Physiology of Reproduction. Knobil E, Neill J D, editors. New York: Raven; 1994. pp. 221–302. [Google Scholar]
- 7.Russo J, Russo I. Lab Invest. 1987;57:112–137. [PubMed] [Google Scholar]
- 8.Thordarson G, Jin E, Guzman R C, Swanson S M, Nandi S, Talamantes F. Carcinogenesis. 1995;16:2847–2853. doi: 10.1093/carcin/16.11.2847. [DOI] [PubMed] [Google Scholar]
- 9.Huggins C, Moon R C, Morii S. Proc Natl Acad Sci USA. 1962;48:379–386. doi: 10.1073/pnas.48.3.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McCormick G M, Moon R C. Eur J Cancer. 1973;9:483–486. doi: 10.1016/0014-2964(73)90131-x. [DOI] [PubMed] [Google Scholar]
- 11.Grubbs C J, Peckham J C, McDonough K D. Carcinogenesis. 1983;4:495–497. doi: 10.1093/carcin/4.4.495. [DOI] [PubMed] [Google Scholar]
- 12.Grubbs C J, Juliana M M, Whitaker L M. Anticancer Res. 1988;8:113–117. [PubMed] [Google Scholar]
- 13.Sivaraman L, Stephens L C, Markaverich B M, Clark J A, Krnacik S, Conneely O M, O’Malley B W, Medina D. Carcinogenesis. 1998;19:1573–1582. doi: 10.1093/carcin/19.9.1573. [DOI] [PubMed] [Google Scholar]
- 14.Srivsatava P, Russo J, Russo I H. Carcinogenesis. 1997;18:1799–1808. doi: 10.1093/carcin/18.9.1799. [DOI] [PubMed] [Google Scholar]
- 15.Wolf D M, Jordan V C. Recent Res Cancer Res. 1993;127:23–33. doi: 10.1007/978-3-642-84745-5_4. [DOI] [PubMed] [Google Scholar]
- 16.Jordan, V. C. (1983) Breast Cancer Res. Treat.3, Suppl. 1, S73–S76. [DOI] [PubMed]
- 17.Thompson H J, Adlakha H. Cancer Res. 1992;51:3411–3415. [PubMed] [Google Scholar]
- 18.Ben David M. Endocrinology. 1968;83:1217–1223. doi: 10.1210/endo-83-6-1217. [DOI] [PubMed] [Google Scholar]
- 19.Tokunaga, M., Land, C. E. & Tokuoka, S. (1991) J. Radiat. Res. (Tokyo)32, Suppl., 201–211. [DOI] [PubMed]
- 20.Hildreth N, Shore R, Dvuretski P. N Engl J Med. 1989;321:1285–1288. doi: 10.1056/NEJM198911093211901. [DOI] [PubMed] [Google Scholar]
- 21.Huggins C. Cancer Res. 1965;25:1163–1167. [PubMed] [Google Scholar]
- 22.Zhu B T, Conney A H. Carcinogenesis. 1998;19:1–27. doi: 10.1093/carcin/19.1.1. [DOI] [PubMed] [Google Scholar]