

Abstract
Wax esters, ester-linked fatty acids and long-chain alcohols, are important energy storage compounds in select bacteria. The synthesis of wax esters from fatty acids is proposed to require the action of a four-enzyme pathway. An essential step in the pathway is the reduction of a fatty aldehyde to the corresponding fatty alcohol, although the enzyme responsible for catalyzing this reaction has yet to be identified in bacteria. We report here the purification and characterization of an enzyme from the wax ester-accumulating bacterium Marinobacter aquaeolei VT8, which is a proposed fatty aldehyde reductase in this pathway. The enzyme, a 57-kDa monomer, was expressed in Escherichia coli as a fusion protein with the maltose binding protein on the N terminus and was purified to near homogeneity by using amylose affinity chromatography. The purified enzyme was found to reduce a number of long-chain aldehydes to the corresponding alcohols coupled to the oxidation of NADPH. The highest specific activity was observed for the reduction of decanal (85 nmol decanal reduced/min/mg). Short-chain and aromatic aldehydes were not substrates. The enzyme showed no detectable catalysis of the reverse reaction, the oxidation of decanol by NADP+. The mechanism of the enzyme was probed with several site-specific chemical probes. The possible uses of this enzyme in the production of wax esters are discussed.
Wax esters, long-chain fatty acids linked to long-chain alcohols, serve many different functions in biological systems (4, 14). In plants, wax esters are a primary constituent of the outer waxy layer of leaves, where they function as a hydrophobic barrier to prevent water loss. Wax esters are also found in high concentration in the spermaceti organ of the sperm whale, where they are thought to aid in regulating buoyancy. The unique properties of this family of compounds have made them valuable as additives in cosmetic and medical formulations, as well as in high-grade lubricants and food additives. A small group of microbes has been shown to accumulate wax esters, probably as energy storage compounds (3, 5). There is interest in understanding the pathways and enzymes utilized in bacterial wax ester synthesis as a possible way to produce wax esters of interest to biotechnology (4, 6, 7).
Early work on bacterial wax synthesis indicates a four-enzyme pathway starting from long-chain fatty acids (Fig. 1) (7, 21). The first step in the pathway involves the formation of fatty acyl coenzyme A (CoA) from a fatty acid by the action of a fatty acid:CoA ligase that also utilizes CoA and MgATP. The fatty acyl-CoA is reduced to the corresponding fatty aldehyde by the NADPH-dependent acyl-CoA reductase (11). It is proposed that the fatty aldehyde is then reduced to a fatty alcohol by a fatty aldehyde reductase (FALDR), although the enzyme responsible has yet to be identified and confirmed in any bacteria. The final step in wax ester formation is catalyzed by a wax ester synthase/acyl-CoA:diacylglycerol acyltransferase (WS/DGAT) (5). This enzyme has been purified from bacteria and partially characterized (5, 16, 19). The enzyme shows a broad substrate range for alcohols, ranging from ethanol to triacontanol and for acyl-CoAs of various lengths. Branched and aromatic alcohols are also substrates (8, 16, 19). The wide substrate range of this enzyme offers the possibility of producing a number of wax esters biologically, including diesel substitutes (6).
FIG. 1.

Proposed bacterial pathway for wax ester synthesis from fatty acids.
A significant obstacle in utilizing the WS/DGAT enzymes has been the inability to identify an FALDR in a bacterial system that would supply the fatty alcohol required for wax ester synthesis (7). With such an enzyme in hand, a multienzyme expression system could be developed that would allow biological production of wax esters as high-value oils and waxes, all derived from renewable resources.
In this work, we have cloned and expressed a gene from the marine bacterium Marinobacter aquaeolei that was predicted to be an FALDR in the pathway for wax ester synthesis, based on similarity analysis. The gene for this putative FALDR was identified by searching known bacterial genomes with a proposed FALDR gene (CER4) sequence from the plant Arabidopsis (13). Marinobacter species are known to accumulate branched wax esters when grown in a medium supplemented with phytol, but detailed reports regarding the production of wax esters from simple sugars have not been reported (1). The product of the putative FALDR gene in M. aquaeolei shares primary sequence identity with that of the CER4 gene from Arabidopsis and is thus predicted to serve a similar function in wax ester synthesis (13). The product of the FALDR gene from M. aquaeolei has been cloned and purified as a fusion protein with the maltose binding protein (MBP). A description of this enzyme and its substrate specificity is reported.
MATERIALS AND METHODS
Materials.
All reagents were purchased from Sigma-Aldrich Company (St. Louis, MO) unless otherwise specified. Restriction enzymes, T4 DNA ligase, and Escherichia coli strain TB1 were obtained from New England Biolabs (Ipswich, MA). Bovine serum albumin was fraction V (Sigma P/N A2153) and was prepared fresh daily in the same buffer as that used for the assay. NADP+-dependent alcohol dehydrogenase from Thermoanaerobium brokii (Sigma P/N A8435) was prepared fresh the day of use for control experiments.
Strains and plasmid constructions.
M. aquaeolei VT8 was obtained from the American Type Culture Collection (ATCC) and was grown initially on ATCC medium 2084 (Halomonas medium [2]) at 30°C. Plates of the 2084 medium were prepared by adding 1.5% Bacto agar (BD, Franklin Lakes, NJ). Genomic DNA was isolated by first growing 1 liter of cells to an optical density at 600 nm of 0.6 and then collecting the cells by centrifugation at 7,000 × g. The cell pellet was washed once with 50 mM phosphate buffer (pH 7.2). The cells were then suspended in 5 ml of 50 mM Tris-HCl buffer at pH 7.8 with 2% Triton X-100 and 5 mg of lysozyme. The suspension was allowed to sit for 10 min at room temperature and was then incubated in boiling water for 5 min. The solution was centrifuged at 10,000 × g for 10 min. The supernatant was retained, and an equal volume of isopropanol was added to precipitate the DNA. The DNA was washed once with 10 ml of isopropanol and then twice with 10 ml of ice-cold ethanol. The DNA was allowed to air dry and was then subjected to restriction digest and purification following the desalting protocol from the Qiaex II kit (Qiagen, Valencia, CA).
A gene (accession number NC_008740.1) proposed to code for an FALDR from Marinobacter aquaeolei VT8 was amplified from genomic DNA by using the primers BBP244, 5′ GATGAGGATCCATGGAGCAATACAGCAGGTACATCACGCTGAC, and BBP245, 5′ GACTGGAATTCAGGCAGCTTTTTTGCGCTGGCGCGC (where the underlined sequences are the NcoI and EcoRI sites, respectively), following the Failsafe protocol (Epicenter, Madison, WI), using buffer G and an annealing temperature of 60°C. The PCR product was purified using the Qiaex II desalting protocol (Qiagen, Valencia, CA) and was digested with EcoRI and NcoI along with the plasmid pBB052 (a pUC19 derivative with kanamycin in place of ampicillin for selection, and an N-terminal His tag followed by an NcoI site). The reaction was terminated by heat inactivation at 65°C, followed by ligation. Plasmids were maintained in E. coli strain JM109 unless specified otherwise. The gene encoding the N-terminal His-tagged protein was then transferred to the pET-30A vector to express the FALDR protein in E. coli strain BL21. Additionally, the EcoRI site following the FALDR gene in the original vector was removed by digestion with EcoRI, filling in with T4 DNA polymerase, and ligation of the blunt-end product. A new EcoRI site was then introduced just upstream of the second codon of the gene by PCR amplification, removing the methionine start codon and preparing the gene to be placed in-frame with an EcoRI site following an MBP from the pMAL-c2x plasmid (New England Biolabs, Ipswich, MA). The modified gene was then transferred to the pMAL-c2x plasmid and sequenced to confirm that it contained no mistakes, prior to transferring this plasmid to the E. coli strain TB1 for expression of the MBP fused to the FALDR.
Enzyme purification.
One liter of LB medium supplemented with ampicillin (100 μg/ml) in a 4-liter flask was inoculated with 16 ml of an overnight culture of E. coli TB1 transformed with the pMAL-c2x vector expressing the MBP-FALDR fusion and was grown for approximately 5 hours at 37°C prior to induction by the addition of 50 mg of IPTG (isopropyl β-d-1-thiogalactopyranoside), after which the culture was grown for an additional 2 hours at room temperature. Cells were harvested by centrifugation and were immediately frozen for later use. The cells were suspended in 30 ml of column buffer (20 mM Tris-HCl [pH 7.4], 200 mM NaCl, 1 mM EDTA) supplemented with 0.1 mM phenylmethylsulfonyl fluoride. Cells were lysed by passing the suspended cells through a French pressure cell (SLM Aminco) three times in the presence of DNase (P/N DN-25; Sigma-Aldrich). Soluble protein was collected by centrifuging the cell lysate at 17,500 × g for 10 min. The supernatant was diluted threefold with column buffer and was then applied to a column containing a 10-ml column bed of amylose resin (New England Biolabs, Ipswich, MA), and the column was washed with 30 ml of column buffer containing 1 M NaCl, followed by a second wash with 30 ml of column buffer. The fusion protein was eluted with 15 ml of column buffer containing 10 mM maltose.
Preliminary assays of FALDR activity.
For initial assessment of the activity of the purified FALDR enzyme, 50 μg of purified FALDR protein was added to a reaction mixture containing 100 mM of Tris buffer at pH 7.9, 100 mM NaCl, 2.4 mM of either NADPH or NADH as a reductant, and decanal, oleic acid, and hexadecanol as possible substrates. The assays were run under an argon atmosphere in septum-sealed vials overnight at room temperature with constant gentle mixing. The products of the reactions were then extracted from the buffer by adding an equal volume of hexane, and organic layer components were analyzed by gas chromatography equipped with a flame ionization detector (30-m by 0.32-mm inner diameter with 0.5-μm film thickness, with argon as a carrier and a temperature ramp from 60°C to 360°C, increasing at 10°C per minute) (Forte HT5 column; SGE Analytical Science, Austin, TX). Samples containing new peaks not present in a control sample were also run through a gas chromatograph equipped with a mass spectrometer (Shimadzu GC-2010 and GCMS-QP2010S) to assign the identity of the product.
Fatty aldehyde continuous spectrophotometric assay development.
All assays were performed in a sealed quartz cuvette with a 1-cm path length. The cuvette and solutions were first degassed with argon on a manifold to remove oxygen from the headspace and solutions. Substrates were initially disbursed into a buffer solution containing bovine serum albumin at a concentration of 0.5 mg/ml by sonication for three 10-s intervals, using a microtip sonicator (Branson, Danbury, CT). The initial assay for pH optimization included a buffer of 100 mM 3-(N-morpholino)propanesulfonic acid (MOPS), 100 mM 2-(N-morpholino)ethanesulfonic acid (MES), and 100 mM [(2-hydroxy-1,1-bis(hydroxymethyl)ethyl)amino]-1-propanesulfonic acid (TAPS). For reduction assays, 75 μl of a 2 mg/ml NADPH stock was added to bring the initial concentration of NADPH in the cuvette to approximately 200 μM and the approximate absorbance at 340 nm to 1.2, based on the reported extinction coefficient of 6,220 M−1 cm−1 for NADPH (22). In all assays, the reaction was run for at least 2 minutes with the NADPH and substrate present, prior to the addition of any enzyme, to obtain a background oxidation rate for the NADPH. The absorbance reading at 340 nm was read every 0.5 s, and the reaction was run until a steady rate was achieved for at least 60 s. A linear fit of the data was then used to establish the rate, and the initial background rate was subtracted, to determine the rate associated with substrate reduction by the enzyme.
When the assay was used to check for activity with NADH in place of NADPH, this compound was simply substituted at the same concentration and the assay run as described above. Reactions using either NADP+ or NAD+ used the same concentration but, instead, followed the increase in absorbance monitored at 340 nm. Once the pH optimum was established for the enzyme, TAPS and MOPS were removed from the buffer, and only MES was used.
RESULTS AND DISCUSSION
Expression of a proposed FALDR.
A previous report by Rowland et al. proposed that the gene CER4 from Arabidopsis thaliana encodes a wax ester biosynthetic enzyme (13). When this gene was disrupted in Arabidopsis, a phenotype resulted, with significant decreases in concentration of measured primary alcohols and wax esters and slightly elevated levels of aldehydes found in the waxy cuticle that coats the aerial surfaces of the plant (13). Based on this evidence, the CER4 gene was tentatively assigned as an FALDR gene. In an effort to find a similar enzyme in bacteria, this gene was used here in a BLAST search of several completed bacterial genomes that are known to contain the wax ester synthase (WS/DGAT) gene. Two species of marine bacteria, including Marinobacter aquaeolei VT8, were found to contain an open reading frame with moderate similarity (48% positive and 27% identical for one species) to the CER4 gene.
The gene for the putative FALDR enzyme from M. aquaeolei VT8 was first cloned into a pUC19 derivative vector with an N-terminal His8 tag. This gene was further modified to remove various restriction sites through silent mutations while maintaining the integrity of the protein sequence, and the modified gene was inserted into a Novagen pET vector (EMD Chemicals, Inc., San Diego, CA) to include the N-terminal histidine tag. While initial expression experiments using the pET vector showed a high level of expression of the protein, initial attempts to purify the protein were hampered by low solubility. In all cases, the majority of the expressed protein associated with the cell debris following cell disruption and centrifugation (Fig. 2). Efforts to improve solubility by inclusion of a variety of common detergents {Tween 20, Triton X-100, dodecyl maltoside, and 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate (CHAPS)} in varied concentrations were met with limited success and were eventually abandoned for other approaches, as the majority of the FALDR seemed to remain in the insoluble pellet.
FIG. 2.

FALDR SDS-PAGE. (A) Solubility of N-terminal histidine-tagged FALDR. Lane 1, molecular-weight markers (sizes as indicated); lane 2, soluble fraction of total cell lysate; lane 3, insoluble fraction of total cell lysate. (B) Purification of MBP-FALDR fusion protein. Lane 1, molecular-weight markers (sizes as indicated); lane 2, insoluble fraction of total cell lysate; lane 3, soluble fraction of total cell lysate; lane 4, amylose column flowthrough; lane 5, protein eluted with 10 mM maltose.
Several approaches were undertaken to improve the solubility of the FALDR enzyme. Eventually, the solubility problem was overcome by creating a fusion protein utilizing the highly soluble MBP. This was accomplished by inserting the modified gene into the multiple cloning site of the pMAL-c2x vector (New England Biolabs), which when expressed in the proper E. coli host strain, produced a protein that remained predominantly in the soluble fraction, even without the inclusion of any detergents (Fig. 2). This approach allowed a quick purification by using an amylose resin (New England Biolabs, Ipswich, MA), to bind the MBP portion of the fusion protein, and maltose for elution, resulting in a relatively pure protein (approximately 90% pure by sodium dodecyl sulfate-polyacrylamide gel electrophoresis [SDS-PAGE] analysis). The other minor protein components seen in the preparation are presumed to be related to the FALDR, resulting from either proteolytic degradation or premature termination of expression by the host system, as the associated bands were specific to the expression of the FALDR protein.
Initial assessment of the activity of the FALDR.
Preliminary experiments were run with the FALDR enzyme to probe possible substrates by mixing either NADH or NADPH with an aldehyde (decanal), an alcohol (hexadecanol), or a fatty acid (oleic acid) and then looking for changes in concentration of one or more of the potential substrates by gas chromatography analysis. The assays were allowed to run overnight in a sealed vial under an atmosphere of argon to maintain the reduced forms of the nicotinamide coenzymes. Of the possible substrates included in this experiment, only the vial containing NADPH showed a decrease in the decanal peak and the generation of a new peak, which correlated with the retention time for decanol, and was confirmed by mass spectrometry. The reverse reaction using NADP+ and hexadecanol or decanol showed no detectable levels of hexadecanal or decanal production. This initial result pointed to the likelihood that the enzyme is an NADPH-dependent FALDR, and further experiments were run to characterize this activity. While none of the experiments performed as part of this work indicated that the enzyme is capable of oxidizing the product decanol using NADP+, it is possible that this reaction is extremely slow compared to the reduction of the aldehyde and, thus, is below the level of detection. The activity of medium-chain alcohol dehydrogenases (MCADH) from a broad range of species shows rates of oxidation of alcohols that are 10% of the rates of the reduction of the corresponding aldehyde, so it is possible that this rate is too low to detect (10).
Continuous assay of FALDR activity.
Having established that the FALDR would utilize NADPH as a substrate, it was possible to employ a continuous spectrophotometric assay to monitor substrate reduction rates based on the loss of absorbance at 340 nm when NADPH is oxidized to NADP+. Using this assay, it was possible to establish the pH dependence of the reduction reaction. The rate of reaction was highest at pH 6.3, with a steep increase in activity from pH 8.0 to 6.3. The rate at pH 6.3 was only slightly higher than that at pH 6.5 (30% higher). Several factors had to be considered in establishing the optimal pH of the assay. The first was the background oxidation of NADPH without any enzyme present. NADPH naturally degrades to NADP+ at neutral and acidic pH values and is most stable under alkaline conditions (12, 23). The assays were run under an argon atmosphere to minimize oxidation of the NADPH by O2. Another consideration was the rapid degradation that occurs at the lowest pH values. While the highest enzyme rates were observed at pH 6.3, the rates were highly sensitive to slight pH variations at this value. So, for standard assays used here, a pH of 6.5 was selected, where the activity was minimally affected by slight variations in pH.
Substrate specificity and kinetic parameters.
Six commercially available substrates were examined for reduction by the FALDR enzyme. The long-chain aldehydes decanal and dodecanal were examined, as well as the smaller aldehydes butanal, hexanal, and octanal. The larger, unsaturated aldehyde cis-11-hexadecenal was also tested. Testing of other long-chain aldehydes will require those to be synthesized and purified. In addition to these straight-chain aldehydes, activity was also tested with the aromatic aldehyde benzaldehyde.
Utilizing the continuous assay for FALDR activity, the dependence of rates on substrate concentration was determined for all of the commercially available substrates described above. Each assay was repeated multiple times to confirm the results from each single determination. The results obtained for cis-11-hexadecenal are shown in Fig. 3 and are representative of the results obtained for repeated determinations and for the other substrates. The data were fit to the Michaelis-Menten equation, revealing a Km of approximately 177 μM and a maximum velocity of 63 nmol/min/mg. The activity obtained for this substrate was slightly lower than that obtained for decanal. It is difficult to benchmark these rates, as no comparable enzyme has been purified. In addition, several factors could limit the observed activity. All of the substrates tested have limited solubility in aqueous solutions and must first be suspended in solution via sonication. Even under these conditions, it is likely that the substrates exist inside micelles. The sonication process utilized here could result in the partial degradation of the substrate over time. Additionally, the suspension of the substrate in solution is only temporary, and the substrate slowly separates from aqueous solution. Due to these limitations, the measured activity is likely to be an underestimation of the actual activity. In the cell, the enzyme and substrate are likely associated with the hydrophobic membranes. The Km values obtained for each of the substrates that showed activity were in the micromolar range, indicating that the enzyme should be active in the cell even at low substrate concentrations. It was also observed that the enzyme was unstable over time, even when stored at 4°C, losing as much as 50% of the activity over a period of a week. Though the purification is quite rapid, it is uncertain how much activity is lost during preparation.
FIG. 3.
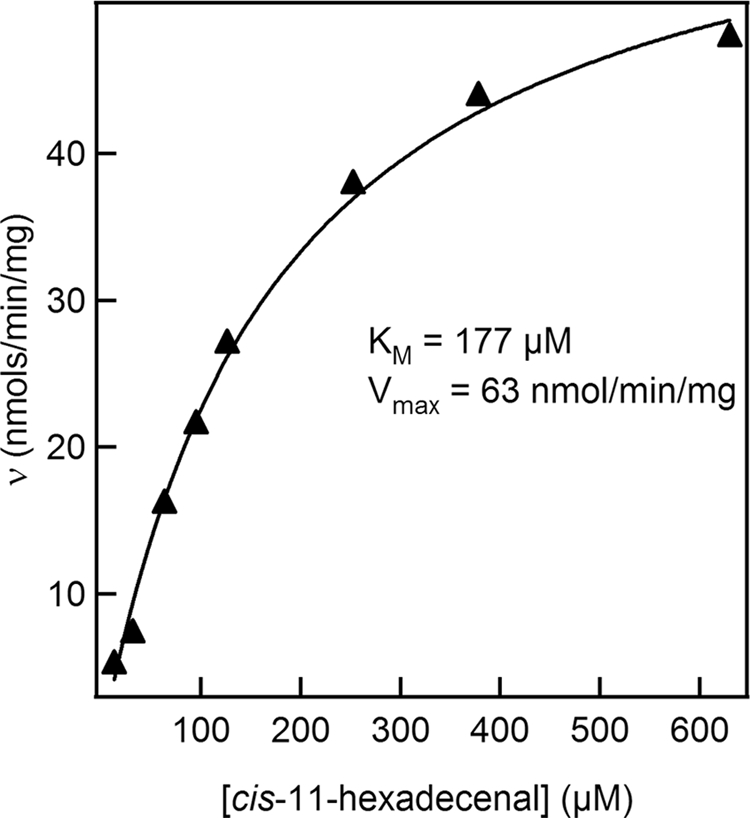
Substrate saturation for the MBP-FALDR fusion protein. Aldehyde reductase activity of MBP-FALDR was measured spectrophotometrically by monitoring the oxidation of NADPH continuously at 340 nm. Absorbance-versus-time measurements were used to determine initial rates of aldehyde reduction at each concentration of cis-11-hexadecenal. The initial rates were fit to the Michaelis-Menten equation to determine Vmax and Km. Experiments were repeated three times to optimize and confirm reproducibility. The results shown here represent the data obtained from a single set of data. The curve fit and standard deviation for this set of data are 177 ± 17 μM for Km and 63 ± 3 nmol/min/mg for Vmax.
As can be seen in Table 1, the FALDR enzyme required a minimal-chain-length C8 aldehyde to show significant activity. The shorter substrates, butanal (C4) and hexanal (C6), showed no apparent activity under these conditions, while the activity for octanal (C8) was approximately half the activity obtained for decanal (C10), which had the highest activity of the substrates tested (85 nmol/min/mg). A further investigation of the octanal activity revealed that the apparent Km for this substrate may be very close to the concentration tested here (∼750 μM), so that the activity would be greater with higher concentrations of substrate. The activity was lower with dodecanal (C12), though the apparent Km was lower (∼200 μM) than that for octanal and similar to that for decanal (∼100 μM). In this instance, the solubility may play a more important role, as dodecanal was the longest saturated aldehyde commercially available that was used in this work. The substrate cis-11-hexadecenal (C16) showed activity comparable to that for decanal, though this substrate does contain a site of unsaturation, which could change accessibility to the active site versus dodecanal. It is also possible that the results here are related more to substrate solubility and availability to the enzyme. Because a primary wax ester found in M. aquaeolei VT8 grown under nitrogen-deficient growth conditions is hexadecyl hexadecanoate (unpublished data), it would be expected that hexadecanal would be a likely natural substrate of this enzyme. Future work will include efforts to produce adequate quantities of purified hexadecanal for kinetic studies. Finally, the aromatic ring containing aldehyde benzaldehyde showed no apparent activity when assayed at similar concentrations, indicating that the active site may be specific for straight-chain aldehydes (saturated or unsaturated).
TABLE 1.
FALDR substrate comparisons
 |
aAll activity assays were measured at a substrate concentration of 100 μg/ml, following the assay protocol described in Materials and Methods, using 160 μg of enzyme. The percentage of activity reported here is the result from a single sample analysis with a specific set of data. The measurements were repeated several times to confirm reproducibility.
bThe activity for decanal was 85 nmol/min/mg for this set of data, and all other activities were divided by this result and multiplied by 100 to obtain the percentage.
Since all the assays were conducted with the MBP-FALDR fusion protein, a principal concern was whether the fusion protein accurately represented the activity of the wild-type enzyme. To examine whether the MBP affected the activity of the wild-type enzyme, the MBP was removed from the N terminus of the FALDR protein by factor Xa cleavage. The near complete cleavage of MBP was verified by SDS-PAGE (see the supplemental material). This cleavage is facilitated by the incorporation of a factor Xa cleavage site (Ile-Glu-Gly-Arg) just upstream of the EcoRI site of the pMAL-c2x vector. Assays conducted with the cleaved FALDR protein and decanal did not reveal any loss or improvement in the rate of substrate reduction from the rate exhibited by the MBP-FALDR fusion protein.
Reversibility of the enzyme with fatty alcohols.
To test the possible reversibility of the FALDR, the oxidation of a number of alcohols to the corresponding aldehyde, using NADP+ as the oxidant, was tested. In these assays, increases in absorbance at 340 nm were monitored. Under no conditions did any reduction of NADP+ occur in the presence of the fatty alcohols tested. As a control, the reduction of NADP+ by the alcohol dehydrogenase from Thermoanaerobium brokii was assayed with 2-propanol and the same stock of NADP+ to confirm the integrity of the assay. These results showed no evidence that FALDR is capable of catalyzing the reverse reaction, the oxidation of a fatty alcohol to a fatty aldehyde. Similarly, reactions were followed in the same manner by substituting NADH or NAD+ for NADPH or NADP+ using decanal and decanol, respectively, with no indication of activity over background with any combination except with decanal and NADPH. Based on this, it is proposed that the enzyme exhibits activity only for the reduction of fatty aldehydes in an NADPH-dependent reaction.
Inhibition studies.
To probe the possible mechanism of the FALDR, some potential chemical inhibitors were tested for effects on the reduction of decanal by the enzyme in the presence of NADPH. In all cases, the compound was tested initially at concentrations of 1.0 mM, and if a significant inhibition was found, it was also tested at lower concentrations (Table 2). The metal chelator EDTA, which is used during purification of the enzyme to limit the activity of metalloproteases, had little effect on the activity. Reductants, such as ascorbic acid, dithiothreitol, and β-mercaptoethanol, also showed little effect (less than 25% decrease in activity). Only dithionite showed a significant decrease in activity. At higher concentrations, this was difficult to assess fully, as the dithionite interferes with the absorbance at 340 nm where activity is measured. At the lower concentration of 250 μM, the interference is lower, and the inhibition is more pronounced. This could be an indication of an active site residue or cofactor that is susceptible to reduction. The two metal chelators dipyridyl and diethyldithiocarbamate showed only a moderate inhibition of activity at elevated concentrations of 1.0 mM. This would indicate that if a transition metal is involved in the catalysis, it is not readily accessible to such chelators. Finally, the ability of decanol to inhibit reduction of decanal was also tested. Here, inhibition of almost 45% at the two concentrations tested was observed, indicating a possibility that product inhibition can regulate activity, even though the enzyme is apparently not reversible. Future studies will investigate the nature of this inhibition further and will also utilize group-specific reagents to determine the nature of the active site chemistry utilized by this enzyme.
TABLE 2.
Inhibition of decanal reductiona
| Substrate | % Activity at inhibitor concn of:
|
|
|---|---|---|
| 1.0 mM | 0.25 mMb | |
| EDTA (disodium salt) | 81 | ND |
| Ascorbic acid | 76 | ND |
| Dithiothreitol | 76 | ND |
| β-Mercaptoethanol | 75 | ND |
| Dithionite | 25 | 8 |
| Diethyldithiocarbamate | 50 | 61 |
| Dipyridyl | 46 | 59 |
| Dodecanol | 54 | 55 |
Assays were run in 100 mM MES buffer (pH 6.5) with 200 μM NADPH, 250 μM decanal, and 125 μg of FALDR fusion protein. All compounds were dissolved in buffer, except dipyridyl, which was dissolved in dimethyl sulfoxide. Inclusion of dimethyl sulfoxide did not result in decreased activity at the same concentration. The percentage of activity reported here is the result from a single sample analysis with a specific set of data. The measurements were repeated several times to confirm reproducibility. Activity is reported as the percentage of activity remaining in the presence of each compound based on the activity without any inhibitor present.
ND, not determined.
Contrasting the FALDR and alcohol dehydrogenases.
As a means of comparison, MCADH can reduce a broad range of aldehydes, including propanal, butanal, hexanal, octanal, and benzaldehyde (9, 17, 20). Studies conducted with the MCADH enzyme from Saccharomyces cerevisiae did not test the activity of the enzyme with longer-chain substrates such as decanal and dodecanal. For the substrate octanal, the largest saturated aldehyde tested with this MCADH, the activity was reported to be approximately 150 μmol/min/mg (9), which is higher than the activities found here for FALDR. An MCADH from Acinetobacter, however, was shown to be active with the long-chain aldehyde tetradecanal (17). However, this enzyme exhibited, with the substrate tetradecanal, only 7.2% of its maximal activity obtained with the substrate heptanal (17). Larroy et al. demonstrated, with a multiple sequence alignment, that the MCADH enzyme from both S. cerevisiae and Acinetobacter exhibited strict conservation of residues, typical of MCADH (9). Using the FALDR amino acid sequence from M. aquaeolei (YP_959486), a BLAST 2 sequence (18) search was conducted (using the BLOSUM62 matrix with a default expect value of 10.0) against the amino acid sequences for the MCADH from both S. cerevisiae (NP_014051) and Acinetobacter (BAB12270). No significant similarity was found.
Even though the MCADH from Acinetobacter exhibited activity toward the fatty aldehyde tetradecanal, its effective participation in the formation of wax esters would likely be limited, as MCADH enzymes are generally soluble and would be partitioned from the lipophilic substrates necessary for wax synthesis. Additionally, these enzymes are highly active with other smaller aldehydes, such as butanal and hexanal, which were poor substrates for the FALDR. M. aquaeolei does contain a gene with high similarity to that of the reported MCADH enzyme class, and efforts to clone and characterize this gene are also underway. Those efforts will include characterization with longer aldehydes (decanal, etc.) that were not tested for the MCADH enzyme from Saccharomyces cerevisiae (9) but were tested for the Acinetobacter MCADH (17).
Summary.
This work has demonstrated that M. aquaeolei contains a gene that encodes an FALDR. This enzyme is specific for NADPH and does not appear to be reversible. In the native form, the enzyme is relatively insoluble and partitions with the membrane fraction, where substrates would also likely accumulate. It is possible that this enzyme may be involved in the synthesis and accumulation of wax esters under conditions of nutrient deficiency. Previous reports have identified genes involved in the synthesis of fatty aldehydes from species of Acinetobacter (11, 15), but to our knowledge, this is the first report of a bacterial enzyme that is capable of specifically reducing fatty aldehydes to the corresponding alcohol. The enzyme is able to reduce a range of aldehydes, although a minimum size is required (larger than hexanal), and no activity was found for the reduction of the aromatic compound benzaldehyde. The highest activity found here was for the substrate decanal. The activity, while lower than that found for MCADH, is significantly higher than that reported for the acyl-CoA reductase from Acinetobacter (11), which is believed to catalyze the preceding step in the pathway, the reduction of a fatty acid CoA substrate to the fatty aldehyde. The FALDR enzyme activity was not impacted significantly by reductants such as ascorbic acid but was dramatically lowered when dithionite was included in the buffer. The discovery of this FALDR in M. aquaeolei VT8 now paves the way for inclusion of this enzyme along with others in the pathway as a way to synthesize wax esters of specified lengths in bacteria.
Supplementary Material
Acknowledgments
We thank the Sustainable Energy Research Center at Utah State University and the Utah Science, Technology, and Research Initiative for support.
Footnotes
Published ahead of print on 6 March 2009.
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1.Holtzapple, E., and C. Schmidt-Dannert. 2007. Biosynthesis of isoprenoid wax ester in Marinobacter hydrocarbonoclasticus DSM 8798: identification and characterization of isoprenoid coenzyme A synthetase and wax ester synthases. J. Bacteriol. 189:3804-3812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huu, N. B., E. B. M. Denner, D. T. C. Ha, G. Wanner, and H. Stan-Lotter. 1999. Marinobacter aquaeolei sp. nov., a halophilic bacterium isolated from a Vietnamese oil-producing well. Int. J. Syst. Bacteriol. 49:367-375. [DOI] [PubMed] [Google Scholar]
- 3.Ishige, T., A. Tani, Y. R. Sakai, and N. Kato. 2003. Wax ester production by bacteria. Curr. Opin. Microbiol. 6:244-250. [DOI] [PubMed] [Google Scholar]
- 4.Jetter, R., and L. Kunst. 2008. Plant surface lipid biosynthetic pathways and their utility for metabolic engineering of waxes and hydrocarbon biofuels. Plant J. 54:670-683. [DOI] [PubMed] [Google Scholar]
- 5.Kalscheuer, R., and A. Steinbüchel. 2003. A novel bifunctional wax ester synthase/acyl-CoA: diacylglycerol acyltransferase mediates wax ester and triacylglycerol biosynthesis in Acinetobacter calcoaceticus ADP1. J. Biol. Chem. 278:8075-8082. [DOI] [PubMed] [Google Scholar]
- 6.Kalscheuer, R., T. Stolting, and A. Steinbüchel. 2006. Microdiesel: Escherichia coli engineered for fuel production. Microbiology 152:2529-2536. [DOI] [PubMed] [Google Scholar]
- 7.Kalscheuer, R., T. Stöveken, H. Luftmann, U. Malkus, R. Reichelt, and A. Steinbuchel. 2006. Neutral lipid biosynthesis in engineered Escherichia coli: jojoba oil-like wax esters and fatty acid butyl esters. Appl. Environ. Microbiol. 72:1373-1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.King, A., J. W. Nam, J. Han, J. Hilliard, and J. G. Jaworski. 2007. Cuticular wax biosynthesis in petunia petals: cloning and characterization of an alcohol-acyltransferase that synthesizes wax-esters. Planta 226:381-394. [DOI] [PubMed] [Google Scholar]
- 9.Larroy, C., M. R. Fernández, E. González, X. Parés, and J. A. Biosca. 2002. Characterization of the Saccharomyces cerevisiae YMR318C (ADH6) gene product as a broad specificity NADPH-dependent alcohol dehydrogenase: relevance in aldehyde reduction. Biochem. J. 361:163-172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Manríquez, D., I. El-Sharkawy, F. B. Flores, F. El-Yahyaoui, F. Regad, M. Bouzayen, A. Latché, and J. C. Pech. 2006. Two highly divergent alcohol dehydrogenases of melon exhibit fruit ripening-specific expression and distinct biochemical characteristics. Plant Mol. Biol. 61:675-685. [DOI] [PubMed] [Google Scholar]
- 11.Reiser, S., and C. Somerville. 1997. Isolation of mutants of Acinetobacter calcoaceticus deficient in wax ester synthesis and complementation of one mutation with a gene encoding a fatty acyl coenzyme A reductase. J. Bacteriol. 179:2969-2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rover, L., Jr., J. C. B. Fernandes, G. de Oliveira Neto, L. T. Kubota, E. Katekawa, and S. H. P. Serrano. 1998. Study of NADH stability using ultraviolet-visible spectrophotometric analysis and factorial design. Anal. Biochem. 260:50-55. [DOI] [PubMed] [Google Scholar]
- 13.Rowland, O., H. Zheng, S. R. Hepworth, P. Lam, R. Jetter, and L. Kunst. 2006. CER4 encodes an alcohol-forming fatty acyl-coenzyme A reductase involved in cuticular wax production in Arabidopsis. Plant Physiol. 142:866-877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Samuels, L., L. Kunst, and R. Jetter. 2008. Sealing plant surfaces: cuticular wax formation by epidermal cells. Annu. Rev. Plant Biol. 59:683-707. [DOI] [PubMed] [Google Scholar]
- 15.Singer, M. E., and W. R. Finnerty. 1985. Fatty aldehyde dehydrogenases in Acinetobacter sp. strain Ho1-N: role in hexadecane and hexadecanol metabolism. J. Bacteriol. 164:1011-1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stöveken, T., R. Kalscheuer, U. Malkus, R. Reichelt, and A. Steinbüchel. 2005. The wax ester synthase/acyl coenzyme A:diacylglycerol acyltransferase from Acinetobacter sp. strain ADP1: characterization of a novel type of acyltransferase. J. Bacteriol. 187:1369-1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tani, A., Y. Sakai, T. Ishige, and N. Kato. 2000. Thermostable NADP+-dependent medium-chain alcohol dehydrogenase from Acinetobacter sp. strain M-1: purification and characterization and gene expression in Escherichia coli. Appl. Environ. Microbiol. 66:5231-5235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tatusova, T. A., and T. L. Madden. 1999. BLAST 2 S, a new tool for comparing protein and nucleotide sequences. FEMS Microbiol. Lett. 174:247-250. [DOI] [PubMed] [Google Scholar]
- 19.Uthoff, S., T. Stöveken, N. Weber, K. Vosmann, E. Klein, R. Kalscheuer, and A. Steinbüchel. 2005. Thio wax ester biosynthesis utilizing the unspecific bifunctional wax ester synthase/acyl coenzyme A:diacylglycerol acyltransferase of Acinetobacter sp. strain ADP1. Appl. Environ. Microbiol. 71:790-796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Valencia, E., C. Larroy, W. F. Ochoa, X. Parés, I. Fita, and J. A. Biosca. 2004. Apo and Holo structures of an NADP(H)-dependent cinnamyl alcohol dehydrogenase from Saccharomyces cerevisiae. J. Mol. Biol. 341:1049-1062. [DOI] [PubMed] [Google Scholar]
- 21.Wältermann, M., T. Stöveken, and A. Steinbüchel. 2007. Key enzymes for biosynthesis of neutral lipid storage compounds in prokaryotes: properties, function and occurrence of wax ester synthases/acyl-CoA:diacylglycerol acyltransferases. Biochimie 89:230-242. [DOI] [PubMed] [Google Scholar]
- 22.Windholz, M. (ed.). 1983. The Merck index, 10th ed. Merck & Co., Inc., Rahway, NJ.
- 23.Wu, J. T., L. H. Wu, and J. A. Knight. 1986. Stability of NADPH: effect of various factors on the kinetics of degradation. Clin. Chem. 32:314-319. (Erratum, 33:724.) [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.