

Abstract
Vibrio parahaemolyticus is a common food-borne pathogen that is normally associated with seafood. In 1996, a pandemic O3:K6 strain abruptly appeared and caused the first pandemic of this pathogen to spread throughout many Asian countries, America, Europe, and Africa. The role of temperate bacteriophages in the evolution of this pathogen is of great interest. In this work, a new temperate phage, VP882, from a pandemic O3:K6 strain of V. parahaemolyticus was purified and characterized after mitomycin C induction. VP882 was a Myoviridae bacteriophage with a polyhedral head and a long rigid tail with a sheath-like structure. It infected and lysed high proportions of V. parahaemolyticus, Vibrio vulnificus, and Vibrio cholerae strains. The genome of phage VP882 was sequenced and was 38,197 bp long, and 71 putative open reading frames were identified, of which 27 were putative functional phage or bacterial genes. VP882 had a linear plasmid-like genome with a putative protelomerase gene and cohesive ends. The genome does not integrate into the host chromosome but was maintained as a plasmid in the lysogen. Analysis of the reaction sites of the protelomerases in different plasmid-like phages revealed that VP882 and ΦHAP-1 were highly similar, while N15, ΦKO2, and PY54 made up another closely related group. The presence of DNA adenine methylase and quorum-sensing transcriptional regulators in VP882 may play a specific role in this phage or regulate physiological or virulence-associated traits of the hosts. These genes may also be remnants from the bacterial chromosome following transduction.
Vibrio parahaemolyticus is a common gram-negative food-borne enteropathogenic bacterium in Taiwan, Japan, and other Asian Pacific countries. The high incidence of this pathogen is undoubtedly the result of the frequent consumption of raw seafood in these countries. Clinical symptoms include diarrhea, abdominal cramps, nausea, vomiting, headache, fever, and chills, and incubation periods range from 4 to 96 h (26). Most clinical isolates are hemolytic on Wagatsuma agar (Kanagawa phenomenon positive) and produce a thermostable direct hemolysin, which is one of the main virulence factors in this pathogen (51).
Since 1996, gastroenteritis that is caused by particular O3:K6 strains (designated as pandemic O3:K6) of V. parahaemolyticus has been widespread in Southeast and East Asia (55), North America (27), South America (20), Europe (34), and Africa (3). It has been regarded as the first pandemic of this pathogen (43). These widespread pandemic O3:K6 strains have been found to be genetically closely related based on various molecular methods and appear to constitute a clone that differs markedly from the nonpandemic O3:K6 strains that were isolated before 1996 (4, 43, 55). The acquisition of the traits associated with these strains that account for the pandemic is of great concern.
In pandemic Vibrio cholerae, virulence factors are encoded in the genome of an integrated filamentous phage, CTXΦ (53). The integration of the filamentous phage CTXΦ into the bacterial genome conveys pathogenicity to V. cholerae and to Vibrio mimicus (8). Filamentous phage f237, which is similar to the previously described phage VF33, has been identified in a pandemic O3:K6 V. parahaemolyticus strain (38, 39). ORF8 in this phage is unique; no homologous sequence is present in the databases, and no biological function has hitherto been identified (39). This unique sequence has been used as a genetic marker of the presence of this filamentous phage (25), and the homologous sequence has been demonstrated in other pandemic O3:K6 and closely related strains (12, 37).
The role of this filamentous phage in the virulence of the pandemic O3:K6 strain has not been established. Along with the filamentous phage, the presence of temperate phages (VP882 and VP1092) in pandemic O3:K6 strains 882 and 1092, respectively, was demonstrated in our laboratory. In this study, VP882 was characterized using electron microscopy and genomic sequencing to provide more information on the possible roles of temperate phages on lysogenic conversion and on the diversity of this pathogen. Specific traits of the VP882 phage were identified by comparative genomic analysis with other vibriophages and plasmid-like prophages.
MATERIALS AND METHODS
Bacterial strains and media.
Table 1 presents the V. parahaemolyticus, V. cholerae, and Vibrio vulnificus strains used in this work. These strains were stored in culture broth with 10% glycerol at −80°C. The V. parahaemolyticus, V. cholerae, and V. vulnificus strains were cultured in tryptic soy broth (TSB) (Difco Laboratories, Sparks, NJ) with 3% NaCl, Luria-Bertani broth (LB) (Difco) with 2% NaCl, and brain heart infusion (BHI) (Difco) with 0.85% NaCl, respectively. All these bacteria were cultured at 37°C and shaken at 120 rpm.
TABLE 1.
V. parahaemolyticus, V. cholerae, and V. vulnificus strains used in this study
| Species and strain | Serotypea | Source | Yr isolated | Plaque formation by VP882 on strainb |
|---|---|---|---|---|
| V. parahaemolyticus | ||||
| A6 | O1:K25 | Thailand, clinical | 1999 | 0 |
| VP300 | O1:K38 | India, clinical | 1999 | 0 |
| 1261 | O1:K56 | Taiwan, clinical | 1998 | 2 |
| VP318 | O1:K56 | India, clinical | 1999 | 1 |
| 50707 | O2:K3 | Taiwan, clinical | 1999 | 2 |
| 50708 | O2:K3 | Taiwan, clinical | 1999 | 2 |
| 10603 | O2:K3 | Taiwan, clinical | 1998 | 0 |
| 10604 | O2:K3 | Taiwan, clinical | 1998 | 0 |
| AQ4733 | O3:K6 | Singapore, clinical | 1992 | 1 |
| AQ4853 | O3:K6 | Hong Kong, clinical | 1993 | 2 |
| 1134 | Pandemic O3:K6 | Taiwan, clinical | 1997 | 1 |
| 1167 | Pandemic O3:K6 | Taiwan, clinical | 1997 | 0 |
| 1662 | Pandemic O3:K6 | Korea, clinical | 1999 | 0 |
| VP81(2053) | Pandemic O3:K6 | India, clinical | 1996 | 0 |
| AN-8373(3146) | Pandemic O3:K6 | Bangladesh, clinical | 1998 | 1 |
| VP47(3505) | Pandemic O3:K6 | Thailand, clinical | 1998 | 0 |
| VP2(3724) | Pandemic O3:K6 | Korea, clinical | 1998 | 2 |
| 1256 | Pandemic O3:K6 | Taiwan, clinical | 1999 | 1 |
| VP45 | Pandemic O3:K6 | India, clinical | 1996 | 2 |
| VP155 | Pandemic O3:K6 | India, clinical | 1996 | 2 |
| 50396 | O4:K8 | Taiwan, clinical | 1999 | 0 |
| 50400 | O4:K8 | Taiwan, clinical | 1999 | 2 |
| 50401 | O4:K8 | Taiwan, clinical | 1999 | 1 |
| AN-16000(3145) | O4:K68 | Bangladesh, clinical | 1998 | 1 |
| A18 | O4:K68 | Singapore, clinical | 1998 | 2 |
| VP301 | O4:K68 | India, clinical | 1999 | 1 |
| VP289 | O5:K15 | India, clinical | 1999 | 2 |
| A17 | O6:K18 | Singapore, clinical | 1998 | 0 |
| 10701 | O7:K10 | Taiwan, clinical | 1998 | 2 |
| 10710 | O7:K10 | Taiwan, clinical | 1998 | 0 |
| V. cholerae | ||||
| 236-93 | O139 | Japan, clinical | 1993 | 1 |
| 294-94 | O139 | Japan, clinical | 1994 | 2 |
| AQ97853-1 | O139 | Taiwan, environmental | 1997 | 2 |
| AQ97811-5 | O139 | Taiwan, environmental | 1977 | 2 |
| 783 | O139 | Taiwan, clinical | 1997 | 0 |
| V. vulnificus | ||||
| CGJ05 | ND | Taiwan, clinical | 1993 | 0 |
| CGJ11 | ND | Taiwan, clinical | 1993 | 0 |
| CGJ64 | ND | Taiwan, clinical | 1993 | 2 |
| NV42 | ND | Taiwan, environmental | 1997 | 1 |
| NV48 | ND | Taiwan, environmental | 1997 | 0 |
Pandemic O3:K6 strains are those pandemic strains that were isolated after 1996. ND, not determined.
The characteristics of the plaques formed are given as follows: 1, clear plaque; 2, turbid plaque; 0, no plaque.
Mitomycin C treatment.
The growth of pandemic O3:K6 strains was monitored by measuring the absorbance of the culture at 550 nm. Mitomycin C (Sigma Chemical Co., St. Louis, MO) was added to the culture to a final concentration of 0.5 μg/ml in the early exponential phase when the absorbance of the culture was about 0.22 to 0.25 and incubated for another 3 h to burst the cells by the method of Koga and Kawata (28). Changes in cell morphology and viability during the mitomycin C treatment were examined by microscopy with BacLight kit staining (56).
Isolation of phage particles and viral DNA.
The lysed bacterial culture was centrifuged at 5,500 × g for 15 min, and the supernatant was filtered through a 0.22-μm membrane (Millipore Co., Billerica, MA). Sodium chloride was added to the filtrate to a final concentration of 1 M, and the filtrate was stored statically for 30 min on ice. Next, the phage particles were precipitated by adding 10% polyethylene glycol 6000 (Sigma) to the filtrate and incubating it at 4°C for at least 24 h (47). The phage particles were recovered by centrifugation at 5,500 × g for 20 min at 4°C, washed with phosphate-buffered saline containing 3% NaCl, and resuspended in a buffer that contained 10 mM of Tris-HCl and 1 mM of EDTA at pH 8.0, and the viral DNA was isolated and purified by the procedure of Sambrook et al. (47).
Electron microscopy.
The phage particles were fixed with 5% formaldehyde, washed in distilled water, and then negatively stained with 2% phosphotungstic acid, before being examined using a Phillips CM100 (FEI Company, Hillsboro, OR) transmission electron microscope (19).
Molecular techniques.
Quantification, electrophoresis, enzyme restriction, nucleotide sequencing, and other general manipulations of DNA were performed by the standard procedures as described by Sambrook et al. (47). The size of the phage genome was determined by pulsed-field gel electrophoresis (PFGE), following published methods (55). Briefly, the purified genome DNA was resolved using a CHEF II apparatus (Bio-Rad) at 200 V with a 0.5- to 1.5-min pulse for 18 h. Lambda DNA monocut mix (New England BioLabs, Beverly, MA) was used as a size marker in PFGE. 2-Log DNA ladder (New England BioLabs) and HindIII-digested lambda DNA were used as size markers in agarose gel electrophoresis.
The primers used for PCR were designed using Primer Design Assistant software (14). PCR was conducted using a GeneAmp PCR system 9700 (Applied Biosystems, Foster City, CA) as follows: an initial step of 2 min at 94°C; followed by 30 cycles, with 1 cycle consisting of 1 min at 94°C, 1 min at 60°C, and 2 min at 72°C; and a final step of 10 min at 72°C.
Whole-genome shotgun sequencing.
Genome sequencing was conducted at the VGH National Yang-Ming University Genome Research Center by the shotgun sequencing method (13). A 10-fold genome coverage was achieved using procedures briefly described herein. The genomic DNA of phage VP882 was fragmented by sonication, the single strands were digested by BalIII, and blunt-end fragments were made using T4 DNA polymerase and separated by agarose gel electrophoresis. Fragments (2.5 to 3.0 kilobase pairs [kbp]) were eluted, digested by SmaI, ligated to the pUC18 vector, and transformed into Max Efficiency DH5α competent cells (Invitrogen, Carlsbad, CA). Ampicillin-resistant transformants were selected and sequenced using a BigDye terminator cycle sequencing ready reaction kit (Applied Biosystems, Foster City, CA) with an automated sequencer (Applied Biosystems 3730). Sequences were jointly assembled using Phred/Phrap/Consed software (obtained from the University of Washington, Seattle). The accuracy, order, and orientation of contigs were examined based on linking information from forward and reverse sequence ends of each clone. Sequence gaps were closed by editing the end sequences of each contig, by primer walking on linking clones, and by sequencing PCR products from shotgun clones (13).
Gene prediction and annotation.
A combination of three gene prediction programs, Glimmer (version 3.02), GeneMark HMM (version 2.0), and Zcurve_V (version 1.0), were used to predict the protein-coding regions (17, 22, 32). The final designation of protein-coding sequence (CDS) is a unified set of the three prediction results. A protein sequence homology search of the predicted coding regions was performed using NCBI BLAST (version 2.2.15) against NCBI nonredundant protein, KEGG genes, and Uniprot Trembl protein databases to assign a hypothetical role or function to every CDS and thus generate a putative gene map of VP882 genome. CDSs with no blast hit were designated as unknown, and those with blast hits but unknown function were designated as putative phage-related proteins.
Bioinformatics analysis.
Inverted repeat sequence (IRS) analysis of the six plasmid-like prophage genomes was carried out with the Inverted Repeat Finder program (version 3.05) (54). Nucleotide sequence comparison of the six prophage genomes was analyzed with the DOTTER program (50). These prophage genomes included the Vibrio phage VP882 (GenBank accession no. EF057797), Halomonas phage ΦHAP-1 (NC_010342), Vibrio phage VHML (NC_004456), Yersinia phage PY54 (NC_005069), enterobacterial phage N15 (NC_001901), and Klebsiella phage ΦKO2 (NC_005857) (NC numbers in parentheses are GenBank nucleotide database accession numbers). IRSs were aligned and visualized by using the Weblogo program (version 2.8.2) (16). The multiple-sequence alignment program Clustalw2 (29) was used to align protelomerase protein sequences. The aligned sequences were taken from prophages VP882 (YP_001039865), ΦHAP-1 (YP_001686770), VHML (NP_758894), PY54 (NP_892077), N15 (NP_046924), and ΦKO2 (YP_006606) (YP and NP numbers are GenBank protein database accession numbers). Comparison of the important residues of the protelomerase catalytic domain was also performed.
Plaque formation.
The susceptibility of various strains of V. parahaemolyticus, V. cholerae, and V. vulnificus to phage VP882 was determined by spot assay. Fifty microliters of bacterial culture (about 104 CFU/ml) was spread on different agar plates (TSB with 3% NaCl for V. parahaemolyticus, LB with 2% NaCl for V. cholerae, and BHI with 0.85% NaCl for V. vulnificus) on which 20 μl of viral suspension (about 104 PFU/ml) was spotted. The plates were incubated at 37°C for 16 h, and the characteristics of the plaques were observed.
Nucleotide sequence accession number.
The genome sequence of the VP882 phage was deposited in the GenBank database under accession number EF057797.
RESULTS AND DISCUSSION
Isolation and general features of VP882.
Several phages have been isolated and characterized in V. parahaemolyticus—the Myoviridae VP16T, VP16C (48), and KVP241 (35), the Podoviridae VpV262 (23), and the Inoviridae f237 (11) and VF33 (12). In this study, mitomycin C was added to the cultures of 11 V. parahaemolyticus pandemic O3:K6 strains (strains 872, 882, 938, 943, 957, 1078, 1092, 1100, 1119, and 1163 from Taiwan and strain 1189 from Thailand), and the turbidity of strain 882 and 1092 cultures increased over 1 hour, before falling rapidly, suggesting a typical burst of the cells caused by phage particles that were formed within the cells (42). Cells of other strains were enlarged and elongated without lysis, as observed under a microscope, and identified as dead by BacLight kit staining (data not shown) (56). The phages from strains 882 and 1092 were named VP882 and VP1092, respectively.
Electron microscopy revealed that the VP882 and VP1092 particles were morphologically similar; only VP882 was further characterized in this work. VP882 phage exhibited a polyhedral head (48 ± 2 nm) with a rigid long tail (156 ± 8 nm), which was loosely associated with a special sheath-like structure (length, 67 ± 5 nm; width, 20 ± 1 nm) (Fig. 1). VP882 was identified as a Myoviridae phage based on the morphological characteristics (a polyhedral head, a neck/collar region, and a sheathed rigid tail) (Fig. 1). The tail sheath of VP882 was probably loosely associated with the tail and attached to it at various positions or even dislodged in the viral particles (Fig. 1). The heads of VP882 (48 nm) were smaller than those previously described for Myoviridae, and they had diameters of 53 to 160 nm (21, 41, 46). The shape and size of VP882 were similar to those of the VHML phage of Vibrio harveyi (41), while VP882 had a shorter tail than ΦHAP-1, a similar marine phage, did (36).
FIG. 1.

Electron micrograph of the VP882 particles. Bar, 50 nm.
Susceptibility to lysis by VP882.
Thirty clinical V. parahaemolyticus strains (10 pandemic O3:K6 strains, 2 nonpandemic O3:K6 strains isolated before 1996; 18 other serotypes), 5 V. cholerae O139 strains, and 5 V. vulnificus strains were assayed for lysis by phage VP882. High percentages of these Vibrio species were infected and yielded clear or turbid plaques (Table 1). Nineteen V. parahaemolyticus strains (63%) were lysed and formed clear plaques (8 strains) or turbid plaques (11 strains). The formation of clear or turbid plaques was unrelated to the serotypes of these V. parahaemolyticus strains. Four out of five V. cholerae O139 strains formed plaques, while only one formed a clear plaque. Two out of five V. vulnificus strains formed plaques, and only one formed a clear plaque.
Phages are commonly found in marine environments and have been isolated from 28% of marine molluscan shellfish, crustaceans, seawater, and sediments. The predominant phage types are specific for some strains of V. parahaemolyticus and some other agar-digesting and psychrophilic Vibrio species (6). These marine phages do not infect Vibrio alginolyticus or V. cholerae (5, 6). Recently, a survey of the water column in northwestern America revealed that V. parahaemolyticus phages, mostly siphoviruses, are frequently isolated and infect high percentages of the host strains, especially those isolated from oysters and sediments, and half of these phages are not species specific and can infect V. alginolyticus strains (15). This investigation demonstrated that VP882 was probably a marine phage that exhibits interspecies infection in marine vibrios and V. cholerae, which proliferates well in freshwater and grows well in lake water samples amended with different concentrations of NaCl (52).
Genome properties.
The size of the genomic DNA molecule was determined by PFGE to be about 38 kbp. The genome sequencing of phage VP882 revealed that the genome size was 38,197 base pairs (bp) (GenBank accession no. EF057797), which was close to the size determined by PFGE. The G+C percentage was 56.95%.
Analysis of the genome sequence identified 71 putative open reading frames (ORFs). Based upon translated sequence homology, 27 are putative phage or bacterial genes with hypothetical functions; 15 are putative phage-related genes or other genes with unknown functions (ORF5, -14, -17, -22, -25, -26, -30, -38, -45, -51, -52, -65, -69, -70, and -71), and 29 are unknown (exhibited no BLASTp hit). The table in the supplemental material presents details of position, size, and nearest match with the results of a BLASTp search.
Structural module.
The ORFs of the VP882 genome are arranged in a typical modular structure. The major structural genes are arranged in the order of portal protein, capsid protein, tail plate assembly genes, tail fiber, tail sheath, tail tube, and tail length protein. ORF6 to ORF34 encode the structural proteins (Fig. 2), including portal protein (ORF6), capsid (ORF9), tail proteins (ORF12, -20, -21, -29, -31, -32, and -34), baseplate and assembly proteins (ORF16, -18, and -19), and tail sheath protein (ORF28). These structural genes are organized in a manner similar to those of lambdoid phages and other plasmid-like phages, with head genes near the left end and tail genes clustered immediately to their right. All of these structural genes are transcribed in a rightward direction (9).
FIG. 2.

Schematic genome map of phage VP882. ORFs were numbered arbitrarily starting from the left end. The orientation of each gene is denoted by the direction of the arrow.
The terminase consists of a small subunit and a large subunit and packages viral DNA into the capsid. The putative terminase small-subunit protein gene (ORF3) and large-subunit protein gene (ORF4) are located at the left end of the structural protein module of VP882.
ORF35 putatively encodes a late-control gene D protein which is similar to late-control proteins of many phages. ORF35 locates at the end of the ORFs for tail proteins, and it also shares amino acid identity with a few tail proteins, such as ΦHAP-1 (54% identity) (36) and a phage of Xylella fastidiosa (GenBank accession no. NP_779294; 29% identity). No other lytic genes have been identified in VP882.
Genome maintenance module.
ORFs associated with maintenance of the VP882 genome in V. parahaemolyticus are similar to those of other plasmid-like temperate phages. The putative ParA protein of VP882 (ORF53) exhibits high identity with those of marine phage ΦHAP-1 (60%) (36) and V. harveyi phage VHML (65%) (40). ParA, working with ParB and ParS, is a partition protein associated with the movement of the correct copy number of bacterial chromosome and plasmids into daughter cells (49).
Protelomerase (ORF54) was identified, and it is responsible for the maintenance of the linear plasmid-like phages. The IRS, reaction site of protelomerase, in phage VP882 was searched by the Inverted Repeat Finder program (54), and an inverted palindromic repeat sequence was found between the regions encoding ParA (ORF53) and the protelomerase (ORF54) (Table 2). The IRS of VP882 (region of the genome from positions 26135 to 26247) consists of 113 bp and is located 121 bp upstream from the protelomerase (ORF54).
TABLE 2.
Inverted repeat sequences in phage VP882 and five other phages with a plasmid-like genome
| Phagea | Location of the left arm (bp) | Length of the left arm (bp) | Location of the right arm (bp) | Length of the right arm (bp) | Total length of the IRS (bp) | Sequence of the center hairpin |
|---|---|---|---|---|---|---|
| VP882 | 26135-26191 | 57 | 26192-26247 | 56 | 113 | TACGTA |
| ΦHAP-1 | 26847-26904 | 58 | 26905-26962 | 58 | 116 | TACGTA |
| N15 | 24775-24802 | 28 | 24803-24830 | 28 | 56 | CGCGCG |
| ΦKO2 | 29114-29145 | 32 | 29146-29179 | 34 | 66 | CGCGCG |
| PY54 | 24439-24463 | 25 | 24464-24488 | 25 | 50 | CGCGCG |
| VHML | 43116-43198 | 83 | 1-80 | 80 | 163 | GTATAC |
The presence of the IRS was determined by the Inverted Repeat Finder program (version 3.05) (54).
Replication protein (ORF57) is responsible for the replication of viral DNA, and an alternative translational initiation site (positions 32125 to 32127) is identified. This replication protein is similar to those of other plasmid-like prophages, such as the RepA proteins of ΦHAP-1 (51% identity) (36), PY54 (30% identity) (24), and N15 (30% identity) (45). These replication proteins exhibit multifunctional activities of primase and helicase, similar to Rep protein of plasmids replicating by the θ-mechanism (44).
Lysogeny module.
Putative lysogeny-related genes were identified in phage VP882. Repressor protein (ORF59) will bind to the phage DNA; it physically inhibits the binding of any transcription machinery and may be responsible for the lysogeny of VP882. The antirepressor (ORF60) probably binds to and acts against the main phage repressor (33). Antitermination protein Q (ORF62) is involved in late expression and allows RNA polymerase to override the termination signal and transcribe the lysis genes, which are the capsid protein and processing genes.
The putative conjugative transfer protein of VP882 (ORF61) is similar to TraR of Salmonella sp. (47% identity) (GenBank accession no. NP_490571), Escherichia coli (45% identity) (GenBank accession no. YP_538713) and other species, and it regulates genes that are required for conjugation (10) and may also associate with the transfer of the plasmid-like genome of VP882.
Other ORFs.
Some of the putative functional genes of VP882, namely, the DNA adenine methylase (DAM) (ORF37), exonuclease (ORF46), and the transcriptional regulator (LuxR family; ORF56), are often present in bacterial genomes and also in some phages. These genes may be the remains of a bacterial genome after viral transduction or play specific roles in the phages. DAM has been identified in many bacteria, and the putative DAM of phage VP882 (ORF37) exhibits high identity with similar functional proteins of Halomonas aquarmarina phage ΦHAP-1 (72%) (36), Methylobacillus flagellatus (67%) (GenBank accession no. YP_546786), Chromobacterium violaceum ATCC 12472 (67%) (GenBank accession no. NP_901781), Burkholderia cepacia AMMD (66%) (GenBank accession no. YP_773740) and other Burkholderia species, and low identity with the DAMS of Vibrio species, such as V. parahaemolyticus (24%) (GenBank accession no. NP_799121), V. vulnificus YJ016 (24%) (GenBank accession no. NP_935779), V. harveyi (24%) (GenBank accession no. ABO14793), V. alginolyticus (23%) (GenBank accession no. ZP_01261704) and others. DAM has multiple roles in cell physiology. DAM modifies the affinity of regulatory proteins toward DNA and is a transcriptional regulator. DAM regulates the expression of pilus-adhesins and other virulence-associated factors in E. coli, Salmonella spp., Yersinia pseudotuberculosis and V. cholerae (31).
The putative transcriptional regulator of VP882 (LuxR family; ORF56) exhibits high identity with those of V. cholerae (43%) (GenBank accession no. NP_233459) and Burkholderia oklahomensis (42%) (GenBank accession no. ZP_02358706) and with the DNA-binding helix-turn-helix domain-containing protein of V. vulnificus (41%) (GenBank accession no. NP_763032). Similar helix-turn-helix or LuxR proteins are present in the VHML phage of V. harveyi (40), VP16C and VP16T and in the VpV262 phages of V. parahaemolyticus (23, 48). LuxR is a quorum sensor and regulates the expression of virulence genes (1, 30).
The DAM and quorum-sensing transcriptional regulators in phage VP882 may regulate physiological or virulence-associated traits of the host, V. parahaemolyticus. VP882 infected and lysed several strains of V. parahaemolyticus, V. vulnificus, and V. cholerae (Table 1); may probably be involved in horizontal genetic shift among these three Vibrio species.
The putative exonuclease of VP882 (ORF46) exhibits a high identity with exonuclease RNase T and DNA polymerase III of Yersinia pseudotuberculosis (58%) (GenBank accession no. ACC88872), Thauera sp. (53%) (GenBank accession no. EDS56474), and Vibrio harveyi (48%) (GenBank accession no. ZP_01987444) and also in some phages, including Klebsiella ΦKO2 (41%) (GenBank accession no. YP_006619) and enterobacterial phage N15 (39%) (GenBank accession no. NP_046936). These putative exonucleases are associated with the processing of single-stranded DNA or RNA and in the replication of bacterial DNA.
Demonstration of plasmid-like genome.
Based on the presence of typical ORFs of linear plasmid-like phages like those of N15 of E. coli (44) and ΦHAP-1 of H. aquarmarina (36), VP882 most likely belongs to the same group of viruses. After infection of its host, the phage molecule will become circularized via its cohesive ends, and it is then digested by protelomerase to form a linear molecule with covalently closed ends (Fig. 3) (44).
FIG. 3.
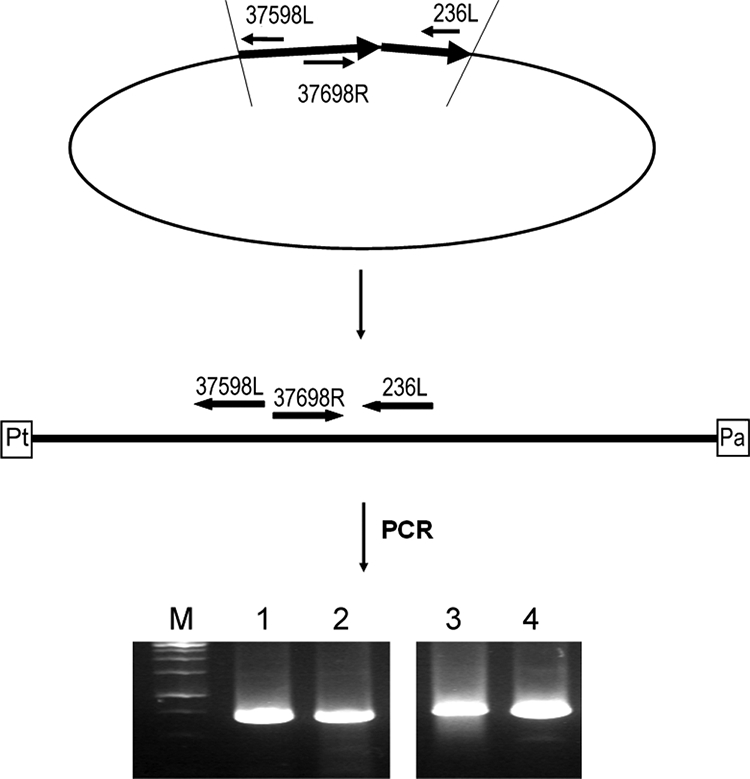
Demonstration of the plasmid-like genome in phage VP882. The locations of the primers used in demonstrating the plasmid-like genomes are indicated, and details of the experimental procedures are addressed in Materials and Methods. (Top) After infection of the host, the virion genome of a linear plasmid-like phage will become circularized via its cohesive ends. (Middle) The protelomerase will then cut at the hairpin region and form covalently closed ends (36, 44). Pt, protelomerase; Pa, ParA. (Bottom) In addition, the host chromosomal DNA preparation (lanes 1 and 2) and the VP882 viral DNA (lanes 3 and 4) were separately amplified with the 37698R and 236L primers, and the amplicons were separated with (lanes 2 and 4) or without (lanes 1 and 3) prior digestion with BfucI or SalI. Lane M contains molecular size markers (Fermentas 1-kb DNA ladder; BioLabTech, Kyiv, Ukraine).
For demonstrating the plasmid-like genome of phage VP882, the commonly used inverse PCR method was adopted in this study (7). Briefly, the chromosomal preparation of V. parahaemolyticus strain 882 was partially digested with BfuCI or SalI, and the digested fragments were circularized by ligation. The ligated fragments were amplified by PCR using the primers 37598L (5′-TCGAACTCAATCAACTGCTCAGGGT) and 37698R (5′-TACCCTGTCTAAGCGTTTCGTCTATTACTTC) which are located at the regions of the VP882 genome from bp 37598 to 37574 and 37698 to 37728, respectively (Fig. 3). BfuCI and SalI have 84 and 30 cutting sites, respectively, on the VP882 genome, but there is no cutting site between the primer annealing sites (bp 37574 to 37728). The amplicons were directly sequenced, or the amplicons were cloned by the TA cloning method (57), and the insert fragments of these clones were then sequenced. All the sequencing results revealed that the ends of VP882 genome were self-linked by cohesive ends of nine base pairs, GGCGGCAAA (Fig. 3, top and middle).
For further confirmation, the bacterial chromosomal DNA preparation and the purified VP882 phage DNA were separately amplified by PCR using primers 37698R and 236L (5′-CAGGGTTTTCTTTGTGACAGTGATACCCAT, located at the region of the genome from positions 236 to 208). The amplified fragments were separated directly or subjected to digestion with BfuCI or SalI before separation by agarose gel electrophoresis. Both the host chromosomal DNA and phage DNA preparations yielded a single band of 735 bp (Fig. 3, bottom). This result showed the presence of a circular DNA (Fig. 3, top) or a linearized molecule (Fig. 3, middle) in these preparations.
Comparative analysis of protelomerases and their reaction sites.
Protelomerase is in a dimer form, and the core of its monomer consists of an N-terminal domain, a catalytic domain, and a C-terminal DNA binding domain named the stirrup which is required for the resolution of hairpin telomeres (50). In this study, the amino acid sequences of the protelomerases in VP882 and five other plasmid-like prophages, namely, ΦHAP-1, VHML, ΦKO2, N15, and PY54, were analyzed.
The putative protelomerases of phages VP882, ΦHAP-1, VHML, ΦKO2, N15, and PY54 consist of 538, 520, 509, 640, 631, and 628 amino acid residues, respectively. The entire amino acid sequence of the putative protelomerase of VP882 was aligned with the protelomerases of five other prophages. Alignment analysis revealed that the protelomerase of VP882 has 54, 26, 33, 33, and 32% identity with that of ΦHAP-1, VHML, N15, ΦKO2, and PY54, respectively. The sequence conservation, quality, and consensus of these six aligned protelomerases revealed that they could be divided into two groups, except for VHML prophage. The VP882 and ΦHAP-1 phages were highly similar, while the other three were in another group (data not shown).
The active site residues of the protelomerases have been analyzed and showed mixed features of the tyrosine recombinases and type IB topoisomerases (50). Our results showed that the Arg223, Lys248, Lys325, Arg328, His361, and Tyr370 residues of the catalytic active site of VP882 protelomerase are characteristic residues also identified in five other plasmid-like prophages (Fig. 4). These results suggested that these plasmid-like prophages probably share a common mechanism in using protelomerase to resolve the telomere in the lysogenic cycle.
FIG. 4.

Conserved active site of the protelomerase catalytic domain from six plasmid-like prophages. The protelomerase sequences were aligned and presented by Clustalw2 (29). The asterisk below the sequences means that the residues in that column are identical in all sequences in the alignment. Residues with 70% or more sequence identity are shown on a gray background. The numbers above the sequences indicate the corresponding amino acid residue number in the VP882 protelomerase. Gaps introduced to maximize alignment are indicated by dashes.
Diverse IRS patterns identified in VP882 genome.
Previous investigations showed that IRSs are present in plasmid-like prophages and are related to the lysogenic cycle. Also, conserved patterns, such as the hairpin structure, catalytic binding domain, and stirrup binding domain, are present in the IRSs of phages N15, ΦKO2, and PY54 (2, 18). In this study, we examined the IRSs in VP882 and five other plasmid-like prophages. Our results revealed that VP882 phage possesses a diverse IRS pattern, which is more similar to that of ΦHAP-1 than the traditional IRS pattern of N15 phage (2, 36). The hairpin structure and catalytic binding domain of the IRS region of VP882 are identical to those of ΦHAP-1 (Fig. 5B and Table 2), whereas the IRS pattern of the catalytic active site of VP882 is only similar to that of N15 phage (Fig. 5B). Phage VHML also exhibits a diverse pattern compared with five other prophages (Fig. 5B).
FIG. 5.

Diverse IRS patterns found in the VP882 phage genome compared with those of five other plasmid-like phages. (A) Illustration of IRS patterns by using the Weblogo tool (16). The predicted cutting site (center hairpin region) of the protelomerase is indicated by a short black vertical line below the graphs. The nucleotide sequences shown inside the boxes are the consensus binding sites of protelomerase genes. (B) Using the binding site of the protelomerase from N15 phage as a reference template (2, 18) to compare the consensus binding patterns among the six plasmid-like phages. The solid arrow lines and the broken arrow lines indicate that the regions have the same sequence or exhibit a different sequence from that of the reference template, respectively. The hairpin region (H) and the binding regions for the stirrup domain (S) and the catalytic domain (C) are indicated.
The conserved sequences of these IRSs were analyzed by the Weblogo tool (16). Result showed that these IRSs could be divided into two groups which have different lengths and sequence patterns. One group consists of phage N15, ΦKO2, and PY54, which have three conserved patterns of hairpin structure and catalytic [(c/g)CAT(t/a)(a/c)TACGC] and stirrup binding domains [CAC(c/a)(t/c)A(t/a)(t/c)]. The other group consisted of phage VP882 and ΦHAP-1, which have only two conserved sequences in hairpin structure and catalytic binding domains, CCCATACTATAC (Fig. 5A and Table 2).
Comparative genomics of six plasmid-like prophages.
Following the addition of VP882, a total of 18 completed genome sequences of vibriophages are available in the NCBI genome database; these are 10 phages from V. cholerae (fs1, VSKK, VSK, KSF-1Φ, VGJΦ, fs2, K139, VP4, VP5, and VP2), seven phages from V. parahaemolyticus (VfO4K68, Vf12, Vf33, VfO3K6, VP882, VpV262, and KVP40), and one phage (VHML) from V. harveyi. While 10 of them are filamentous phages (Inoviridae), 4 are Myoviridae, and another 4 are Podoviridae.
The genome of phage VP882 has certain specific properties; for example, its G+C percentage (56.95) is among the highest of these vibriophages with complete genome sequences. The average G+C percentage of these vibriophages is 46.61, which is close to the values of their hosts. The G+C percentages of V. cholerae and V. parahaemolyticus are 47.48 (GenBank accession no. AE003852.1 and AE003853.1, respectively) and 46% (GenBank accession no. BA000032.2 and BA000031.2, respectively), respectively. Phages VP16T (GenBank accession no. AY328852) and VP16C (GenBank accession no. AY328853) with incomplete genome sequence both have G+C content of 59% (48). A difference of about 11% exists between the VP882 genome and the genome DNA of V. parahaemolyticus. The sequence variations in these vibriophages were further analyzed using the DOTTER program, which is a pair-wise sequence comparison tool (50). Results revealed that the vibriophage VP882 does not exhibited relatedness with other vibriophages (data not shown).
Since there is little sequence similarity of VP882 genome with other complete sequenced vibriophages, we investigated whether VP882 has sequence similarity with five selected plasmid-like prophage genomes by the DOTTER program. The results indicated that the genomic sequence VP882 phage is more similar to the ΦHAP-1 genome, while the N15, ΦKO2, and PY54 genomes are more similar to each other than to VP882 (Fig. 6). Moreover, ΦHAP-1 was the only BLASTN match when the whole VP882 genome was used to search against the NCBI genomic refseq database. From the genomic sequence and IRS and protelomerase sequence comparative analysis, both VP882 and ΦHAP-1 are rare plasmid-like marine prophages, unlike N15 and other typical plasmid-like prophages, and may be classified into a new subtype of the plasmid-like phages.
FIG. 6.

Genomic alignment of the VP882 and five other plasmid-like phage genomes. The sequence similarity was visualized with the DOTTER program (50) by using a sliding window of 25 bp. The arrangement of the phages was ordered according to their genome sizes, from smallest to largest.
In conclusion, a new temperate polyhedral phage, VP882, in V. parahaemolyticus pandemic O3:K6 strain 882 was isolated and characterized. Morphological and genomic analysis indicated that this was a Myoviridae phage with plasmid-like genome and characteristic protelomerase and cohesive ends. It may be involved in horizontal genetic movement and may be important in establishing the genetic diversity of this pathogen.
Supplementary Material
Acknowledgments
We thank the Department of Health and National Science Council of the Republic of China for financially supporting this research under contract no. DOH91-DC-1006 and NSC96-2313-B-031-001.
Footnotes
Published ahead of print on 13 March 2009.
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1.Ahmer, B. M., J. van Reeuwijk, C. D. Timmers, P. J. Valentine, and F. Heffron. 1998. Salmonella typhimurium encodes an SdiA homolog, a putative quorum sensor of the LuxR family, that regulates genes on the virulence plasmid. J. Bacteriol. 180:1185-1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aihara, H., W. M. Huang, and T. Ellenberger. 2007. An interlocked dimer of the protelomerase TelK distorts DNA structure for the formation of hairpin telomeres. Mol. Cell 27:901-913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ansaruzzaman, M., M. Lucas, J. L. Deen, N. A. Bhuiyan, X. Y. Wang, A. Safa, M. Sultana, A. Chowdhury, G. B. Nair, D. A. Sack, L. von Seidlein, M. K. Puri, M. Ali, C. L. Chaignat, J. D. Clemens, and A. Barreto. 2005. Pandemic serovars (O3:K6 and O4:K68) of Vibrio parahaemolyticus associated with diarrhea in Mozambique: spread of the pandemic into the African continent. J. Clin. Microbiol. 43:2559-2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bag, P. K., S. Nandi, R. K. Bhadra, T. Ramamurthy, S. K. Bhattacharya, M. Nishibuchi, T. Hamabata, S. Yamasaki, Y. Takeda, and G. B. Nair. 1999. Clonal diversity among recently emerged strains of Vibrio parahaemolyticus O3:K6 associated with pandemic spread. J. Clin. Microbiol. 37:2354-2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baross, J. A., J. Liston, and R. Y. Morita. 1978. Incidence of Vibrio parahaemolyticus bacteriophages and other Vibrio bacteriophages in marine samples. Appl. Environ. Microbiol. 36:492-499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baross, J. A., J. Liston, and R. Y. Morita. 1978. Ecological relationship between Vibrio parahaemolyticus and agar-digesting vibrios as evidenced by bacteriophage susceptibility patterns. Appl. Environ. Microbiol. 36:500-505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bernhardt, T. G., W. D. Roof, and R. Young. 2000. Genetic evidence that the bacteriophage phi X174 lysis protein inhibits cell wall synthesis. Proc. Natl. Acad. Sci. USA 97:4297-4302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boyd, E. F., K. E. Moyer, L. Shi, and M. K. Waldor. 2000. Infectious CTXΦ and the vibrio pathogenicity island prophage in Vibrio mimicus: evidence for recent horizontal transfer between V. mimicus and V. cholerae. Infect. Immun. 68:1507-1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Casjens, S. R., E. B. Gilcrease, W. M. Huang, K. L. Bunny, M. L. Pedulla, M. E. Ford, J. M. Houtz, G. F. Hatfull, and R. W. Hendrix. 2004. The pKO2 linear plasmid prophage of Klebsiella oxytoca. J. Bacteriol. 186:1818-1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chai, Y., and S. C. Winans. 2005. Amino-terminal protein fusions to the TraR quorum-sensing transcription factor enhance protein stability and autoinducer-independent activity. J. Bacteriol. 187:1219-1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chan, B., H. Miyamoto, H. Taniguchi, and S. Yoshida. 2002. Isolation and genetic characterization of a novel filamentous bacteriophage, a deleted form of phage f237, from a pandemic Vibrio parahaemolyticus O4:K68 strain. Microbiol. Immunol. 46:565-569. [DOI] [PubMed] [Google Scholar]
- 12.Chang, B., S. Yoshida, H. Miyamoto, M. Ogawa, K. Horikawa, M. Nishibuchi, and H. Taniguchi. 2000. A unique and common restriction fragment pattern of the nucleotide sequences homologous to the genome of Vf33, a filamentous bacteriophage, in pandemic strains of Vibrio parahaemolyticus O3:K6 O4:K68, and O1:K untypeable. FEMS Microbiol. Lett. 192:231-236. [DOI] [PubMed] [Google Scholar]
- 13.Chen, C. Y., K. M. Wu, Y. C. Chang, C. H. Chang, H. C. Tsai, T. L. Liao, Y. M. Liu, H. J. Chen, A. B. Shen, J. C. Li, T. L. Su, C. P. Shao, C. T. Lee, L. I. Hor, and S. F. Tsai. 2003. Comparative genome analysis of Vibrio vulnificus, a marine pathogen. Genome Res. 13:2577-2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen, S. H., C. Y. Lin, C. S. Cho, C. Z. Lo, and C. A. Hsiung. 2003. Primer Design Assistant (PDA): a web-based primer design tool. Nucleic Acids Res. 31:3751-3754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Comeau, A. M., A. M. Chan, and C. A. Suttle. 2006. Genetic richness of vibriophages isolated in a coastal environment. Environ. Microbiol. 8:1164-1176. [DOI] [PubMed] [Google Scholar]
- 16.Crooks, G. E., J.-M. Chandonia, and S. E. Brenner. 2004. WebLogo: a sequence logo generator. Genome Res. 14:1188-1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Delcher, A. L., K. A. Bratke, E. C. Powers, and S. L. Salzberg. 2007. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 23:673-679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deneke, J., G. Ziegelin, R. Lurz, and E. Lanka. 2002. Phage N15 telomere resolution. Target requirements for recognition and processing by the protelomerase. J. Biol. Chem. 277:10410-10419. [DOI] [PubMed] [Google Scholar]
- 19.DePaola, A., M. L. Motes, A. M. Chan, and C. A. Suttle. 1998. Phages infecting Vibrio vulnificus are abundant and diverse in oysters (Crassostrea virginica) collected from the Gulf of Mexico. Appl. Environ. Microbiol. 64:346-351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gonzalez-Escalona, N., V. Cachicas, C. Acevedo, M. L. Rioseco, J. A. Vergara, F. Cabello, J. Romero, and R. T. Espejo. 2005. Vibrio parahaemolyticus diarrhea, Chile, 1998 and 2004. Emerg. Infect. Dis. 11:129-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goodridge, L., A. Gallaccio, and M. W. Griffiths. 2003. Morphological, host range, and genetic characterization of two coliphages. Appl. Environ. Microbiol. 69:5364-5371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guo, F. B., and C. T. Zhang. 2006. ZCURVE_V: a new self-training system for recognizing protein-coding genes in viral and phage genomes. BMC Bioinform. 7:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hardies, S. C., A. M. Comeau, P. Serwer, and C. A. Suttle. 2003. The complete sequence of marine bacteriophage VpV262 infecting Vibrio parahaemolyticus indicates that an ancestral component of a T7 viral supergroup is widespread in the marine environment. Virology 310:359-371. [DOI] [PubMed] [Google Scholar]
- 24.Hertwig, S., I. Klein, V. Schmidt, S. Beck, J. A. Hammerl, and B. Appel. 2003. Sequence analysis of the genome of the temperate Yersinia enterocolitica phage PY54. J. Mol. Biol. 331:605-622. [DOI] [PubMed] [Google Scholar]
- 25.Iida, T., A. Hattori, K. Tagomori, H. Nasu, R. Naim, and T. Honda. 2001. Filamentous phage associated with recent pandemic strains of Vibrio parahaemolyticus. Emerg. Infect. Dis. 7:477-478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Joseph, S. W., R. R. Colwell, and J. B. Kaper. 1983. Vibrio parahaemolyticus and related halophilic vibrios. CRC Crit. Rev. Microbiol. 10:77-123. [DOI] [PubMed] [Google Scholar]
- 27.Khan, A. A., S. McCarthy, R. F. Wang, and C. E. Cerniglia. 2002. Characterization of United States outbreak isolates of Vibrio parahaemolyticus using enterobacterial repetitive intergenic consensus (ERIC) PCR and development of a rapid PCR method for detection of O3:K6 isolates. FEMS Microbiol. Lett. 206:209-214. [DOI] [PubMed] [Google Scholar]
- 28.Koga, T., and T. Kawata. 1991. Comparative characterization of inducible and virulent Vibrio parahaemolyticus bacteriophages having unique head projections. Microbiol. Immunol. 35:49-58. [DOI] [PubMed] [Google Scholar]
- 29.Larkin, M. A., G. Blackshields, N. P. Brown, R. Chenna, P. A. McGettigan, H. McWilliam, F. Valentin, I. M. Wallace, A. Wilm, R. Lopez, J. D. Thompson, T. J. Gibson, and D. G. Higgins. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23:2947-2948. [DOI] [PubMed] [Google Scholar]
- 30.Latifi, A., M. K. Winson, M. Foglino, B. W. Bycroft, G. S. Stewart, A. Lazdunski, and P. Williams. 1995. Multiple homologues of LuxR and LuxI control expression of virulence determinants and secondary metabolites through quorum sensing in Pseudomonas aeruginosa PAO1. Mol. Microbiol. 17:333-343. [DOI] [PubMed] [Google Scholar]
- 31.Low, D. A., N. J. Weyand, and M. J. Mahan. 2001. Roles of DNA adenine methylation in regulating bacterial gene expression and virulence. Infect. Immun. 69:7197-7204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lukashin, A. V., and M. Borodovsky. 1998. GeneMark.hmm: new solutions for gene finding. Nucleic Acids Res. 26:1107-1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mardanov, A. V., and N. V. Ravin. 2007. The antirepressor needed for induction of linear plasmid-prophage N15 belongs to the SOS regulon. J. Bacteriol. 189:6333-6338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martinez-Urtaza, J., L. Simental, D. Velasco, A. DePaola, M. Ishibashi, Y. Nakaguchi, M. Nishibuchi, D. Carrera-Flores, C. Rey-Alvarez, and A. Pousa. 2005. Pandemic Vibrio parahaemolyticus O3:K6, Europe. Emerg. Infect. Dis. 11:1319-1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matsuzaki, S., T. Inoue, S. Tanaka, T. Koga, M. Kuroda, S. Kimura, and S. Imai. 2000. Characterization of a novel Vibrio parahaemolyticus phage, KVP241, and its relatives frequently isolated from seawater. Microbiol. Immunol. 44:953-956. [DOI] [PubMed] [Google Scholar]
- 36.Mobberley, J. M., R. N. Authement, A. M. Segall, and J. H. Paul. 2008. The temperate marine phage ΦHAP-1 of Halomonas aquamarina possesses a linear plasmid-like prophage genome. J. Virol. 82:6618-6630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Myers, M. L., G. Panicker, and A. K. Bej. 2003. PCR detection of a newly emerged pandemic Vibrio parahaemolyticus O3:K6 pathogen in pure cultures and seeded waters from the Gulf of Mexico. Appl. Environ. Microbiol. 69:2194-2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nakasone, N., M. Ikema, N. Higa, T. Yamashiro, and M. Iwanaga. 1999. A filamentous phage of Vibrio parahaemolyticus O3:K6 isolated in Laos. Microbiol. Immunol. 43:385-388. [DOI] [PubMed] [Google Scholar]
- 39.Nasu, H., T. Iida, T. Sugahara, Y. Yamaichi, K. S. Park, K. Yokoyama, K. Makino, H. Shinagawa, and T. Honda. 2000. A filamentous phage associated with recent pandemic Vibrio parahaemolyticus O3:K6 strains. J. Clin. Microbiol. 38:2156-2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oakey, H. J., B. R. Cullen, and L. Owens. 2002. The complete nucleotide sequence of the Vibrio harveyi bacteriophage VHML. J. Appl. Microbiol. 93:1089-1098. [DOI] [PubMed] [Google Scholar]
- 41.Oakey, H. J., and L. Owens. 2000. A new bacteriophage, VHML, isolated from a toxin-producing strain of Vibrio harveyi in tropical Australia. J. Appl. Microbiol. 89:702-709. [DOI] [PubMed] [Google Scholar]
- 42.Ohnishi, T., and K. Nozu. 1986. Induction of phage-like particles from a pathogenic strain of Vibrio parahaemolyticus by mitomycin C. Biochem. Biophys. Res. Commun. 141:1249-1253. [DOI] [PubMed] [Google Scholar]
- 43.Okuda, J., M. Ishibashi, E. Hayakawa, T. Nishino, Y. Takeda, A. K. Mukhopadhyay, S. Garg, S. K. Bhattacharya, G. B. Nair, and M. Nishibuchi. 1997. Emergence of a unique O3:K6 clone of Vibrio parahaemolyticus in Calcutta, India, and isolation of strains from the same clonal group from Southeast Asian travelers arriving in Japan. J. Clin. Microbiol. 35:3150-3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ravin, N. V. 2003. Mechanisms of replication and telomere resolution of the linear plasmid prophage N15. FEMS Microbiol. Lett. 221:1-6. [DOI] [PubMed] [Google Scholar]
- 45.Ravin, V., N. Ravin, S. Casjens, M. E. Ford, G. F. Hatfull, and R. W. Hendrix. 2000. Genomic sequence and analysis of the atypical temperate bacteriophage N15. J. Mol. Biol. 299:53-73. [DOI] [PubMed] [Google Scholar]
- 46.Sails, A. D., D. R. Wareing, F. J. Bolton, A. J. Fox, and A. Curry. 1998. Characterisation of 16 Campylobacter jejuni and C. coli typing bacteriophages. J. Med. Microbiol. 47:123-128. [DOI] [PubMed] [Google Scholar]
- 47.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 48.Seguritan, V., I. W. Feng, F. Rohwer, M. Swift, and A. M. Segall. 2003. Genome sequences of two closely related Vibrio parahaemolyticus phages, VP16T and VP16C. J. Bacteriol. 185:6434-6447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sergueev, K., A. Dabrazhynetskaya, and S. Austin. 2005. Plasmid partition system of the P1par family from the pWR100 virulence plasmid of Shigella flexneri. J. Bacteriol. 187:3369-3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sonnhammer, E. L., and R. Durbin. 1995. A dot-matrix program with dynamic threshold control suited for genomic DNA and protein sequence analysis. Gene 167:GC1-GC10. [DOI] [PubMed] [Google Scholar]
- 51.Takeda, Y. 1988. Thermostable direct hemolysin of Vibrio parahaemolyticus. Methods Enzymol. 165:189-193. [DOI] [PubMed] [Google Scholar]
- 52.Vital, M., H. P. Fuchslin, F. Hammes, and T. Egli. 2007. Growth of Vibrio cholerae O1 Ogawa Eltor in freshwater. Microbiology 153:1993-2001. [DOI] [PubMed] [Google Scholar]
- 53.Waldor, M. K., and J. J. Mekalanos. 1996. Lysogenic conversion by a filamentous phage encoding cholera toxin. Science 272:1910-1914. [DOI] [PubMed] [Google Scholar]
- 54.Warburton, P. E., J. Giordano, F. Cheung, Y. Gelfand, and G. Benson. 2004. Inverted repeat structure of the human genome: the X-chromosome contains a preponderance of large, highly homologous inverted repeats that contain testes genes. Genome Res. 14:1861-1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wong, H. C., S. H. Liu, T. K. Wang, C. L. Lee, C. S. Chiou, D. P. Liu, M. Nishibuchi, and B. K. Lee. 2000. Characteristics of Vibrio parahaemolyticus O3:K6 from Asia. Appl. Environ. Microbiol. 66:3981-3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wong, H. C., and P. Wang. 2004. Induction of viable but nonculturable state in Vibrio parahaemolyticus and its susceptibility to environmental stresses. J. Appl. Microbiol. 96:359-366. [DOI] [PubMed] [Google Scholar]
- 57.Zhou, M. Y., and C. E. Gomez-Sanchez. 2000. Universal TA cloning. Curr. Issues Mol. Biol. 2:1-7. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.