

Abstract
Colonization of the upper respiratory tract is an initial step that may lead to disease for many pathogens. To prevent compromise of the epithelial barrier, the host must monitor and tightly control bacterial levels on the mucosa. Here we show that innate immune functions of respiratory epithelial cells control colonization by Streptococcus pneumoniae and Haemophilus influenzae in a Toll-like receptor (TLR)-dependent manner. Activation of inflammatory pathways, including mitogen-activated protein kinase signaling, in respiratory epithelial cells was accompanied by the induction of the transforming growth factor β signaling cascade during early colonization. Thus, colonization resulted in upregulation of factors involved in a proinflammatory response (e.g., interleukin-6) as well as factors known to modulate the epithelial barrier (e.g., Snail-1). These in vivo data provided a link between inflammation control and maintenance of the mucosal barrier function during infection and emphasized the importance of TLR-dependent inflammatory responses of the respiratory epithelium.
The epithelial surface of the upper respiratory tract is constantly exposed to commensals and potentially pathogenic microbes. For pathogens, colonization of the upper respiratory tract is often the first step in a multistep process leading to disease. Thus, even though the mucosa of the upper respiratory tract is permissive for colonization by a variety of different bacterial microbes, the density and composition of colonization need to be controlled by the host (33). In recent years, it has become evident that the epithelium of the upper respiratory tract not only provides a physical barrier that is highly effective at blocking penetration by most microbes but also has the ability to recognize microbes and to initiate an inflammatory response. However, to prevent excessive colonization and invasion by potential pathogens, the epithelial barrier needs to be a dynamic structure, allowing the egress of immune cells and antimicrobial factors. Insights into the signaling events of the respiratory epithelium in response to bacterial infection, therefore, may provide a better understanding of the initial events of host-pathogen interaction.
Numerous in vitro studies have been performed to gain insight into the immune functions of respiratory epithelial cells (9, 10, 26, 30, 37). Respiratory epithelial cells in culture are capable of recognizing microbes through pattern recognition receptors such as Toll-like receptors (TLRs). Haemophilus influenzae, for instance, has been reported to induce immune responses through TLR2 and TLR4, which bind its lipoproteins and lipopolysaccharide (LPS), respectively (6, 40, 43). Streptococcus pneumoniae activates respiratory epithelial cells through the activity of TLR2 via its lipid-modified membrane components and TLR4 via the toxin pneumolysin (22, 37, 38). Recognition of bacterial products by TLRs leads to signaling events that result in activation of NF-κB through the activity of, for instance, mitogen-activated protein kinase (MAPK) signaling cascades, including p38 MAPK (15). Furthermore, in vitro studies suggest that activated respiratory epithelial cells are directly involved in initiating an immune response by releasing cytokines, chemokines, and factors such as antimicrobial peptides that directly mitigate the infectious process (1).
Host factors that modulate the barrier function of the respiratory epithelium are less well defined. Transforming growth factor β (TGF-β) signaling through the TGF-β receptor is involved in the remodeling and repair of epithelial surfaces but also mediates epithelial-to-mesenchymal transition, thereby contributing to the progression of complex diseases such as pulmonary fibrosis and cancer (28, 29, 44). Moreover, H. influenzae and S. pneumoniae have been shown to stimulate TGF-β signaling in respiratory cells in vitro (2, 29). We have demonstrated that stimulation of TGF-β signaling in polarized respiratory epithelial cells in culture results in the opening of tight junctions and loss of barrier integrity. These events could be required for the trafficking of immune cells and inflammatory mediators such as neutrophils and complement to the airway lumen, where microbes reside. Opening of the barrier, however, may also result in invasion by bacteria that are not adequately controlled by these host responses (2).
Despite the multitude of studies dealing with the immune functions of respiratory epithelial cells in vitro, little is known about whether microbial pathogens activate these pathways during colonization in vivo under physiological conditions. As relatively high numbers of bacteria or concentrations of TLR ligands or other pathogen-associated molecular pattern recognition receptors are needed to activate respiratory tissue in vitro, it is arguable whether colonization of the upper airway by bacterial pathogens is sufficient to evoke an inflammatory response in the respiratory epithelium in vivo. In particular, it is not clear whether the TGF-β signaling pathway is engaged upon bacterial colonization of the respiratory epithelium in vivo.
The purpose of this study was to characterize the role of respiratory epithelial cells during bacterial colonization in vivo. We demonstrate innate immune functions of respiratory epithelial cells in vivo by taking advantage of physiologically relevant murine models of colonization of the upper respiratory tract by H. influenzae and S. pneumoniae, the leading gram-negative and gram-positive bacterial pathogens of the human respiratory tract, respectively. We show that epithelial cells not only produce factors that promote an inflammatory response but also activate pathways that modulate barrier and immune functions of the mucosal surface. Furthermore, TLRs are central to both these functions, and deficiency in their signaling leads to decreased control of initial colonization by bacterial pathogens and increased susceptibility to invasive disease.
MATERIALS AND METHODS
Bacterial strains and products.
H636, a spontaneously streptomycin-resistant clinical isolate of H. influenzae type b, was grown in brain heart infusion media supplemented with 2% Fildes enrichment and 2 μg/ml NAD (Difco Laboratories). S. pneumoniae strain P1121 (type 23F clinical isolate) was selected because of its ability to efficiently colonize the murine nasopharynx without causing invasive infection. S. pneumoniae strain P303 (type 6A clinical isolate) and a pneumolysin-deficient derivative of P303 were selected because of their ability to cause invasive murine infection following intranasal inoculation of mice. The S. pneumoniae strains used were previously described and were grown as previously reported (34). Pam3CSK4 was purchased from InvivoGen. LPS was purified from H. influenzae strain H636 by hot phenol-water extraction as previously described (24).
Mouse model of nasopharyngeal colonization and sepsis.
Six- to eight-week-old female C57BL/6J mice were obtained from Taconic Laboratories. Wild-type and TLR2- and TLR4-deficient animals were housed in accordance with Institutional Animal Care and Use Committee protocols. The TLR2-deficient animals were described previously (41). TLR4-deficient animals (C57BL10ScNJ) were obtained from Jackson Laboratories. Wild-type and TLR2- and TLR4-deficient mice were colonized using a previously described model of nasopharyngeal colonization (35). Briefly, mice were inoculated intranasally without anesthesia with 10 μl of a mixture containing 1 × 107 to 5 × 107 CFU of phosphate-buffered saline (PBS)-washed, mid-log-phase H. influenzae or S. pneumoniae applied atraumatically to the nares. Septic-appearing animals with decreased activity were sacrificed and their blood and spleen evaluated to confirm the presence of bacteremia. At the time indicated, the animals were sacrificed, the trachea was cannulated, and 200 μl of PBS was instilled. Lavage fluid was collected from the nares for determination of viable counts of bacteria in serial dilutions plated on selective medium containing neomycin (20 μg/ml) to allow the growth of S. pneumoniae or streptomycin (100 μg/ml) to allow the growth of H. influenzae without contaminants. Survival of animals colonized with S. pneumoniae strain P303 was monitored over a period of 4 days. To characterize the inflammatory response to TLR ligands, mice were intranasally inoculated with 7.5 μl of purified LPS (8 μg/ml) or Pam3CSK4 (8 μg/ml; InvivoGen) per nostril. At 3 h after challenge, RNA from the upper respiratory tract was obtained as described below.
Immunohistochemistry and immunofluorescence.
Animal tissues were obtained after decapitation, fixed, and decalcified by serial overnight incubations in 4% paraformaldehyde-PBS-Decal decalcification agent (Decal Corporation). Tissue was embedded in paraffin, and immunohistochemistry was performed as described earlier (2). p-p38 MAPK (Cell Signaling), ATF-2 (Cell Signaling), and p-Smad2 and -3 (Abcam) were detected with primary antibodies diluted 1:100 in PBT (1× PBS, 0.1% bovine serum albumin, 0.2% Triton X-100). A biotinylated anti-rabbit secondary antibody (Vector Laboratories) was added, followed by avidin-horseradish peroxidase ABC reagent (Vector Laboratories). Negative controls included uninfected animals, mixtures lacking primary antibody, and irrelevant isotype-matched primary antibodies. Signal was developed using a DAB kit (Vector Laboratories). Imaging was performed using a Nikon E600 Eclipse microscope equipped with a high-resolution charge-coupled-device digital camera (CoolSnap CF; Roper Scientific).
For immunofluorescence detection, the head was extensively decalcified for further processing by serial incubations in 0.12 M EDTA (pH 7.0) during 1 month before being frozen in Tissue-Tek O.C.T. embedding medium (Miles) and a Tissue-Tek Cryomold. Five-micrometer-thick sections were cut and stored at −80°C. Tissue was postfixed with 1:1 acetone-methanol, followed by blocking with protein blocking reagent before the addition of primary antibody (diluted 1:500 in PBT). S. pneumoniae P1121 was detected using typing sera (Statens Serum Institut, Copenhagen, Denmark) (diluted 1:500 in PBT) for 2 h at room temperature. H. influenzae was detected using a primary antibody to the cell surface protein P5 (diluted 1:500 in PBT). Signal was detected with Cy3-conjugated species-specific secondary antibodies (Jackson ImmunoResearch, West Grove, PA) incubated in a 1:400 dilution of PBT for 2 h at room temperature. After washes with PBS followed by distilled water, sections were counterstained with DAPI (4′,6-diamidino-2-phenylindole) (Molecular Probes, Invitrogen, Carlsbad, CA) diluted 1:10,000 in distilled water. All image analysis was carried out using IPLAB software (Scanalytics, Fairfax, VA).
Western blotting.
Lavage fluid (15 μl) was mixed with 5× sample buffer, treated at 100°C for 5 min, and directly separated on a denaturing 10% Tris-HCl polyacrylamide gel. Proteins were transferred to a polyvinylidene difluoride membrane (Millipore) and probed for the heavy chain of mouse immunoglobulin G (IgG) by the use of a peroxidase-labeled anti-mouse IgG antibody (Amersham).
Quantitative reverse transcription-PCR (qRT-PCR).
To obtain total RNA from tissue lining the upper respiratory tract, the animal was sacrificed, the trachea cannulated, and 400 μl of a lysis buffer (Qiagen) instilled. This treatment has previously been shown to be sufficient to lyse the entire epithelium (32). Lavage fluid was collected from the nares, and RNA was isolated using an isolation kit (Qiagen). A 1.5-μg volume of total RNA preparation was reverse transcribed using a cDNA synthesis kit (Stratagene) applying oligo(dT)18 primers. cDNA was diluted at a 1-to-5 ratio, and 5 μl was used as a template in a mixture with 25 μl of SYBR green for PCR, according to the protocol of the manufacturer (Bio-Rad). Mouse GAPDH (glyceraldehyde-3-posphate dehydrogenase) primers (sense, 5′-TGT GTC CGT CGT GGA TCT GA-3′; antisense, 5′-CCT GCT TCA CCA CCT TCT TGA-3′), mouse interleukin-6 (IL-6) primers (sense, 5′-GCC TCC TTG GGA CTG ATG CT-3′; antisense, 5′-AGT CTC CTC TCC GGA CTT GTG-3′), mouse TLR4 primers (sense, 5′-AAG AGC CGG AAG GTT ATT GTG-3′; antisense, 5′-CCC ATT CCA GGT AGG TGT TTC-3′), mouse TLR2 primers (sense, 5′-TAT CCG GAG GTT GCA TAT CCC-3′; antisense, 5′-CCA TCA GAT TTT CGC TGA GGG TC-3′), mouse Muc5AC primers (sense, 5′-CCA TGC AGA GTC CTC AGA ACA A-3′; antisense, 5′-TTA CTG GAA AGG CCC AAG CA-3′), mouse siderocalin primers (sense, 5′-GGC CTC AAG GAC GAC AAC A-3′; antisense, 5′-GCATCCCAGTCAGCCACACT-3′), Snail1 primers (sense, 5′-TCC AAA CCC ACT CGG ATG TGA AGA-3′; antisense, 5′-GGA TGT GAA GAG ATA CCA GTG CC-3′), and CD45 primers (sense, 5′-CAG AGC ATT CCA CGG GTAT T-3′; antisense, 5′-GGA CCC TGC ATC TCC ATT TA-3′) were purchased from Integrated DNA Technologies. The specificity of the RT-PCR was controlled by omission of the template or of the RT. Quantitative PCR values were calculated using the ΔΔCT method; results are expressed relative to those obtained with the corresponding uninfected control group. Because the PCR efficiencies for all four reactions were similar, the threshold values were normalized to GAPDH data.
Statistical analysis.
Comparisons between experimental groups were performed using Student's t test or analysis of variance as appropriate. The Mann-Whitney U test was used to evaluate differences in colonization density, and the Kaplan-Meier log-rank test was used to compare differences in mouse survival rates. All statistics were evaluated using Prism 4 software (GraphPad Software).
RESULTS
TLRs mediate clearance of bacterial pathogens during early colonization.
To examine the physiological relevance of TLRs during the early course of bacterial colonization of the upper respiratory tract, we used a well-characterized murine model of colonization (27) and the availability of TLR2- and TLR4-deficient animals. Mice were inoculated intranasally with either H. influenzae or S. pneumoniae, and the levels of colonization and the survival rates of the animals were monitored. Tissue sections showed that at 24 h postchallenge, S. pneumoniae or H. influenzae bacteria were found at the mucosal surface in direct contact with the respiratory epithelium (Fig. 1). By 24 h postchallenge, both TLR2- and TLR4-deficient animals showed higher H. influenzae colonization levels than wild-type mice (Fig. 2A). In the case of S. pneumoniae infection performed with P1121, a noninvasive strain, TLR2 but not TLR4 deficiency resulted in higher colonization levels (Fig. 2B). Moreover, TLR2-deficient animals infected with the invasive S. pneumoniae P303 strain showed a higher rate of mortality than wild-type and TLR4-deficient animals within the first 4 days of infection (Fig. 2C).
FIG. 1.
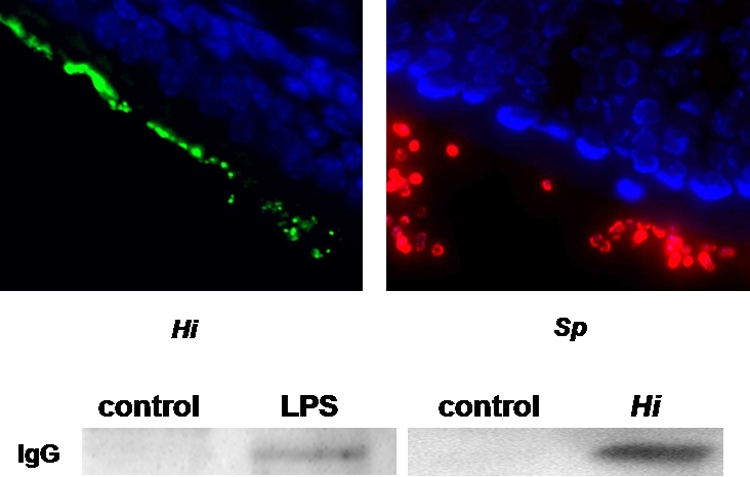
Colonization of the upper respiratory tract leads to disruption of the epithelial barrier. Mice were infected intranasally with H. influenzae (Hi [green]) or S. pneumoniae (Sp [red]). The results of immunofluorescence staining (DAPI [blue]) of frozen nasal tissue at 24 h postinfection (magnification, ×1,000) are shown. A Western blot performed with lavage samples from mice colonized with H. influenzae (Hi [at 24 h]) or challenged with LPS (60 μg/naris [at 3 h]) shows increased levels of total IgG.
FIG. 2.

TLRs mediate the early clearance of colonizing bacterial pathogens. (A) At 24 h postchallenge, TLR4- and TLR2-deficient animals showed enhanced levels of colonization by H. influenzae (Hi) in the upper respiratory tract compared to wild-type (WT) animals. (B) At 24 h postchallenge, TLR2-deficient animals, but not TLR4-deficient animals, showed enhanced levels of colonization by noninvasive S. pneumoniae (Sp) strain P1121 in the upper respiratory tract compared to wild-type animals. A box-and-whiskers plot indicates high and low values and median and interquartile ranges (n ≥ 8 for each group). (C) TLR2 deficiency results in significantly higher mortality within the first 4 days after challenge with invasive S. pneumoniae isolate P303. *, P < 0.05 (Kaplan-Meier log-rank test; n ≥ 9 for each group).
To determine whether bacterial colonization resulted in opening of the epithelial barrier and in accumulation of inflammatory mediators from the bloodstream on the mucosal surface, lavage samples from control mice and animals challenged with H. influenzae were analyzed for the presence of IgG by Western blotting. Figure 1 shows that bacterial colonization resulted in increased accumulation of nonsecretory IgG at the mucosa. Moreover, administration of a TLR ligand (highly purified H. influenzae LPS) was sufficient to cause loss of epithelial barrier function.
Together, these results demonstrated that TLRs were involved in maintenance of the epithelial barrier and the clearance of bacterial pathogens in the upper respiratory tract during the early course of colonization and disease.
Bacterial pathogens activate host tissue in a TLR-dependent manner.
Next, we sought to determine whether initial colonization of the upper respiratory tract results in activation of host tissue. Animals were intranasally inoculated with H. influenzae or S. pneumoniae. At 24 h after inoculation, a lavage of the upper respiratory tract was performed using a lysis buffer to obtain RNA from the respiratory epithelium, and qRT-PCR analyses of several inflammatory markers that are representative of an early inflammatory response were performed.
We chose to look for expression of IL-6 and TLR2, two factors that are known to be upregulated in a TLR-dependent manner (17, 25). Additionally, siderocalin was chosen as a control because its transcription has previously been shown to be upregulated in response to colonization by either H. influenzae or S. pneumoniae (32). The mucin Muc5AC that contributes to mucociliary clearance served as a marker that is expressed exclusively by the epithelium (36).
Figure 3A, B, and C show that colonization by H. influenzae resulted in an approximately 15- to -30-fold induction of IL-6 and siderocalin and in an approximately fivefold induction of TLR2 in the mucosal tissue of wild-type animals. In contrast, TLR4-deficient animals did not demonstrate induction of IL-6, siderocalin, or TLR2 at 24 h postchallenge (Fig. 3A, B, and C). TLR2-deficient animals showed the same levels of induction of IL-6 and siderocalin as wild-type animals. The level of TLR4 expression was not affected by colonization by bacteria in wild-type or TLR2-deficient animals (Fig. 3B). In addition, there was approximately twofold-enhanced expression of Muc5AC in wild-type and TLR4-deficient mice, whereas TLR2-deficient animals did not show any induction of Muc5AC expression (Fig. 3D).
FIG. 3.

H. influenzae activates host tissue via TLRs. (A to D) Wild-type (WT) and TLR4- and TLR2-deficient mice were inoculated with H. influenzae. At 24 h postchallenge, the upper respiratory tract was flushed with lysis buffer to obtain total RNA. qRT-PCR was performed to measure the induction of IL-6 (A), TLR2 (open boxes) and TLR4 (gray boxes) (B), siderocalin (SnC) (C), and Muc5AC (D). n ≥ 4; *, P < 0.05; **, P < 0.005; n.s, not significant.
To determine whether S. pneumoniae also has the ability to induce an inflammatory response in the upper respiratory tract, mice were infected with S. pneumoniae. qRT-PCR analysis revealed a significant upregulation of IL-6 in challenged mice at 3 h postinoculation compared to the results seen with noninfected littermates (Fig. 4A), whereas at 24 h after inoculation only a fraction of infected animals showed an upregulation of IL-6 (data not shown). Pneumolysin-deficient S. pneumoniae also induced upregulation of IL-6, indicating that pneumolysin was not required to provoke an inflammatory response during early colonization (data not shown).
FIG. 4.

Colonization by S. pneumoniae (Sp) results in an inflammatory response in the respiratory epithelium. (A and B) At 3 h postchallenge, the upper respiratory tract of wild-type mice was flushed with lysis buffer to obtain total RNA. qRT-PCR was performed to measure the induction of IL-6 (A) and Snail-1 (B). n ≥ 4; *, P < 0.05; **, P < 0.005. (C) Immunohistochemistry of upper respiratory tract sections of wild-type (WT) and TLR2-deficient mice stained for phosphorylated p38 MAPK at 24 h postchallenge. Magnification, ×600.
These proinflammatory events occurred well before neutrophils were first observed to have appeared in the nasal lumen in response to colonizing bacteria (13). To determine whether the observed upregulation of inflammatory markers was of epithelial origin rather than a result of the influx of leukocytes, the expression of CD45 was assessed. CD45 is a marker expressed exclusively by leukocytes and not by the epithelium. A lack of enhanced CD45 expression in the lavage samples (data not shown) indicated that the contribution of leukocytes to these effects at 24 h postchallenge was negligible.
These results suggested that during the early course of colonization of the upper respiratory tract by H. influenzae and S. pneumoniae, TLRs mediate inflammatory response and activate the respiratory epithelium.
Respiratory epithelial cells are activated by bacterial pathogens through TLRs.
Signaling factors known to be involved in mediating the activation of host tissue after stimulation with stressors such as bacteria were examined in respiratory epithelial cells in vivo. One such pathway involves activation of MAPK p38 by phosphorylation as a consequence of the presence of the TLR signaling cascade (15). ATF-2 is a transcription factor that is in turn phosphorylated by MAPKs, including p38 MAPK (11, 15). To determine whether infection of the upper respiratory tract with H. influenzae resulted in p38 MAPK and ATF-2 activation in vivo, immunohistochemical staining of paraffin-embedded tissue sections from infected and mock-infected wild-type animals was performed at 24 h postchallenge. Figure 5A and B show that H. influenzae infection of the upper respiratory tract resulted in activation of p38 MAPK and ATF-2 in respiratory epithelial cells. This was detected as strong staining for phosphorylated p38 MAPK and ATF-2 in the nuclei of olfactory respiratory epithelial cells in animals infected with H. influenzae (Fig. 5A and B). To examine the role of TLRs in the activation of respiratory epithelium, TLR2- and TLR4-deficient animals were infected with H. influenzae. Both TLR2 and TLR4 deficiency resulted in decreased activation of p38 MAPK and ATF-2 signaling at 24 h postchallenge (Fig. 5A and B). In this case, minimal levels of phosphorylated p38 MAPK and ATF-2 were seen in the nuclei of TLR-deficient animals compared to wild-type mice. Similarly, Fig. 4C shows that S. pneumoniae infection of the upper respiratory tract resulted in phosphorylation and nuclear translocation of p38 MAPK in the respiratory epithelium at 24 h postchallenge. TLR2-deficient animals showed a defect in p38 MAPK activation when challenged with S. pneumoniae (Fig. 4C). These results confirmed that respiratory epithelial cells are activated during early colonization by H. influenzae and S. pneumoniae in a TLR-dependent manner. Furthermore, the results were consistent with the colonization data described above, showing a role for TLRs in the early clearance of H. influenzae and S. pneumoniae during colonization of the upper airway.
FIG. 5.

H. influenzae (Hi) activates p38 MAPK and ATF-2 signaling in respiratory epithelial cells in a TLR-dependent manner. (A and B) Immunohistochemistry of upper airway sections stained for phosphorylated p38 MAPK (A) and phosphorylated ATF-2 (B) at 24 h postchallenge. Magnification, ×600. WT, wild type.
TGF-β signaling pathways are activated in the respiratory epithelium in vivo.
Having shown that bacterial colonization resulted in an early inflammatory response in the host, we examined whether it also induces pathways that are involved in the regulation of inflammation and the barrier function of the epithelium, e.g., TGF-β signaling. TGF-β signaling is mediated by the transcription factors Smad2 and -3. As a consequence of TGF-β receptor activation, Smad2 and -3 are phosphorylated and are localized to the nuclei (12). To determine whether infection of the upper respiratory tract with H. influenzae resulted in Smad2 and -3 activation in vivo, immunohistochemical staining of paraffin-embedded tissue sections from infected and mock-infected wild-type animals was performed using a primary antibody specific for phosphorylated Smad2 and -3. Figure 6A shows that H. influenzae infection of the upper respiratory tract resulted in phosphorylation and nuclear translocation of Smad2 and -3 in respiratory epithelial cells in wild-type animals as early as 3 h postchallenge. To examine whether TLRs contribute to the activation of the TGF-β signaling cascade, TLR2- and TLR4-deficient mice were infected with H. influenzae. No Smad2 or -3 phosphorylation was observed in TLR4-deficient animals infected with H. influenzae, whereas in TLR2-deficient mice H. influenzae infection resulted in the activation of the TGF-β signaling cascade (Fig. 6A).
FIG. 6.

H. influenzae (Hi) activates the TGF-β signaling cascade. (A) Immunohistochemistry of upper respiratory tract sections stained for phosphorylated Smad2 and -3 at 3 h postchallenge. Magnification, ×600. WT, wild type. (B) At 3 h postchallenge, the upper respiratory tract was flushed with lysis buffer to obtain total RNA. qRT-PCR was performed to measure the induction of Snail-1. n ≥ 4; *, P < 0.05; **, P < 0.005; n.s, not significant.
Recent studies have shown that the transcription factor Snail-1 is induced via the TGF-β signaling cascade (14). Snail-1 is involved in complex processes such as epithelial-to-mesenchymal transition by suppressing epithelial markers (5, 44). We used qRT-PCR analysis to examine the expression of Snail-1 in respiratory cells in vivo. Infection of the upper respiratory tract with either H. influenzae or S. pneumoniae resulted in the induction of Snail-1 in the respiratory epithelium of wild-type mice at 3 h postchallenge (Fig. 6B and Fig. 4B). In addition, Fig. 6B shows that there was a defect in Snail-1 upregulation in TLR4-deficient animals, but not in TLR2-deficient animals, when they were infected with H. influenzae.
To evaluate whether ligands for TLRs alone are sufficient to induce IL-6 and Snail-1 expression in respiratory cells in vivo, animals were intranasally inoculated with highly purified H. influenzae LPS or Pam3CSK4, a synthetic ligand for TLR1 and -2, and lavage samples were analyzed by RT-PCR. Figure 7A and B show that the presence of purified H. influenzae LPS resulted in upregulation of Snail-1 or IL-6 in the respiratory epithelium of wild-type mice. There was no upregulation of Snail-1 or IL-6 in TLR4-deficient animals. In addition, there was a defect in the induction of IL-6 and Snail-1 in the respiratory epithelium of TLR2-deficient animals inoculated with Pam3CSK4 compared to wild-type animals, as shown by qRT-PCR analysis (Fig. 7C and D).
FIG. 7.

Specific ligands for TLRs are sufficient to induce an inflammatory response and TGF-β signaling. (A to D) At 3 h after intranasal challenge of wild-type (WT) and TLR4- and TLR2-deficient mice with 15 μg of LPS (A and B) or Pam3CSK4 (C and D), the upper respiratory tract was flushed with lysis buffer to obtain total RNA. qRT-PCR was performed to measure the induction of IL-6 (A and C) and Snail-1 (B and D). n ≥ 3; *, P < 0.05.
These results showed that the activation of the TGF-β signaling cascade depended on TLR signaling in vivo and that the presence of TLR ligands was sufficient to elicit these inflammatory responses.
DISCUSSION
This report focuses on the mucosal surface of the upper respiratory tract, since it is at this site that the host encounters colonizing pathogens for the first time. It is shown that respiratory epithelial cells are activated during bacterial colonization and orchestrate the initial immune response to colonizing pathogens in a TLR-dependent manner. The innate immune response of the respiratory epithelium includes the elaboration of inflammatory mediators as well as the activation of signaling pathways known to modulate the epithelial barrier. These responses are required to control the density of colonizing bacterial pathogens and prevent invasive disease.
In recent years, a variety of in vitro studies have shown that various microbes are capable of activating epithelial cells in a TLR-dependent manner (1, 9). Despite the multitude of studies dealing with bacterium-host interaction in vitro, little is known about the function of epithelial TLRs during bacterial colonization of the upper respiratory tract in vivo. An important goal of this study was to gain direct insight into immune functions of respiratory cells in vivo. Data from immunohistochemistry staining of paraffin-embedded tissue sections presented in this report reveal that MAPK signaling is induced in respiratory epithelial cells in a TLR-dependent manner. In the case of H. influenzae, TLR2 and TLR4 are needed to evoke a full response of the respiratory cells, whereas for S. pneumoniae TLR2 serves as the dominant receptor to elicit an immune response in the epithelial surface. This is consistent with in vitro studies showing a role for both receptors in recognizing H. influenzae (6, 19). Furthermore, we show that the induction of inflammatory markers such as IL-6, siderocalin, and Muc5AC in the respiratory epithelium depends on the presence of TLR signaling pathways. This is in line with in vitro studies showing that these factors are induced by the presence of bacterial pathogens in respiratory epithelial cells in a TLR-dependent manner (3, 23, 37). In addition, we could identify H. influenzae LPS as a ligand for TLR4 able to induce an inflammatory response in the upper respiratory tract, whereas the activation of the respiratory epithelium by a synthetic ligand for TLR1 and -2 was largely dependent on the presence of TLR2.
An important result of this study is the observation that the inflammatory response of respiratory cells to the presence of bacterial pathogens is accompanied by activation of the TGF-β signaling pathway. The finding that TGF-β signaling depends on detection of colonizing pathogens via TLRs in respiratory epithelial cells provides a direct link between the innate immune response of respiratory cells and signaling pathways known to regulate inflammation and to affect the barrier function of the respiratory epithelium (2). We show that bacterial colonization, in addition to activating the TGF-β signaling cascade, results in the induction of Snail-1, a transcription factor that is under the control of the TGF-β signaling pathway (28). In epithelial cells, Snail-1 downregulates epithelial markers such as E-cadherin, a factor required for functional tight junctions (5, 14). We show that Snail-1 induction depends on the activation of TLR and that activation of TLR2 or TLR4 with specific ligands is sufficient to induce the expression of Snail-1. Our results are consistent with recent studies showing that TLR signaling can result in enhanced TGF-β signaling (7, 39).
Even though the epithelial barrier is highly effective in preventing the invasion of pathogens, it is evident that it needs to be a dynamic structure, allowing the egress of immune cells and inflammatory mediators from the tissues to the mucosal surface, where bacteria reside (2, 21). Our data indicate that bacterial infection results in the opening of the epithelial barrier through recognition of pathogen-associated molecular patterns by TLRs, as demonstrated by the efflux of antimicrobial factors, including IgG, at the mucosal surface. The data presented in this study, together with those from previous studies, strongly suggest that the proinflammatory response of respiratory cells and the activation of the TGF-β signaling cascade mediate the loss of tight junctions and the subsequent opening of the epithelial barrier. In addition, it has been suggested that TGF-β signaling, together with that of IL-6, might be important in shifting the immune response toward Th17, which is now known to contribute to eventual clearance (18, 20). Even though innate immune functions and TGF-β signaling are needed for an appropriate immune response, they may, in some cases, be permissive for invasion of those microbial pathogens that are not adequately controlled by these inflammatory events and are able to take advantage of the gaps in the epithelial barrier. Furthermore, aberrant TGF-β signaling caused by bacterial infection within the respiratory tract may contribute to epithelial dysfunction and the progression of complex diseases, such as, for example, the development of chronic H. influenzae infections in patients with chronic obstructive pulmonary disease (4, 8, 31).
The colonization and survival data presented in this study underline the importance of the innate immune response of the upper respiratory tract during the early course of colonization in controlling colonization by opportunistic mucosal pathogens and in prevention of invasive disease caused by these microbes. Whereas murine pneumonia models did not show any contribution of TLR2 to the antimicrobial defense against S. pneumoniae in the lung (16), our colonization model revealed that activation of the mucosa via TLR2 provides protection from colonization and invasive infection in the upper airway. The different outcomes of the two models might be explained by the fact that S. pneumoniae is a pathogen that can directly invade the host at the mucosal surface of the upper respiratory tract without disseminating into the lung. Furthermore, our findings extend those of a study published by van Rossum et al. (42) that showed a defect of TLR2-deficient animals with respect to the clearance of S. pneumoniae at 3 weeks after colonization of the upper respiratory tract. In the case of H. influenzae, both TLR2 and TLR4 contribute to clearance of the bacteria in the upper respiratory tract during the early course of colonization. This adds to data showing that multiple pattern recognition receptors are needed to control H. influenzae colonization in the course of bacterial colonization (unpublished data). These findings are also consistent with a study published by Wang et al. that showed that TLR4 deficiency results in reduced clearance of H. influenzae in the lung (43).
In summary, our observations provide insight into the contributions of respiratory epithelial cells during bacterial colonization in vivo. Colonization of the upper respiratory tract by bacterial pathogens results in an early activation of proinflammatory and TGF-β signaling pathways of the respiratory epithelium in a TLR-dependent manner. Our study underlines the importance of innate immune functions of respiratory epithelial cells in controlling colonization and dissemination of bacterial pathogens in vivo.
Acknowledgments
We thank Kerstin Kandler for careful reading of the manuscript. Immunohistochemical studies were performed at the Morphology Core of the Center for the Molecular Studies of Liver and Digestive Disease (Center grant P30 DK50306).
This work was supported by grants from the U.S. Public Health Service to J.N.W. (AI 44231 and AI 138446).
Editor: A. Camilli
Footnotes
Published ahead of print on 2 March 2009.
REFERENCES
- 1.Bals, R., and P. S. Hiemstra. 2004. Innate immunity in the lung: how epithelial cells fight against respiratory pathogens. Eur. Respir. J. 23327-333. [DOI] [PubMed] [Google Scholar]
- 2.Beisswenger, C., C. B. Coyne, M. Shchepetov, and J. N. Weiser. 2007. Role of p38 MAP kinase and transforming growth factor-beta signaling in transepithelial migration of invasive bacterial pathogens. J. Biol. Chem. 28228700-28708. [DOI] [PubMed] [Google Scholar]
- 3.Birchler, T., R. Seibl, K. Buchner, S. Loeliger, R. Seger, J. P. Hossle, A. Aguzzi, and R. P. Lauener. 2001. Human Toll-like receptor 2 mediates induction of the antimicrobial peptide human beta-defensin 2 in response to bacterial lipoprotein. Eur. J. Immunol. 313131-3137. [DOI] [PubMed] [Google Scholar]
- 4.Bonniaud, P., P. J. Margetts, K. Ask, K. Flanders, J. Gauldie, and M. Kolb. 2005. TGF-beta and Smad3 signaling link inflammation to chronic fibrogenesis. J. Immunol. 1755390-5395. [DOI] [PubMed] [Google Scholar]
- 5.Cano, A., M. A. Perez-Moreno, I. Rodrigo, A. Locascio, M. J. Blanco, M. G. del Barrio, F. Portillo, and M. A. Nieto. 2000. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 276-83. [DOI] [PubMed] [Google Scholar]
- 6.Chen, R., J. H. Lim, H. Jono, X. X. Gu, Y. S. Kim, C. B. Basbaum, T. F. Murphy, and J. D. Li. 2004. Nontypeable Haemophilus influenzae lipoprotein P6 induces MUC5AC mucin transcription via TLR2-TAK1-dependent p38 MAPK-AP1 and IKKβ-IκBα-NF-κB signaling pathways. Biochem. Biophys. Res. Commun. 3241087-1094. [DOI] [PubMed] [Google Scholar]
- 7.Chow, E. K., R. M. O'Connell, S. Schilling, X. F. Wang, X. Y. Fu, and G. Cheng. 2005. TLR agonists regulate PDGF-B production and cell proliferation through TGF-beta/type I IFN crosstalk. EMBO J. 244071-4081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gauldie, J., M. Kolb, K. Ask, G. Martin, P. Bonniaud, and D. Warburton. 2006. Smad3 signaling involved in pulmonary fibrosis and emphysema. Proc. Am. Thorac. Soc. 3696-702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gómez, M. I., and A. Prince. 2008. Airway epithelial cell signaling in response to bacterial pathogens. Pediatr. Pulmonol. 4311-19. [DOI] [PubMed] [Google Scholar]
- 10.Greene, C. M., and N. G. McElvaney. 2005. Toll-like receptor expression and function in airway epithelial cells. Arch. Immunol. Ther. Exp. (Warsaw) 53418-427. (In Polish.) [PubMed] [Google Scholar]
- 11.Gupta, S., D. Campbell, B. Derijard, and R. J. Davis. 1995. Transcription factor ATF2 regulation by the JNK signal transduction pathway. Science 267389-393. [DOI] [PubMed] [Google Scholar]
- 12.Itoh, S., F. Itoh, M. J. Goumans, and D. P. Ten. 2000. Signaling of transforming growth factor-beta family members through Smad proteins. Eur. J. Biochem. 2676954-6967. [DOI] [PubMed] [Google Scholar]
- 13.Kadioglu, A., J. N. Weiser, J. C. Paton, and P. W. Andrew. 2008. The role of Streptococcus pneumoniae virulence factors in host respiratory colonization and disease. Nat. Rev. Microbiol. 6288-301. [DOI] [PubMed] [Google Scholar]
- 14.Kaimori, A., J. Potter, J. Y. Kaimori, C. Wang, E. Mezey, and A. Koteish. 2007. Transforming growth factor-beta1 induces an epithelial-to-mesenchymal transition state in mouse hepatocytes in vitro. J. Biol. Chem. 28222089-22101. [DOI] [PubMed] [Google Scholar]
- 15.Kawai, T., and S. Akira. 2007. TLR signaling. Semin. Immunol. 1924-32. [DOI] [PubMed] [Google Scholar]
- 16.Knapp, S., C. W. Wieland, C. van't Veer, O. Takeuchi, S. Akira, S. Florquin, and T. van der Poll. 2004. Toll-like receptor 2 plays a role in the early inflammatory response to murine pneumococcal pneumonia but does not contribute to antibacterial defense. J. Immunol. 1723132-3138. [DOI] [PubMed] [Google Scholar]
- 17.Kobayashi, K., L. D. Hernandez, J. E. Galan, C. A. Janeway, Jr., R. Medzhitov, and R. A. Flavell. 2002. IRAK-M is a negative regulator of Toll-like receptor signaling. Cell 110191-202. [DOI] [PubMed] [Google Scholar]
- 18.Li, M. O., Y. Y. Wan, and R. A. Flavell. 2007. T cell-produced transforming growth factor-β1 controls T cell tolerance and regulates Th1- and Th17-cell differentiation. Immunity 26579-591. [DOI] [PubMed] [Google Scholar]
- 19.Lorenz, E., D. C. Chemotti, A. L. Jiang, and L. D. McDougal. 2005. Differential involvement of Toll-like receptors 2 and 4 in the host response to acute respiratory infections with wild-type and mutant Haemophilus influenzae strains. Infect. Immun. 732075-2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu, Y. J., J. Gross, D. Bogaert, A. Finn, L. Bagrade, Q. Zhang, J. K. Kolls, A. Srivastava, A. Lundgren, S. Forte, C. M. Thompson, K. F. Harney, P. W. Anderson, M. Lipsitch, and R. Malley. 2008. Interleukin-17A mediates acquired immunity to pneumococcal colonization. PLoS. Pathog. 4e1000159. doi: 10.1371/journal.ppat.1000159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lysenko, E. S., A. J. Ratner, A. L. Nelson, and J. N. Weiser. 2005. The role of innate immune responses in the outcome of interspecies competition for colonization of mucosal surfaces. PLoS. Pathog. 1e1. doi: 10.1371/journal.ppat.0010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Malley, R., P. Henneke, S. C. Morse, M. J. Cieslewicz, M. Lipsitch, C. M. Thompson, E. Kurt-Jones, J. C. Paton, M. R. Wessels, and D. T. Golenbock. 2003. Recognition of pneumolysin by Toll-like receptor 4 confers resistance to pneumococcal infection. Proc. Natl. Acad. Sci. USA 1001966-1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martin, F. J., and A. S. Prince. 2008. TLR2 regulates gap junction intercellular communication in airway cells. J. Immunol. 1804986-4993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Masoud, H., E. R. Moxon, A. Martin, D. Krajcarski, and J. C. Richards. 1997. Structure of the variable and conserved lipopolysaccharide oligosaccharide epitopes expressed by Haemophilus influenzae serotype b strain Eagan. Biochemistry 362091-2103. [DOI] [PubMed] [Google Scholar]
- 25.Matsuguchi, T., T. Musikacharoen, T. Ogawa, and Y. Yoshikai. 2000. Gene expressions of Toll-like receptor 2, but not Toll-like receptor 4, is induced by LPS and inflammatory cytokines in mouse macrophages. J. Immunol. 1655767-5772. [DOI] [PubMed] [Google Scholar]
- 26.Mayer, A. K., M. Muehmer, J. Mages, K. Gueinzius, C. Hess, K. Heeg, R. Bals, R. Lang, and A. H. Dalpke. 2007. Differential recognition of TLR-dependent microbial ligands in human bronchial epithelial cells. J. Immunol. 1783134-3142. [DOI] [PubMed] [Google Scholar]
- 27.McCool, T. L., and J. N. Weiser. 2004. Limited role of antibody in clearance of Streptococcus pneumoniae in a murine model of colonization. Infect. Immun. 725807-5813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Medici, D., E. D. Hay, and D. A. Goodenough. 2006. Cooperation between snail and LEF-1 transcription factors is essential for TGF-beta1-induced epithelial-mesenchymal transition. Mol. Biol. Cell 171871-1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mikami, F., J. H. Lim, H. Ishinaga, U. H. Ha, H. Gu, T. Koga, H. Jono, H. Kai, and J. D. Li. 2006. The transforming growth factor-beta-Smad3/4 signaling pathway acts as a positive regulator for TLR2 induction by bacteria via a dual mechanism involving functional cooperation with NF-kappaB and MAPK phosphatase 1-dependent negative cross-talk with p38 MAPK. J. Biol. Chem. 28122397-22408. [DOI] [PubMed] [Google Scholar]
- 30.Muir, A., G. Soong, S. Sokol, B. Reddy, M. I. Gomez, H. A. Van Heekeren, and A. Prince. 2004. Toll-like receptors in normal and cystic fibrosis airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 30777-783. [DOI] [PubMed] [Google Scholar]
- 31.Murphy, T. F., A. L. Brauer, A. T. Schiffmacher, and S. Sethi. 2004. Persistent colonization by Haemophilus influenzae in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 170266-272. [DOI] [PubMed] [Google Scholar]
- 32.Nelson, A. L., J. M. Barasch, R. M. Bunte, and J. N. Weiser. 2005. Bacterial colonization of nasal mucosa induces expression of siderocalin, an iron-sequestering component of innate immunity. Cell Microbiol. 71404-1417. [DOI] [PubMed] [Google Scholar]
- 33.Pamer, E. G. 2007. Immune responses to commensal and environmental microbes. Nat. Immunol. 81173-1178. [DOI] [PubMed] [Google Scholar]
- 34.Ratner, A. J., E. S. Lysenko, M. N. Paul, and J. N. Weiser. 2005. Synergistic proinflammatory responses induced by polymicrobial colonization of epithelial surfaces. Proc. Natl. Acad. Sci. USA 1023429-3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roche, A. M., S. J. King, and J. N. Weiser. 2007. Live-attenuated Streptococcus pneumoniae induce serotype-independent mucosal and systemic protection in mice. Infect. Immun. 752469-2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rubin, B. K. 2002. Physiology of airway mucus clearance. Respir. Care 47761-768. [PubMed] [Google Scholar]
- 37.Schmeck, B., S. Huber, K. Moog, J. Zahlten, A. C. Hocke, B. Opitz, S. Hammerschmidt, T. J. Mitchell, M. Kracht, S. Rosseau, N. Suttorp, and S. Hippenstiel. 2006. Pneumococci induced TLR- and Rac1-dependent NF-kappaB-recruitment to the IL-8 promoter in lung epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 290:L730-L737. [DOI] [PubMed] [Google Scholar]
- 38.Schröder, N. W., S. Morath, C. Alexander, L. Hamann, T. Hartung, U. Zahringer, U. B. Gobel, J. R. Weber, and R. R. Schumann. 2003. Lipoteichoic acid (LTA) of Streptococcus pneumoniae and Staphylococcus aureus activates immune cells via Toll-like receptor (TLR)-2, lipopolysaccharide-binding protein (LBP), and CD14, whereas TLR-4 and MD-2 are not involved. J. Biol. Chem. 27815587-15594. [DOI] [PubMed] [Google Scholar]
- 39.Seki, E., M. S. De, C. H. Osterreicher, J. Kluwe, Y. Osawa, D. A. Brenner, and R. F. Schwabe. 2007. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat. Med. 131324-1332. [DOI] [PubMed] [Google Scholar]
- 40.Shuto, T., H. Xu, B. Wang, J. Han, H. Kai, X. X. Gu, T. F. Murphy, D. J. Lim, and J. D. Li. 2001. Activation of NF-kappa B by nontypeable Haemophilus influenzae is mediated by toll-like receptor 2-TAK1-dependent NIK-IKK alpha /beta-I kappa B alpha and MKK3/6-p38 MAP kinase signaling pathways in epithelial cells. Proc. Natl. Acad. Sci. USA 988774-8779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Takeuchi, O., K. Hoshino, T. Kawai, H. Sanjo, H. Takada, T. Ogawa, K. Takeda, and S. Akira. 1999. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity 11443-451. [DOI] [PubMed] [Google Scholar]
- 42.van Rossum, A. M., E. S. Lysenko, and J. N. Weiser. 2005. Host and bacterial factors contributing to the clearance of colonization by Streptococcus pneumoniae in a murine model. Infect. Immun. 737718-7726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang, X., C. Moser, J. P. Louboutin, E. S. Lysenko, D. J. Weiner, J. N. Weiser, and J. M. Wilson. 2002. Toll-like receptor 4 mediates innate immune responses to Haemophilus influenzae infection in mouse lung. J. Immunol. 168810-815. [DOI] [PubMed] [Google Scholar]
- 44.Willis, B. C., and Z. Borok. 2007. TGF-beta-induced EMT: mechanisms and implications for fibrotic lung disease. Am. J. Physiol. Lung Cell Mol. Physiol. 293L525-L534. [DOI] [PubMed] [Google Scholar]