

Abstract
Listeria monocytogenes σB and positive regulatory factor A (PrfA) are pleiotropic transcriptional regulators that coregulate a subset of virulence genes. A positive regulatory role for σB in prfA transcription has been well established; therefore, observations of increased virulence gene expression and hemolytic activity in a ΔsigB strain initially appeared paradoxical. To test the hypothesis that L. monocytogenes σB contributes to a regulatory network critical for appropriate repression as well as induction of virulence gene expression, genome-wide transcript profiling and follow-up quantitative reverse transcriptase PCR (qRT-PCR), reporter fusion, and phenotypic experiments were conducted using L. monocytogenes prfA*, prfA* ΔsigB, ΔprfA, and ΔprfA ΔsigB strains. Genome-wide transcript profiling and qRT-PCR showed that in the presence of active PrfA (PrfA*), σB is responsible for reduced expression of the PrfA regulon. σB-dependent modulation of PrfA regulon expression reduced the cytotoxic effects of a PrfA* strain in HepG2 cells, highlighting the functional importance of regulatory interactions between PrfA and σB. The emerging model of the role of σB in regulating overall PrfA activity includes a switch from transcriptional activation at the P2prfA promoter (e.g., in extracellular bacteria when PrfA activity is low) to posttranscriptional downregulation of PrfA regulon expression (e.g., in intracellular bacteria when PrfA activity is high).
Listeria monocytogenes is a food-borne pathogen capable of transitioning from saprotrophic survival in the environment (55) to intracellular infection in a wide range of hosts (21, 65). While cellular L. monocytogenes infection and the systemic stages of listeriosis have been studied extensively (14, 29, 74), less attention has been directed to the preceding phases of the infection process (e.g., bacterial survival in foods and in the human gastrointestinal tract). Mounting evidence indicates that the transition of L. monocytogenes from saprotroph to pathogen relies upon regulatory networks that fine-tune virulence factor expression in response to environmental signals (29). These networks include genes that regulate the bacterial stress response and survival, therefore contributing to transmission of L. monocytogenes, including that during the gastrointestinal and systemic stages of infection (12, 26). One important network that links stress response and virulence in L. monocytogenes is coregulated by σB and positive regulatory factor A (PrfA) (12, 49, 56).
σB, an alternative sigma factor, regulates genes that are important for environmental stress survival in gram-positive bacteria (36, 59, 75). In L. monocytogenes, σB contributes to survival under low pH, oxidative stress, and carbon starvation (13, 22, 23, 35, 76). In addition to stress response genes, the L. monocytogenes σB regulon also includes some genes that are important for virulence (36, 59). Examples of σB-dependent virulence-associated genes include inlAB, encoding internalins A and B (38, 39, 42), which are important for invasion of human epithelial cells, as well as hfq, encoding the Hfq RNA binding protein (13), and bsh, encoding bile salt hydrolase (18). Phenotypically, an L. monocytogenes ΔsigB strain is less invasive in human tissue culture cells and has reduced virulence in guinea pigs following intragastric infection relative to an otherwise isogenic parent strain (26, 38).
PrfA is a member of the Crp (cyclic AMP receptor protein)/Fnr (fumarate and nitrate reduction regulator) family of transcriptional regulators (41). PrfA positively regulates the expression of virulence genes essential for intracellular survival of L. monocytogenes (e.g., hly, mpl, plcA, actA, and plcB) (28, 73). PrfA recognizes a 14-bp palindromic “PrfA box” sequence, which is typically located ∼40 nucleotides upstream of a target transcriptional start site (7, 24, 25, 69). Regulation of prfA expression and PrfA activity is complex, occurring at transcriptional and posttranscriptional levels. At the posttranscriptional level, PrfA activity is influenced by a number of environmental factors and physiological states, including the presence of fermentable carbohydrates (5, 16, 61, 63) or activated charcoal (20, 61, 63), intracellular status (43, 50), growth in minimal medium (5, 71), and entry into stationary phase (47, 64, 71). Several prfA mutations that yield constitutively active PrfA (PrfA*) have been identified (70). Compared to wild-type PrfA, PrfA* has a greater affinity for the PrfA box (19), which appears to result in increased relative expression of PrfA-regulated target genes. As a consequence, PrfA* strains are useful tools for in vitro simulation of the high PrfA activity levels typical of intracellular L. monocytogenes cells, i.e., transcript levels of the PrfA-dependent gene plcA are similar in a PrfA* strain grown in a liquid medium (46) and in intracellular L. monocytogenes.
At the transcriptional level, prfA is transcribed from three different promoter sites (Fig. 1). One promoter site (PplcA), which is located upstream of plcA, can initiate synthesis of a bicistronic mRNA comprising plcA and prfA (8, 24). Two promoter sites (P1prfA and P2prfA) are located immediately upstream of prfA (25). The P1prfA promoter is recognized by the L. monocytogenes housekeeping sigma factor σA (48). The P2prfA promoter region consists of two overlapping promoters, one recognized by σA and one recognized by σB (53, 60, 67), indicating a positive regulatory role for σB in prfA expression. This positive regulatory role for σB initially appeared to be contradicted by observations of increased virulence gene expression and hemolytic activity in a ΔsigB strain (10, 33, 53, 59). However, in the gram-positive pathogen Staphylococcus aureus, σB appears to have both positive and negative roles in regulating expression of virulence factors, positively influencing expression of a number of adhesins and negatively influencing expression of numerous exoenzymes and toxins (3). Therefore, to test the hypothesis that as with S. aureus σB, L. monocytogenes σB contributes to a regulatory network critical for appropriate induction and repression of virulence gene expression, comprehensive microarray-based genome-wide transcript profiling was conducted using L. monocytogenes prfA*, prfA* ΔsigB, ΔprfA, and ΔprfA ΔsigB strains. To gain further insight into the functional relevance of the emerging σB-PrfA regulatory network suggested by the transcriptional profiling results, microarray analyses were followed by quantitative reverse transcriptase PCR (qRT-PCR) and phenotypic studies.
FIG. 1.

L. monocytogenes plcA-prfA region (not drawn to scale). The line represents the DNA sequence, with the plcA and prfA coding sequences indicated as open boxes. Promoter regions contributing to prfA transcription are indicated by brackets above the line. Regulatory elements for each promoter region are indicated below the line. P2prfA −35 and −10 promoter sites represent sequences for two overlapping promoters, one of which is σB dependent and one of which is σA dependent.
MATERIALS AND METHODS
Bacterial growth.
To enable identification of PrfA- and σB-coregulated genes, growth conditions and strains were selected to provide high levels of activity for both proteins. To illustrate, because entry into stationary phase induces σB-dependent transcription (10, 59), early-stationary-phase cells were used to ensure high σB activity. An L. monocytogenes strain with a prfA*(G155S) allele was used to constitutively express high levels of PrfA-regulated virulence genes (46, 52, 70). L. monocytogenes stock cultures of the strains used in this study (Table 1) were stored at −80°C in brain heart infusion broth (BHI broth; Difco, Detroit, MI) plus 15% glycerol. Cultures were streaked from stocks onto BHI agar and incubated at 37°C for ∼24 h to obtain single colonies. Individual colonies of each stock culture were inoculated into 5 ml BHI broth and incubated at 37°C with aeration (shaking at 250 rpm) for 12 h, followed by serial passages as described below to maximize the relative proportion of viable cells present in a similar growth state. Specifically, 50 μl of a 12-h culture was inoculated into 5 ml fresh, prewarmed BHI broth. Cells were then grown to mid-logarithmic phase (optical density at 600 nm = 0.4), diluted 1:100 in 50 ml prewarmed BHI broth in 300-ml Nephelo culture flasks (Bellco Glass Co., Vineland, NJ), and grown to early stationary phase (i.e., growth was monitored to an optical density at 600 nm of 1.0, and then the culture was incubated for an additional 3 h).
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Relevant genotype | Reference or constructiona |
|---|---|---|
| pNF771 | prfA*(G155S) | 70 |
| pBMB54 | P1P2prfA-gus | This work |
| pBMB6 | ΔP1(−10)P2prfA-gus | 67 |
| pBMB7 | P1ΔP2(−10)prfA-gus | 67 |
| pBMB8 | ΔP1(−10)ΔP2(−10)prfA-gus | 67 |
| 10403S | Parent strain; serotype 1/2a | 4 |
| FSL A1-254 | ΔsigB | 76 |
| FSL B2-046 | ΔprfA | 47 |
| FSL B2-068 | ΔsigB ΔprfA | 47 |
| FSL B2-237 | prfA*(G155S) | pNF771→10403S |
| FSL B2-238 | prfA*(G155S) ΔsigB | pNF771→FSL A1-254 |
| FSL B2-220 | P1P2prfA-gus ΔprfA | pBMB54→FSL B2-046 |
| FSL B2-153 | P1ΔP2(−10)prfA-gus ΔprfA | pBMB7→FSL B2-046 |
| FSL B2-149 | ΔP1(−10)P2prfA-gus ΔprfA | pBMB6→FSL B2-046 |
| FSL B2-165 | ΔP1(−10)ΔP2(−10)prfA-gus ΔprfA | pBMB8→FSL B2-046 |
| FSL B2-221 | P1P2prfA-gus ΔsigB ΔprfA | pBMB54→FSL B2-068 |
| FSL B2-152 | P1ΔP2(−10)prfA-gus ΔsigB ΔprfA | pBMB7→FSL B2-068 |
| FSL B2-148 | ΔP1(−10)P2prfA-gus ΔsigB ΔprfA | pBMB6→FSL B2-068 |
| FSL B2-164 | ΔP1(−10)ΔP2(−10)prfA-gus ΔsigB ΔprfA | pBMB8→FSL B2-068 |
| FSL B2-243 | P1P2prfA-gus prfA* | pBMB54→FSL B2-237 |
| FSL B2-241 | P1ΔP2(−10)prfA-gus prfA* | pBMB7→FSL B2-237 |
| FSL B2-240 | ΔP1(−10)P2prfA-gus prfA* | pBMB6→FSL B2-237 |
| FSL B2-242 | ΔP1(−10)ΔP2(−10)prfA-gus prfA* | pBMB8→FSL B2-237 |
| FSL B2-252 | P1P2prfA-gus prfA* ΔsigB | pBMB54→FSL B2-238 |
| FSL B2-250 | P1ΔP2(−10)prfA-gus prfA* ΔsigB | pBMB7→FSL B2-238 |
| FSL B2-249 | ΔP1(−10)P2prfA-gus prfA* ΔsigB | pBMB6→FSL B2-238 |
| FSL B2-251 | ΔP1(−10)ΔP2(−10)prfA-gus prfA* ΔsigB | pBMB8→FSL B2-238 |
Arrows denote transformation of the plasmid into the recipient strain.
RNA extraction.
RNA extraction from L. monocytogenes was conducted as previously described (59). Briefly, 2 volumes of RNAprotect bacterial reagent (Qiagen, Valencia, CA) was added to each bacterial culture just prior to harvest. The mixture was vortexed and incubated for 5 min at room temperature. Cells were harvested by centrifugation for 5 min at 5,000 × g and stored at −80°C. RNA isolation was performed using an RNeasy Midi kit (Qiagen, Valencia, CA). Samples were treated with DNase for 1 h at 37°C, using 40 units RQ1 DNase enzyme (Promega, Madison, WI) in the presence of 400 units of RNasin Plus RNase inhibitor (Promega). Each lysate was extracted with phenol-chloroform, and then RNA was precipitated using a 1/10 volume of 3 M sodium acetate and 2.5 volumes of ice-cold 100% ethanol. Precipitated RNA was stored at −80°C. Prior to use, RNA quality was assessed using agarose gel electrophoresis and measurement of 260/280 and 260/230 absorption ratios, using a NanoDrop-1000 instrument (NanoDrop Technologies, Wilmington, DE).
cDNA labeling and microarray hybridization.
cDNA labeling was performed as previously described (59), with minor modifications. Briefly, cDNA synthesis and labeling of total RNA were performed using the SuperScript Plus indirect cDNA labeling system for DNA microarrays (Invitrogen, Carlsbad, CA). For cDNA synthesis, 10 μg total RNA was mixed with 5 μg random hexamers in a total volume of 18 μl RNase-free water. The RNA-primer mix was incubated for 10 min at 70°C, with a subsequent chill on ice for at least 5 min. After the addition of Superscript III RT, amino-modified deoxynucleoside triphosphates, dithiothreitol, RNaseOUT, and buffer, the reaction mix was incubated at 42°C for approximately 17 h to allow cDNA synthesis. RNA was hydrolyzed with the addition of 10 μl 1 N NaOH and 10 μl 0.5 M EDTA, followed by incubation at 65°C for 15 min. Prior to cDNA purification using a Qiagen PCR purification kit (Qiagen), the mixture was neutralized using 10 μl 1 N HCl. cDNA labeling reactions with Alexa Fluor 555 or Alexa Fluor 647 fluorescent dyes were performed for 2 h at room temperature according to the manufacturer's protocol. Differentially labeled cDNAs from the two strains to be cohybridized were combined, dried in a Savant SVC100 Speed-Vac (Farmingdale, NY), and stored at −80°C until hybridization.
Microarrays were constructed as previously described (9, 59). Briefly, 70-mer probes targeting 2,857 L. monocytogenes open reading frames (ORFs) (Array-Ready Oligo set; Operon Technologies, Huntsville, AL) (designed from the L. monocytogenes EGD-e genome sequence [27]) were spotted onto Corning UltraGAPS slides (Corning Inc., Corning, NY) at the Microarray Core Facility at Cornell University (Ithaca, NY). EGD-e and L. monocytogenes 10403S both represent the same L. monocytogenes lineage (II), serotype (1/2a), and ribotype (DUP-1039C), and therefore probes designed using the EGD-e genome were expected to hybridize well with 10403S genes (9, 59). Cross-hybridization identities between the EGD-e probes and the target genes in strain 10403S were determined using the unfinished genome sequence for strain 10403S (9, 59; Listeria monocytogenes Database [http://www.broad.mit.edu/annotation/genome/listeria_group/MultiHome.html]). The array used here allows comprehensive identification of differentially expressed genes in strain 10403S, with the possibility of some false-negative results (9, 59).
Microarray hybridization was performed as previously described (59), with minor modifications. Prior to hybridization, spotted microarray slides were incubated for 1 h in a 1% bovine serum albumin-5× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate)-0.1% sodium dodecyl sulfate (SDS) solution prewarmed to 42°C. Subsequently, slides were washed twice for 5 min in 0.1× SSC and twice for 1 min in filtered water and then dried by centrifugation at 1,800 rpm for 3 min. The combined cDNA targets were reconstituted in 55 μl hybridization buffer and denatured at 95°C for 5 min. Targets were applied to microarray slides and overlaid with mSeries LifterSlips (Erie Scientific, Portsmouth, NH), followed by overnight hybridization at 42°C. Posthybridization washes were performed for (i) 5 min in 42°C prewarmed 2× SSC plus 0.1% SDS, (ii) 5 min in 2× SSC, and (iii) 2.5 min in 0.2× SSC. After a final wash in filtered water, slides were dried by centrifugation and scanned with a GenePix 4000B scanner (Molecular Devices, Sunnyvale, CA) at the Cornell University Microarray Core Facility.
Statistical analysis of microarray data.
The microarray experiments were designed as a balanced double loop (Fig. 2) to allow direct comparison of results for any given strain to those for any other strain used in this study (37). This design allowed us to identify genes regulated by σB, PrfA, or both proteins. Raw intensity values for all probes on each array were normalized using pin-tip LOWESS (58) in R v.2.2.1 with the MAANOVA (v. 0.98-8) package (R/MAANOVA [http://www.bioconductor.org/packages/bioc/1.7/src/contrib/html/maanova.html]). Signals from two replicate probes on each array were averaged, and log2 transformations were performed after normalization. Differences in transcript levels between strains were determined using a mixed-model one-way analysis of variance (ANOVA) in R/MAANOVA, where Y (the log2-transformed intensity data) = array + dye + strain + sample (biological replicate) + E (error). Differences in transcript levels were considered meaningful if they met both adjusted P values of <0.05 and changes of >1.5-fold.
FIG. 2.
Schematic of the double loop design used to compare transcript levels in L. monocytogenes prfA*, prfA* ΔsigB, ΔprfA, and ΔprfA ΔsigB strains. Each pairwise comparison was performed for four independent biological replicates. Each arrow represents two different biological replicates. Arrowheads represent Alexa Fluor 555-labeled RNA, and arrow bases represent Alexa Fluor 647-labeled RNA. Factors used for two-way ANOVA are indicated above or below the strain designation.
Contributions of both PrfA and σB (SigB) to changes in transcript levels were determined using a mixed-model two-way ANOVA, where Y (the log2-transformed intensity data) = array + dye + PrfA + SigB + SigB* PrfA + sample + E. In this model, the factors “PrfA” and “SigB” can have one of two levels, determined by the presence or absence of the protein in the tested strain (Fig. 2). ANOVA modeling allows for consideration of appropriate error structures for experiments with multiple sources of variation in microarray measurements (37). The random effects of the models were biological replicate and array effects, whereas the fixed effects were PrfA, SigB, and dye effects. The Fs statistic, a shrinkage estimator for gene-specific variance components that makes no assumptions about the distribution of variances across genes, was estimated (15). Significant differences in expression between strains were determined by calculating the P values for the Fs statistic for each gene, using 1,000 random permutations. The P values were adjusted to correct for type I error with the Benjamini-Hochberg (B-H) linear step-up correction implemented in R/MAANOVA, with a cutoff adjusted P value of <0.05. Pairwise contrasts of individual mutants were estimated by the t test in R/MAANOVA. Contrast P values were corrected for multiple testing by using the Benjamini-Hochberg step-up correction.
Expression profiles of the 607 ORFs with significant differences (adjusted P values of <0.05) in at least one strain-to-strain comparison (as determined by one-way ANOVA) were analyzed using quality threshold (QT) clustering, which groups genes with similar expression profiles based on jackknife correlations (32). QT clustering of significant ORFs was conducted in MeV 4.0 (www.tigr.org), with a diameter of 0.5 and a minimum cluster size of 5.
TaqMan qRT-PCR.
TaqMan primer and probe sets (see Table S1 in the supplemental material) were designed using Primer Express 2.0 software (Applied Biosystems, Foster City, CA). qRT-PCR was performed as described by Sue et al. (72), using TaqMan one-step RT-PCR master mix reagent, Multiscribe RT, and an ABI Prism 7000 sequence detection system (all from Applied Biosystems). qRT-PCRs excluding Multiscribe RT were run in parallel to quantify genomic DNA contamination. DNA standard curves for each gene were included to allow for absolute quantification of cDNA levels. Each qRT-PCR was run in duplicate on each of the four RNA samples that were used for the microarray experiments. The L. monocytogenes housekeeping genes rpoB and gap were included to allow for normalization of absolute transcript levels as described previously (11, 36). Data analysis was conducted with ABI Prism 7000 SDS software, and significant differences in transcript levels between the different strains were determined by ANOVA as described previously (72). To confirm the two-way ANOVA results for the microarray data, transcript levels determined by qRT-PCR were analyzed using a two-way ANOVA model similar to that described above, with the following linear model: transcript level Y = PrfA + SigB + SigB* PrfA + sample + E.
Hemolytic activity assays.
Hemolytic activity assays using purified sheep red blood cells were performed as previously described (10) to assess hemolytic activity in the supernatants of bacterial cultures. Hemolytic activity assays were performed on strains grown independently three times; resulting hemolytic units were averaged.
Invasion assays.
Invasion assays were performed using the human hepatic epithelial cell line HepG2 (ATCC HB-8065) as described previously (39), with minor modifications. Briefly, HepG2 cells were grown in Dulbecco's modified Eagle's medium (DMEM). Tissue culture cells were seeded 2 days before infection, at a density of 7.5 × 104 cells per well, in 24-well tissue culture plates and incubated at 37°C (5% CO2). Infection of HepG2 cells was performed using approximately 2 × 107 CFU of L. monocytogenes cells grown to early stationary phase as described above. At 30 min postinfection, cells were washed three times using prewarmed sterile phosphate-buffered saline to remove extracellular bacteria. Subsequently, 1 ml of fresh prewarmed DMEM was added to each well, plates were incubated for an additional 15 min to allow attached L. monocytogenes cells to enter HepG2 cells, and then the DMEM was removed and replaced with 1 ml prewarmed DMEM containing 150 μg/ml gentamicin to kill remaining extracellular bacteria. The 150-μg/ml gentamicin concentration was selected to reduce the risk of survival and (false-positive) detection of extracellular L. monocytogenes, particularly as we used a short incubation period with gentamicin (i.e., 45 min). While the gentamicin concentration used is higher than that in some other studies, it is well within the range of concentrations previously reported, including the use of 150 μg/ml gentamicin by Kim et al. (38) for invasion assays with Henle 2 cells, the recommended use of 150 μg/ml gentamicin for Caco-2 cell invasion assays (44), and the use of an even higher concentration (i.e., 250 μg/ml) by Moroni et al. (51). After a final incubation of 45 min, HepG2 cells were lysed by the addition of ice-cold sterile distilled water. Numbers of intracellular L. monocytogenes cells were determined by plating the cell suspensions on BHI agar, using a spiral plater (Autoplate 4000; Spiral Biotech, Bethesda, MD). Plates were incubated at 37°C for ∼24 h before enumerating colonies by use of QCount (Spiral Biotech). The invasion efficiency was calculated as the number of bacteria recovered from a HepG2 cell suspension (in CFU/ml) divided by the initial bacterial number (in CFU/ml) used for the inoculation. Three independent biological replicates were performed for each strain (triplicate wells were infected with a given strain).
Cytotoxicity assay.
As a measure of the cytotoxic interactions between L. monocytogenes strains and HepG2 cells, lactate dehydrogenase (LDH) release from HepG2 cells following infection was measured using a CytoTox 96 nonradioactive cytotoxicity assay kit (Promega, Madison, WI) as previously described by Decatur and Portnoy (17), with minor modifications. HepG2 cells were grown, seeded, and infected as described previously (17). The experimental design included three wells containing only DMEM to account for background absorption as well as three wells containing uninfected HepG2 cells to measure spontaneous LDH release. At 30 min postinfection, DMEM was removed and replaced with DMEM containing 150 μg/ml gentamicin, followed by incubation at 37°C for 1 h. To determine maximum LDH release, 100 μl of lysis buffer was added to triplicate infected wells 45 min prior to LDH measurement. At 90 min postinfection, the 24-well plates were centrifuged at 250 × g for 4 min, using a Sorvall RT600B swing-bucket centrifuge (Kendro, Asheville, NC). A 50-μl aliquot of the supernatant was removed and used for the LDH assay. The supernatant was incubated for 30 min with 50 μl substrate mix prior to the addition of 50 μl stop solution. Absorption at 490 nm was then measured using a Packard fusion instrument (Perkin-Elmer Inc., Waltham, MA). After background correction, the percent cytotoxicity was calculated as follows: % cytotoxicity = [(experimental LDH release − spontaneous LDH release)/(maximum LDH release − spontaneous LDH release)] × 100. Three independent biological replicates of the cytotoxicity assay were performed for each strain.
GUS activity assay.
Previously described GUS reporter fusion (GUS-RF) plasmids (67) were introduced into L. monocytogenes ΔprfA, ΔsigB ΔprfA, prfA*, and prfA* ΔsigB strains (Table 1) to monitor transcription from the individual promoters that contribute to prfA transcription (Fig. 1); one fusion plasmid (i.e., P1P2prfA-gus) was reconstructed to have identical 5′ and 3′ junctions to those of all other fusions (Table 1). While the use of reporter fusions on multicopy plasmids can cause problems in interpreting reporter fusion data (e.g., due to plasmid copy number variation), all reporter fusion strains contained very similar inserts (which differed by only one or two 20-nucleotide deletions) and were grown under identical standardized conditions, making significant differences in copy numbers between the different strains unlikely in our assay. GUS activity was measured by monitoring cleavage of the β-glucuronidase substrate 4-methylumbelliferyl β-d-glucuronide (MUG) (34) in a 96-well-plate format. To maintain the extrachromosomal GUS-RF plasmid, all growth experiments were performed using BHI supplemented with 10 μg/ml chloramphenicol. L. monocytogenes GUS-RF strains were grown to early stationary phase as described above. Bacterial cells (1 ml) were harvested by centrifugation and washed with 1 ml 0.1 M potassium phosphate buffer (60 mM K2HPO4, 40 mM KH2PO4, 0.1 M NaCl), and cell pellets were then flash frozen in liquid nitrogen and stored at −80°C. Bacterial numbers were determined by plating sample aliquots on BHI-chloramphenicol agar plates. Prior to GUS measurements, cell pellets were thawed and suspended in 1 ml potassium phosphate buffer. Cells were lysed by the addition of 135 μl CellLytic B reagent (Sigma-Aldrich, St. Louis, MO), followed by incubation for 10 min at room temperature. Duplicate samples of bacterial lysates (80 μl) and appropriate dilutions (in potassium phosphate buffer) were pipetted into 96-well flat-bottomed black polystyrene plates (Corning Inc., Corning, NY). The enzymatic reaction was initiated by addition of 20 μl of 0.4-mg/ml MUG (Sigma-Aldrich) in dimethyl sulfoxide. A standard curve corresponding to 3.75, 1.88, 0.94, 0.47, and 0.09 μM 4-methylumbelliferone (MU; Sigma-Aldrich) was included with every plate. The enzymatic reaction was stopped after 10 min by the addition of 1 M Na2CO3 stop solution. Immediately after the addition of stop solution, fluorescence was measured at 460 nm (with an excitation wavelength of 365 nm), using a Packard fusion instrument (Perkin-Elmer). The amount of background fluorescence determined for a given sample was subtracted from the fluorescence measurement in the corresponding experimental well, and the concentration of liberated MU was calculated using the standard curve. The GUS activity for each strain was measured in three independent biological replicates and reported as nM MU/log CFU/min.
Statistical analysis of data from phenotypic experiments.
A mixed-model ANOVA was performed in SAS (SAS v 9.1; SAS Institute Inc., Cary, NC) to determine significant differences in invasion efficiency, LDH release, and GUS activity among the L. monocytogenes strains. The significance of differences in invasion efficiencies was evaluated using the following linear model: invasion efficiency = strain + biological replicate. The significance of differences in percent LDH release was determined using the following linear model: percent LDH release = strain + biological replicate. To determine the statistical significance of differences in GUS activity among the strains, a one-way ANOVA was performed, comparing the GUS activities of a given promoter fusion among the different genetic backgrounds by using the following model: GUS activity = strain (genetic background strain) + rep (biological replicate). A two-way ANOVA was also used to determine if the GUS activity was significantly affected by the presence or absence of SigB and/or PrfA, using the following model: GUS activity = SigB (presence or absence of σB) + PrfA (presence or absence of PrfA) + SigB* PrfA (interaction effect) + rep (biological replicate). Tukey's multiple comparison correction was applied to all ANOVA results to determine significant differences among the strains. Adjusted P values of <0.05 were considered statistically significant.
Microarray data accession number.
Microarray data were deposited at the Gene Expression Omnibus under accession number GSE 11347.
RESULTS
σB- and PrfA-dependent L. monocytogenes genes represent eight clusters of genes with distinct transcription profiles.
L. monocytogenes whole-genome microarrays were used to compare global gene expression of L. monocytogenes prfA*, prfA* ΔsigB, ΔprfA, and ΔprfA ΔsigB strains, allowing identification of genes regulated by σB, PrfA, or both proteins (Fig. 2). One-way ANOVA (see Table S2 in the supplemental material) with the microarray data identified 607 genes that were differentially expressed (adjusted P value of <0.05; ≥1.5-fold change) in at least one strain-to-strain comparison (e.g., PrfA* versus ΔprfA strains). Subsequently, t tests were used to determine significant differences in transcript levels between all possible pairs of strains. QT cluster analysis of expression profiles grouped 603 differentially expressed genes into eight clusters, with each cluster containing genes with similar expression profiles (Fig. 3), as determined by jackknife correlation (35). The expression profiles for four genes could not be placed into any of the clusters due to a lack of similarity to other identified profiles. The majority of genes (518/603 genes [86%]) were assigned to clusters 1 and 2. Relative transcript levels for genes in these clusters were not significantly different between the prfA* and ΔprfA strains or between the prfA* ΔsigB and ΔprfA ΔsigB strains (as determined by t tests), indicating that transcription of the genes in these two clusters is PrfA independent. Genes assigned to cluster 1 (n = 274) had higher relative transcript levels in ΔsigB strains (ΔprfA ΔsigB and prfA* ΔsigB strains) than in strains carrying an intact sigB gene (ΔprfA and prfA* strains), suggesting that σB negatively influences expression of these genes. Conversely, genes in cluster 2 (n = 244) had higher relative transcript levels in ΔprfA and prfA* strains than in ΔprfA ΔsigB and prfA* ΔsigB strains, suggesting that transcription of these genes is positively influenced by σB. σB-dependent transcription has been reported previously for 114 of the cluster 2 genes (59).
FIG. 3.

QT clusters for 603 differentially expressed genes with significant changes in relative transcript levels. (A) Heat map of the log2 relative expression values for each gene in each strain. The QT clusters are separated by a gray line, and each cluster is denoted by a number on the side of the heat map. (B) For each QT cluster, the log2 relative expression values are plotted for each gene, as well as the average log2 relative expression values. For a given cluster, differences in transcript levels between strains can be determined by comparing their average log2 relative expression values. For example, while cluster 3 gene transcript levels do not differ for ΔprfA and ΔsigB ΔprfA strains, transcripts levels differ by ∼2.3 log2 between the ΔsigB ΔprfA (average log2 expression = −1.3) and prfA* (average log2 expression = 1.0) strains.
Cluster 3 contained 24 genes, including all members of the L. monocytogenes virulence gene cluster (see Table S2 in the supplemental material). Transcript levels of the genes in this cluster were higher in strains with constitutively active PrfA* (prfA* and prfA* ΔsigB strains) than in ΔprfA strains (ΔprfA and ΔprfA ΔsigB strains) (see Table S2 in the supplemental material), indicating that the presence of PrfA* increased transcription of these genes (Fig. 3). For genes in this cluster, the average difference in transcript levels between the PrfA* and ΔprfA strains was 5.2-fold, while the average difference in transcript levels between the prfA* ΔsigB and ΔprfA ΔsigB strains was 12.5-fold. On average, genes in cluster 3 had 2.4-fold higher transcript levels in the prfA* ΔsigB strain than in the PrfA* mutant strain, indicating that PrfA*-mediated increases in transcript levels for genes in cluster 3 are higher in the absence of σB than in its presence.
A total of 51 genes were assigned to clusters 4, 5, 6, 7, and 8 (Fig. 3). For genes in these clusters, the observed average difference in transcript levels between strains was generally low, with the largest differences in transcript levels observed for cluster 5 genes. Transcript levels for genes within this cluster were, on average, twofold higher in the prfA* ΔsigB strain than those in the ΔprfA strain. This result suggests that genes in this cluster are positively regulated by PrfA* and negatively regulated by σB, as the highest transcript levels were observed in the presence of PrfA* and the absence of σB.
Transcript levels for a number of genes are significantly affected by both PrfA* and σB.
To identify potential interactions between σB and PrfA* that affect gene expression, we used a two-way ANOVA model to assess the effects of the presence or absence of PrfA* or σB on transcript levels. This model tested whether PrfA* significantly influenced transcript levels by comparing transcript levels observed in the presence of PrfA* to those observed in the absence of PrfA* (Fig. 2). Likewise, the effect of σB was tested by comparing transcript levels in the presence of σB to those in the absence of σB (see Table S3 in the supplemental material). A significant PrfA*-σB statistical interaction effect does not enable inferences regarding potential direct or indirect regulation by the proteins, nor does it imply physical interaction between the proteins, but it simply indicates that the effect of either PrfA* or σB on transcript levels is influenced by the presence or absence of the other protein (Table 2).
TABLE 2.
L. monocytogenes 10403S genes coregulated by PrfA* and σB, as determined by microarray and qRT-PCR analyses
| Gene | QTCc | Microarray result (fold change)f
|
qRT-PCR result (fold change)f
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Two-way ANOVA main effecta
|
Two-way ANOVA interaction effect with PrfA* presentb
|
Two-way ANOVA main effecta
|
Two-way ANOVA interaction effect with PrfA* presentb
|
||||||||
| PrfA* | SigB | SigB present | SigB absent | SigB absent/SigB presentd | PrfA* | SigB | SigB present | SigB absent | SigB absent/SigB presentd | ||
| hly | 3 | 160 | −1.8 | 620 | 3.7 × 103 | 6.0 | |||||
| actAe | 3 | 30 | −2.3 | 3.4 × 106 | 1.8 × 107 | 5.3 | |||||
| plcA | 3 | 180 | NS | 2.6 × 103 | 8.3 × 103 | 3.2 | |||||
| plcB | 3 | 110 | −1.7 | ND | ND | ND | ND | ND | |||
| lmo0207 | 3 | 8.5 | −2.2 | ND | ND | ND | ND | ND | |||
| lmo2219 | 3 | 7.4 | NS | 6.9 | 30 | 4.3 | |||||
| lmo2684 | 5 | 1.5 | −1.6 | ND | ND | ND | ND | ND | |||
| inlAe | 3 | 4.5 | NS | ND | ND | ND | ND | ||||
| mpl | 3 | 1.2 | 4.5 | 3.8 | 60 | 300 | 5 | ||||
| inlB | 3 | −1.4 | 4.7 | 6.6 | 5.5 | 85 | 15 | ||||
| inlC | 3 | −1.3 | 5.9 | 7.7 | 7.9 | 57 | 7.2 | ||||
| lmo0090 | −1.5 | 2.1 | 3.1 | NS | 2.0 | ||||||
| lmo0443 | 6 | 1.2 | −1.7 | 0.5 | 1.1 | 1.7 | 1.5 | ||||
| lmo0484e | 8 | 1.3 | −1.5 | 0.5 | ND | ND | ND | ND | ND | ||
| lmo2261 | 8 | 1.4 | −1.6 | 0.4 | ND | ND | ND | ND | ND | ||
The column titled PrfA* reports the ratio of transcript levels in the presence of PrfA* to transcript levels in the absence of PrfA*; the column titled SigB reports the ratio of transcript levels in the presence of σB to transcript levels in the absence of σB. For genes that show a statistical interaction effect, main effects cannot be reported.
For genes that show no significant statistical interaction effect, no values are shown; for genes that show a significant statistical interaction effect, transcript ratios are reported for (i) the presence of PrfA* compared to the absence of PrfA* in the presence of σB (column “SigB present”) and (ii) the presence of PrfA* compared to the absence of PrfA* in the absence of σB (column “SigB absent”).
QT cluster, as determined from one-way ANOVA results of microarray data (see Fig. 3).
Ratio of gene expression in a PrfA* background in the absence of SigB to that in the presence of SigB. Ratios were calculated by dividing the “SigB absent” statistical interaction effect by the “SigB present” statistical interaction effect. Negative values were first converted to positive values by taking the reciprocal before calculating the ratio.
Cross-hybridization identities between the 10403S genes listed in this table and the EGD-e-based probes on the microarray were 100%, except for actA (94%), lmo0484 (98%), and inlA (98%).
NS, not significant (change of <1.5-fold or adjusted P value of ≥0.05); ND, not determined using qRT-PCR.
Two-way ANOVA showed a significant main effect of PrfA* (i.e., significantly different transcript levels in the presence of PrfA* compared to those in its absence) on expression patterns for 17 genes. Specifically, for 13 genes (including the virulence genes plcA, hly, actA, plcB, uhpT, and inlA) (see Table S4 in the supplemental material), transcript levels were higher in the presence of PrfA* than in its absence, with transcript levels for plcA, hly, and plcB being >100-fold higher in the presence of PrfA*. For four genes (ftsH, gltX, cysS, and lmo0208), transcript levels were lower in the presence of PrfA* than in its absence, with 1.5- to 1.7-fold lower transcript levels in the presence of PrfA*, suggesting a negative PrfA* effect on transcript levels for these genes. However, a negative role for PrfA* in regulation of two of these genes (ftsH and gltX) was not confirmed by qRT-PCR; cysS and lmo0208 transcript levels were not assessed by qRT-PCR. The absence of confirmatory evidence of a negative role for PrfA in the present study is consistent with previous reports (56, 68), suggesting that PrfA's role in negative or indirect gene regulation may be weak and therefore difficult to reproduce. Among the 17 genes with a significant effect of PrfA* on transcript levels, 6 also showed a significant effect of σB on transcript levels. Five of these genes (i.e., hly, actA, plcB, lmo0207, and lmo2684) had higher transcript levels in the presence of PrfA* and lower transcript levels in the presence of σB (Table 2). Differences in hly, actA, and plcB transcript levels in the presence of σB than in its absence ranged from 1.7- to 2.3-fold.
In addition to the independent effects of PrfA* and σB on transcript levels for the 17 genes described above, two-way ANOVA of transcript levels for 7 genes (i.e., mpl, inlB, inlC, lmo0090, lmo0443, lmo2261, and lmo0484) identified significant statistical interaction effects for these genes, indicating that the effect of PrfA* on transcript levels was dependent on the presence or absence of σB or vice versa (Table 2). The effect of the presence or absence of PrfA* on transcript levels of these genes in the presence of σB appeared to be small, with absolute differences ranging from 1.2- to 1.5-fold. In contrast, the effect of the presence or absence of PrfA* on transcript levels of these genes was larger in the absence of σB, with absolute differences in transcript levels ranging from 1.5- to 5.9-fold, suggesting that PrfA* has a greater effect on transcript levels of these seven genes when σB is absent. For four genes, transcript levels were higher in the presence of PrfA* (indicating positive regulation), while for three genes, transcript levels were lower in the presence of PrfA*.
Comparison of positively PrfA*-regulated genes identified in this study with PrfA-dependent genes identified in previous studies.
Overall, our microarray data identified 17 genes (including the virulence genes actA, hly, plcA, plcB, and mpl) (see Table S4 in the supplemental material) that are positively regulated by PrfA*. Specifically, a total of 13 genes showed significantly higher transcript levels in the presence of PrfA* than in the absence of PrfA* (as determined by two-way ANOVA), while 4 genes showed significant interaction effects between σB and PrfA*, with higher transcript levels in the presence of PrfA*. The genes identified in this study as positively regulated by PrfA* in strain 10403S were consistent with genes previously identified as positively regulated by PrfA in strains EGD-e (49) and EGD (45), with the exceptions of lmo0107, lmo0206, lmo0217, lmo1536, lmo2219, and lmo2684, which were not identified as positively regulated by PrfA* in previous microarray studies (45, 49) (see Table S4 in the supplemental material). lmo2219 was previously identified as PrfA dependent in a proteomic study with 10403S (57).
qRT-PCR data confirm PrfA*-σB interaction effects on transcript levels for selected genes.
qRT-PCR was used as a sensitive, quantitative approach for measuring transcript levels of genes identified as being regulated by either or both PrfA* and σB. Coregulation of two genes identified by microarray analysis as positively regulated by PrfA* and negatively regulated by σB (i.e., hly and actA) was confirmed by qRT-PCR (Table 2). qRT-PCR results also indicated an interaction effect of PrfA* and σB on hly, actA, plcA, and lmo2219 transcript levels. Microarray analyses suggested positive regulation of these four genes by PrfA and negative regulation of these genes by σB, although negative regulation of plcA and lmo2219 by σB did not meet our microarray cutoff criteria (adjusted P value of <0.05; change of >1.5-fold): lmo2219 had a change of −1.87-fold (adjusted P value = 0.084), and plcA had a change of −1.28-fold (adjusted P value = 0.013). For all four genes (i.e., hly, actA, plcA, and lmo2219), qRT-PCR data showed higher PrfA*/ΔprfA transcript ratios in the absence of σB (e.g., 3.7 × 103 for hly) than the PrfA*/ΔprfA transcript ratios in the presence of σB (e.g., 617 for hly) (Table 2), supporting negative regulation of these genes by σB in the presence of PrfA*. Together with the microarray data, the qRT-PCR results confirm an overall negative effect of σB on transcript levels for hly, actA, plcA, and lmo2219.
qRT-PCR confirmed the significant PrfA*-σB interaction effects on transcript levels for mpl, inlC, inlB, and lmo0443 that were identified in our microarray analyses. For mpl, qRT-PCR results were consistent with the microarray data; both data sets showed higher PrfA*/ΔprfA transcript ratios in the absence of σB (i.e., 4.5) than the transcript ratios in the presence of σB (i.e., 1.2) (Table 2). qRT-PCR also confirmed the microarray data for inlB and inlC. For example, in the presence of PrfA*, microarray and qRT-PCR results showed that inlC transcript levels were 7.7 and 7.2 times higher, respectively, in the absence of σB than in the presence of σB (Table 2), thus supporting negative regulation of inlC by σB in the presence of PrfA*. qRT-PCR confirmed the significant PrfA*-σB interaction effect on transcript levels for lmo0443 and found a slight negative effect of σB on transcript levels in the presence of PrfA* (1.5-fold) (Table 2). The negative effect of σB had not been observed in the microarray data, possibly reflecting the overall low changes observed (<2-fold for all comparisons) for lmo0443 (Table 2). Finally, although positive regulation of lmo0090 by σB was confirmed by qRT-PCR, the significant PrfA*-σB interaction effect suggested by the microarray data was not confirmed. Overall, significant PrfA*-σB interaction effects on transcript levels were identified by qRT-PCR for eight genes, including four genes for which both microarray and qRT-PCR results showed significant PrfA*-σB interaction effects (Table 2).
Phenotypic assays confirm the importance of PrfA*-σB interactions on L. monocytogenes virulence gene expression.
To further characterize the effects of prfA and sigB deletions on virulence-associated characteristics, we performed HepG2 invasion and LDH release assays as well as hemolytic activity assays with the L. monocytogenes 10403S parent strain and the prfA*, prfA* ΔsigB, ΔprfA, and ΔprfA ΔsigB strains. In the HepG2 invasion assay, 10403S and the ΔprfA strain showed similar recoveries of gentamicin-protected bacteria (Fig. 4A), consistent with previous data showing that PrfA is not critical for host cell invasion (29). In contrast, the ΔprfA ΔsigB strain was recovered at lower numbers than those for 10403S (adjusted P value = 0.07), consistent with previous data showing that σB is important for transcription of genes important for invasion (42, 43). Interestingly, the prfA* and prfA* ΔsigB strains were recovered at lower numbers than those for both the parent and ΔprfA strains, indicating that L. monocytogenes strains expressing constitutively active PrfA* were less protected from the gentamicin treatment than strains lacking PrfA* (Fig. 4A). Although these results could be interpreted as resulting from reduced invasion of the PrfA* strain, visual inspection of HepG2 monolayers revealed that the host cells appeared to be damaged when infected with the prfA* or prfA* ΔsigB strain, suggesting possible increased cytotoxicity of the L. monocytogenes PrfA* strains relative to the 10403S and ΔprfA strains. Therefore, to test this hypothesis and to quantify relative cytotoxicities of the prfA* and prfA* ΔsigB strains to HepG2 cells, we performed an LDH release assay on infected HepG2 cells (Fig. 4B). HepG2 monolayers infected with the prfA* or prfA* ΔsigB strain showed 28% or 49% LDH release, respectively, which was significantly higher than the amount of LDH released from HepG2 cells infected with the 10403S, ΔprfA, or ΔprfA ΔsigB strain (adjusted P values of <0.0001) (Fig. 4B). The low release of LDH (<2%) from HepG2 cells infected with the 10403S, ΔprfA, or ΔprfA ΔsigB strain indicated little host cell damage resulting from infection with these strains. Interestingly, LDH release was significantly higher in HepG2 cells infected with the prfA* ΔsigB strain than in those infected with the prfA* strain (adjusted P value of <0.0001), suggesting that the presence of σB modulates PrfA-mediated gene expression to reduce host cell damage induced by intracellular L. monocytogenes.
FIG. 4.
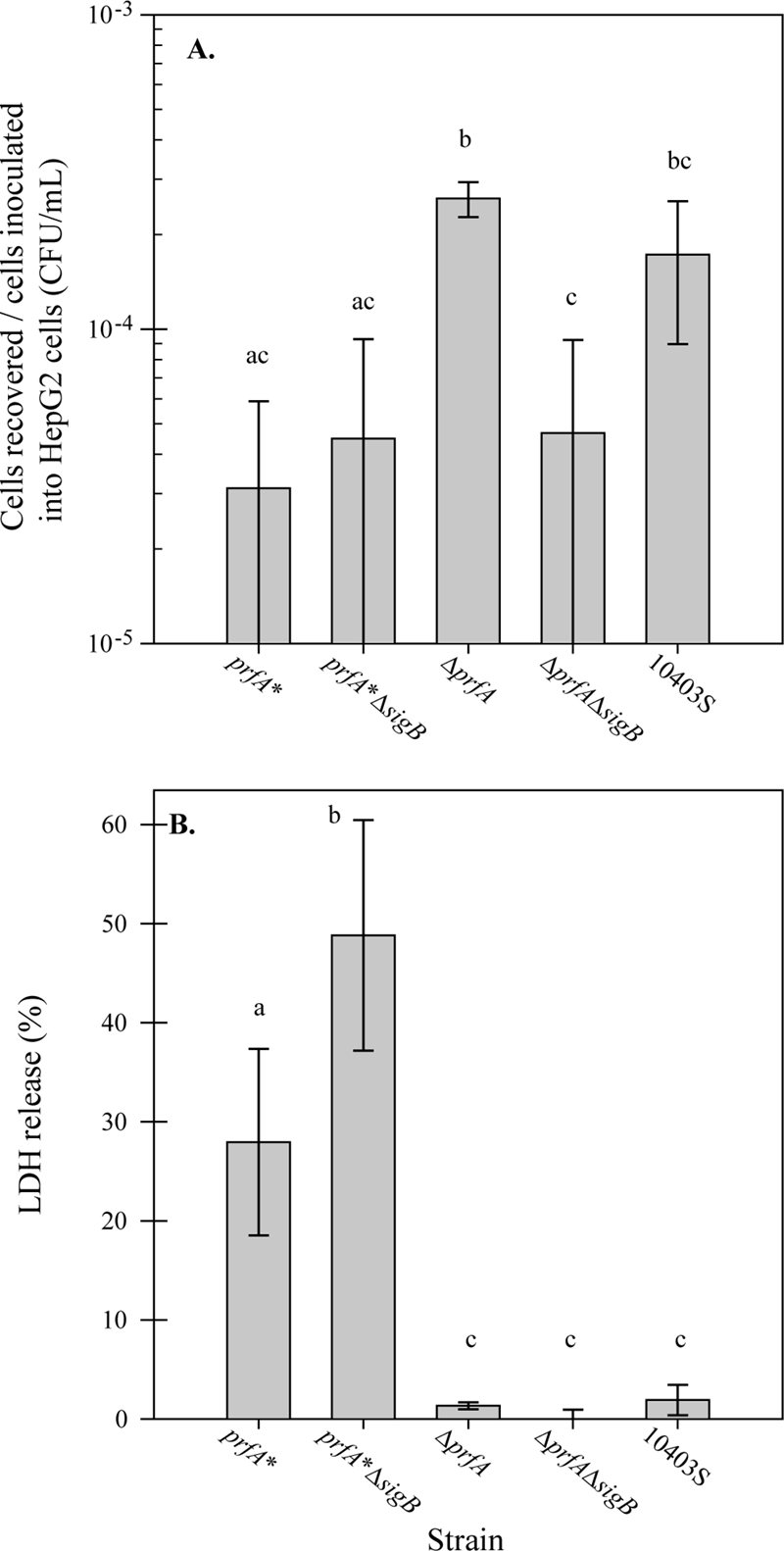
Ratios of L. monocytogenes cells recovered to L. monocytogenes cells inoculated into HepG2 cells (in CFU/ml) (A) and percent LDH release from HepG2 cells infected with L. monocytogenes (B). The average and standard deviation for three independent biological replicates are plotted for each strain. Significant differences (adjusted P values of <0.05) between strains in either number of cells recovered from HepG2 cells after gentamicin treatment or LDH release are indicated by lowercase letters above the bars. Strains with the same letter are not significantly different.
Measurement of hemolytic activities among the various strains further supported the observation that σB modulates gene expression to reduce expression of virulence genes in a PrfA* background (Table 3). Specifically, we showed that the prfA* ΔsigB strain had higher hemolytic activity (232 hemolytic units [HU]) than a prfA* strain (213.3 HU) (Table 3). Low hemolytic activities were observed for the ΔprfA and ΔprfA ΔsigB strains, consistent with the PrfA-dependent nature of hly transcription (6, 54, 57, 71).
TABLE 3.
Hemolytic activities of L. monocytogenes strains used in this study
| Strain | HU of extracellular hemolysina |
|---|---|
| PrfA* | 213.3 |
| PrfA* ΔsigB | 232 |
| ΔprfA | 2 |
| ΔsigB ΔprfA | 2 |
| 10403S | 18.7 |
Values represent the averages for three independent biological replicates.
Reporter fusion assays show that σB regulates prfA transcription in a PrfA* background.
A number of PrfA-dependent virulence genes had higher transcript levels in the prfA* ΔsigB strain than in the prfA* strain in both our microarray and qRT-PCR studies (Table 2; Fig. 3). Therefore, to measure specific contributions of σB and PrfA* to prfA transcription, we introduced a series of extrachromosomal plasmids containing GUS-RF fused to the prfA promoter region into the prfA*, prfA* ΔsigB, ΔprfA, and ΔprfA ΔsigB strains (Fig. 5). Reporter fusion measurements in bacterial cells grown to stationary phase showed that all strains with P1P2prfA-gus and ΔP1(−10)P2prfA-gus fusions had higher activities than strains with reporter fusions that contained P1ΔP2(−10)prfA-gus (Fig. 5), indicating that the P2prfA promoter region plays the predominant role in activating prfA transcription under these conditions. In addition, the strains with wild-type σB (i.e., prfA* and ΔprfA strains) showed higher GUS activities with both the P1P2prfA-gus fusion and the ΔP1(−10) P2prfA-gus fusion than the corresponding ΔsigB strains (i.e., prfA* ΔsigB and ΔprfA ΔsigB strains) did, indicating that σB positively influences prfA transcription, most likely due to the previously described σB-dependent transcriptional activation of the P2prfA promoter (53). Specifically, results from two-way ANOVA indicated significantly higher GUS activity in the presence of σB than in its absence for the P1P2prfA-gus fusion (1.3-fold higher; P = 0.027). Strains with PrfA* (i.e., prfA* and prfA* ΔsigB strains) showed lower GUS activity from the ΔP1(−10)P2prfA-gus fusion than did the corresponding ΔprfA strains (i.e., ΔprfA, ΔsigB ΔprfA, and ΔsigB strains), indicating that PrfA* negatively influences prfA transcription in this fusion, consistent with previous data suggesting that PrfA-dependent repression of prfA transcription occurs under some conditions (25). Conversely, strains with PrfA* (i.e., prfA* and prfA* ΔsigB strains) always showed higher GUS activity for all reporter fusions with the P2prfA −10 deletion [i.e., the P1ΔP2(−10)prfA-gus and ΔP1(−10)ΔP2(−10)prfA-gus fusions] than did strains with ΔprfA, indicating that PrfA* positively regulates prfA transcript levels in strains with a P2prfA −10 promoter region deletion. Taken together, our results indicate that complex interactions between different promoter elements, PrfA*, and σB contribute to fine-tuning prfA transcript levels. This conclusion is supported by results from two-way ANOVA analyses, which showed a significant statistical interaction indicating that GUS fusion activities due to PrfA* were different depending on the presence/absence of σB for both the ΔP1(−10)P2prfA-gus and ΔP1(−10)ΔP2(−10)prfA-gus fusion strains (P = 0.0257 and P = 0.0005, respectively). Overall, our GUS reporter fusion results show that σB itself does not negatively influence prfA transcription. These results suggest that the negative effects of σB on expression of PrfA-regulated genes are not a consequence of direct σB-dependent downregulation of prfA transcription but rather occur at a subsequent stage (i.e., likely through a σB-dependent gene product).
FIG. 5.

GUS activities, in nM MU/log CFU/min, for prfA promoter-gus reporter fusions expressed in different L. monocytogenes genetic backgrounds. Diagrams of prfA-gus promoter fusion constructs show the promoter elements present in each reporter fusion plasmid. L. monocytogenes strains bearing extrachromosomal plasmids with a prfA promoter-gus reporter fusion were grown to early stationary phase. Data represent the mean quantities of liberated MU measured in three independent biological replicates; standard deviations are reported in parentheses. Different superscript letters indicate significantly different GUS activities (adjusted P values of <0.05) within a given row (i.e., in a comparison among strains containing the same gus reporter fusion).
DISCUSSION
Previous studies have clearly established that σB plays an important role in regulating L. monocytogenes gene expression during gastrointestinal survival and invasion (26, 38, 39, 47, 72), while PrfA is well established as a critical transcriptional regulator of L. monocytogenes virulence gene expression during intracellular survival and cell-to-cell spread (reviewed in reference 40). Increasing evidence suggests that σB and PrfA coregulate genes important for the transition of L. monocytogenes from the extracellular to the intracellular environment (12, 56). To gain further insight into the σB-PrfA regulatory network, we evaluated genome-wide transcription patterns for L. monocytogenes 10403S strains with and without PrfA* and σB, using both single (ΔsigB and ΔprfA strains) and double (ΔsigB ΔprfA strain) mutant strains (Fig. 2). A PrfA* strain was selected for these studies to ensure constitutively high PrfA activity and, consequently, to increase the likelihood of identifying regulatory interactions between highly active PrfA (i.e., PrfA*) and σB. L. monocytogenes strains with the PrfA*(G155S) allele have PrfA activity levels similar to those found in intravacuolar bacteria, as determined by hly and actA transcript levels, even when the PrfA* strain is grown under in vitro conditions (36, 46). While PrfA* strains are important tools for studying PrfA-dependent transcription outside the mammalian host (45, 49, 62), one cannot exclude the possibility that intracellular L. monocytogenes organisms differ from extracellular PrfA* strains in some aspects of PrfA-dependent gene regulation (e.g., levels and activity status of PrfA).
In addition to microarray analyses, the σB-PrfA regulatory network was characterized using our set of isogenic mutant strains in a series of qRT-PCR, reporter fusion, and phenotypic assays. Overall, our data indicate that (i) σB downregulates expression of the PrfA regulon in the presence of PrfA*, reducing the host-cell-damaging potential of PrfA-mediated gene expression in intracellular L. monocytogenes; and (ii) the L. monocytogenes 10403S PrfA regulon is small (<20 positively regulated genes) and includes a complex regulatory system of prfA transcription involving autoregulation by PrfA* and positive regulation of prfA transcription by σB. Combined with results from previous studies (e.g., see references 26, 36, and 56), these findings suggest a complex σB-PrfA regulatory network that includes a switch from σB-dependent upregulation of genes important during the gastrointestinal stage of infection, including initial upregulation of prfA, to σB-mediated downregulation of the PrfA regulon in intracellular L. monocytogenes.
The core PrfA regulon is downregulated by σB in the presence of PrfA*.
Our results conclusively establish a new role for L. monocytogenes σB, i.e., its importance in downregulating PrfA-regulated transcription in the presence of active PrfA (PrfA*). Data supporting this conclusion include (i) higher transcript levels for members of the PrfA regulon in the prfA* ΔsigB strain than in the prfA* strain in our microarray analyses, suggesting a negative effect of σB on the transcription of certain genes; (ii) qRT-PCR confirmation of the microarray transcription pattern for selected genes in the PrfA regulon, with higher actA and hly transcript levels in the prfA* ΔsigB strain than in the prfA* strain; (iii) higher hemolytic activity in the prfA* ΔsigB strain than in the prfA* strain; and (iv) higher HepG2 cytotoxicity for the prfA* ΔsigB strain than for the prfA* strain, consistent with the observed increased hemolytic activity. Importantly, these observations, obtained using a PrfA* strain, are consistent with observations previously reported for L. monocytogenes strains with wild-type PrfA. To illustrate, an L. monocytogenes ΔsigB strain had significantly higher hly transcript levels under growth conditions similar to those used in this study (59). In another study (33), the same ΔsigB strain also showed slightly but significantly higher plcA transcript levels than those in the parent strain when bacteria were exposed to σB-inducing conditions (salt stress). The ΔsigB strain has also been shown to have higher hemolytic activity than 10403S, supporting increased virulence gene expression in the absence of σB (10, 53).
The phenotypic importance of σB-mediated downregulation of the PrfA regulon in intracellular L. monocytogenes was clearly demonstrated through hemolytic activity and tissue culture assays. Therefore, we propose that in addition to the previously described biochemical and regulatory mechanisms that appear to protect the host cell from L. monocytogenes-mediated cytotoxic damage, such as the low pH necessary for optimal hemolysin activity (i.e., hemolysin activity is reduced at cytoplasmic pH) (2) and control of hly translation (66), our observations provide evidence for the existence of a transcriptional fine-tuning mechanism that acts to control the expression of genes encoding cytotoxic proteins in cytoplasmic L. monocytogenes.
The L. monocytogenes 10403S PrfA regulon appears to be small and includes a complex regulatory system of prfA transcription involving autoregulation by PrfA* and positive regulation of prfA transcription by σB.
Our microarray data indicate that a small number of genes (<20) are positively regulated by PrfA*, consistent with microarray data reported by Milohanic et al. (49), suggesting that PrfA predominantly regulates a small set of genes through direct transcriptional activation via a PrfA binding site. Marr et al. (45) reported a much larger number of genes as positively regulated by PrfA*, including many genes without a preceding PrfA binding site. These genes were identified when PrfA* was overexpressed, so it is possible that regulation of these genes by PrfA may be of limited importance under most conditions in wild-type strains. The positively regulated PrfA-dependent genes identified in our study were generally consistent with genes previously identified as positively regulated by PrfA in EGD-e (49), EGD (45), and 10403S (57). While previous studies have shown that inlA and inlB transcription is both σB and PrfA dependent in a wild-type strain under in vitro conditions (38, 39), in our experiments with a PrfA* strain these genes were predominantly PrfA* dependent. Taken together, these results suggest that transcription of inlAB may switch from primarily σB dependent under extracellular conditions (e.g., in intestinal bacteria) to PrfA-dependent transcription in intracellular bacteria.
A number of studies have shown that regulation of prfA transcription includes an autoregulatory feedback loop through a bicistronic plcA-prfA transcript initiating from the PrfA-dependent plcA promoter (8, 24, 25). In addition to regulation mediated through the plcA promoter, our data indicate that PrfA*, σB, and the presence of both P1prfA and P2prfA promoters play complex roles in fine-tuning prfA transcription. The P2prfA promoter appears to contribute the majority of prfA transcripts, consistent with previous reports (36). In the absence of either intact P1prfA or intact P2prfA, PrfA* appears to upregulate or downregulate prfA transcription in a manner that is dependent on the existing promoter configuration. Specifically, in the ΔP1(−10) P2prfA-gus fusion strain, the presence of PrfA* appears to repress prfA transcription, consistent with data reported by Freitag and Portnoy (24, 30) which suggested that binding of PrfA to the P2prfA PrfA box might inhibit overall prfA transcription. On the other hand, for prfA reporter fusions that lack the P2(−10)prfA region (but retain the PrfA box in the P2prfA region), higher GUS activities were found in strains containing PrfA* than in ΔprfA strains, suggesting that PrfA* also activates prfA transcription at the P2prfA promoter and that the PrfA box in the P2prfA region may be sufficient for initiation of low levels of PrfA*-mediated transcription, even in the absence of a P2prfA −10 region, possibly due to the high affinity of PrfA*(G155S) for its target DNA (79). In contrast with results for the ΔP1(−10)P2prfA-gus fusion strain, in the absence of the P2prfA −10 region the loss of PrfA* does not enhance transcription at P2prfA, likely because of the overall reduced transcription initiation at P2prfA.
Alternatively, it is possible that the role of PrfA* in prfA transcription switches from activator to repressor depending on the level of active prfA transcription. To illustrate, our data are consistent with a model in which PrfA* negatively affects prfA transcription under conditions when prfA transcript levels are high [e.g., in the ΔP1(−10)P2prfA-gus fusion strain] but positively affects prfA transcription when prfA transcript levels are low [e.g., in the P1ΔP2(−10)prfA-gus and ΔP1(−10)ΔP2(−10)prfA-gus fusion strains]. Results from statistical analyses of our reporter fusion experiments, which show a significant statistical interaction effect between σB and PrfA on prfA transcript levels, lend further credence to the notion of complex interactions among PrfA*, σB, and prfA promoter elements, and possibly prfA transcript levels. While Rauch et al. (60) did not report a statistically significant effect of the presence of wild-type PrfA on prfA transcription initiation from the P1 or the P2 promoter, in vitro assays with PrfA* (shown in Fig. 4A in reference 60) did show numerically higher transcript levels initiating from P2prfA in the presence of PrfA* (compared to wild-type PrfA or the absence of PrfA), supporting positive regulation of prfA transcription from P2prfA by PrfA*.
Our reporter fusion experiments provide independent evidence of prfA transcription activation by σB at the P2prfA promoter, consistent with previous findings using a variety of different strategies (36, 53, 60). Our data do not support a model in which σB directly downregulates prfA transcription. Therefore, the negative effect of σB on expression of the PrfA regulon does not occur at the level of prfA transcription. Rather, in combination, our gene expression and reporter fusion data suggest that σB likely regulates transcription of an as yet unrecognized protein(s) or noncoding RNA(s) that is responsible for regulating PrfA at the posttranscriptional or posttranslational level to yield reduced expression of the PrfA regulon. This hypothesis is supported by the fact that the L. monocytogenes σB regulon includes >140 genes, including those encoding proteins with importance in stress response, virulence, transcriptional regulation, metabolism, and transport (59). σB also transcribes genes that encode other regulators, including small RNAs (12, 54) and proteins that affect RNA stability (e.g., Hfq) (13).
Conclusions.
Our data support the existence of a complex σB-PrfA regulatory network that contributes to L. monocytogenes virulence and transmission. This network includes a large σB regulon, which is comprised of a large range of stress response and virulence genes (31, 35, 59), and a smaller PrfA regulon that is comprised predominantly of virulence genes, including some genes that are regulated by both σB and PrfA (e.g., inlA) (35, 39, 47). While σB-dependent transcription appears to be important predominantly under environmental or host-induced stress conditions (e.g., acid stress or osmotic stress) (1, 22, 72), PrfA appears to be switched “on” after L. monocytogenes enters the host cell, thus activating transcription of a number of virulence genes required for bacterial escape from the phagocytic vacuole and for cell-to-cell spread (40). The σB-PrfA regulatory network involves two autoregulatory feedback loops, including (i) PrfA-dependent transcription of prfA (e.g., through PrfA-dependent readthrough transcripts from the plcA promoter) (8) and (ii) σB-dependent transcription of a portion of the sigB operon (1), as well as σB-dependent activation of prfA transcription from P2prfA (36, 53, 60, 67). In addition to PrfA- and σB-dependent upregulation of prfA transcription, under some conditions these two regulators both also appear to downregulate and fine-tune prfA expression and/or PrfA activity, and hence expression of the PrfA regulon. While PrfA appears to be able to directly downregulate prfA transcription (25), σB appears to downregulate expression of the PrfA regulon by an as yet undefined mechanism. We thus propose a model for PrfA activity regulation that requires careful fine-tuning of PrfA regulon expression through complex PrfA and σB interactions to ensure the rapid induction of regulon expression to facilitate infection and cell-to-cell spread when needed, as well as subsequent downregulation to avoid overexpression of virulence genes, which could result in excessive host cell damage and thus impair the success of the organism in establishing an infection (17, 66).
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (grant 5 R01 AI052151-04 to K.J.B.).
The contents of the manuscript are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Editor: A. Camilli
Footnotes
Published ahead of print on 2 March 2009.
Supplemental material for this article may be found at http://iai.asm.org/.
REFERENCES
- 1.Becker, L. A., M. S. Cetin, R. W. Hutkins, and A. K. Benson. 1998. Identification of the gene encoding the alternative sigma factor σB from Listeria monocytogenes and its role in osmotolerance. J. Bacteriol. 1804547-4554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Behari, J., and P. Youngman. 1998. Regulation of hly expression in Listeria monocytogenes by carbon sources and pH occurs through separate mechanisms mediated by PrfA. Infect. Immun. 663635-3642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bischoff, M., P. Dunman, J. Kormanec, D. Macapagal, E. Murphy, W. Mounts, B. Berger-Bachi, and S. Projan. 2004. Microarray-based analysis of the Staphylococcus aureus σB regulon. J. Bacteriol. 1864085-4099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bishop, D. K., and D. J. Hinrichs. 1987. Adoptive transfer of immunity to Listeria monocytogenes. The influence of in vitro stimulation on lymphocyte subset requirements. J. Immunol. 1392005-2009. [PubMed] [Google Scholar]
- 5.Bohne, J., H. Kestler, C. Uebele, Z. Sokolovic, and W. Goebel. 1996. Differential regulation of the virulence genes of Listeria monocytogenes by the transcriptional activator PrfA. Mol. Microbiol. 201189-1198. [DOI] [PubMed] [Google Scholar]
- 6.Brehm, K., M. T. Ripio, J. Kreft, and J. A. Vazquez-Boland. 1999. The bvr locus of Listeria monocytogenes mediates virulence gene repression by beta-glucosides. J. Bacteriol. 1815024-5032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bubert, A., H. Kestler, M. Gotz, R. Bockmann, and W. Goebel. 1997. The Listeria monocytogenes iap gene as an indicator gene for the study of PrfA-dependent regulation. Mol. Gen. Genet. 25654-62. [DOI] [PubMed] [Google Scholar]
- 8.Camilli, A., L. G. Tilney, and D. A. Portnoy. 1993. Dual roles of plcA in Listeria monocytogenes pathogenesis. Mol. Microbiol. 8143-157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chan, Y. C., S. Raengpradub, K. J. Boor, and M. Wiedmann. 2007. Microarray-based characterization of the Listeria monocytogenes cold regulon in log- and stationary-phase cells. Appl. Environ. Microbiol. 736484-6498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chaturongakul, S., and K. J. Boor. 2004. RsbT and RsbV contribute to σB-dependent survival under environmental, energy, and intracellular stress conditions in Listeria monocytogenes. Appl. Environ. Microbiol. 705349-5356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chaturongakul, S., and K. J. Boor. 2006. σB activation under environmental and energy stress conditions in Listeria monocytogenes. Appl. Environ. Microbiol. 725197-5203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chaturongakul, S., S. Raengpradub, M. Wiedmann, and K. J. Boor. 2008. Modulation of stress and virulence in Listeria monocytogenes. Trends Microbiol. 16388-396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Christiansen, J. K., M. H. Larsen, H. Ingmer, L. Sogaard-Andersen, and B. H. Kallipolitis. 2004. The RNA-binding protein Hfq of Listeria monocytogenes: role in stress tolerance and virulence. J. Bacteriol. 1863355-3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cossart, P., J. Pizarro-Cerda, and M. Lecuit. 2003. Invasion of mammalian cells by Listeria monocytogenes: functional mimicry to subvert cellular functions. Trends Cell Biol. 1323-31. [DOI] [PubMed] [Google Scholar]
- 15.Cui, X., J. T. Hwang, J. Qiu, N. J. Blades, and G. A. Churchill. 2005. Improved statistical tests for differential gene expression by shrinking variance components estimates. Biostatistics 659-75. [DOI] [PubMed] [Google Scholar]
- 16.Datta, A. R., and M. H. Kothary. 1993. Effects of glucose, growth temperature, and pH on listeriolysin O production in Listeria monocytogenes. Appl. Environ. Microbiol. 593495-3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Decatur, A. L., and D. A. Portnoy. 2000. A PEST-like sequence in listeriolysin O essential for Listeria monocytogenes pathogenicity. Science 290992-995. [DOI] [PubMed] [Google Scholar]
- 18.Dussurget, O., D. Cabanes, P. Dehoux, M. Lecuit, C. Buchrieser, P. Glaser, and P. Cossart. 2002. Listeria monocytogenes bile salt hydrolase is a PrfA-regulated virulence factor involved in the intestinal and hepatic phases of listeriosis. Mol. Microbiol. 451095-1106. [DOI] [PubMed] [Google Scholar]
- 19.Eiting, M., G. Hageluken, W. D. Schubert, and D. W. Heinz. 2005. The mutation G145S in PrfA, a key virulence regulator of Listeria monocytogenes, increases DNA-binding affinity by stabilizing the HTH motif. Mol. Microbiol. 56433-446. [DOI] [PubMed] [Google Scholar]
- 20.Ermolaeva, S., S. Novella, Y. Vega, M. T. Ripio, M. Scortti, and J. A. Vazquez-Boland. 2004. Negative control of Listeria monocytogenes virulence genes by a diffusible autorepressor. Mol. Microbiol. 52601-611. [DOI] [PubMed] [Google Scholar]
- 21.Farber, J. M., and P. I. Peterkin. 1991. Listeria monocytogenes, a food-borne pathogen. Microbiol. Rev. 55476-511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ferreira, A., C. P. O'Byrne, and K. J. Boor. 2001. Role of σB in heat, ethanol, acid, and oxidative stress resistance and during carbon starvation in Listeria monocytogenes. Appl. Environ. Microbiol. 674454-4457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ferreira, A., D. Sue, C. P. O'Byrne, and K. J. Boor. 2003. Role of Listeria monocytogenes σB in survival of lethal acidic conditions and in the acquired acid tolerance response. Appl. Environ. Microbiol. 692692-2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Freitag, N. E., and D. A. Portnoy. 1994. Dual promoters of the Listeria monocytogenes prfA transcriptional activator appear essential in vitro but are redundant in vivo. Mol. Microbiol. 12845-853. [DOI] [PubMed] [Google Scholar]
- 25.Freitag, N. E., L. Rong, and D. A. Portnoy. 1993. Regulation of the prfA transcriptional activator of Listeria monocytogenes: multiple promoter elements contribute to intracellular growth and cell-to-cell spread. Infect. Immun. 612537-2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garner, M. R., B. L. Njaa, M. Wiedmann, and K. J. Boor. 2006. σB contributes to Listeria monocytogenes gastrointestinal infection but not to systemic spread in the guinea pig infection model. Infect. Immun. 74876-886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Glaser, P., L. Frangeul, C. Buchrieser, C. Rusniok, A. Amend, F. Baquero, P. Berche, H. Bloecker, P. Brandt, T. Chakraborty, A. Charbit, F. Chetouani, E. Couve, A. de Daruvar, P. Dehoux, E. Domann, G. Dominguez-Bernal, E. Duchaud, L. Durant, O. Dussurget, K. D. Entian, H. Fsihi, F. G. Portillo, P. Garrido, L. Gautier, W. Goebel, N. Gomez-Lopez, T. Hain, J. Hauf, D. Jackson, L. M. Jones, U. Kaerst, J. Kreft, M. Kuhn, F. Kunst, G. Kurapkat, E. Madueno, A. Maitournam, J. M. Vicente, E. Ng, H. Nedjari, G. Nordsiek, S. Novella, B. de Pablos, J. C. Perez-Diaz, R. Purcell, B. Remmel, M. Rose, T. Schlueter, N. Simoes, A. Tierrez, J. A. Vazquez-Boland, H. Voss, J. Wehland, and P. Cossart. 2001. Comparative genomics of Listeria species. Science 294849-852. [DOI] [PubMed] [Google Scholar]
- 28.Gouin, E., J. Mengaud, and P. Cossart. 1994. The virulence gene cluster of Listeria monocytogenes is also present in Listeria ivanovii, an animal pathogen, and Listeria seeligeri, a nonpathogenic species. Infect. Immun. 623550-3553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gray, M. J., N. E. Freitag, and K. J. Boor. 2006. How the bacterial pathogen Listeria monocytogenes mediates the switch from environmental Dr. Jekyll to pathogenic Mr. Hyde. Infect. Immun. 742505-2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Greene, S. L., and N. E. Freitag. 2003. Negative regulation of PrfA, the key activator of Listeria monocytogenes virulence gene expression, is dispensable for bacterial pathogenesis. Microbiology 149111-120. [DOI] [PubMed] [Google Scholar]
- 31.Hain, T., H. Hossain, S. S. Chatterjee, S. Machata, U. Volk, S. Wagner, B. Brors, S. Haas, C. T. Kuenne, A. Billion, S. Otten, J. Pane-Farre, S. Engelmann, and T. Chakraborty. 2008. Temporal transcriptomic analysis of the Listeria monocytogenes EGD-e σB regulon. BMC Microbiol. 820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Heyer, L. J., S. Kruglyak, and S. Yooseph. 1999. Exploring expression data: identification and analysis of coexpressed genes. Genome Res. 91106-1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hu, Y., S. Raengpradub, U. Schwab, C. Loss, R. H. Orsi, M. Wiedmann, and K. J. Boor. 2007. Phenotypic and transcriptomic analyses demonstrate interactions between the transcriptional regulators CtsR and σB in Listeria monocytogenes. Appl. Environ. Microbiol. 737967-7980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jefferson, R. A., T. A. Kavanagh, and M. W. Bevan. 1987. GUS fusions: beta-glucuronidase as a sensitive and versatile gene fusion marker in higher plants. EMBO J. 63901-3907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kazmierczak, M. J., S. C. Mithoe, K. J. Boor, and M. Wiedmann. 2003. Listeria monocytogenes σB regulates stress response and virulence functions. J. Bacteriol. 1855722-5734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kazmierczak, M. J., M. Wiedmann, and K. J. Boor. 2006. Contributions of Listeria monocytogenes σB and PrfA to expression of virulence and stress response genes during extra- and intracellular growth. Microbiology 1521827-1838. [DOI] [PubMed] [Google Scholar]
- 37.Kerr, M. K. 2003. Linear models for microarray data analysis: hidden similarities and differences. J. Comput. Biol. 10891-901. [DOI] [PubMed] [Google Scholar]
- 38.Kim, H., K. J. Boor, and H. Marquis. 2004. Listeria monocytogenes σB contributes to invasion of human intestinal epithelial cells. Infect. Immun. 727374-7378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim, H., H. Marquis, and K. J. Boor. 2005. σB contributes to Listeria monocytogenes invasion by controlling expression of inlA and inlB. Microbiology 1513215-3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kreft, J., and J. A. Vazquez-Boland. 2001. Regulation of virulence genes in Listeria. Int. J. Med. Microbiol. 291145-157. [DOI] [PubMed] [Google Scholar]
- 41.Lampidis, R., R. Gross, Z. Sokolovic, W. Goebel, and J. Kreft. 1994. The virulence regulator protein of Listeria ivanovii is highly homologous to PrfA from Listeria monocytogenes and both belong to the Crp-Fnr family of transcription regulators. Mol. Microbiol. 13141-151. [DOI] [PubMed] [Google Scholar]
- 42.Lingnau, A., E. Domann, M. Hudel, M. Bock, T. Nichterlein, J. Wehland, and T. Chakraborty. 1995. Expression of the Listeria monocytogenes EGD inlA and inlB genes, whose products mediate bacterial entry into tissue culture cell lines, by PrfA-dependent and -independent mechanisms. Infect. Immun. 633896-3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Makino, M., M. Kawai, I. Kawamura, M. Fujita, F. Gejo, and M. Mitsuyama. 2005. Involvement of reactive oxygen intermediate in the enhanced expression of virulence-associated genes of Listeria monocytogenes inside activated macrophages. Microbiol. Immunol. 49805-811. [DOI] [PubMed] [Google Scholar]
- 44.Marquis, H. 2006. Tissue culture cell assays used to analyze Listeria monocytogenes. Curr. Protoc. Microbiol. Chapter 9Unit 9B.4. [DOI] [PubMed] [Google Scholar]
- 45.Marr, A. K., B. Joseph, S. Mertins, R. Ecke, S. Muller-Altrock, and W. Goebel. 2006. Overexpression of PrfA leads to growth inhibition of Listeria monocytogenes in glucose-containing culture media by interfering with glucose uptake. J. Bacteriol. 1883887-3901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McGann, P., S. Raengpradub, R. Ivanek, M. Wiedmann, and K. J. Boor. 2008. Differential regulation of Listeria monocytogenes internalin and internalin-like genes by σB and PrfA as revealed by subgenomic microarray analyses. Foodborne Pathog. Dis. 5417-435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McGann, P., M. Wiedmann, and K. J. Boor. 2007. The alternative sigma factor σB and the virulence gene regulator PrfA both regulate transcription of Listeria monocytogenes internalins. Appl. Environ. Microbiol. 732919-2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mengaud, J., S. Dramsi, E. Gouin, J. A. Vazquez-Boland, G. Milon, and P. Cossart. 1991. Pleiotropic control of Listeria monocytogenes virulence factors by a gene that is autoregulated. Mol. Microbiol. 52273-2283. [DOI] [PubMed] [Google Scholar]
- 49.Milohanic, E., P. Glaser, J. Y. Coppee, L. Frangeul, Y. Vega, J. A. Vazquez-Boland, F. Kunst, P. Cossart, and C. Buchrieser. 2003. Transcriptome analysis of Listeria monocytogenes identifies three groups of genes differently regulated by PrfA. Mol. Microbiol. 471613-1625. [DOI] [PubMed] [Google Scholar]
- 50.Moors, M. A., B. Levitt, P. Youngman, and D. A. Portnoy. 1999. Expression of listeriolysin O and ActA by intracellular and extracellular Listeria monocytogenes. Infect. Immun. 67131-139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moroni, O., E. Kheadr, Y. Boutin, C. Lacroix, and I. Fliss. 2006. Inactivation of adhesion and invasion of food-borne Listeria monocytogenes by bacteriocin-producing Bifidobacterium strains of human origin. Appl. Environ. Microbiol. 726894-6901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mueller, K. J., and N. E. Freitag. 2005. Pleiotropic enhancement of bacterial pathogenesis resulting from the constitutive activation of the Listeria monocytogenes regulatory factor PrfA. Infect. Immun. 731917-1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nadon, C. A., B. M. Bowen, M. Wiedmann, and K. J. Boor. 2002. σB contributes to PrfA-mediated virulence in Listeria monocytogenes. Infect. Immun. 703948-3952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nielsen, J. S., A. S. Olsen, M. Bonde, P. Valentin-Hansen, and B. H. Kallipolitis. 2008. Identification of a σB-dependent small noncoding RNA in Listeria monocytogenes. J. Bacteriol. 1906264-6270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Oliver, H. F., M. Wiedmann, and K. J. Boor. 2007. Environmental reservoir and transmission into the mammalian host, p. 111-137. In H. Goldfine and H. Shen (ed.), Listeria monocytogenes: pathogenesis and host response. Springer-Verlag, New York, NY.
- 56.Ollinger, J., M. Wiedmann, and K. J. Boor. 2008. σB- and PrfA-dependent transcription of genes previously classified as putative constituents of the Listeria monocytogenes PrfA regulon. Foodborne Pathog. Dis. 5281-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Port, G. C., and N. E. Freitag. 2007. Identification of novel Listeria monocytogenes secreted virulence factors following mutational activation of the central virulence regulator, PrfA. Infect. Immun. 755886-5897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Quackenbush, J. 2002. Microarray data normalization and transformation. Nat. Genet. 32(Suppl.)496-501. [DOI] [PubMed] [Google Scholar]
- 59.Raengpradub, S., M. Wiedmann, and K. J. Boor. 2008. Comparative analysis of the σB-dependent stress responses in Listeria monocytogenes and Listeria innocua strains exposed to selected stress conditions. Appl. Environ. Microbiol. 74158-171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rauch, M., Q. Luo, S. Muller-Altrock, and W. Goebel. 2005. SigB-dependent in vitro transcription of prfA and some newly identified genes of Listeria monocytogenes whose expression is affected by PrfA in vivo. J. Bacteriol. 187800-804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ripio, M. T., K. Brehm, M. Lara, M. Suarez, and J. A. Vazquez-Boland. 1997. Glucose-1-phosphate utilization by Listeria monocytogenes is PrfA dependent and coordinately expressed with virulence factors. J. Bacteriol. 1797174-7180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ripio, M. T., G. Dominguez-Bernal, M. Lara, M. Suarez, and J. A. Vazquez-Boland. 1997. A Gly145Ser substitution in the transcriptional activator PrfA causes constitutive overexpression of virulence factors in Listeria monocytogenes. J. Bacteriol. 1791533-1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ripio, M. T., G. Dominguez-Bernal, M. Suarez, K. Brehm, P. Berche, and J. A. Vazquez-Boland. 1996. Transcriptional activation of virulence genes in wild-type strains of Listeria monocytogenes in response to a change in the extracellular medium composition. Res. Microbiol. 147371-384. [DOI] [PubMed] [Google Scholar]
- 64.Ripio, M. T., J. A. Vazquez-Boland, Y. Vega, S. Nair, and P. Berche. 1998. Evidence for expressional crosstalk between the central virulence regulator PrfA and the stress response mediator ClpC in Listeria monocytogenes. FEMS Microbiol. Lett. 15845-50. [DOI] [PubMed] [Google Scholar]
- 65.Schlech, W. F. 1996. Pathogenesis and immunology of Listeria monocytogenes. Pathol Biol. (Paris) 775-782. [PubMed]
- 66.Schnupf, P., J. Hofmann, J. Norseen, I. J. Glomski, H. Schwartzstein, and A. L. Decatur. 2006. Regulated translation of listeriolysin O controls virulence of Listeria monocytogenes. Mol. Microbiol. 61999-1012. [DOI] [PubMed] [Google Scholar]
- 67.Schwab, U., B. Bowen, C. Nadon, M. Wiedmann, and K. J. Boor. 2005. The Listeria monocytogenes prfAP2 promoter is regulated by σB in a growth phase dependent manner. FEMS Microbiol. Lett. 245329-336. [DOI] [PubMed] [Google Scholar]
- 68.Scortti, M., H. J. Monzo, L. Lacharme-Lora, D. A. Lewis, and J. A. Vazquez-Boland. 2007. The PrfA virulence regulon. Microbes Infect. 91196-1207. [DOI] [PubMed] [Google Scholar]
- 69.Sheehan, B., A. Klarsfeld, T. Msadek, and P. Cossart. 1995. Differential activation of virulence gene expression by PrfA, the Listeria monocytogenes virulence regulator. J. Bacteriol. 1776469-6476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shetron-Rama, L. M., K. Mueller, J. M. Bravo, H. G. Bouwer, S. S. Way, and N. E. Freitag. 2003. Isolation of Listeria monocytogenes mutants with high-level in vitro expression of host cytosol-induced gene products. Mol. Microbiol. 481537-1551. [DOI] [PubMed] [Google Scholar]
- 71.Sokolovic, Z., J. Riedel, M. Wuenscher, and W. Goebel. 1993. Surface-associated, PrfA-regulated proteins of Listeria monocytogenes synthesized under stress conditions. Mol. Microbiol. 8219-227. [DOI] [PubMed] [Google Scholar]
- 72.Sue, D., D. Fink, M. Wiedmann, and K. J. Boor. 2004. σB-dependent gene induction and expression in Listeria monocytogenes during osmotic and acid stress conditions simulating the intestinal environment. Microbiology 1503843-3855. [DOI] [PubMed] [Google Scholar]
- 73.Vazquez-Boland, J. A., C. Kocks, S. Dramsi, H. Ohayon, C. Geoffroy, J. Mengaud, and P. Cossart. 1992. Nucleotide sequence of the lecithinase operon of Listeria monocytogenes and possible role of lecithinase in cell-to-cell spread. Infect. Immun. 60219-230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vazquez-Boland, J. A., M. Kuhn, P. Berche, T. Chakraborty, G. Dominguez-Bernal, W. Goebel, B. Gonzalez-Zorn, J. Wehland, and J. Kreft. 2001. Listeria pathogenesis and molecular virulence determinants. Clin. Microbiol. Rev. 14584-640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Volker, U., S. Engelmann, B. Maul, S. Riethdorf, A. Volker, R. Schmid, H. Mach, and M. Hecker. 1994. Analysis of the induction of general stress proteins of Bacillus subtilis. Microbiology 140741-752. [DOI] [PubMed] [Google Scholar]
- 76.Wiedmann, M., T. J. Arvik, R. J. Hurley, and K. J. Boor. 1998. General stress transcription factor σB and its role in acid tolerance and virulence of Listeria monocytogenes. J. Bacteriol. 1803650-3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.