

Abstract
Because coinfections can alter helicobacter gastritis, we investigated whether enterohepatic Helicobacter bilis modulates Helicobacter pylori gastritis in C57BL/6 mice. Thirty mice per group were sham dosed, H. bilis or H. pylori infected, or H. bilis infected followed in 2 weeks by H. pylori and then evaluated at 6 and 11 months postinfection (mpi) for gastritis and premalignant lesions. Compared to H. pylori-infected mice, H. bilis/H. pylori-infected mice at 6 and 11 mpi had less severe gastritis, atrophy, mucous metaplasia and hyperplasia (P < 0.01) and, additionally, at 11 mpi, less severe intestinal metaplasia and dysplasia (P < 0.05). H. bilis/H. pylori-infected mice at 11 mpi exhibited less Ki67 labeling of proliferating epithelial cells, reduced numbers of FoxP3+ T-regulatory (TREG) cells, and lower FoxP3+ mRNA levels than did H. pylori-infected mice (P < 0.05). Proinflammatory interleukin-1β (IL-1β), gamma interferon, and tumor necrosis factor alpha mRNA levels were attenuated in H. bilis/H. pylori-infected mice at 6 and 11 mpi (P < 0.01), although anti-inflammatory IL-10, IL-13, and transforming growth factor β1 mRNA levels were not consistently impacted by H. bilis coinfection. Decreased pathology in H. bilis/H. pylori-infected mice correlated with higher gastric H. pylori colonization at 6 mpi (P < 0.001) and lower Th1-associated immunoglobulin G2c responses to H. pylori at 6 and 10 mpi (P < 0.05). We hypothesized that reduced pathology in H. bilis/H. pylori-infected mice was due to H. bilis-primed TREG cells in the lower bowel that migrated to the gastric compartment and inhibited Th1 responses to subsequent H. pylori infection. Thus, H. pylori-induced gastric lesions may vary in mouse models of unknown enteric helicobacter infection status and, importantly, variable sequelae to human H. pylori infection, particularly in developing countries, may occur where coinfection with lower bowel helicobacters and H. pylori may be common.
Helicobacter pylori, first isolated by Warren and Marshall, induces a persistent infection and gastritis and is known to colonize the stomach of over 50% of the human population (2). In a subset of infected individuals, H. pylori is linked to the development of peptic ulcer disease, gastric adenocarcinoma, and gastric mucosa-associated lymphoid tissue lymphoma. It has been classified by the World Health Organization as a class I carcinogen (25). It is not clear why some individuals infected with H. pylori develop serious disease, while others do not. Host and environmental factors, as well as the virulence properties of H. pylori, appear to play an important role in determining disease outcome (17, 52, 60). Poor socioeconomic conditions promote early acquisition and infection with H. pylori and infection rates often approach >90% in these populations. Interestingly, some African countries with especially high prevalence rates of infection have lower-than-expected rates of gastric cancer. This paradox has been referred to as “the African Enigma” (24). The low incidence of gastric cancer has been linked to endemic parasites, diet, poor cancer registry data, and the low pathogenicity of some H. pylori strains (3a, 12, 31a).
Like humans, mice respond immunologically to infectious agents with a repertoire of memory T cells that respond most efficiently after antigen priming (28) but also appear to modulate host responses to unrelated infections and likely are also effective in disease caused by organisms sharing common antigens. These cross-reactive T cells, when activated, not only modulate the immune response but also determine the eventual outcome of heterologous infections. This host immune response is often referred to as heterologous immunity (47). This phenomenon has been studied to a limited extent in mouse models of gastric helicobacter pathogenesis that have had varied pathological outcomes. In a C57BL/6 mouse model of H. felis gastritis, coinfection with an enteric helminth, Heligmosomoides polygyrus, stimulated a Th2 response that attenuated Th1-promoted gastric pathology (11). In contrast, in BALB/c mice which have a Th2-biased response to gastric helicobacter infection resulting in no discernible gastritis, coinfection with Toxoplasma gondii promoted a robust Th1 immune response, resulting in a progressive helicobacter-associated gastritis, gastric atrophy, and metaplasia (50). Recently, we have demonstrated that the colitis induced by Citrobacter rodentium resulted in a prolonged recovery of the disease in C57BL/6 mice when the animals were coinfected with H. hepaticus (34). It is also known that host immune responses resulting from infections with atypical mycobacteria can influence how mice or humans respond immunologically to BCG vaccination (9, 62). These examples of heterologous immunity suggest that disease outcomes can be impacted by modulation of Th1 and Th2 inflammatory responses (37).
Subclinical lower bowel helicobacter infections are prevalent worldwide in mouse colonies; however, the persistent infection in certain inbred strains of mice often elicits demonstrable pathology (53). In susceptible mouse strains, enterohepatic helicobacters cause inflammatory bowel disease, colonic adenocarcinoma, hepatitis, cholecystitis, and hepatocellular carcinoma (8, 31, 33, 57). Non-H. pylori helicobacters are increasingly cited in association with human diarrheal disease, particularly in developing countries, as well as with hepatobiliary diseases in humans (10, 12, 13, 22, 38). These observations of enterohepatic helicobacter-associated disease in humans and the common occurrence of enteric helicobacter infections in mice suggest that helicobacter coinfections could impact murine studies involving H. pylori pathogenesis, vaccine strategies, and antimicrobial modalities. Thus, we initiated an experiment to ascertain whether coinfection with H. bilis, an enterohepatic helicobacter with a wide host range (10), could impact the progression of H. pylori-induced gastric disease and inflammatory responses in C57BL/6 mice (10, 12, 19, 46).
MATERIALS AND METHODS
Experimental infections.
Five-week-old, female C57BL/6 mice obtained from Taconic Farms (Germantown, NY) were housed in groups of five in polycarbonate microisolator cages on hardwood bedding (PharmaServ, Framingham, MA) under specific-pathogen-free conditions (free of Helicobacter spp., Citrobacter rodentium, Salmonella spp., endoparasites, ectoparasites, and known murine viral pathogens) in an Association for the Assessment and Accreditation of Laboratory Animal Care International accredited facility. Mouse rooms were maintained at constant temperature and humidity on a 12/12-h light-dark cycle, and mice were provided standard rodent chow (Purina Mills, St. Louis, MO) and water ad libitum. All protocols were approved by the MIT Committee on Animal Care.
Groups of 30 mice were either uninfected, sham-dosed controls, orally inoculated with H. bilis only, with H. pylori only, or with H. bilis followed in 2 weeks by H. pylori. Mice were dosed with 0.2 ml of 108 CFU ml of H. pylori SS1 or H. bilis Missouri strain in brucella broth every other day for a total of three doses. At 6 and 11 months postinoculation (mpi), 15 mice from each group were euthanized with CO2 and necropsied.
Necropsy and histopathology.
At necropsy, stomach samples from the lesser curvature extending from the squamous forestomach through the duodenum were collected and processed as described previously (18, 44). Tissues were graded by a comparative pathologist (A.B.R.) blinded to sample identity for inflammation, epithelial defects, atrophy, hyperplasia, pseudopyloric metaplasia, dysplasia, hyalinosis and mucous metaplasia as defined elsewhere (15, 18, 44). Gastric lesions were scored on an ascending scale from 0 to 4 using criteria previously described (15, 44). Briefly, inflammation is defined as the extent of leukocyte infiltration in the mucosa and submucosa. Epithelial defects were scored on the basis of degeneration of the surface lining epithelium and underlying oxyntic glands, as well as the presence of dilated glands and presence of mucosal erosion/ulceration. Mucous metaplasia is the expansion of gastric mucous neck cells secreting a mixture of neutral and acidic mucins in the oxyntic mucosa. Hyalinosis is defined as the accumulation of brightly eosinophilic droplets and/or crystals composed of Ym2 protein in the cytoplasm of the surface epithelium and within the glandular lumen. Atrophy represents the loss of oxyntic glands (parietal/chief cell loss) and is usually accompanied by compensatory hyperplasia of the foveolar epithelium. Pseudopyloric metaplasia is a preneoplastic change and represents the replacement of normal corpus with a mucosa resembling the pyloric antrum with regard to glandular phenotype and mucin expression. Defining characteristics for dysplasia were adapted from consensus guidelines on mouse models of intestinal cancer (3). Dysplasia is defined as the degree of cellular and glandular atypia with scores of three and four representing gastric intraepithelial neoplasia and unequivocal invasive carcinoma, respectively. For each sample, a gastric histologic activity index (HAI) was generated by combining scores for all criteria except hyalinosis and mucous metaplasia which may develop spontaneously irrespective of helicobacter infection.
Immunohistochemistry.
Immunohistochemistry for a variety of targets was performed according to a previously described protocol (43). For assessment of epithelial cell proliferation, Ki67 (BD Biosciences, San Jose, CA) labeling indices were determined as described previously (15) with slight modifications. Briefly, formalin-fixed stomach samples from three randomly selected animals per treatment group at 11 mpi were assessed for Ki67 immunolabeling. The number of positively stained nuclei from five well-oriented proximal corpus glands was enumerated, and the mean value was defined as the epithelial cell proliferation labeling index (LI). Transforming growth factor β (TGF-β; R&D Systems, Minneapolis, MN) and the NF-κB subunit p65 (Zymed/Invitrogen, Carlsbad, CA) were labeled as previously described (45) using gastric tissues from five mice per group. FoxP3+ immunohistochemistry was performed using FoxP3 antibody (FJK-16S; eBiosciences, San Diego, CA). Stomach sections from six uninfected control mice, six H. bilis-infected mice, and fifteen mice each from H. pylori-infected and H. bilis/H. pylori-infected mice were evaluated at 11 mpi. Cells expressing FoxP3+ in the gastric corpus were counted from both the mucosa and submucosa at a magnification of ×20 (one field = 1.00 mm2). Nuclear labeling was considered specific for T-regulatory (TREG) cells, whereas granular cytoplasmic staining of oxyntic cells, if any, was considered as nonspecific background staining. Ten fields were counted per stomach, and the results are presented as the average number of FoxP3+ cells/mm2 of stomach.
Q-PCR for H. pylori SS1 and H. bilis.
To quantify colonization levels of H. pylori strain SS1 within the gastric mucosa, a real-time quantitative PCR assay (Q-PCR) was utilized (18, 33). A standard curve was generated by using serial 10-fold dilutions (from 5 × 105 to 5) of H. pylori SS1 genome copies, estimated from an average H. pylori genome size of 1.66 Mb (1, 54). By using the same approach, the levels of H. bilis Missouri strain in cecal or gastric samples were also measured. The H. bilis 16S rRNA gene-based primers and the probe for Q-PCR were previously described (6). The numbers of the H. bilis genome in these samples were calculated based on a genomic size of 1.73 Mb, which is an average size of two sequenced H. pylori genomes and the H. hepaticus 3B1 genome (1, 51, 54). The copy numbers of the gastric H. pylori or H. bilis genome were standardized using micrograms of murine chromosomal DNA determined by Q-PCR using a mammalian 18S rRNA gene-based primer and probe mixture (Applied Biosystems, Foster City, CA) as described previously (22, 59).
H. bilis PCR.
Bacterial DNA was extracted from fecal and cecal contents and amplified as previously described using the H. bilis-specific primers C62 and C12 (16).
Gastric cytokines.
Total RNA from stomachs of C57BL/6 mice was prepared using TRIzol reagent according to the recommendations of the manufacturer (Invitrogen). For cytokine mRNA quantification, 5 μg of total RNA was converted into cDNA using a high capacity cDNA archive kit (Applied Biosystems). Levels of interleukin 1β (IL-1β), gamma interferon (IFN-γ), tumor necrosis factor alpha (TNF-α), p65/RelA, IL-10, IL-13, TGF-β1, and FoxP3+ mRNA were measured by Q-PCR using TaqMan gene expression assays for use in the ABI Prism sequence detection system 7500 Fast (Applied Biosystems). Transcript levels were normalized to the endogenous control glyceraldehyde-3-phosphate dehydrogenase mRNA (GAPDH) and expressed as the fold change compared to samples from sham-dosed control mice using the Comparative CT method (Applied Biosystems User Bulletin No. 2).
ELISA for anti-H. pylori IgG2c and IgG1 in serum.
Sera were collected from 58 mice at necropsy at 6 mpi (13 control animals, 15 H. pylori-infected animals, 15 H. bilis-infected animals, and 15 animals coinfected with H. bilis and H. pylori) and from 58 mice under isoflurane anesthesia at 10 mpi (14 control animals, 14 H. pylori-infected animals, 15 H. bilis-infected animals, and 15 coinfected animals; 1 month prior to necropsy). Serum was evaluated for helicobacter-specific immunoglobulin G2c (IgG2c) and IgG1 by enzyme-linked immunosorbent assay (ELISA) using an outer membrane protein (OMP) preparation from H. pylori SS1 strain and H. bilis Missouri strain as described previously (40).
Statistics.
Gastric HAI scores were compared across groups by using the Kruskal-Wallis one-way analysis of variance with Dunn's post-test and between groups by the Mann-Whitney U-test using Prism software (GraphPad, San Diego, CA). Quantitative data from Ki67 and FoxP3+ immunolabeling experiments were analyzed by one-way analysis of variance followed by the Bonferroni post test. The data on the levels of H. pylori and H. bilis colonization, cytokine mRNA in the tissues, and serology were analyzed by using a two-tailed Student t test. P values of <0.05 were considered significant.
RESULTS
Coinfection of H. pylori and H. bilis attenuated gastritis and gastric premalignant lesions.
In agreement with previous findings by our group and others, C57BL/6 mice infected with H. pylori exhibited moderate gastritis at 6 mpi and severe gastritis with early dysplasia at 11 mpi (Fig. 1 and 2) (12, 30). Lesions were characterized by lymphocyte-predominant mucosal and submucosal infiltrates, multifocal surface erosions and glandular ectasia, oxyntic atrophy, hyperplasia, pseudopyloric metaplasia, and dysplasia (Fig. 2A). The progressive nature of disease was reflected by a higher mean HAI in infected mice at 11 mpi versus 6 mpi (n = 15, P < 0.01) (Fig. 2B). H. pylori-infected mice also exhibited mucous metaplasia of the oxyntic mucosa that contributed to parietal cell atrophy, although this was not included in the gastric HAI because it is not a helicobacter-specific lesion (15, 44). As expected, monoinfection with the enterohepatic bacterium H. bilis did not produce gastritis. H. bilis colonization of the lower bowel, as expected, did not result in lower bowel inflammation (not shown). Nevertheless, mice colonized with H. bilis/H. pylori exhibited a significantly lower gastric HAI at both 6 and 11 mpi than mice infected with H. pylori alone (n = 15, P < 0.01 and P < 0.02, respectively). Lesions in the coinfected group were of a similar character to those induced by H. pylori alone but were uniformly less severe. Therefore, concurrent H. bilis colonization of the lower bowel significantly abrogated the histologic severity of stomach lesions induced by H. pylori.
FIG. 1.

Gastric pathology scores. (A) Gastric pathology scores at 6 mpi. The data are shown as means ± the standard errors of the mean (n = 15). At 6 mpi, H. pylori-infected mice generally show more severe pathology than H. bilis/H. pylori-coinfected mice with significantly greater gastritis, mucous metaplasia, oxyntic gland atrophy, and foveolar hyperplasia (*, P < 0.01). No significant pathology was observed in either the uninfected or the H. bilis-infected groups. (B) Gastric pathology scores at 11 mpi. The data are shown are means ± the standard errors of the mean (n = 15). At 11 mpi, there is increased gastric pathology in the H. bilis/H. pylori-infected mice compared to the H. bilis/H. pylori-infected mice at 6 mpi; however, H. bilis/H. pylori-infected mice still show significantly less gastritis, oxyntic gland atrophy, foveolar hyperplasia, intestinal metaplasia, and dysplasia compared to the H. pylori-infected mice (*, P < 0.05). H. bilis mice had no significant pathology compared to uninfected mice.
FIG. 2.

Gastric histology. (A) Representative histopathology of gastric tissues from mice infected with H. bilis or H. pylori or coinfected with H. bilis and H. pylori at 6 to 11 mpi. Lesions were characterized by lymphocyte-predominant mucosal and submucosal infiltrates, multifocal surface erosions and glandular ectasia, oxyntic atrophy, hyperplasia, pseudopyloric metaplasia, and dysplasia. (B) Gastric HAI. Tissues from mice infected with H. pylori, H. bilis, or both H. bilis and H. pylori (n = 15 for all groups) were graded for inflammation, epithelial defects, atrophy, hyperplasia, pseudopyloric metaplasia, dysplasia, hyalinosis, and mucous metaplasia. A gastric HAI was generated by combining scores for all criteria except hyalinosis and mucous metaplasia, which may develop irrespective of helicobacter infection. The HAI was higher in H. pylori-infected mice compared to H. bilis/H. pylori-infected mice (6 mpi, P < 0.01; 11 mpi, P < 0.02).
H. bilis coinfection modulated gastric epithelial responses induced by H. pylori.
The gastric epithelial cell proliferation index, as assessed by Ki67 nuclear labeling, was similarly mild to moderate in extent within the basal glands of the antral mucosa in all mice examined at 11 mpi (n = three per group and five counts/mouse). This LI in the gastric corpus was significantly increased in both the H. pylori-infected and the H. bilis/H. pylori-infected mice compared to uninfected and H. bilis-infected mice (P < 0.001) (Fig. 3). Ki67 nuclear labeling was mostly localized to the neck and isthmus region and occasionally observed in the basal aspects of the glands. H. bilis/H. pylori-infected mice exhibited a significant reduction in the LI compared to H. pylori-infected mice (P < 0.05). As reported above, a significantly reduced HAI score in the H. bilis/H. pylori-infected mice correlated with the reduction in the epithelial LI.
FIG. 3.

Immunohistochemical detection of Ki-67 in the gastric proximal corpus at 11 mpi. (a to d) Representative Ki-67 staining of gastric epithelial cells in uninfected mice (a), H. bilis-infected mice (b), H. pylori-infected mice (c), and H. bilis/H. pylori-infected mice (d). (e) Ki67 LI in the proximal corpus of three mice per group at 11 mpi. The gastric epithelial LIs in H. pylori-infected and H. bilis/H. pylori-infected groups were significantly higher than in both uninfected and H. bilis-infected mice (*, P < 0.001), but coinfection in H. bilis/H. pylori-infected mice significantly decreased the LI (*, P < 0.05).
In addition to epithelial cell proliferation, select gastric tissues (n = 5 per group) were evaluated using immunohistochemistry for anti-inflammatory TGF-β and the proinflammatory NF-κB subunit p65, which typically is expressed by tissue-infiltrating leukocytes. In mice with H. pylori-associated severe gastritis accompanied by glandular metaplasia and dysplasia, TGF-β epithelial staining was moderately decreased compared to the more normal mucosa sampled from infected mice with mild or no lesions. Qualitative assessment did not suggest a difference between H. pylori-infected and H. bilis/H. pylori-infected mice (data not shown). Further, some of the H. pylori-infected stomachs also exhibited a concomitant moderate increase in TGF-β cytoplasmic immunoreactivity within the hyperplastic foveolar epithelium. In addition, upregulated cytoplasmic expression and nuclear translocation of p65 labeling was observed in regions of glandular epithelium affected by severe inflammation, hyperplasia, and dysplasia, with rare staining in normal gastric glands. Again, qualitative assessment of p65 labeling did not discern a difference between the H. pylori-infected and H. bilis/H. pylori-infected groups (data not shown), suggesting increased p65 labeling was most associated with H. pylori infection and was not sensitive to the effects of H. bilis coinfection.
The number of FoxP3+ cells associated with H. pylori infection was reduced by H. bilis coinfection.
A significant decrease in the numbers of FoxP3+ TREG cells in correlation with the decreased inflammation was observed in the stomachs of H. bilis/H. pylori-coinfected mice (26 ± 19 FoxP3+ cells/mm2) compared to those of H. pylori-infected mice (42 ± 14 FoxP3+ cells/mm2) at 11 mpi (n = 15, P < 0.05) (Fig. 4c, d, and e). FoxP3+ TREG cells were multifocally distributed among the mucosal and submucosal inflammatory aggregates in both H. pylori-infected and H. bilis/H. pylori-infected mice. In the absence of any significant gastric inflammation, FoxP3+ TREG cells were also sparse in the stomach of both the uninfected (2 ± 1 FoxP3+ cells/mm2) and H. bilis-infected mice (4 ± 3 FoxP3+ cells/mm2) (Fig. 4a, b, and e).
FIG. 4.

Quantification of FoxP3+ TREG cells in the gastric corpus at 11 mpi. (a to d) Representative immunohistochemistry for FoxP3, a regulatory T-cell marker, in the gastric corpus in uninfected mice (a), H. bilis-infected mice (b), H. pylori-infected mice (c), and H. bilis/H. pylori-coinfected mice (d). (e) Average numbers of FoxP3+ cells in the stomach assessed from 10 fields (magnification, ×200; 1.00 mm2) per mouse gastric corpus. Foxp3+ cells were significantly higher in the mucosa and submucosa of both the H. pylori-infected mice (n = 15) and H. bilis/H. pylori-infected mice (n = 15) compared to uninfected controls (n = 6) and H. bilis-infected mice (n = 6) (*, P < 0.001). H. bilis/H. pylori-infected animals showed a significant quantitative decrease in gastric FoxP3+ TREG cells compared to H. pylori-infected animals (*, P < 0.05). Bar, 80 μm.
Taken together, immunohistochemistry observations confirmed the histopathology results and demonstrated that H. bilis colonization of the lower bowel in H. bilis/H. pylori-infected mice significantly reduced H. pylori-associated intragastric cell proliferation and numbers of FoxP3+ TREG cells, a finding consistent with decreased H. pylori-induced gastritis and associated premalignant lesions.
Coinfection with H. bilis attenuated upregulation of gastric proinflammatory cytokine mRNAs induced by H. pylori infection.
It has been reported that IL-1β polymorphisms are associated with increased risk of gastric cancer in humans, presumably via enhancing production of IL-1β (7). In addition, expression of proinflammatory gastric Th1 cytokines such as IFN-γ, TNF-α, and IL-1β were also enhanced in H. pylori-infected patients, as well as in mice infected with gastric helicobacters (11, 15, 18). Since concurrent H. bilis/H. pylori infection in the present study had attenuated gastric pathology compared to mice infected with H. pylori alone, we measured and compared the mRNA levels of proinflammatory IFN-γ, TNF-α, IL-1β, and NF-κB subunit 65, as well as the anti-inflammatory mediators IL-10, IL-13, and TGF-β1 and the regulatory T-cell marker of FoxP3 (Fig. 5). H. bilis infection alone did not enhance the transcription of any selected pro or anti-inflammatory genes in the stomach compared to the sham control mice (P > 0.1). H. pylori infection significantly increased gastric mRNA levels of IL-1β (6 mpi, P < 0.003; 11 mpi, P < 0.009), IFN-γ (6 mpi, P < 0.0003; 11 mpi, P < 0.015), TNF-α (both time points, P < 0.0001), and p65 (6 mpi, P < 0.002; 11 mpi, P = 0.08) compared to H. bilis/H. pylori-infected mice. Compared to sham-dosed mice, H. pylori infection also increased expression of IL-10 at 6 and 11 mpi (P < 0.0003); however, the levels in H. pylori- and H. bilis/H. pylori-infected mice were similar. Higher mRNA levels of gastric IL-13 (P < 0.05) were observed in H. pylori-infected mice at 6 mpi compared to H. bilis/H. pylori-infected mice, but at 11 mpi IL-13 mRNA expression was similar among groups.
FIG. 5.

Gastric cytokine, Foxp3, and NF-κB p65 mRNA expression levels. Gastric tissues (n = 15 per group) from mice infected with H. pylori, H. bilis, or both helicobacters (H. bilis/H. pylori) were evaluated by Q-PCR for expression levels of mRNA for pro- and anti-inflammatory cytokines, FoxP3, a regulatory T-cell marker, and NF-κB p65, all normalized to the expression of the housekeeping gene GAPDH. The y axis represents the mean fold change (± the standard deviation) of the mRNA levels in reference to uninfected controls. H. bilis/H. pylori-infected mice expressed lower levels of proinflammatory mediators (IL-1β, IFN-γ, and TNF-α) at both time points. Lower levels of mRNA for p65, IL-13, and TGF-β were noted at 6 mpi, but only FoxP3 was lower at 11 mpi (P values are given in the figure).
The levels of TGF-β1 mRNA at 6 mpi were lower in H. bilis/H. pylori-infected than in H. pylori-infected mice (P < 0.05) with a similar lower trend at 11 mpi (P = 0.08). Compared to H. bilis-infected mice, the levels of FoxP3+ mRNA expression in the gastric tissue were upregulated by H. pylori infection at both 6 and 11 mpi (P < 0.0009, 0.0001). FoxP3+ mRNA levels at 11 mpi were lower in H. bilis/H. pylori-infected than in H. pylori-infected mice (P < 0.05), a finding consistent with lower numbers of these cells, as shown by immunohistochemistry (Fig. 4).
H. bilis coinfection lowered the proinflammatory IgG2c response to H. pylori infection.
Mice infected with H. pylori alone produced significant Th1-promoted serum IgG2c responses to H. pylori OMP antigens by 6 mpi (P < 0.001), which increased through 10 mpi (P < 0.001) (Fig. 6). The IgG2c responses of H. bilis/H. pylori-infected mice to H. pylori antigens were significantly lower than the IgG2c responses of H. pylori-infected mice at both time points (6 mpi, P < 0.05; 10 mpi, P < 0.001). As expected, Th2-associated IgG1 responses to H. pylori infection were lower in magnitude than the IgG2c responses but were lower in H. bilis/H. pylori-infected mice compared to H. pylori-infected mice only at 6 mpi (P < 0.05).
FIG. 6.

Serum IgG isotype responses to H. pylori (Hp) and H. bilis (Hb). (A) H. pylori-infected and H. bilis/H. pylori-infected mice produced significant Th1-promoted IgG2c responses to H. pylori by 6 mpi (P < 0.001), which increased through 10 mpi (P < 0.001). IgG2c levels were lower in H. bilis/H. pylori-infected mice at 6 mpi (P < 0.05) and 10 mpi (P < 0.001). (B) IgG1 responses to H. pylori followed the same trends. (C) H. bilis/H. pylori-infected mice produced similar IgG2c responses to H. bilis at 6 and 10 mpi (P = 0.28) and were equivalent to the responses of H. bilis-infected mice (P = 0.86). (D) IgG1 responses to H. bilis in H. bilis-infected mice were higher than in H. bilis/H. pylori-infected mice at 6 mpi (P < 0.03) but not at 10 mpi (P = 0.22). The data represent mean values ± the standard errors from 13 to 15 sera per infection status and time point.
In contrast to the results from the H. pylori OMP-based assay, H. bilis/H. pylori-infected mice produced similar IgG2c responses to H. bilis OMP at 6 and 10 mpi (P = 0.28) and were equivalent to the responses of H. bilis-infected mice (P = 0.86). IgG1 responses to H. bilis OMP in mice infected with H. bilis were higher than in coinfected mice at 6 mpi (P < 0.03) but not at 10 mpi (P = 0.22). Mice infected only with H. bilis or with H. pylori did not develop serum IgG2c or IgG1 that was cross-reactive with the alternate (i.e., H. bilis or H. pylori) antigen.
H. bilis established persistent infection in the lower bowel.
Successful H. bilis infection was confirmed 1 mpi by fecal PCR of fresh feces pooled from each cage housing H. bilis-dosed mice, using the H. bilis specific primers C62 and C12. At 6 mpi, there was no significant difference in colonization levels of H. bilis in the cecum between coinfected and H. bilis-infected mice (Fig. 7A, P = 0.2). At 11 mpi, the H. bilis-infected mice contained cecal H. bilis levels similar to those of both groups at 6 mpi (P > 0.1); however, there was significantly less H. bilis colonization in the coinfected mice compared to the H. bilis-infected group (P < 0.0001). No H. bilis was detected in selected cecal DNA of H. pylori-infected mice (four at each time point).
FIG. 7.
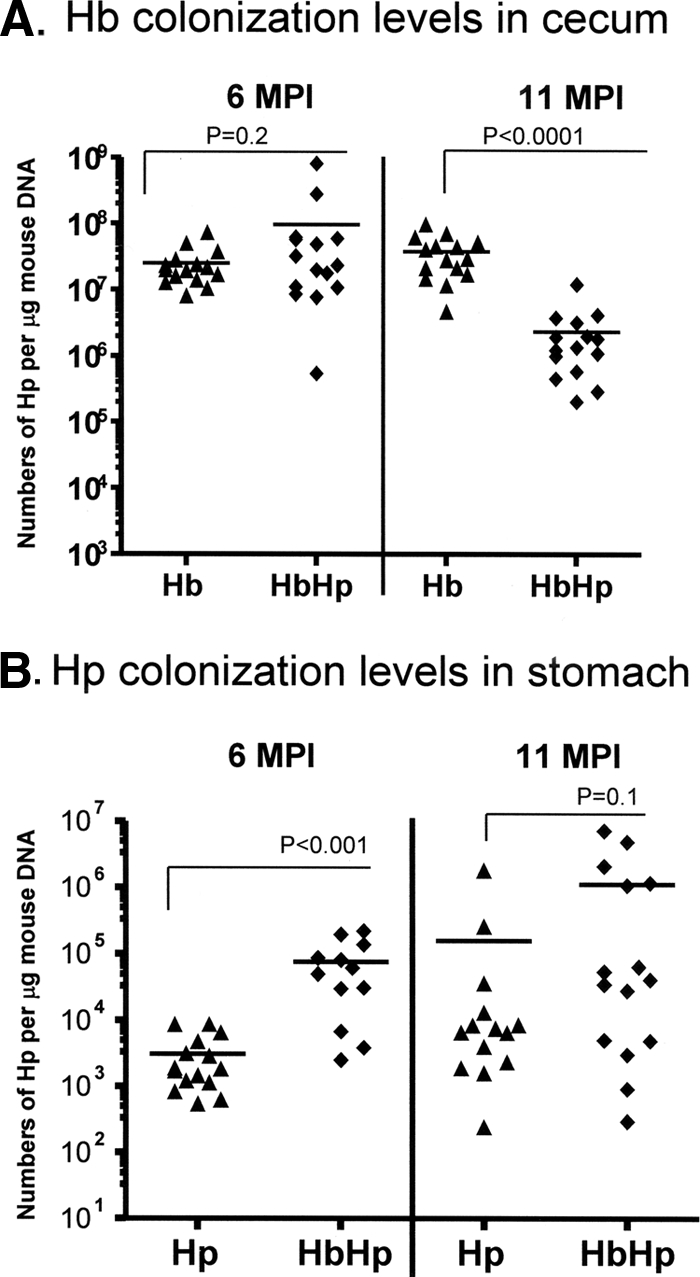
Quantitation of cecal H. bilis and gastric H. pylori. (A) Copy numbers of H. bilis (Hb) in the ceca were estimated by Q-PCR analysis of cecal samples from mice infected with H. pylori or coinfected with H. bilis and H. pylori (HbHp). H. bilis colonization levels were significantly lower at 11 mpi (P < 0.0001). (B) Copy numbers of H. pylori estimated by Q-PCR of gastric samples from H. pylori-infected (Hp) or H. bilis/H. pylori-infected (HbHp) mice were higher in H. bilis/H. pylori-coinfected mice at 6 mpi (P < 0.001), with a similar trend at 11 mpi (P = 0.1).
H. bilis was detected by Q-PCR in the gastric DNA samples from the H. bilis-infected group (11/15 at 6 mpi, 6/15 at 11 mpi) and H. bilis/H. pylori-infected group (6/15 at 6 mpi, 8/15 at 11 mpi). The average copy numbers of the H. bilis genome in the H. bilis-positive gastric tissues were 1,645 at 6 mpi and 827 at 11 mpi in the H. bilis-infected mice and 1,389 at 6 mpi and 2,341 at 11 mpi in the H. bilis/H. pylori-infected mice. There was no significant difference in H. bilis levels between the H. bilis/H. pylori-infected and H. bilis-infected groups.
Colonization of gastric H. pylori was enhanced in mice concurrently infected with H. bilis.
Coinfected mice were colonized with H. pylori in gastric tissues at significantly higher levels than H. pylori monoinfected mice at 6 mpi (P < 0.0001) as determined by Q-PCR (Fig. 7B). At 11 mpi, the mean H. pylori colonization level in coinfected mice was sevenfold higher than H. pylori-infected mice, a finding consistent with attenuated gastric pathology in the H. bilis/H. pylori-infected mice.
DISCUSSION
It is well established in both humans and C57BL/6 mice that H. pylori induces a robust Th1 proinflammatory response associated with gastric inflammation, atrophy, epithelial hyperplasia, and dysplasia (17). For the first time, we have demonstrated that H. pylori-induced gastric premalignant lesions were significantly attenuated in mice coinfected with H. bilis, despite chronic inflammation and a high density of H. pylori colonization. The reduction in gastritis severity in H. bilis/H. pylori-infected mice was associated with a reduction in mRNA expression levels of the proinflammatory cytokines IL-1β, IFN-γ, and TNF-α and subunit p65 of NF-κB in gastric tissues. Systemic effects in coinfected mice were reflected in an attenuated, proinflammatory Th1-associated IgG2c response to H. pylori. Although low levels of gastric H. bilis were detected in some of the H. bilis- or H. bilis/H. pylori-infected mice, reduction of H. pylori-induced gastric pathology in the H. bilis/H. pylori-infected mice apparently resulted from intestinal and not gastric colonization by H. bilis. Lesion severity and proinflammatory and anti-inflammatory mediator mRNA levels were similar between H. bilis/H. pylori-infected mice that were either positive or negative for the presence of H. bilis in the stomach by PCR. The attenuated gastritis in H. bilis/H. pylori-infected mice was associated with lower numbers of FoxP3+ TREG cells compared to H. pylori-infected mice. This observation suggests that the timing and chronic nature of H. bilis primary infection before H. pylori challenge was important in creating an anti-inflammatory bias to H. pylori infection at the outset of coinfection, with relatively lower demand for TREG cells observed at more chronic time points because the Th1 response to H. pylori was suppressed by prior H. bilis infection.
Immunocompromised mice infected with H. bilis often develop typhlocolitis and hepatitis (20, 49). C57BL/6 mice chronically infected with H. bilis do not develop inflammatory bowel disease or hepatitis (25, 26, 55, 57). The absence of a clinically significant inflammatory response in the gut of C57BL/6 mice implies that a sufficient regulatory anti-inflammatory response develops in response to H. bilis infection, similar to C57BL/6 mice infected with H. hepaticus (28, 42). In the present study, the numbers of FoxP3+ TREG cells in the stomach were higher in H. pylori-infected mice compared to H. bilis/H. pylori-infected mice in direct correlation with decreased gastric inflammation. This suggests that mice previously exposed to shared helicobacter antigens (i.e., H. bilis) could have had helicobacter-primed native TREG cells originate in the lower bowel that then quickly migrated to the stomach upon experimental infection with H. pylori, resulting in attenuated H. pylori gastritis. Other investigators have shown that CD45RBlo CD25− TREG cells transferred from H. hepaticus-infected mice were more efficacious than naive TREG cells in protecting Rag mice against H. hepaticus-induced T-effector-mediated colitis (28). In an analogous gastrointestinal tumor model system, we have shown that TREG cells isolated from H. hepaticus-exposed donors were significantly more effective at suppressing H. hepaticus-induced intestinal tumors with Rag2-deficient C57BL/6 Apcmin/+ mice compared to TREG cells from naive donors (42). These data are also consistent with mouse studies using splenocyte transfer from donor mice to H. pylori-infected SCID mice (39). Gastritis scores in H. pylori-infected SCID C57BL/6 recipients of naive splenocytes were statistically higher than those in H. pylori SCID recipients of immune splenocytes from H. pylori-infected donors. The authors of that study suggested that the lack of H. pylori-sensitized TREG cells in SCID recipients of naive splenocytes was likely to be responsible for the more aggressive gastritis noted compared to the mice receiving immune splenocytes. In our study, it appears important that H. bilis infection was established 2 weeks before H. pylori, allowing time for helicobacter-primed cells TREG cells to further differentiate and be available for a robust host response at a new site of helicobacter antigen exposure in the stomach. H. bilis colonization was lower in H. bilis/H. pylori-infected mice at 11 mpi for unexplained reasons, but the levels were still sufficient for maintaining attenuation of H. pylori gastritis.
Further studies using TREG-cell transfer are needed to determine whether H. bilis-primed TREG cells do in fact originate in the lower bowel and have activity against H. pylori-induced gastritis in H. bilis-free mice. An important consideration is whether the H. bilis-associated attenuation of H. pylori gastritis reflects a phenomenon known as heterologous immunity. Our data are consistent with emerging evidence that innate and adaptive immune responses to infectious agents can prime TREG cells capable of responding to subsequent infections with unrelated pathogens such as H. pylori, potentially as a by-product of degenerative T-cell recognition of peptide-major histocompatibility complexes (56). Support for T-cell-mediated heterologous immunity in the present study is demonstrated by the ELISA data revealing a lack of IgG2c or IgG1 cross-reactivity in sera from mice monoinfected with H. bilis or H. pylori to the alternate (i.e., H. bilis or H. pylori) antigen. Furthermore, our lab has previously reported that outer membrane proteins of H. pylori and H. bilis lacked significant cross-reactivity when used in Western blots (21). In addition, the ability of H. bilis infection to prime the immune response to unrelated commensal flora was evident in gnotobiotic C3H/HeN mice that developed immune responsiveness to Altered Schaedler Flora when also infected with H. bilis (27). A recent study reported that mice lacking intestinal Peyer's patches do not develop H. pylori gastritis (36). It was observed that H. pylori coccoid forms, phagocytosed by dendritic cells in the Peyer's patches, primed CD4+ T cells to H. pylori antigens. These cells then migrated to the infected stomach and were responsible for induction and modulation of an inflammatory response. In mice infected with H. bilis, H. bilis-specific antigens may be similarly processed by Peyer's patch dendritic cells, which in turn prime TREG cells. These activated CD4+ regulatory lymphocytes could then migrate to the stomach and downregulate the H. pylori-associated gastric inflammatory response.
Natural TREG cells play an important role in suppressing pathological, as well as maintaining physiological, host immune responses. Microbial infections increase the number and activity of natural TREG cells. FoxP3 expression is currently considered the most specific marker for these cells (4). The role of these TREG cells in controlling inflammation and progression of helicobacter-induced gastric disease has been recently recognized (41). Large numbers of FoxP3+ cells were noted in H. pylori-infected mice but not in uninfected controls. In addition, H. pylori-infected C57BL/6 mice, treated with C61 monoclonal antibody to deplete FoxP3+ T cells, developed a severe gastritis with elevated proinflammatory cytokines and decreased levels of H. pylori colonization. In agreement with these findings, our results have shown a significant reduction in both the numbers of FoxP3+ TREG cells and the FoxP3 mRNA levels in the stomachs of H. bilis/H. pylori-infected mice compared to H. pylori-infected mice, findings bearing a strong correlation to their respective degrees of gastric inflammation and premalignant lesions. We surmise that the increased levels of the FoxP3 mRNA noted in H. pylori-infected mice in the present study resulted from an attempt by the host to modulate the robust Th1 response, whereas lower levels of FoxP3 cells and Foxp3 mRNA levels in H. bilis/H. pylori-infected mice reflected a dampened Th1 response with lower proinflammatory cytokine levels and concomitant greater colonization levels of H. pylori.
As a corollary to our study, investigators recently reported that splenocytes recovered from mice orally infected with Mycobacterium avium prior to M. bovis BCG vaccination (which induces a potent innate immune response) had decreased ability to produce IFN-γ when subsequently stimulated with mycobacterium antigen (62). M. avium infection also induced a mycobacterial antigen-specific M. avium serological response in M. avium-sensitized BCG-immunized mice compared to the minimal serological responses in mice immunized only with BCG. This downregulation of IFN-γ responses and the upregulation of Th2 IgG1 antibody responses are characteristic of modulating a Th1 response to one more characteristic of a Th2-biased immune response (62). The present study supports epidemiological evidence in Africa, where the efficacy of M. bovis BCG vaccine against tuberculosis correlates with the geographic distance from the equator; greater protection has been observed at high latitudes (9). Greater exposure to atypical mycobacteria commonly found in warm climates is thought to influence subsequent responses to BCG (9).
Our current data demonstrating the profound effect of heterologous immunity are also supportive of our previous findings where an intestinal helminth infection in H. felis-infected C57BL/6 mice significantly reduced gastric atrophy, a premalignant lesion, despite high gastric helicobacter colonization (11). Reduced gastric atrophy was also associated with decreased levels of mRNA for proinflammatory cytokines (11). However, in the helminth coinfection studies, mice were only infected with gastric helicobacter for 16 weeks. In the present study, we extended the time points to 6 and 11 months after H. pylori infection, showing that persistent coinfections with H. bilis and H. pylori sustain long-term downregulation of proinflammatory responses. However, unlike the mice coinfected with H. felis and H. polygyrus (11), the H. bilis/H. pylori-coinfected mice in the present study did not produce mRNA levels of gastric Th2-type cytokines IL-10 and IL-13 that were consistently different from H. pylori-infected mice. The increased levels of IL-10 in H. pylori-infected mice compared to uninfected mice, despite helicobacter-associated gastritis, is consistent with our earlier findings in TFF2−/− C57BL/6 mice, where IL-10 levels were elevated in infected TFF2−/− mice versus infected wild-type mice at 6 mpi, even though corpus inflammation was more severe in infected TFF2−/− mice (15). IL-10 is a known downstream target of IL-1β and other proinflammatory cytokines, and the IL-10 elevation may in part reflect a host response intended to dampen the inflammatory response to H. pylori infection (61).
We recently described that inhabitants of Tumaco, Colombia, living at a costal location with a high prevalence of H. pylori but a low rate of gastric cancer, were heavily parasitized with intestinal helminths, whereas the population living in mountainous areas had similar infection rates of H. pylori, but low intestinal parasite burdens, and higher gastric cancer rates (58). Populations residing at higher elevations also had a higher H. pylori-specific IgG2 antibody response, indicative of a proinflammatory Th1 humoral immune response, than their counterparts living in the coastal areas of Colombia. Others reported similar serological results in South African natives; these H. pylori-infected individuals had higher Th2-associated IgG1 immune responses to H. pylori than populations in developed parts of Europe with higher gastric cancer rates (35). Populations in the developing world are subjected not only to endemic intestinal parasitism but also to a myriad of intestinal microbial pathogens. Among other potential pathogens, enteric Helicobacter spp. have also been identified in the stools of diarrheic African patients, as well as children with diarrhea in Mexico (22, 29). Indeed, at least seven species of enteric helicobacters, including “H. rappini,” which belongs to the taxa H. bilis, have been identified in the gastrointestinal tracts of humans, and an increasing number of novel enteric helicobacters have been cultured from a variety of mammals and birds (5, 10, 23, 46). It is not known whether these enteric helicobacters can persistently infect asymptomatic humans. Interestingly, several enteric Helicobacter spp., including H. bilis, have been identified recently in children with Crohn's disease (32). It is reasonable to speculate that coinfection with lower bowel helicobacters and gastric H. pylori could be common in these populations in the developing world, and their presence capable of modulating long-term sequelae to persistent H. pylori infection.
The present study also has relevance in experimental in vivo settings as well. Mice in particular are known to be persistently colonized with enterohepatic helicobacters (48). In some cases, these organisms can cause serious hepatobiliary and lower-bowel diseases in susceptible mouse strains (10, 57). H. hepaticus was isolated and characterized in the early 1990s in association with hepatitis and hepatic tumors (14, 55). The natural habitat for this organism and related enterohepatic helicobacters in clinically normal mice is the cecum and colon, where these bacteria persistently colonize the intestinal crypts (14). Soon thereafter, H. bilis was isolated and identified from the lower intestines and livers of aged, inbred and, more recently, outbred mice (16, 19). Indeed, in a recently published report, more than 85% of academic-maintained mouse colonies surveyed worldwide were colonized with enteric Helicobacter species (53). Given the large numbers of studies conducted in mice exploring the pathogenesis of H. pylori-induced disease, as well as studies involved in prophylactic and therapeutic vaccines, the data presented here illustrate the ability of enteric helicobacters to attenuate gastric premalignant lesions by modulating the host's immune response by downregulating the proinflammatory Th1 response. The results obtained when using mice with enteric helicobacter infections should be interpreted with this in mind.
Acknowledgments
This study was supported by National Institutes of Health grants R01CA67529, R01AI51404, T32RR07036, and P30 ES02109 to J.G.F.
Editor: A. J. Bäumler
Footnotes
Published ahead of print on 17 February 2009.
REFERENCES
- 1.Alm, R. A., L. S. Ling, D. T. Moir, B. L. King, E. D. Brown, P. C. Doig, D. R. Smith, B. Noonan, B. C. Guild, B. L. deJonge, G. Carmel, P. J. Tummino, A. Caruso, M. Uria-Nickelsen, D. M. Mills, C. Ives, R. Gibson, D. Merberg, S. D. Mills, Q. Jiang, D. E. Taylor, G. F. Vovis, and T. J. Trust. 1999. Genomic-sequence comparison of two unrelated isolates of the human gastric pathogen Helicobacter pylori. Nature 397176-180. [DOI] [PubMed] [Google Scholar]
- 2.Amieva, M. R., and E. M. El-Omar. 2008. Host-bacterial interactions in Helicobacter pylori infection. Gastroenterology 134306-323. [DOI] [PubMed] [Google Scholar]
- 3.Boivin, G. P., K. Washington, K. Yang, J. M. Ward, T. P. Pretlow, R. Russell, D. G. Besselsen, V. L. Godfrey, T. Doetschman, W. F. Dove, H. C. Pitot, R. B. Halberg, S. H. Itzkowitz, J. Groden, and R. J. Coffey. 2003. Pathology of mouse models of intestinal cancer: consensus report and recommendations. Gastroenterology 124762-777. [DOI] [PubMed] [Google Scholar]
- 3a.Bravo, L. E., L. J. van Doom, J. L. Realpe, and P. Correa. 2002. Virulence-associated genotypes of Helicobacter pylori: do they explain the African enigma? Am. J. Gastroenterol. 972839-2842. [DOI] [PubMed] [Google Scholar]
- 4.Demengeot, J., S. Zelenay, M. F. Moraes-Fontes, I. Caramalho, and A. Coutinho. 2006. Regulatory T cells in microbial infection. Springer Semin. Immunopathol. 2841-50. [DOI] [PubMed] [Google Scholar]
- 5.Dewhirst, F. E., J. G. Fox, E. N. Mendes, B. J. Pater, C. E. Gates, C. A. Kirkbride, and K. A. Eaton. 2000. Flexispira rappini strains represent at least 10 Helicobacter taxa. Int. J. Syst. Evol. Microbiol. 501781-1787. [DOI] [PubMed] [Google Scholar]
- 6.Drazenovich, N. L., C. L. Franklin, R. S. Livingston, and D. G. Besselsen. 2002. Detection of rodent Helicobacter spp. by use of fluorogenic nuclease polymerase chain reaction assays. Comp. Med. 52347-353. [PubMed] [Google Scholar]
- 7.El-Omar, E. M., M. Carrington, W. H. Chow, K. E. McColl, J. H. Bream, H. A. Young, J. Herrera, J. Lissowska, C. C. Yuan, N. Rothman, G. Lanyon, M. Martin, J. F. Fraumen, and C. S. Rabkin. 2000. Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature 404398-402. [DOI] [PubMed] [Google Scholar]
- 8.Erdman, S. E., V. P. Rao, T. Poutahidis, M. Ihrig, Z. Ge, Y. Zeng, M. Tomczak, A. B. Rogers, B. H. Horwitz, and J. G. Fox. 2003. CD4+ CD25+ regulatory lymphocytes require interleukin 10 to interrupt colon carcinogenesis in mice. Cancer Res. 636042-6050. [PubMed] [Google Scholar]
- 9.Fine, P. E. 1995. Variation in protection by BCG: implications of and for heterologous immunity. Lancet 3461339-1345. [DOI] [PubMed] [Google Scholar]
- 10.Fox, J. G. 2002. The non-Helicobacter pylori helicobacters: their expanding role in gastrointestinal and systemic diseases. Gut 50273-283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fox, J. G., P. Beck, C. A. Dangler, M. T. Whary, T. C. Wang, H. N. Shi, and C. Nagler-Anderson. 2000. Concurrent enteric helminth infection modulates inflammation and gastric immune responses and reduces helicobacter-induced gastric atrophy. Nat. Med. 6536-542. [DOI] [PubMed] [Google Scholar]
- 12.Fox, J. G., C. A. Dangler, N. S. Taylor, A. King, T. J. Koh, and T. C. Wang. 1999. High-salt diet induces gastric epithelial hyperplasia and parietal cell loss, and enhances Helicobacter pylori colonization in C57BL/6 mice. Cancer Res. 594823-4828. [PubMed] [Google Scholar]
- 13.Fox, J. G., F. E. Dewhirst, Z. She, Y. Feng, N. S. Taylor, B. J. Paster, R. L. Ericson, C. N. Lau, P. Correa, J. C. Araya, and I. Roa. 1998. Hepatic Helicobacter species identified in bile and gallbladder from Chileans with chronic cholecystitis. Gastroenterology 114755-763. [DOI] [PubMed] [Google Scholar]
- 14.Fox, J. G., F. E. Dewhirst, J. G. Tully, B. J. Pater, l. Yan, N. S. Taylor, M. J. Collins, P. L. Gorelick, and J. M. Ward. 1994. Helicobacter hepaticus sp. nov., a microaerophilic bacterium isolated from livers and intestinal mucosal scrapings from mice. J. Clin. Microbiol. 321238-1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fox, J. G., A. B. Rogers, M. T. Whary, Z. Ge, M. Ohtani, E. K. Jones, and T. C. Wang. 2007. Accelerated progression of gastritis to dysplasia in the pyloric antrum of TFF2−/− C57BL/6 × Sv129 Helicobacter pylori-infected mice. Am. J. Pathol. 1711520-1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fox, J. G., A. B. Rogers, M. T. Whary, N. S. Taylor, S. Xu, Y. Feng, and S. Keys. 2004. Helicobacter bilis-associated hepatitis in outbred mice. Comp. Med. 54541-577. [PubMed] [Google Scholar]
- 17.Fox, J. G., and T. C. Wang. 2007. Inflammation, atrophy, and gastric cancer. J. Clin. Investig. 11760-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fox, J. G., T. C. Wang, A. B. Rogers, T. Poutahidis, Z. Ge, N. Taylor, C. A. Dangler, D. A. Israel, W. Krishna, K. Gaus, and R. M. Peek. 2003. Host and microbial constituents influence Helicobacter pylori-induced cancer in a murine model of hypergastrinemia. Gastroenterology 1241879-1890. [DOI] [PubMed] [Google Scholar]
- 19.Fox, J. G., L. L. Yan, F. E. Dewhirst, B. J. Paster, B. Shames, J. C. Murphy, A. Hayward, J. C. Belcher, and E. N. Mendes. 1995. Helicobacter bilis sp. nov., a novel Helicobacter species isolated from bile. J. Clin. Microbiol. 33445-454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Franklin, C. L., L. K. Riley, R. S. Livingston, C. S. Beckwith, C. L. Besch-Williford, and R. R. Hook. 1998. Enterohepatic lesions in SCID mice infected with Helicobacter bilis. Lab. Anim. Sci. 48334-339. [PubMed] [Google Scholar]
- 21.Ge, Z., P. Doig, and J. G. Fox. 2001. Characterization of proteins in the outer membrane preparation of a murine pathogen, Helicobacter bilis. Infect. Immun. 693502-3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haggerty, T. D., S. Perry, L. Sanchez, G. Perez-Perez, and J. Parsonnet. 2005. Significance of transiently positive enzyme-linked immunosorbent assay results in detection Helicobacter pylori in stool samples from children. J. Clin. Microbiol. 432220-2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hänninen, M. L., R. I. Kärenlampi, J. M. Koort, T. Mikkonen, and K. J. Björkroth. 2005. Extension of the species Helicobacter bilis to include the reference strains of Helicobacter sp. flexispira taxa 2, 3, and 8 and Finnish canine and feline flexispira strains. Int. J. Syst. Evol. Microbiol. 55891-898. [DOI] [PubMed] [Google Scholar]
- 24.Holcombe, C. 1992. Helicobacter pylori: the African enigma. Gut 33429-431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. 1994. Schistosomes, liver flukes, and Helicobacter pylori. IARC Monogr. Eval. Carcinog. Risks Humans 611-241. [PMC free article] [PubMed] [Google Scholar]
- 26.Ihrig, M., M. T. Whary, C. A. Dangler, and J. G. Fox. 2005. Gastric helicobacter infection induces a Th2 phenotype but does not elevate serum cholesterol in mice lacking inducible nitric oxide synthase. Infect. Immun. 731664-1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jergens, A. E., J. H. Wilson-Welder, A. Dorn, A. Henderson, Z. Liu, R. B. Evans, J. Hostetter, and M. J. Wannemuehler. 2007. Helicobacter bilis triggers persistent immune reactivity to antigens derived from the commensal bacteria in gnotobiotic C3H/HeN mice. Gut 56934-940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kullberg, M., D. Jankovic, P. Gorelick, P. Caspar, J. Letterio, A. Cheever, and A. Sher. 2002. Bacteria-triggered CD4+ T regulatory cells suppress Helicobacter hepaticus-induced colitis. J. Exp. Med. 196505-515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lastovica, A. J., and E. le Roux. 2000. Efficient isolation of campylobacteria from stools. J. Clin. Microbiol. 382798-2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee, A., J. O'Rourke, M. C. De Ungria, B. Robertson, G. Daskalopoulos, and M. F. Dixon. 1997. A standardized mouse model of Helicobacter pylori infection: introducing the Sydney strain. Gastroenterology 1121386-1397. [DOI] [PubMed] [Google Scholar]
- 31.Maggio-Price, L., P. Treuting, W. Zeng, M. Tsang, H. Bielefeldt-Ohmann, and B. M. Iritani. 2006. Helicobacter infection is required for inflammation and colon cancer in SMAD3-deficient mice. Cancer Res. 66828-838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31a.Maizels, R. M., and M. Yazdanbakhsh. 2003. Immune regulation by helminth parasites: cellular and molecular mechanisms. Nat. Rev. Immunol. 3733-744. [DOI] [PubMed] [Google Scholar]
- 32.Man, S. M., L. Zhang, A. S. Day, S. Leach, and H. Mitchell. 2008. Detection of enterohepatic and gastric Helicobacter species in fecal specimens of children with Crohn's disease. Helicobacter 13234-238. [DOI] [PubMed] [Google Scholar]
- 33.Maurer, K. J., A. B. Rogers, Z. Ge, A. J. Wiese, M. C. Carey, and J. G. Fox. 2006. Helicobacter pylori and cholesterol gallstone formation in C57L/J. mice: a prospective study. Am. J. Physiol. Gastrointest. Liver Physiol. 290G175-G182. [DOI] [PubMed] [Google Scholar]
- 34.McBee, M. E., P. Z. Zheng, A. B. Rogers, J. G. Fox, and D. B. Schauer. 2008. Modulation of acute diarrheal illness by persistent bacterial infection. Infect. Immun. 764851-4858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mitchell, M. H., R. Ally, A. Wadee, M. Wiseman, and I. Segal. 2002. Major differences in the IgG subclass response to Helicobacter pylori in the first and third worlds. Scand. J. Gastroenterol. 37517-522. [DOI] [PubMed] [Google Scholar]
- 36.Nagai, S., H. Mimuro, T. Yamada, Y. Baba, K. Moro, T. Nochi, H. Kiyono, T. Suzuki, C. Sasakawa, and S. Koyasu. 2007. Role of Peyer's patches in the induction of Helicobacter pylori-induced gastritis. Proc. Natl. Acad. Sci. USA 1048971-8976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Page, K. R., A. L. Scott, and Y. C. Manabe. 2006. The expanding realm of heterologous immunity: friend or foe? Cell Microbiol. 8185-196. [DOI] [PubMed] [Google Scholar]
- 38.Pellicano, R., V. Mazzaferro, W. F. Grigioni, M. A. Cutufia, S. Fagoonee, L. Silengo, M. Rizzetto, and A. Ponzetto. 2004. Helicobacter species sequences in liver samples from patients with and without hepatocellular carcinoma. World J. Gastroenterol. 10598-601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Peterson, R. A., T. Hoepf, and K. A. Eaton. 2003. Adoptive transfer of splenocytes in SCID mice implicates CD4+ T cells in apoptosis and epithelial proliferation associated with Helicobacter pylori-induced gastritis. Comp. Med. 53498-509. [PubMed] [Google Scholar]
- 40.Pronovost, A., S. Rose, J. Pawlak, H. Robin, and R. Schneider. 1994. Evaluation of a new immunodiagnostic assay for Helicobacter pylori antibody detection: correlation with histopathological and microbiological results. J. Clin. Microbiol. 3246-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rad, R., L. Brenner, S. Bauer, S. Schwendy, L. Layland, C. P. da Costa, W. Reindl, A. Dossumbekova, M. Friedrich, D. Saur, H. Wagner, R. M. Schmid, and C. Prinz. 2006. CD25+/Foxp3+ T cells regulate gastric inflammation and Helicobacter pylori colonization in vivo. Gastroenterology 131525-537. [DOI] [PubMed] [Google Scholar]
- 42.Rao, V. P., T. Poutahidis, Z. Ge, P. R. Nambiar, B. H. Horwitz, J. G. Fox, and S. E. Erdman. 2006. Proinflammatory CD4+ CD45RBhi lymphocytes promote mammary and intestinal carcinogenesis in ApcMin/+ mice. Cancer Res. 6657-61. [DOI] [PubMed] [Google Scholar]
- 43.Rogers, A. B., K. S. Cormier, and J. G. Fox. 2006. Thiol-reactive compounds prevent nonspecific antibody binding in immunohistochemistry. Lab. Investig. 86526-533. [DOI] [PubMed] [Google Scholar]
- 44.Rogers, A. B., N. S. Taylor, M. T. Whary, E. D. Stefanich, T. C. Wang, and J. G. Fox. 2005. Helicobacter pylori but not high salt induces gastric intraepithelial neoplasia in B6129 mice. Cancer Res. 6510709-10715. [DOI] [PubMed] [Google Scholar]
- 45.Rogers, A. B., E. J. Theve, Y. Feng, R. C. Fry, K. Taghizadeh, K. M. Clapp, C. Boussahmain, K. S. Cormier, and J. G. Fox. 2007. Hepatocellular carcinoma associated with liver-gender disruption in male mice. Cancer Res. 6711536-11546. [DOI] [PubMed] [Google Scholar]
- 46.Romero, S., J. R. Archer, M. E. Hamacher, S. M. Bologna, and R. F. Schell. 1988. Case report of an unclassified microaerophilic bacterium associated with gastroenteritis. J. Clin. Microbiol. 26142-143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Selin, L., M. Brehm, Y. Naumov, M. Cornberg, S. Kim, S. Clute, and R. Welsh. 2006. Memory of mice and men: CD8+ T-cell cross-reactivity and heterologous immunity. Immunol. Rev. 211164-181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shames, B., J. G. Fox, F. Dewhirst, L. Yan, Z. Shen, and N. S. Taylor. 1995. Identification of widespread Helicobacter hepaticus infection in feces in commercial mouse colonies by culture and PCR assay. J. Clin. Microbiol. 332968-2972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shomer, N. H., C. A. Dangler, M. D. Schrenzel, and J. G. Fox. 1997. Helicobacter bilis-induced inflammatory bowel disease in SCID mice with defined flora. Infect. Immun. 654858-4864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stoicov, C., M. Whary, A. B. Rogers, F. S. Lee, K. Klucevsek, H. Li, X. Cai, R. Saffari, Z. Ge, I. A. Khan, C. Combe, A. Luster, J. G. Fox, and J. Houghton. 2004. Coinfection modulates inflammatory responses and clinical outcome of Helicobacter felis and Toxoplasma gondii infections. J. Immunol. 1733329-3336. [DOI] [PubMed] [Google Scholar]
- 51.Suerbaum, S., C. Josenhans, T. Sterzenbach, B. Drescher, P. Brandt, M. Bell, M. Droge, B. Fartmann, H. P. Fischer, Z. Ge, A. Horster, R. Holland, K. Klein, J. Konig, L. Macko, G. L. Mendz, G. Nyakatura, D. B. Schauer, Z. Shen, J. Weber, M. Frosch, and J. G. Fox. 2003. The complete genome sequence of the carcinogenic bacterium Helicobacter hepaticus. Proc. Natl. Acad. Sci. USA 1007901-7906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Suerbaum, S., and P. Michetti. 2002. Helicobacter pylori infection. N. Engl. J. Med. 3471175-1186. [DOI] [PubMed] [Google Scholar]
- 53.Taylor, N. S., S. Xu, P. Nambiar, F. E. Dewhirst, and J. G. Fox. 2007. Enterohepatic Helicobacter species are prevalent in mice from commercial and academic institutions in Asia, Europe, and North America. J. Clin. Microbiol. 452166-2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tomb, J. F., O. White, A. R. Kerlavage, R. A. Clayton, G. G. Sutton, R. D. Fleischmann, K. A. Ketchum, H. P. Klenk, S. Gill, B. A. Dougherty, K. Nelson, J. Quackenbush, L. Zhou, E. F. Kirkness, S. Peterson, B. Loftus, D. Richardson, R. Dodson, H. G. Khalak, A. Glodek, K. McKenney, L. M. Fitzgerald, N. Lee, M. D. Adams, E. K. Hickey, D. E. Berg, J. D. Gocayne, T. R. Utterback, J. D. Peterson, J. M. Kelley, M. D. Cotton, J. M. Weidman, C. Fujii, C. Bowman, L. Watthey, E. Wallin, W. Hayes, M. Borodovsky, P. D. Karp, H. O. Smith, C. M. Fraser, and J. C. Venter. 1997. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 388539-547. [DOI] [PubMed] [Google Scholar]
- 55.Ward, J. M., J. G. Fox, M. R. Anver, D. C. Haines, C. V. George, M. J. Collins, P. L. Gorelick, K. Nagashima, M. A. Gonda, and R. V. Gilden. 1994. Chronic active hepatitis and associated liver tumors in mice caused by persistent bacterial infection with a novel Helicobacter species. J. Natl. Cancer Inst. 861222-1227. [DOI] [PubMed] [Google Scholar]
- 56.Welsh, R. M., and L. K. Selin. 2002. No one is naive: the significance of heterologous T-cell immunity. Nat. Rev. Immunol. 2417-426. [DOI] [PubMed] [Google Scholar]
- 57.Whary, M., and J. Fox. 2004. Natural and experimental Helicobacter infections. Comp. Med. 54128-158. [PubMed] [Google Scholar]
- 58.Whary, M., N. Sundina, L. Bravo, P. Correa, F. Quinones, F. Caro, and J. Fox. 2005. Intestinal helminthiasis in Colombian children promotes a Th2 response to Helicobacter pylori: possible implications for gastric carcinogenesis. Cancer Epidemiol. Biomarkers Prev. 141464-1469. [DOI] [PubMed] [Google Scholar]
- 59.Whary, M. T., J. Cline, A. King, Z. Ge, Z. Shen, B. Sheppard, and J. Fox. 2001. Long-term colonization levels of Helicobacter hepaticus in the cecum of hepatitis-prone A/JCr mice are significantly lower than those in hepatitis-resistant C57BL/6 mice. Comp. Med. 51413-417. [PubMed] [Google Scholar]
- 60.Wilson, K. T., and J. E. Crabtree. 2007. Immunology of Helicobacter pylori: insights into the future of the immune response and perspectives on vaccine studies. Gastroenterology 133288-308. [DOI] [PubMed] [Google Scholar]
- 61.Yamaoka, Y., K. Yamauchi, H. Ota, H. Sugiyama, S. Ishizone, D. Y. Graham, F. Maruta, M. Murakami, and T. Katsuyama. 2005. Natural history of gastric mucosal cytokine expression in Helicobacter pylori gastritis in Mongolian gerbils. Infect. Immun. 732205-2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Young, S. L., L. Slobbe, R. Wilson, B. M. Buddle, G. W. de Lisle, and G. S. Buchan. 2007. Environmental strains of Mycobacterium avium interfere with immune responses associated with Mycobacterium bovis BCG vaccination. Infect. Immun. 752833-2840. [DOI] [PMC free article] [PubMed] [Google Scholar]