

Abstract
Cytotoxic necrotizing factor 1 (CNF1) is a protein toxin produced by pathogenic Escherichia coli strains. CNF1 constitutively activates small GTPases of the Rho family by deamidation of a glutamine, which is crucial for GTP hydrolysis. The toxin is taken up into mammalian cells by receptor-mediated endocytosis and is delivered from late endosomes into the cytosol. Here, we show that an approximately 55-kDa fragment of CNF1, which contains the catalytic domain and an additional part of the toxin, is present in the cytosol. The processing of this fragment requires an acidic pH and insertion of the toxin into the endosomal membrane. We define the cleavage site region as the region located between amino acids 532 and 544 of CNF1. The data provide insight into the complex mechanism of uptake of bacterial toxins into mammalian cells.
Cytotoxic necrotizing factor 1 (CNF1) is produced by pathogenic Escherichia coli strains which cause urinary tract infections and neonatal meningitis (4, 10). CNF1 belongs to a family of Rho GTPase-modifying proteins which deamidate the small GTPases at glutamine 63 or 61, leading to their constitutive activation (6, 20). Other members of this toxin family are the E. coli toxins CNF2 and CNF3, which have more than 70% identity to CNF1, and CNFY produced by Yersinia pseudotuberculosis, which has about 60% identity to CNF1 (15, 17, 18). Whereas CNF1 modifies RhoA, Rac, and Cdc42 in HeLa cells, recombinant CNFY selectively activates RhoA (8). All CNFs are 115-kDa single-chain AB toxins with an N-terminal receptor-binding domain and a C-terminal catalytic domain, which contains the deamidase activity (3, 12). The two domains are separated by a putative translocation domain with two hydrophobic helices involved in membrane translocation (for reviews, see references 9 and 13).
CNF1 has been shown to enter cells by receptor-mediated endocytosis, which is independent of clathrin and of sphingolipid-cholesterol-rich membrane microdomains (lipid rafts), including caveolae (5). From late endosomes the toxin enters the cytosol in an acidic pH-dependent manner (1). It was speculated that the toxin reaches the cytosol as an uncleaved protein, because CNF1 could be translocated directly through the plasma membrane by an acidic pulse apparently without a requirement for proteolytic cleavage (19). Using a monoclonal antibody that binds to the catalytic domain of CNF1, we detected an approximately 55-kDa fragment of CNF1 in the cytosolic fraction of intoxicated cells. We show that the appearance of this fragment in the cytosol is dependent on the acidic pH in late endosomes and insertion of the translocation loop into the membrane. We narrow the cleavage site region to a stretch of 13 amino acids (amino acids 532 to 544).
MATERIALS AND METHODS
Preparation of recombinant toxins.
CNF1 and mutants of this protein were purified as N-terminal glutathione S-transferase (GST) fusion proteins, as previously described (14). Glutathione S-transferase-CNF1 is referred to as CNF1 below. CNF1 mutants were generated using a QuikChange site-directed mutagenesis kit (Stratagene). Sequences of all constructs were checked by Dye Terminator cycle sequencing (Applied Biosystems).
Cell culture and preparation of cell lysates.
Caco-2 cells were grown in Dulbecco's modified Eagle's medium (DMEM) (12 mM l-glutamine) supplemented with 10% fetal calf serum, sodium pyruvate (1 mM), 1% nonessential amino acids, penicillin (4 mM), and streptomycin (4 mM). HeLa cells were cultured at 37°C with 5% CO2 in DMEM containing 10% fetal calf serum, 1 mM sodium pyruvate, and 4 mM penicillin-streptomycin. For preparation of cell lysates, subconfluent cells were treated with the appropriate amounts of toxin for 1 to 16 h as indicated below. Subsequently, cells were washed twice with phosphate-buffered saline (PBS), collected by scraping, and lysed by sonication. Lysates were cleared by centrifugation (20 min, 21,000 × g, 4°C). For separation of membrane and cytosolic fractions, lysates were centrifuged (1 h, 100,000 × g, 4°C). The membrane pellet was dissolved in sample buffer (10% sodium dodecyl sulfate [SDS], 100 mM Tris [pH 6.8], 10% glycerol, 100 mM dithiothreitol).
Transfection.
For transfection of HeLa cells, 10 μg of plasmid pCDNA3-RhoA was used in each 10-cm dish using Metafectene (Biontech) according to the manufacturer's protocol. Expression of His-tagged RhoA was analyzed by Western blotting with an anti-RhoA monoclonal antibody (1:1,000; Santa Cruz).
Use of protease inhibitors.
We used E64d (40 μM; Sigma) as a cysteine protease inhibitor. For inhibition of aspartate proteases, we added pepstatin A (10 μM; VWR) to the cells, and to inhibit serine proteases, we used 4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride (Pefabloc; 250 μM; Roche), aprotinin (15 μM; Sigma), elastatinal (10 μM; VWR), or Glu-Gly-Arg-chloromethyl ketone (GGACK) (2 μM; Calbiochem). Moreover, the “Complete” protease inhibitor cocktail (Roche) or a serine protease inhibitor cocktail (set 1; Calbiochem) was added during toxin treatment.
Isolation of endosomes.
After intoxication, HeLa cells were washed twice in PBS and harvested by scraping and centrifugation. The supernatant was removed, and the pellet was washed once with homogenization buffer (250 μM sucrose plus 3 mM imidazole). After additional centrifugation cells were lysed in 200 μl homogenization buffer by mechanical rupturing by moving the cells up and down through a small-gauge needle. The lysates were centrifuged (20 min, 3,000 × g, 4°C), and the supernatant was supplemented with sucrose at a final concentration of 35% and subdivided by using sucrose gradient centrifugation (2 h, 160,000 × g, 4°C; sucrose gradient prepared with homogenization buffer and 25, 35, and 40.6% sucrose). Early endosomes were collected from the 25%-35% interphase and analyzed further by SDS-polyacrylamide gel electrophoresis (PAGE) and Western blotting.
SDS-PAGE and Western blotting.
Cell lysates were subjected to 12.5% SDS-PAGE (for the Rho shift assay, 15% urea-SDS-PAGE), and the proteins were blotted onto a nitrocellulose membrane. Following Ponceau-S staining, the membrane was blocked with 5% nonfat milk. Proteins were detected using anti-CNF1 monoclonal antibody (1:4,000; Santa Cruz), anti-RhoA monoclonal antibody (1:1,000; Santa Cruz), anti-Lamp-1 monoclonal antibody (1:500; Santa Cruz), anti-GM130 polyclonal antibody (1:1,000; abcam), anti-p84 monoclonal antibody; 1:5,000; abcam), anti-glyceraldehyde-3-phosphate dehydrogenase monoclonal antibody (1:20,000; Sigma), anti-Rab5B monoclonal antibody (1:1,000; Santa Cruz), or Rab7 polyclonal antibody (1:1,000; Santa Cruz).
Immunoprecipitation and 2D gel electrophoresis.
HeLa cells (in 40 15-cm-diameter dishes) were intoxicated with 1 μg/ml CNF1 overnight, and the appropriate number of untreated cells was harvested by scraping and centrifugation in the presence of a protease inhibitor cocktail (Roche). Following cell lysis by sonication, the cytosol was obtained by ultracentrifugation (1 h, 100,000 × g, 4°C). Thirty micrograms of anti-CNF1 antibody (Santa Cruz) was bound to 150 μg protein A-Sepharose (GE Healthcare) in PBS (pH 7.4) overnight, washed twice, divided into equal portions, and added to the cytosol preparations. After 2 h of binding at 4°C, beads were washed five times with PBS (pH 7.5). Subsequently the CNF1 fragment was eluted with PBS (pH 3). The eluate was precipitated with 4 volumes of acetone at −20°C for 2 h. After centrifugation the pellet was dried and resolved in rehydration buffer (pH 4 to 7 Immobilin DryStrip; GE Healthcare), and two-dimensional (2D) gel electrophoresis was performed.
Matrix-assisted laser desorption ionization—time of flight (MALDI-TOF) analysis was performed by TopLab (Martinsried, Germany).
pH shift assay.
For direct shift of the toxins through the plasma membrane, cells were treated as described previously (1).
TER assay.
For analysis of the transepithelial resistance (TER) of Caco-2 cells, the cells were grown on filters (Millicell; Millipore). TER was measured using an EVOM epithelial voltohmmeter (World Precision Instruments).
RESULTS
A serine protease is involved in the uptake of CNF1.
In order to study whether CNF1 reaches the cytosol as a full-length protein or has to be processed during its uptake, we incubated HeLa cells with CNF1 in the presence of several protease inhibitors during the intoxication process. The cysteine protease inhibitor that we used was E64d, an inhibitor of the papain family of proteases. For inhibition of aspartate proteases, we added pepstatin A to the cells, and to inhibit serine proteases, we used 4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride, aprotinin, elastatinal, or GGACK. Additionally, we added the “Complete” protease inhibitor cocktail to block several kinds of proteases or a serine protease inhibitor cocktail (set 1) during toxin treatment. To monitor the uptake and action of the toxin, we first studied the induced morphological changes in mammalian cells. Second, we directly analyzed RhoA deamidation in lysates of toxin-treated cells, which can be detected by a shift of deamidated RhoA to a higher molecular weight compared to that of unmodified RhoA (20). The cysteine protease inhibitor, as well as the aspartate protease inhibitor, had no influence on the toxin-induced morphological changes in HeLa cells and did not inhibit the CNF1-catalyzed deamidation of RhoA (not shown). In contrast, the protease inhibitor cocktail (not shown), the serine protease inhibitor cocktail (Fig. 1A and 1B), and Pefabloc among the serine protease inhibitors (not shown) retarded the appearance of typical morphological changes in HeLa cells, like cell flattening and polynucleation, and RhoA deamidation. However, the inhibitors did not completely block intoxication of the cells by CNF1. Aprotinin and GGACK had no influence. The data suggested that a serine protease is involved in uptake and/or processing of CNF1.
FIG. 1.
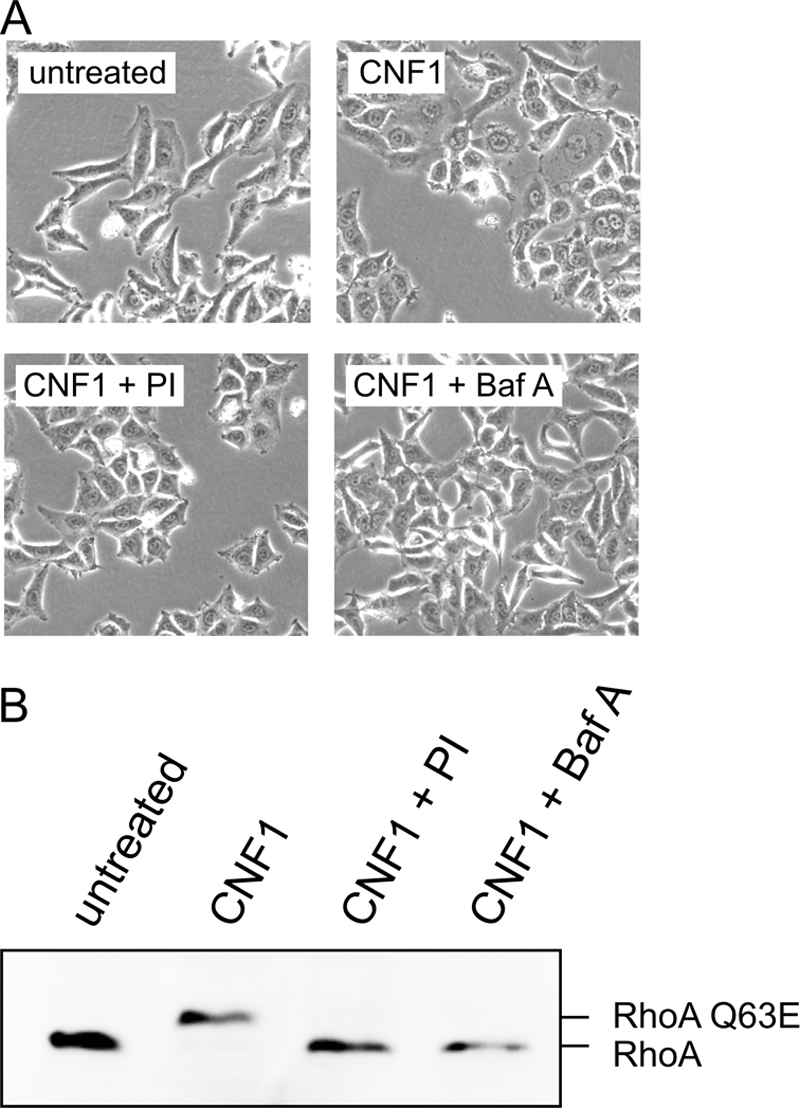
Serine protease inhibitors influence the uptake of CNF1 into the cytosol. The uptake of CNF1 into the cytosol was detected by the CNF1-induced deamidation of RhoA. HeLa cells were treated with 400 ng/ml CNF1 in the presence or absence of a serine protease inhibitor cocktail (PI) for 4 h at 37°C, and photographs were taken (A). As negative control, CNF1 was added to the cells in the presence of bafilomycin A1 (Baf A), which inhibits acidification of the endosomes and thus uptake of CNF1. Subsequently, the cells were lysed. Deamidation of RhoA was detected by the shift of the GTPase in SDS-PAGE in a Western blot with an antibody against the GTPase (B). The experiment was repeated more than three times with similar results.
Identification of an active C-terminal fragment of CNF1 in the cytosol of HeLa cells.
We asked whether cleavage of the toxin is necessary for its action. To monitor whether the toxin itself is cleaved during uptake, we separated the membrane fraction and the cytosol from intoxicated cells and performed Western blotting with a monoclonal antibody against CNF1. As shown in Fig. 2, an approximately 55-kDa protein was detected by the antibody in the cytosolic fraction of CNF1-treated cells, but this protein was not detected in the presence of bafilomycin A1 or in control cytosol without the toxin, indicating that CNF1 is cleaved during uptake. Moreover, cytosol from cells treated with a mutant toxin known to not insert into the endosomal membrane, CNF1(E382/383K) (19), did not contain the toxin fragment (Fig. 2A).
FIG. 2.
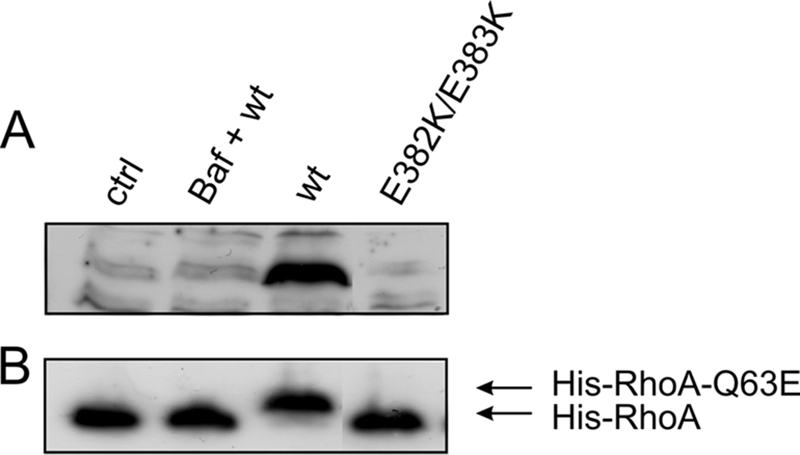
Appearance of a 55-kDa fragment of CNF1 with deamidase activity. (A) HeLa cells were incubated with CNF1 or CNF1(E382/383K) (400 ng/ml each) for 4 h at 37°C in the absence or presence of bafilomycin A1 (Baf) and subsequently lysed. After fractionation of the lysates, the cytosol of toxin-treated cells and the cytosol of untreated control cells were separated by SDS-PAGE, and Western blotting with a monoclonal antibody against CNF1 was performed. A toxin fragment with a molecular mass of about 55 kDa was detected by the antibody only in the cytosol of CNF1-treated cells without bafilomycin A1. The CNF1 fragment has deamidase activity (B). HeLa lysates containing recombinant His-RhoA were incubated with the purified cytosols described above at 37°C overnight. Deamidation of His-RhoA was detected by the shift of the GTPase in SDS-PAGE in a Western blot against the His tag. Only cytosol which contained the CNF1 fragment had deamidase activity. The experiments were repeated more than three times with comparable results. ctrl, control; wt, wild type.
The antibody used for detection of the fragment recognizes an epitope between amino acids 704 and 730 in CNF1, which is located at the beginning of the catalytic domain (amino acids 720 to 1014) (16), indicating that the fragment detected contains the catalytically active part of the toxin. To further analyze whether the fragment in the cytosol contained the complete catalytic domain and had deamidase activity, we incubated the same cytosolic fractions with recombinant His-RhoA, which was expressed in mammalian cells. Figure 2B shows that only incubation of cytosol, which contained the fragment, led to a shift of the GTPase to a higher molecular mass, indicating that deamidation occurred. This shows that the fragment in the cytosol possesses deamidase activity.
The C-terminal fragment of CNF1 encompasses the catalytic domain.
To further characterize the CNF1 fragment, we purified it by immunoprecipitation with the same antibody that was used for Western blotting. Unfortunately, the amount of the precipitated CNF1 fragment was too small for direct measurement of the exact mass of the total fragment in the precipitate or for N-terminal sequencing. Thus, the precipitated proteins from the cytosol of CNF1-treated cells and from the cytosol of control cells were separated by 2D gel electrophoresis. In each case three-quarters of the material was used for Coomassie blue staining and subsequent mass spectrometry, whereas one-quarter was used for Western blotting with an antibody against CNF1. In the Coomassie blue-stained 2D gel containing precipitates from toxin-treated cells, we detected an additional spot at ∼55 kDa and at a pI of approximately 5.6 compared with the gel containing the precipitates of untreated controls. We cut out the spot from the Coomassie blue-stained gel and verified that the protein was a C-terminal fragment of CNF1 by MALDI-TOF analysis. The identified peptides covered the major part of the catalytic domain (Fig. 3B). The corresponding spot was identified as CNF1 by Western blotting (Fig. 3A). On the Western blot, but not on the Coomassie blue-stained SDS gel, a smaller spot at a similar size and a pI of approximately 5.5 was detected, indicating that the fragment may be modified in cell lysates.
FIG. 3.
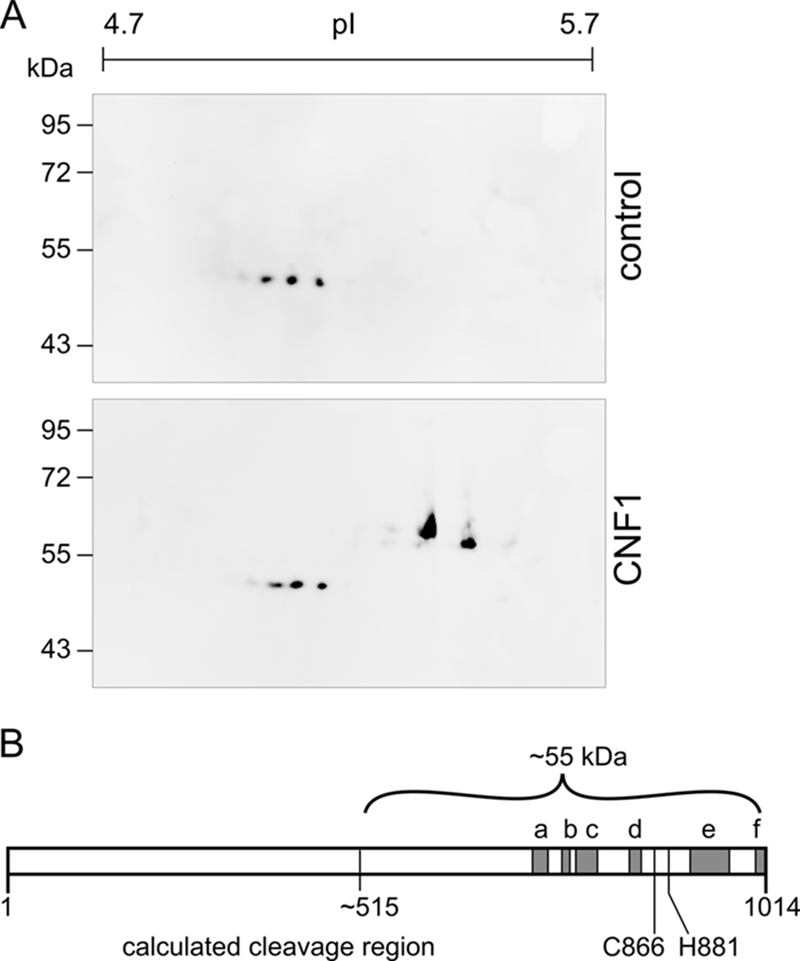
Immunoprecipitation of the CNF1 fragment. HeLa cells (109 cells) were incubated with CNF1 (1 μg/ml) or without the toxin overnight and lysed. Following fractionation of the lysates, cytosols were prepared, and immunoprecipitation with a monoclonal antibody against the C terminus of CNF1 was performed. Part of the precipitate (75%) was separated by 2D gel electrophoresis and stained with Coomassie blue, and the corresponding spot (not present in the control) was prepared for MALDI-TOF analysis. Only peptides with an error between −20 and 20 were considered. The positions of identified peptides which include amino acids 693 to 710 (a), 732 to 741 (b), 753 to 775 (c), 821 to 832 (d), 900 to 944 (e), and 990 to 1012 (f) are indicated by gray boxes in panel B. The estimated cleavage region is located around amino acid 515 (55 kDa, ∼500 amino acids). Part of each precipitate (25%) was separated by 2D gel electrophoresis, and the CNF1 fragment was detected by Western blotting (A).
CNF1 is not cleaved in the cytosol.
The CNF1 fragment, which was detected in the cytosol, may have resulted from cleavage prior to translocation or from proteolysis of CNF1 in the cytosol. To study whether CNF1 is cleaved by a cytosolic serine protease, we took advantage of the fact that the toxin can be pulsed by an acidic pH through the plasma membrane directly into the cytosol (19). If the toxin was cleaved by a cytosolic protease, the fragment should have also been detectable when the endosomal pathway was bypassed. For this purpose CNF1 was bound to its cellular receptor at 4°C, and a pH shift of the culture medium was performed. After the pulse we detected full-length CNF1 in the cytosolic fraction of the cells. We performed additional incubations of the cells at 37°C to study the fate of the pulsed CNF1 in the cytosol. To eliminate the usual uptake of the toxin, we blocked endosome acidification with bafilomycin A1 and thus the release of the toxin from the endosomal compartment into the cytosol. Bafilomycin A1 treatment allowed direct uptake of the toxin exclusively via the plasma membrane.
Even after an additional 4-h incubation period no fragment was detected in the cytosolic fraction of the cells. Instead, the full-length toxin was still present (Fig. 4A). This indicates that the toxin is not cleaved by cytosolic proteases but may be cleaved inside the endosomes.
FIG. 4.
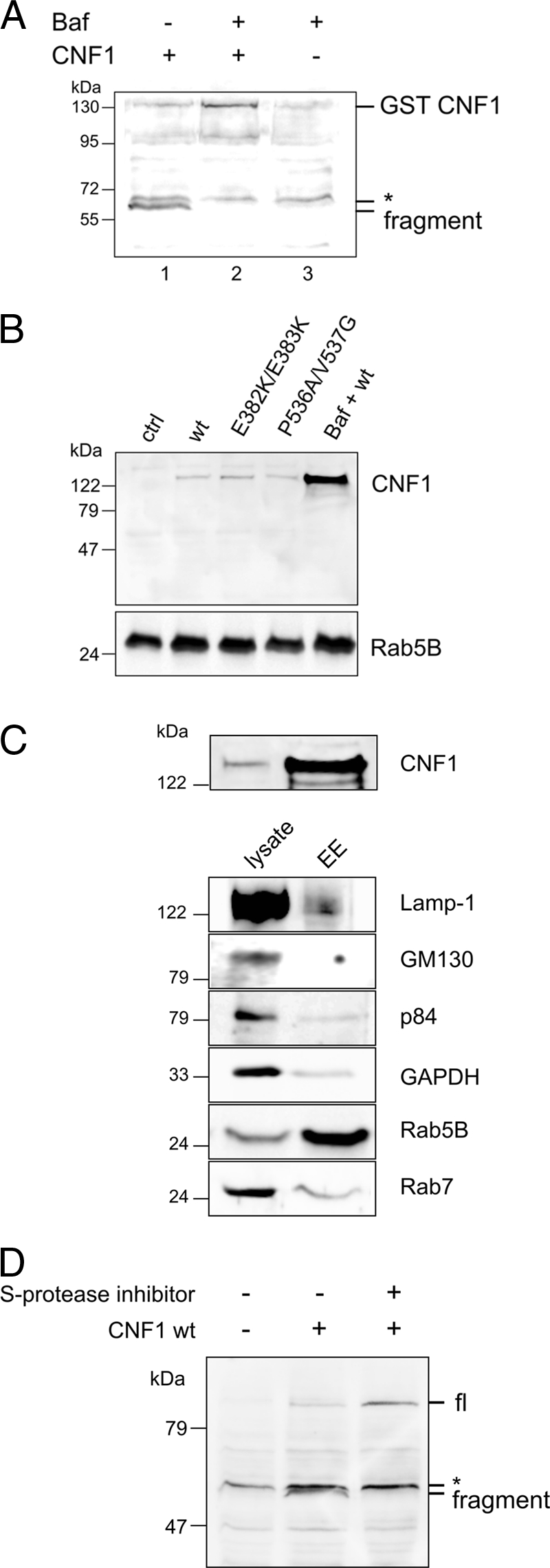
CNF1 is not cleaved in the cytosol (A). CNF1 (1 μg/ml) was bound to HeLa cells for 2 h at 4°C in the absence (lane 1) or in the presence (lane 2) of bafilomycin A1, which blocks endosome-derived uptake of the toxin. After this, cells were washed with Hanks balanced salt solution (pH 4.8) and incubated at 37°C in the buffer for 10 min (lane 2) or left untreated (lane 1). Then cells were incubated in DMEM for 4 h at 37°C in the absence (lane 1) or in the presence (lane 2) of bafilomycin A1 (100 nM). Cell lysate was separated into cytosolic and membrane fractions, and the cytosolic fraction was analyzed for full-length or cleaved CNF1 by Western blotting with an antibody against CNF1. Lane 3 contained a control with bafilomycin A1 but without CNF1. Note that a nonspecific band above the CNF1 fragment band was detected by the antibody (asterisk). In bafilomycin A1-treated cells CNF1 remained uncleaved in endosomes (B). HeLa cells were treated with CNF1 and CNF1 mutants (400 ng/ml) in the absence or presence of bafilomycin A1 (100 nM) overnight. Endosomes of CNF1-treated cells (lanes 2 to 5) and control cells (lane 1) were purified, and the endosomal fraction was separated by SDS-PAGE. CNF1 was detected with an antibody against the C terminus (B). For control of endosome enrichment, the presence of the early endosome marker Rab5B in the corresponding endosomes was analyzed. In endosomes of cells pretreated with bafilomycin A1, large amounts of CNF1 were detected, whereas in the absence of bafilomycin only traces remained in the endosomes of cells intoxicated with wild-type CNF1, CNF1(E382K/E383K) (pore-formation-deficient mutant), and CNF1(P536A/V537G) (cleavage site mutant). (C) Purity of early endosomes from HeLa cells. Fifteen-microgram portions of the lysates of CNF1-treated cells and the corresponding purified endosomes were separated by SDS-PAGE and blotted to determine the presence of CNF1 and specific marker proteins, including Lamp-1 (lysosome), GM130 (Golgi apparatus), p84 (nucleus), glyceraldehyde-3-phosphate dehydrogenase (cytosol), Rab5B (early endosome), and Rab7 (late endosome). (D) In the presence of serine protease inhibitors CNF1 remained uncleaved (full length). HeLa cells were treated with wild-type CNF1 (800 ng/ml) in the absence or presence of a serine protease inhibitor cocktail as indicated for 1 h at 37°C and lysed. Lysates were analyzed to determine the presence of cleaved and uncleaved (full-length) CNF1 in a Western blot against CNF1. Note that a nonspecific band above the CNF1 fragment band was detected by the antibody (asterisk). The experiments were repeated three times with similar results. Baf, bafilomycin A1; GST, glutathione S-transferase; wt, wild-type CNF1; EE, enriched endosomes; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; S-protease inhibitor, serine protease inhibitor cocktail; fl, full length.
In line with these results, bafilomycin A1 (Fig. 4B and 4C) and serine protease inhibitors (Fig. 4D) blocked the intoxication of HeLa cells, as well as the appearance of the CNF1 fragment in the cytosol. Instead, we detected the full-length protein in the lysate (Fig. 4D) or in purified endosomes (Fig. 4B and 4C). We used an anti-Rab5B monoclonal antibody to detect the GTPase as a specific marker of early endosomes. As shown in Fig. 4C, CNF1 and Rab5B were enriched by our purification method, whereas the amount of contaminating vesicles was small.
Our data indicate that an acidic pH and/or insertion of the toxin into the endosomal membrane is required for the cleavage of CNF1. This cleavage may be catalyzed by an endosomal serine protease, or the cleavage may be autocatalytic.
Identification of the cleavage site region.
The size of the C-terminal CNF1 fragment (approximately 55 kDa, 500 amino acids) predicts that there is cleavage around amino acid 515. Therefore, we generated two fragments of CNF1, CNF1(520-1014) and CNF1(510-1014), expressed and purified them, and then compared the sizes of the fragments with the size of the in vivo fragment isolated from cell lysates by Western blotting (not shown). Since even the CNF1(520-1014) fragment ran at a slightly higher molecular weight than the cleaved fragment from lysates, we made further truncations at the end, pointing to a cleavage site around amino acid 540. We decided to generate double mutations in this region of CNF1 (ranging from amino acid 526 or 527 to amino acid 547 or 548) to screen for essential amino acids required for toxin cleavage. Moreover, we used one CNF1 point mutation, CNF1(K540I), that was already available in the laboratory. The possibility that there was misfolding of the proteins was excluded by comparing the velocity of the deamidation of recombinant RhoA. All mutants showed comparable in vitro activity in the Rho shift assay (not shown). We treated HeLa cells with the purified full-length CNF1 mutants and the wild-type toxin for 4 h and analyzed the morphological changes, the appearance of the CNF1 fragment in the lysates, and the shift of RhoA (indicating deamidation). The results of all of the experiments are summarized in Fig. 5A, and representative results of the single experiments are shown in Fig. 5B to D. HeLa cells treated for 4 h with wild-type CNF1 or with the double mutant CNF1(N534S/I535L) had the characteristic morphology induced by the toxin, including spreading and flattening of cells, membrane ruffling, formation of filopodia, and polynucleation. Cells treated with the CNF1(P536A/V537G) mutant had the same morphology as the untreated controls after 4 h of incubation, indicating that this mutant has no or very weak activity with cells. After photographs were taken, the cells were lysed, and the lysates were analyzed for the presence of the 55-kDa CNF1 fragment and for RhoA deamidation by Western blotting against CNF1 and RhoA, respectively. As expected, the fragment was detectable in the lysates of cells treated with the wild-type toxin or with the CNF1(N534S/I535L) double mutant but not in the cell lysates from cells treated with the CNF1(P536A/V537G) mutant or from untreated controls (Fig. 5C). Some mutants [for example, CNF1(F530L/E531A)] showed reduced activity with HeLa cells, resulting in less fragment in the cytosol, whereas some mutations completely blocked cleavage of the toxin [for example, CNF1(D541A/E542A)] (Fig. 5A). Interestingly, the mutants with no detectable fragment in the lysate [for example, CNF1(P536A/V537G)] had a weak effect on HeLa cells when they were incubated for longer times. The cells started to flatten 6 h after addition of the mutant toxin (not shown). Moreover, CNF1(P536A/V537G) was able to slowly deamidate RhoA in the cytosol (there was a partial shift of RhoA after 4 h of intoxication) (Fig. 5D). From these experiments we conclude that the mutants induce deamidation of RhoA more slowly than wild-type CNF1, leading to retarded induction of the typical changes in cell morphology. However, there was no reduced activity of the mutants in an in vitro RhoA shift assay, and the mutants showed the same capacity for binding to the cells, as detected by Western blotting (not shown), indicating that there was retarded uptake of the toxin into the cytosol. This may be due to nonspecific cleavage of CNF1. In endosomes of cells pretreated with bafilomycin A1, large amounts of CNF1 were detected, whereas in the absence of bafilomycin A1 only traces remained in the endosomes of cells intoxicated with wild-type CNF1, CNF1(E382K/E383K) (pore-forming deficient mutant), and CNF1(P536A/V537G) (cleavage site mutant) (Fig. 4B). The data indicate that nonspecific cleavage of CNF1 occurs in the endosomes independent of insertion into the membrane or specific cleavage.
FIG. 5.

Identification of the cleavage site region. (A) Summary of changes in morphology (morphol.), in vitro fragment formation, and Rho shift after treatment of HeLa cells with CNF1 mutants in the putative cleavage site region. The results of at least three independent experiments are summarized. ++, the activity was the same as the activity with wild-type CNF1 for induction of the typical morphology (flattening, ruffling, formation of filopodia, polynucleation) or for the ability to deamidate RhoA (shift) or a comparable amount of the CNF1 fragment was detectable by Western blotting; +, less activity or less fragment than that observed for wild-type CNF1; −, no detectable morphological changes, no ability to shift RhoA, or no fragment in the cytosol. ctrl., control; wt, wild type. (B) Morphological changes in HeLa cells induced by CNF1 and CNF1 mutants. HeLa cells were treated with CNF1 or CNF1 mutants (400 ng/ml each) as indicated. The cells were intoxicated for 4 h, and cytosolic fractions were prepared. (C) C-terminal fragment as revealed by Western blot analysis against the catalytic domain of CNF1. α-CNF1, anti-CNF1. (D) For the in vitro Rho shift, cytosols were separated by urea-SDS-PAGE, and the shift was detected with a monoclonal antibody against RhoA (α-RhoA). (E) TER after CNF1 intoxication in Caco-2 cells grown on filters. Cells were treated with 100 ng/ml CNF1, inactive mutant CNF1(C866S), pore-forming deficient mutant CNF1(E383K/E384K), and two representative cleavage region mutants, CNF1(N534S/I535L) and CNF1(P536A/V537G). TER was measured after 4 h (open bars), 6 h (striped bars), and 8 h (filled bars). The results are expressed as the percentage of the TER at time zero, and the bars and error bars indicate the means ± standard deviations of at least three independent experiments. (F) Amino acid sequence of the cleavage site region of CNF1 (amino acids 526 to 548). Amino acids identified as amino acids necessary for CNF1 processing are indicated by larger type.
We have shown that CNF1 induces a decrease in the TER of human colon carcinoma (Caco-2) cells (7). For a quantitative comparison of the in vivo activities of the toxins, we studied their effects on Caco-2 cells in a TER assay. As shown in Fig. 5E, the TER was decreased to 40% of the time-zero value 4 h after addition of 100 ng/ml wild-type CNF1 to Caco-2 cells. In contrast, there was only a marginal decrease following addition of 10 ng/ml wild-type CNF1 to Caco-2 cells or 100 ng/ml of CNF1(N534S/I535L) within 4 h. The value decreased to 40% of the time-zero value after 6 h of incubation; 100 ng/ml CNF1(P536A/V537G) decreased the TER of Caco-2 cells even more slowly (to 45% of the time-zero value after 8 h incubation). This finding is consistent with the retarded modification of Rho proteins by cleavage site mutants. Also, the pore-formation-deficient mutant CNF1(E383K/E384K) decreased the TER of Caco-2 cells more slowly than the wild-type toxin. The data suggest that membrane insertion, as well as cleavage of CNF1, is required for a fast decrease in the TER of Caco-2 cells.
DISCUSSION
CNF1 is a protein toxin produced by pathogenic E. coli strains. Although the mechanism of action of CNF1 has been known for more than 10 years, it is still not clear how this toxin enters the cytosol to deamidate Rho GTPases. From previous studies it is known that the activity of the toxin and the morphological changes induced by the toxin are comparable for the native toxin and the recombinant protein (for a review see reference 2). Therefore, we do not expect that there are differences in the uptake mechanism. Moreover, the expected concentration of CNF1 produced by a pathogenic E. coli strain is very low and presumably does not allow detection of protein fragments in the cytosol of infected cells. We decided to analyze the uptake of the recombinant protein by using amounts sufficient to detect it in the cytosol.
Here, we show that a fragment of CNF1 which contains the active catalytic domain reached the cytosolic compartment. The toxin fragment appeared as a soluble protein in the cytosol and was not membrane bound. Therefore, CNF1 action may be not restricted to GTPases located in the plasma membranes.
Using the proton pump inhibitor bafilomycin A1 and a CNF1 mutant not able to insert into the endosomal membrane [CNF1(E382/383K) (19)], we showed that toxin cleavage requires acidification of the endosomes, as well as insertion of the toxin into the membrane. CNF1 can be transferred directly into the cytosol by a short acidic pulse (5). Our model therefore suggests that acidification and insertion into the membrane have to occur before the toxin is cleaved. In the purified endosomes we did not find any full-length CNF1 when no bafilomycin A1 was present, indicating that the toxin was already cleaved in this compartment. Moreover, CNF1 mutants not able to insert into the membrane or cleavage site mutants were not present as full-length proteins in the endosomal compartment when acidification occurred, indicating that the toxin was degraded. We cannot rule out the possibility that at least parts of the CNF1 toxin enter the lysosomal compartment. If this occurs, we would expect degradation into undefined fragments rather than specific activation of the toxin upon entrance into the lysosomes. To exclude the possibility that CNF1 is cleaved by lysosomal proteases, we studied toxin uptake with several cell lines of cathepsin B, D, and H knockout mice. All cells were responsive to CNF1 with the same kinetics as wild-type cells, indicating that the cathepsins are not required for uptake of the toxin (data not shown). However, we cannot completely exclude the possibility that lysosomal proteases are involved in CNF1 uptake, because the cathepsins may be redundant with respect to CNF1 cleavage.
It is also not clear whether the toxin is processed before translocation through a putative pore or whether it is translocated and immediately cleaved outside the endosome (Fig. 6). A hint that the second model is correct is that we never detected a toxin fragment in the endosomal fraction of intoxicated cells. Moreover, RhoA modification is retarded but not completely blocked in cells treated with CNF1(P536A/V537G), although the toxin is not cleaved. Interestingly, toxin cleavage did not occur directly in front of the catalytic domain, but the toxin contains an additional approximately 180-amino-acid part which may contain a protease for intrinsic cleavage or have a different function, which we are currently analyzing.
FIG. 6.

Model of uptake and cleavage of CNF1. Following binding of CNF1 to the cellular receptor (probably the p37 laminin receptor precursor [11]) via its N-terminal receptor-binding domain (dark gray line), CNF1 is endocytosed. In late endosomes acidification occurs via an ATP-dependent proton pump (step 1). This is a prerequisite for insertion of two hydrophobic regions (light gray line) located in the central part of CNF1 (step 2). In an unknown manner the C-terminal part of CNF1 may be transported into the cytosol, where cleavage occurs (step 3). A fragment of CNF1 containing an additional part of the toxin with an unknown function (black line) in front of the catalytic domain (broken line) is found in the cytosol.
However, we detected no in vitro autocatalytic cleavage activity of CNF1 at any pH tested. Also, addition of concentrated cell lysates or membranes did not lead to cleavage of recombinant CNF1. There may be more special requirements for autocatalytic cleavage, like special localization of chaperones or a membrane potential. Another possibility is that the toxin is cleaved by a cellular protease which has not been identified. The cleavage region identified shows no homology to any conserved protease cleavage motif known.
Since low toxin concentrations are expected during an E. coli infection, cleavage of CNF1 seems to be essential for efficient intoxication of mammalian cells. For in vivo quantification of the CNF1 activity, we measured the CNF1-induced decrease in the TER of colon carcinoma cells (Caco-2 cells) (7). CNF1(P536A/V537G) did decrease the TER of Caco-2 cell monolayers at a concentration of 100 ng/ml, and the kinetics were even slower than those of a 10-fold-lower concentration of wild-type CNF1 (Fig. 5E). Moreover, CNF1(P536A/V537G) appeared as a full-length protein in endosomes in the presence of bafilomycin A1, indicating that it bound to the cellular receptor, reached the endosomal compartment, and was able to insert into the endosomal membrane. Thus, we conclude that insertion into the endosomal membrane followed by specific cleavage of CNF1 in the center of the protein is required for effective action of the toxins.
Acknowledgments
We thank Gaby Markwirth for practical help. We acknowledge the excellent technical assistance provided by Iris Misicka and Jürgen Dumbach. We thank Thomas Reinheckel (Freiburg, Germany) for providing cathepsin knockout cells. Z.K., B.B., and G.S. conceived and designed the experiments, Z.K. and B.B. performed the experiments, G.S analyzed the data, and Z.K., K.A., and G.S. wrote the paper.
This work was supported by Deutsche Forschungsgemeinschaft (DFG) project SCHM 1385/4-1.
Editor: S. R. Blanke
Footnotes
Published ahead of print on 23 February 2009.
REFERENCES
- 1.Blumenthal, B., C. Hoffmann, K. Aktories, S. Backert, and G. Schmidt. 2007. The cytotoxic necrotizing factors from Yersinia pseudotuberculosis and from Escherichia coli bind to different cellular receptors but take the same route to the cytosol. Infect. Immun. 753344-3353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boquet, P. 2001. The cytotoxic necrotizing factor 1 (CNF1) from Escherichia coli. Toxicon 391673-1680. [DOI] [PubMed] [Google Scholar]
- 3.Buetow, L., G. Flatau, K. Chiu, P. Boquet, and P. Ghosh. 2001. Structure of the Rho-activating domain of Escherichia coli cytotoxic necrotizing factor 1. Nat. Struct. Biol. 8584-588. [DOI] [PubMed] [Google Scholar]
- 4.Caprioli, A., V. Falbo, F. M. Ruggeri, L. Baldassarri, R. Bisicchia, G. Ippolito, E. Romoli, and G. Donelli. 1987. Cytotoxic necrotizing factor production by hemolytic strains of Escherichia coli causing extraintestinal infections. J. Clin. Microbiol. 25146-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Contamin, S., A. Galmiche, A. Doye, G. Flatau, A. Benmerah, and P. Boquet. 2000. The p21 Rho-activating toxin cytotoxic necrotizing factor 1 is endocytosed by a clathrin-independent mechanism and enters the cytosol by an acidic-dependent membrane translocation step. Mol. Biol. Cell 111775-1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Flatau, G., E. Lemichez, M. Gauthier, P. Chardin, S. Paris, C. Fiorentini, and P. Boquet. 1997. Toxin-induced activation of the G protein p21 Rho by deamidation of glutamine. Nature 387729-733. [DOI] [PubMed] [Google Scholar]
- 7.Gerhard, R., G. Schmidt, F. Hofmann, and K. Aktories. 1998. Activation of Rho GTPases by Escherichia coli cytotoxic necrotizing factor 1 increases intestinal permeability in Caco-2 cells. Infect. Immun. 665125-5131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hoffmann, C., M. Pop, J. Leemhuis, J. Schirmer, K. Aktories, and G. Schmidt. 2004. The Yersinia pseudotuberculosis cytotoxic necrotizing factor (CNFY) selectively activates RhoA. J. Biol. Chem. 27916026-16032. [DOI] [PubMed] [Google Scholar]
- 9.Hoffmann, C., and G. Schmidt. 2004. CNF and DNT. Rev. Physiol. Biochem. Pharmacol. 15249-63. [DOI] [PubMed] [Google Scholar]
- 10.Khan, N. A., Y. Wang, K. J. Kim, J. W. Chung, C. A. Wass, and K. S. Kim. 2002. Cytotoxic necrotizing factor-1 contributes to Escherichia coli K1 invasion of the central nervous system. J. Biol. Chem. 27715607-15612. [DOI] [PubMed] [Google Scholar]
- 11.Kim, K. J., J. W. Chung, and K. S. Kim. 2005. 67-kDa laminin receptor promotes internalization of cytotoxic necrotizing factor 1-expressing Escherichia coli K1 into human brain microvascular endothelial cells. J. Biol. Chem. 2801360-1368. [DOI] [PubMed] [Google Scholar]
- 12.Lemichez, E., G. Flatau, M. Bruzzone, P. Boquet, and M. Gauthier. 1997. Molecular localization of the Escherichia coli cytotoxic necrotizing factor CNF1 cell-binding and catalytic domains. Mol. Microbiol. 241061-1070. [DOI] [PubMed] [Google Scholar]
- 13.Lemonnier, M., L. Landraud, and E. Lemichez. 2007. Rho GTPase-activating bacterial toxins: from bacterial virulence regulation to eukaryotic cell biology. FEMS Microbiol. Rev. 31515-534. [DOI] [PubMed] [Google Scholar]
- 14.Lerm, M., J. Selzer, A. Hoffmeyer, U. R. Rapp, K. Aktories, and G. Schmidt. 1999. Deamidation of Cdc42 and Rac by Escherichia coli cytotoxic necrotizing factor 1: activation of c-Jun N-terminal kinase in HeLa cells. Infect. Immun. 67496-503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lockman, H. A., R. A. Gillespie, B. D. Baker, and E. Shakhnovich. 2002. Yersinia pseudotuberculosis produces a cytotoxic necrotizing factor. Infect. Immun. 702708-2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meysick, K. C., M. Mills, and A. D. O'Brien. 2001. Epitope mapping of monoclonal antibodies capable of neutralizing cytotoxic necrotizing factor type 1 of uropathogenic Escherichia coli. Infect. Immun. 692066-2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Orden, J. A., G. Dominguez-Bernal, S. Martinez-Pulgarin, M. Blanco, J. E. Blanco, A. Mora, J. Blanco, J. Blanco, and R. de la Fuente. 2007. Necrotoxigenic Escherichia coli from sheep and goats produce a new type of cytotoxic necrotizing factor (CNF3) associated with the eae and ehxA genes. Int. Microbiol. 1047-55. [PubMed] [Google Scholar]
- 18.Oswald, E., M. Sugai, A. Labigne, H. C. Wu, C. Fiorentini, P. Boquet, and A. D. O'Brien. 1994. Cytotoxic necrotizing factor type 2 produced by virulent Escherichia coli modifies the small GTP-binding proteins Rho involved in assembly of actin stress fibers. Proc. Natl. Acad. Sci. USA 913814-3818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pei, S., A. Doye, and P. Boquet. 2001. Mutation of specific acidic residues of the CNF1 T domain into lysine alters cell membrane translocation of the toxin. Mol. Microbiol. 411237-1247. [DOI] [PubMed] [Google Scholar]
- 20.Schmidt, G., P. Sehr, M. Wilm, J. Selzer, M. Mann, and K. Aktories. 1997. Gln63 of Rho is deamidated by Escherichia coli cytotoxic necrotizing factor 1. Nature 387725-729. [DOI] [PubMed] [Google Scholar]