

Abstract
The sit-encoded iron transport system is present within pathogenicity islands in all Shigella spp. and some pathogenic Escherichia coli strains. The islands contain numerous insertion elements and sequences with homology to bacteriophage genes. The Shigella flexneri sit genes can be lost as a result of deletion within the island. The formation of deletions was dependent upon RecA and occurred at relatively high frequency. This suggests that the sit region is inherently unstable, yet sit genes are maintained in all of the clinical isolates tested. Characterization of the sitABCD genes in S. flexneri indicates that they encode a ferrous iron transport system, although the genes are induced aerobically. The sit genes provide a competitive advantage to S. flexneri growing within epithelial cells, and a sitA mutant is outcompeted by the wild type in cultured epithelial cells. The Sit system is also required for virulence in a mouse lung model. The sitA mutant was able to infect the mice and induce a protective immune response but was avirulent compared to its wild-type parent strain.
The Shigella species are invasive enteric pathogens that cause dysentery in their human host. Following ingestion, the bacteria invade colonic epithelial cells and multiply within the cytoplasm. They spread from cell to cell, eventually causing death of the host cells and provoking an intense inflammatory response (39, 51).
Shigella spp. are closely related to and considered part of the same species as Escherichia coli (40, 41). The patterns that have emerged from analysis of the E. coli and Shigella genomes are that functional genes that are present in two or more members of this enteric group show a high degree of sequence conservation, but the overall organization of the genomes is different. Compared with the E. coli K-12 genome, each of the Shigella genomes contains a significant number of rearrangements, as well as insertions and deletions. Some of the insertions, termed pathogenicity islands (PAI), are quite large and include genes that increase virulence or fitness. Deletions in the chromosome may also affect pathogenicity; for example, the deletion of lysine decarboxylase genes in S. flexneri is associated with increased virulence (25, 26).
One class of genes that shows significant variability among Shigella and E. coli strains are those encoding high-affinity transport systems for iron. Iron is an essential element for Shigella, but the acquisition of iron is complicated by its insolubility in aerobic environments at neutral pH. Bacteria often have multiple pathways for importing iron, allowing the utilization of Fe++, Fe+++, and iron bound to a variety of carriers.
Ferric iron is efficiently transported into E. coli K-12 by the siderophore enterobactin (Ent) (11). This low-molecular-weight iron chelator is synthesized and secreted into the environment, where it binds ferric iron with high affinity (32), and the ferri-siderophore complex is transported back into the cell via a specific transporter system (Fep). Most E. coli, Shigella dysenteriae type 1, and Shigella sonnei strains synthesize and transport enterobactin (11, 34, 36, 38). However, many Shigella boydii and Shigella flexneri strains are Ent− due to deletions and point mutations within the ent/fep operons (36, 52). Shigella strains that fail to produce enterobactin produce a different siderophore, aerobactin, and some strains produce both siderophores (22, 36). Aerobactin is a secondary hydroxamate, and genes for its synthesis (iucABCD) and receptor (iutA) are located within a single operon (4, 22). These genes may be on either the chromosome (22, 24) or a plasmid (4, 8). The chromosomal genes are located within PAI in S. flexneri and S. boydii (31, 42, 57).
The Shigella species have additional iron transport systems that are not found in E. coli K-12. A transport system with homology to the Salmonella enteritidis Sit system is found in all of the Shigella species. Some strains have heme transport systems, the best characterized of which are the S. dysenteriae Shu heme transporter (30, 61) and a nearly identical system in E. coli O157:H7 (54). S. dysenteriae type 1 strains also have the iro genes (43) for the biosynthesis of salmochelin, a modified form of enterobactin first described in Salmonella (13). Additional transport systems for iron are present in enteric pathogens, and some of these are completely uncharacterized.
The only iron transport system that appears to be common to all members of the E. coli/Shigella group is Feo. This is a ferrous iron cytoplasmic membrane transporter encoded by the feoABC genes (5, 19). FeoB is a cytoplasmic membrane protein with GTPase activity (23), but the mechanism of transport and the functions of FeoA and FeoC have not been fully determined.
Despite the apparent variation in the iron transport systems found in Shigella, there are consistent patterns. All express at least one siderophore and have the Feo and Sit transporters. S. flexneri serotype 2a strains SA100 and 2457T have only these three systems and thus were chosen for analysis of iron transport in vitro and within the host cell cytoplasm.
Analysis of isogenic strains lacking one or more of the iron transporters showed that no single mutation eliminated intracellular growth, as the single iucD, feoB, and sitA mutants all produced plaques in cultured cells (48). The double mutants produced smaller plaques, but only the triple mutant was completely defective in growth and plaque formation (48).
Additional information about the roles of these three systems in iron acquisition comes from our previous studies of their regulation. All of the iron transport systems are negatively regulated by Fur (1, 19), an iron-binding repressor protein. However, analysis of the expression of iron transport genes by wild-type S. flexneri growing in the intracellular environment indicated that only the sit genes were highly expressed in this environment (47). The aerobactin genes were downregulated in the intracellular environment (14), and feo expression appeared unchanged. The fact that, of the three iron transport systems, only sit is normally induced intracellularly suggests that it plays an important role when the bacteria inhabit the host cell cytoplasm. The failure of a sit mutant to show a defect in plaque formation may indicate that iron starvation induced by loss of the Sit system resulted in upregulation of the feo and iuc genes when they would not normally have been expressed. Further, since all of these genes appear to be regulated similarly by Fur in vitro, this differential expression of the iron transport genes in the intracellular environment suggested that factors other than iron were contributing to their regulation.
An additional environmental factor that controls the expression of these genes is oxygen (3). As previously noted in E. coli K-12 (19), expression of the feo ferrous iron transport operon was induced under anaerobic conditions, where ferrous iron should predominate over ferric iron. In contrast, the aerobactin genes were induced aerobically, consistent with their role in ferric iron uptake (3). Surprisingly, the S. flexneri sit genes were repressed when the cells were grown anaerobically (3), although the homologous sitABCD genes in Salmonella encode a ferrous iron uptake system (20, 63). Consistent with aerobic induction, the Sit system was sufficient to support plaque formation by S. flexneri under aerobic, but not anaerobic, conditions (3).
Because the Sit iron transport system was found in all of the Shigella species and was induced when the bacteria were growing intracellularly, we undertook a characterization of its genetics and role in the virulence of S. flexneri.
MATERIALS AND METHODS
Strains and media.
Bacterial strains and mutants are described in Table 1. Strains were maintained in Trypticase soy broth (TSB) with 20% glycerol at −80°C. Cultures were routinely grown at 37°C in Luria-Bertani (LB) broth (1% tryptone, 0.5% yeast extract, 1% NaCl) or on LB agar. Congo red agar (35), which contains TSB, 0.01% Congo red, and 1.5% agar, was used to distinguish virulent isolates by their red color. For growth of S. flexneri strains in minimal medium, MM9 (53) without added iron and containing 2 μg of nicotinic acid per ml and 0.4% (wt/vol) glucose was used. The aerobactin for supplementing S. flexneri iron transport mutants was sterile culture supernatant of S. flexneri SA101 prepared as previously described (48). Antibiotics were used at the following concentrations per milliliter: 250 μg of carbenicillin, 50 μg of kanamycin, and 30 μg of chloramphenicol. Expression of T7 RNA polymerase was induced with 500 μM isopropyl-β-d-thiogalactopyranoside (IPTG; Promega, Madison, WI).
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Characteristic(s) | Reference or source |
|---|---|---|
| E. coli strains | ||
| DH5α | endA1 hsdR17 supE44 thi-1 recA1 gyrA96 relA1 Δ(lacZYA-argF)U169 deoR [φ80dlac Δ(lacZ)M15] | 50 |
| HB101 | F− Δ(gpt-proA)62 leu supE44 ara-14 galK2 lacY1 Δ(mcrC-mrr) rpsL20 xyl-5 mtl-1 recA13 | 50 |
| S. flexneri strains | ||
| 2457T | S. flexneri wild-type serotype 2a | 60 |
| SA100 | S. flexneri wild-type serotype 2a | 36 |
| SA101 | SA100, Vir−, source of aerobactin | 21 |
| SA240 | SA100 iucD::kan | 21 |
| SM100 | SA100 Strr | Stefan Seliger |
| SM160 | SM100 recA::cam | Laura Runyen-Janecky |
| SM166 | SM100 sitA::cam | 48 |
| SM193w | SM100 sitA::cam feoB::tmp iucD::kan | 48, 62 |
| SM100 Lac | SM100 with lacZ integrated into chromosome | This study |
| SM166 Lac | SM166 with lacZ integrated into chromosome | This study |
| Plasmids | ||
| pACYC184 | Plasmid cloning vector | 6 |
| pWKS30 | Plasmid cloning vector | 58 |
| pLAFR1 | Cosmid cloning vector | 12 |
| pEG1 | Cosmid vector pLAFR1 carrying SA100 sit operon and surrounding sequence | 48 |
| pEG3 | pWKS30 carrying 8-kb HindIII fragment from pEG1 including sitABCD | This study |
| pAR1219 | T7 RNA polymerase gene under control of lacUV5 promoter in pBR322 | 10 |
| pFZ68 | pACYC184 carrying 4.6-kb EcoRI fragment of pEG1; includes entire sit operon | This study |
| pFZ69 | pACYC184 carrying 6.4-kb EcoRI fragment of pEG1 | This study |
| pFZ70 | pACYC184 carrying 13-kb EcoRI fragment of pEG1; includes T7 promoter region | This study |
| pFZ74 | pACYC184 carrying 5.6-kb EcoRV fragment of pFZ70; includes T7 promoter region | This study |
| pFZ76 | pACYC184 carrying 2-kb EcoRV/DraI fragment of pFZ74; includes T7 promoter region | This study |
| pGRG36 | Tn7 insertion vector | 27 |
| pGRGlac1 | E. coli lacZ cloned into pGRG36 | This study |
Construction of Lac+ S. flexneri strains.
SM100 Lac and SM166 Lac were constructed by inserting the E. coli lacZ gene into the intergenic region between the glmS and pstS genes with Tn7 transposase encoded on plasmid pGRGlac1, which was constructed as follows. E. coli lacZ was amplified from strain ARM110 (29) with Pfx polymerase (Invitrogen, Carlsbad, CA) and primers ec.lacZ.1293 (5′-ATACGCAAACCGCCTCTCCC-3′) and ec.lacZ.5024.rev (5′-CTGACTTTCTCAATAAATGCCTCTACTG-3′). The resulting PCR product was cloned into the SmaI site of pGRG36 (27) to give pGRGlac1. This plasmid was introduced into the indicated S. flexneri strains by electroporation, and colonies that were blue on 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal) plates were selected following growth at 37°C. The proper integration of lacZ was confirmed by PCR with a primer hybridizing within the glmS gene and one hybridizing within lacZ.
PCR detection of sit and T7 promoters.
The presence of the sit operon was detected by amplification with primers sitAB forward (5′-CTCTTGAAGCACTGAAGGAG-3′) and sitAB reverse (5′-CGCACAAATCCCATAATC-3′). The T7 promoter region was detected by PCR with primers SMP028 (5′-GTGTCCCTTCTCCCTATAGTG-3′) and SMP033 (5′-CTTACTACAGACCTGTGTGG-3′).
DNA sequence analysis.
DNA sequencing was performed by the University of Texas Institute for Cellular and Molecular Biology DNA Core Facility with an ABI Prism 3700 DNA sequencer. Analysis of DNA sequences was carried out with MacVector 7.2 and Clone Manager 7.04. BLAST searches and other bioinformatic analyses were done with the National Center for Biotechnology and Enteropathogen Resource Integration Center (www.ERICBRC.org) databases. Pairwise alignments were carried out with ClustalW from within MacVector 7.2, and genomic alignments were done with MAUVE (9).
Iron transport assay.
Cultures of S. flexneri SM193w containing the indicated plasmid were grown overnight in LB broth supplemented with 1/10 volume of sterile MM9 supernatant from S. flexneri strain SA101 as a source of aerobactin. Overnight cultures were diluted 1/25 into MM9 with aerobactin and grown to late exponential phase with aeration at 37°C. Transport assays were performed in triplicate at room temperature in the presence or absence of 5 mM sodium ascorbate, as previously described (62).
T7 polymerase sensitivity assay.
Serial dilutions were made from cultures of SM100/pAR1219, SM160/pAR1219, or E. coli HB101/pEG1/pAR1219, and each dilution was plated on both TSB agar containing carbenicillin (viable count) and the same medium with 1 mM IPTG. The frequency of resistance to T7 RNA polymerase was calculated by dividing the number of colonies on the plates containing IPTG by the viable count. The frequency of loss of the sit operon in T7 RNA polymerase-resistant isolates was determined by screening T7 RNA polymerase-resistant colonies by PCR.
Henle cell plaque assay.
Monolayers of Henle cells (intestinal 407 cells; American Type Culture Collection, Manassas, VA) were maintained in Eagle's minimum essential medium with 2 mM glutamine, 10% fetal bovine serum, tryptose phosphate broth, and minimal essential amino acids in a 5% CO2 atmosphere at 37°C. Plaque assays were performed as described previously (16, 33), with the following modifications. Confluent Henle cell monolayers grown in 35-mm-diameter plates were infected with 2 × 104 bacteria. After 60 min of incubation, the medium overlying the Henle cells was removed and replaced with fresh medium plus 0.45% (wt/vol) glucose and 20 μg of gentamicin per ml. The cells were then incubated for 72 h.
Competition assay.
Henle cell monolayers were infected with a mixture of equal numbers of SM166 Lac and SM100 or SM166 and SM100 Lac bacteria. The exact ratio of the two strains in each experiment was determined by plating dilutions of the inoculum on agar medium with X-Gal and counting the Lac+ and Lac− colonies. After 72 h of incubation, the infected monolayers were detached with 0.025% (wt/vol) trypsin and lysed with 0.5% (wt/vol) sodium deoxycholate as described previously (16) to recover the intracellular bacteria. Serial dilutions of the recovered bacteria were plated on TSB agar with X-Gal, and the ratio of SM100 to SM166 bacteria recovered was determined by counting the Lac+ and Lac− colonies. The competitive index was calculated as the ratio of SM166 to SM100 bacteria recovered from the cells divided by the ratio of SM166 to SM100 bacteria in the inoculum.
Mouse virulence.
Eight-week-old BALB/cJ female mice weighing approximately 25 g (Jackson Laboratory, Bar Harbor, ME) were sedated by intramuscular injection of a mixture of xylazine hydrochloride (40 mg/kg) (Rompun; Mobay Corp., Shawnee, KS) and ketamine hydrochloride (12 mg/kg) (Ketaset; Aveco Co., Fort Dodge, IA) in 50 μl of saline. Groups of five mice were inoculated with 30 μl of a suspension containing 107 wild-type or mutant S. flexneri bacteria, which was applied dropwise to the external nares of each mouse with a 100-μl Hamilton syringe. Mice were observed for 10 days for deaths. Weight loss and rebound were recorded. Three weeks after the initial infection, surviving mice were challenged intranasally with a lethal dose of S. flexneri serotype 2a strain 2457T (107 CFU/30 μl). The mouse challenge dose was prepared from a frozen lot of S. flexneri serotype 2a bacteria that had been harvested during the log phase of growth and then stored in liquid nitrogen.
RESULTS
Sit iron transport.
While the Salmonella Sit system has been shown to be a ferrous iron transporter (20), the iron ligand preference of the related S. flexneri Sit system has not been determined experimentally. Our previous data indicated that expression of the S. flexneri sit genes is induced, and it contributes to intracellular growth most effectively, under aerobic conditions (3) where iron should normally be in the ferric form. Thus, while the S. flexneri genes were classified as sit genes based on amino acid identity (64 to 77%) of the predicted gene products to the Salmonella Sit proteins (48), there could be differences in the structure and function of the proteins and in the metal ligands they transport. Therefore, we determined the uptake of ferric and ferrous iron by SM193w carrying sitABCD cloned into a low-copy-number vector, pWKS30. SM193w has mutations in iucD, sitA, and feoB and does not grow or transport significant amounts of iron in the absence of added siderophore or expression of a cloned iron transport system (48, 62). Thus, there is minimal background iron transport in this strain. Transport by the Sit system was measured in the presence or absence of ascorbate, which keeps the iron in the reduced ferrous form (Fig. 1). There was minimal difference in the uptake of radiolabeled iron in the presence or absence of the sit genes when no ascorbate was present in the transport buffer. This indicates that Sit did not enhance the uptake of ferric iron. In contrast, the sit genes greatly increased the transport of iron when ascorbate was present, showing that the Sit system is a ferrous iron transporter.
FIG. 1.
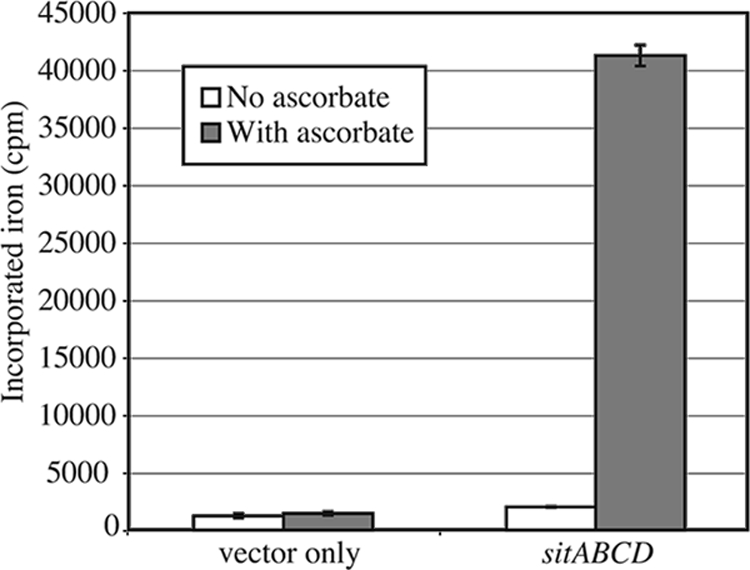
The S. flexneri Sit system transports ferrous iron. S. flexneri strain SM193w carrying either plasmid vector pWKS30 or the cloned sitABCD genes (pEG3) was grown to late exponential phase. The transport of 55Fe was measured as described in Materials and Methods after 5 min of incubation in transport buffer with or without supplementation with 5 mM sodium ascorbate. The data shown are the average of three experiments, and the error bars represent 1 standard deviation.
Genomic location and organization of the sit genes.
The sit genes were present in the chromosomes of all of the Shigella strains that we tested (48), but as noted by Sabri et al. (49), the genes had different locations in different strains. Therefore, we examined the sequenced genomes of additional Shigella strains to determine the location and organization of the sit genes (Fig. 2). The genes are found within islands and are flanked by a variety of insertion sequences and phage-like genes. The locations of the islands relative to the E. coli K-12 genome are different, even in closely related strains such as the two sequenced S. flexneri serotype 2a strains 2457T and 301 (Fig. 2). It is not clear whether the genes were acquired by horizontal transmission multiple times in Shigella lineages or whether the genes have moved in some strains subsequent to their acquisition. In the E. coli/Shigella group, the Sit island insertions fall into five clusters. The uropathogenic and avian pathogenic E. coli strains have the island at a common position, compared to the E. coli K-12 genome, but the sizes of the islands and the orientation of the sit operon within the islands differ (Fig. 2). E. coli SMS-3-5, an environmental isolate that has numerous virulence factors and multiple drug resistance genes (8), has two copies of the sit genes (Fig. 2). One copy is on the chromosome at a unique site, while the other is on a plasmid. A plasmid-encoded Sit system has also been noted in an avian pathogenic E. coli strain (18, 49). There also is considerable variation in the sizes of the islands and the orientation of the genes among sequenced Shigella isolates (Fig. 2). Two of the S. flexneri serotype 2a strains, 2457T and SA100, have the island adjacent to the S. flexneri homolog of E. coli K-12 aspS, a site 500 kb away and in the opposite orientation from the other Shigella isolates. As described by Chen and Schneider (7), comparison of the sit regions in S. flexneri 2457T and 301 suggests that there may have been rearrangements subsequent to the acquisition of the sit island in the ancestral strain, since both strains have the same insertion, which includes ipaH, homologs of recE and intR, and the sit genes (Fig. 3). The uniform presence of the sit genes in Shigella strains, despite the apparent instability of the islands, suggests a selective advantage for their retention.
FIG. 2.
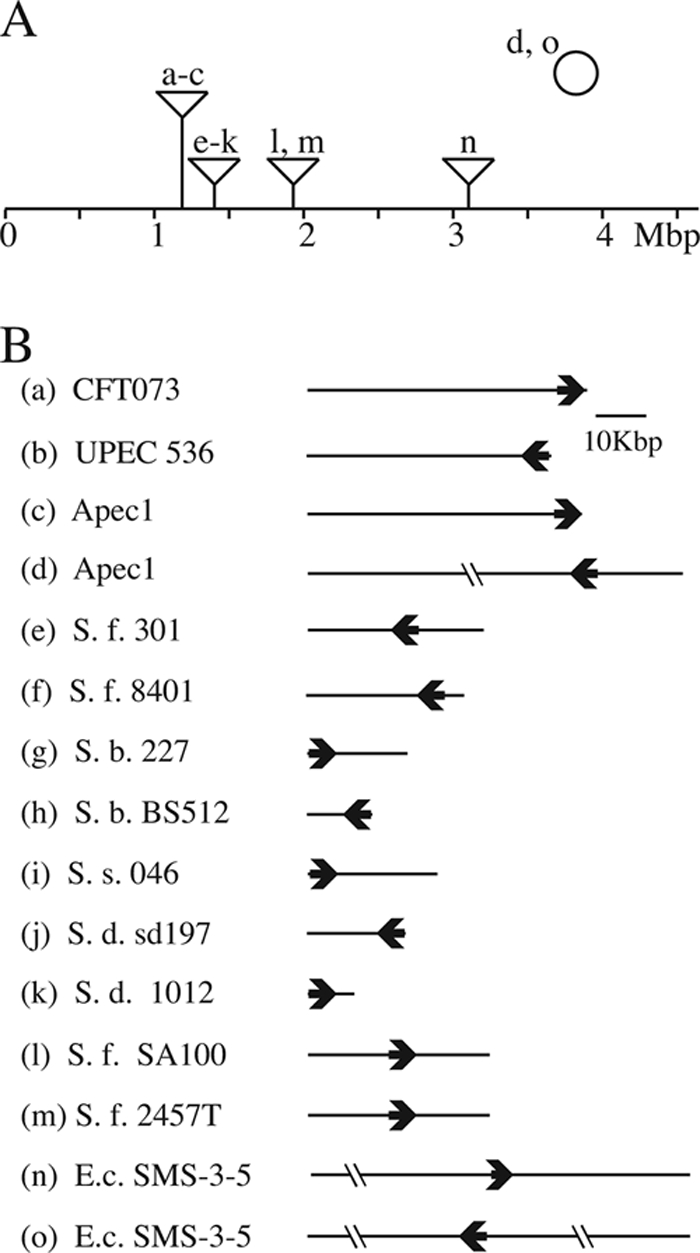
(A) PAI containing sit islands in E. coli and Shigella. The line indicates the E. coli K-12 chromosome with the relative positions of the Sit islands for each sequenced strain shown above the line. Strains a to n contain genomic islands, while strains d and o also have sit genes located on a plasmid, indicated by the circle above the chromosome. The values below the line are distances in megabase pairs. (B) The Sit islands from specific strains are shown, with the length of the line indicating the size of the island and the bold arrow indicating the relative location and direction of transcription of the sitABCD genes. Lines crossed by the short double lines are not to scale. Abbreviations: UPEC, uropathogenic E. coli; Apec, avian pathogenic E. coli; S. f., S. flexneri; S. b., S. boydii; S. s., S. sonnei; S. d., S. dysenteriae; E.c., E. coli.
FIG. 3.

Sit island in S. flexneri SA100 and 2457T. The ORFs within the island are indicated by arrows, and the insertion elements are shown as rectangles. aspS and yecN are the genes with homology to E. coli K-12 that flank the island. The positions of the two (three in SA100) tandem T7 promoters and the insertion sequences are shown. The locations of the deletions in six T7 polymerase-resistant colonies are shown above the map. The approximate endpoints of these deletions were determined by PCR analysis and are indicated by the filled circles. The dashed lines indicate the deleted regions. Three of the clones had the same deletion profile. Below the map are the locations of the fragments of SA100 DNA that were tested for conferring sensitivity to T7 polymerase in E. coli. The arrows on the right of some of these lines indicate that those clones extended past the region shown on the map.
Instability of the sit island.
To measure the stability of the sit island, we took advantage of the fact that the sit genes in S. flexneri serotype 2a strains 2457T and SA100 were linked to tandem repeats of a sequence with a perfect match to a strong T7 polymerase promoter (7). We observed that expression of T7 polymerase in these strains is lethal and that a majority of the T7 polymerase-resistant colonies had deletions in the sit PAI, as determined by DNA hybridization to Shigella genomic microarrays (data not shown). To understand the basis of this lethality, cosmid pEG1, containing much of the sit PAI, was mated into E. coli strain HB101 and found to cause lethality when T7 polymerase was induced from the lacUV5 promoter on plasmid pAR1219 (Fig. 4). Various subclones obtained from pEG1 were tested in this assay, and only fragments containing the T7 promoter repeats caused lethality (Fig. 3). The shortest fragment had no open reading frame (ORF) downstream of the T7 promoters and only a portion of the integrase ORF upstream of the promoters. This, combined with the observation that lethality was only observed upon induction of the T7 polymerase genes, indicates that it is the expression of the T7 promoters themselves, and not the expression of a specific gene, that is responsible for cell death. The mechanism of killing is not known, although it is possible that the high level of activity of these strong promoters leads to depletion of nucleotide pools within the cell.
FIG. 4.
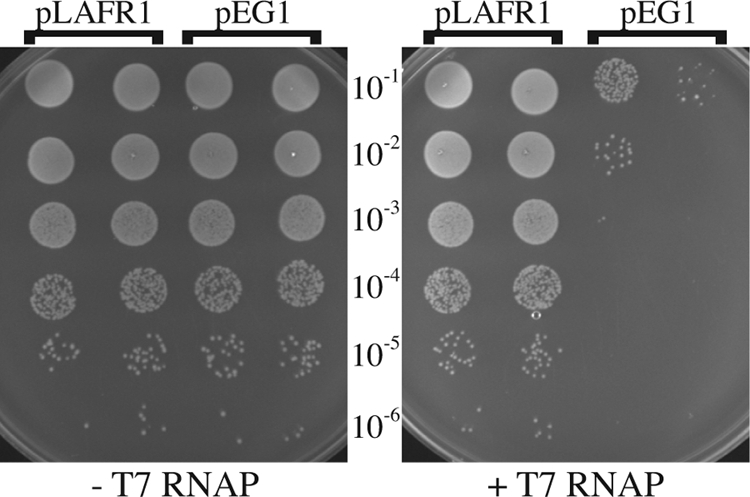
E. coli carrying cloned S. flexneri T7 promoters grown in the presence or absence of IPTG. HB101/pAR1219 containing the vector (pLAFR1) or the cloned sit island (pEG1) was plated on L agar in the presence (+T7 RNAP) or absence (−T7 RNAP) of 500 μM IPTG to induce the expression of T7 polymerase from pAR1219. Ten-microliter spots of the indicated dilutions of fully grown broth cultures were placed on the surfaces of the plates and incubated overnight.
In S. flexneri, sensitivity to T7 polymerase also correlated with deletion of the promoter repeats, and PCR analysis showed that resistant isolates had deletions of the T7 promoters (Table 2 and Fig. 3). To determine the frequency of deletions, the numbers of colonies in the presence and absence of T7 induction were compared (Table 2). The frequency of deletion in either the wild-type strain or a derivative carrying a chloramphenicol resistance cassette in the sit operon was approximately 10−4. Additional PCR analysis showed that many of the T7 promoter deletions included a large portion of the PAI containing the sit genes (Table 2 and Fig. 3).
TABLE 2.
Frequency of T7 RNA polymerase resistance and loss of sitA in RecA+ and RecA− S. flexneri
| Strain (genotype) | Avg frequency of T7 polymerase resistancea | Range | % of resistant isolates with deletion of:
|
|
|---|---|---|---|---|
| T7 promotersb | sit genesc | |||
| SM100/pAR1219 (wild type) | 2.7 × 10−4 | 1.0 × 10−4-4.1 × 10−4 | 100 | 75 |
| SM166/pAR1219 (sit::cam) | 8.9 × 10−3 | 1.6 × 10−3-5.6 × 10−4 | 100 | 60 |
| SM160/pAR1219 (recA::cam) | 2.9 × 10−5 | 2.0 × 10−5-5.7 × 10−6 | 0 | 0 |
Strains were grown in LB broth and plated on medium containing IPTG to induce expression of T7 polymerase from pAR1219. The frequency is the ratio of colonies on IPTG-containing medium to the number of colonies from the same culture plated on medium without IPTG. The average and range of values for 4 to 10 separate experiments are shown.
A minimum of 10 individual colonies was selected and tested by PCR with primers in the T7 promoters.
Individual colonies were selected and tested by PCR with primers in the sitAB genes (SM100 and SM160) or by plating on chloramphenicol (SM166) to determine the presence or absence of the sit operon.
Mapping of several of the deletions showed that they varied in size and were not simply excisions of the entire island (Fig. 3). The large number of insertion sequences and rearrangements within the islands made it difficult to determine the precise endpoints of the deletions, but it appeared that they might result from recombination between two copies of the same insertion sequence. To verify that the deletions occurred by homologous recombination, rather than a site-specific mechanism, the deletion frequencies in RecA+ and RecA− strains were compared. The frequency of T7 polymerase resistance was approximately 10-fold lower in the RecA− strain (Table 2), suggesting that these deletions within the island occur by homologous recombination. T7-resistant colonies from each strain were picked and tested for the presence of the tandem T7 promoters by PCR. The resistant colonies that were obtained in the wild-type strain lacked the T7 promoters, while those in the RecA− strain did not have deletions of the T7 promoter repeats (Table 2). Colonies were also tested for deletions in the sit region with primers within sitAB. The majority of the resistant mutants from the wild type had sit deletions, while no deletions were detected in the recA mutant strain (Table 2). Thus, resistance in the recA isolates is due to a different mechanism, such as mutations in plasmid pAR1219, which encodes the T7 polymerase.
Sit promotes intracellular growth and plaque formation.
The presence of the sit genes in all of the clinical isolates we tested, despite the apparent instability of the island, suggested a selective advantage for the sit genes. Further, the fact that the sit genes are predominately found in intracellular pathogens indicated that the advantage might be related to intracellular growth. A comparison of the wild type and a sitA mutant had not shown a major defect in the ability to invade or form plaques in cultured cell monolayers (2, 48), although the sitA mutant formed slightly smaller plaques. To directly compare the growth of the mutant and that of the wild type in the intracellular environment, a competition assay was performed (Fig. 5). Cultured Henle cells were infected with a mixture of equal numbers of wild-type and sitA mutant cells, and following invasion, gentamicin was added to the medium to kill any extracellular bacteria. S. flexneri is naturally LacZ−, and one of the two competing strains was marked by insertion of the E. coli K-12 lacZ gene to allow discrimination of the two strains on agar containing X-Gal. To avoid any possible bias introduced by lacZ, experiments were performed with the lacZ-marked wild type versus the unmarked sit mutant and with the lacZ-marked sit mutant versus the unmarked wild type. The monolayers were incubated for 72 h, at which time plaques were clearly visible. The bacteria were recovered from the infected Henle cells and plated on medium containing X-Gal to differentiate the two strains. The sitA mutant consistently was recovered in lower numbers than the wild type (Fig. 5), indicating that Sit provides a growth advantage in the intracellular environment. It appeared that there might be some bias with respect to the lac marker, as each strain was recovered in slightly lower numbers when it was Lac+. However, the difference between the ratios of SM100 to SM166 Lac+ and SM100 Lac+ to SM166 bacteria (Fig. 5) was not statistically significant (P = 0.24, two-tailed t test). Thus, the presence of the lacZ gene as a marker did not significantly influence the competitive index of the strains.
FIG. 5.
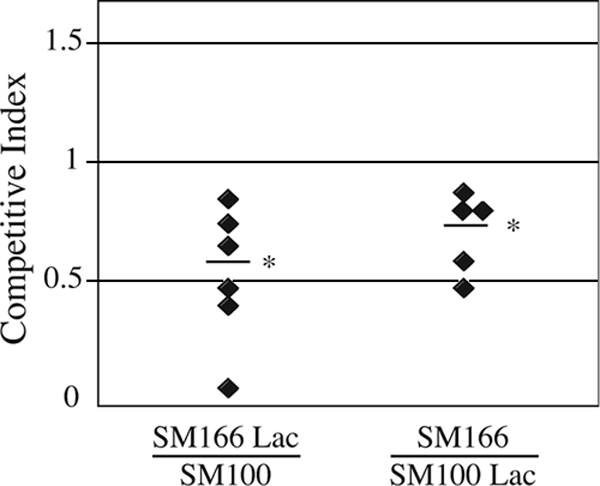
Competition between the wild type and the sitA mutant in a plaque assay. Henle cells were infected with a mixture of equal numbers of wild-type (SM100) and sitA mutant (SM166) S. flexneri bacteria and incubated in the presence of gentamicin to kill extracellular bacteria. In each set of experiments, one of the two strains was marked with lacZ. Bacteria were harvested from the plaques after 72 h of incubation, and the competitive index (ratio of sitA mutant to wild-type bacteria recovered) was determined. Each symbol represents the competitive index in a single experiment. The average competitive index is shown by a horizontal line. The asterisks indicate that the P value (two-tailed t test) for the difference between the observed competitive index and an expected index of 1.0 is <0.02 for SM166 Lac/SM100 and <0.04 for SM166/SM100 Lac.
Sit is required for virulence in the mouse lung model.
To further determine whether Sit plays a role in the virulence of S. flexneri, mice were infected with the wild type or the sitA mutant in a lung model of Shigella virulence (56) (Table 3). In this model, both the wild type and the streptomycin-resistant parent of the mutants were virulent; all five mice in each group died. In contrast, the sitA mutant was attenuated (Table 3). Only one of the five mice died, and the other four recovered within 10 days. The aerobactin (iucD) mutant, which had previously been shown to be indistinguishable from the wild type in plaque and Sereny (37) assays, was virulent in this model. Four of five mice inoculated with the iucD mutant died. This is consistent with our observation that the aerobactin genes are repressed during intracellular growth and do not play a significant role in iron acquisition after the invasion of epithelial cells (15, 21).
TABLE 3.
Virulence of wild type and iron transport mutants of S. flexneri in the mouse lung model
| S. flexneri strain | Primary challenge
|
No. of survivors/total after back challenge with S. flexneri serotype 2a strain 2457T | |
|---|---|---|---|
| Time of death | No. of survivors/total | ||
| SA100 (wild type) | All dead by day 9 | 0/5 | NDa |
| SM100 (Strr) | All dead by day 4 | 0/5 | ND |
| SA240 (iucD) | Last death on day 6 | 1/5 | 1/1 |
| SM166 (sitA) | Last death on day 5 | 4/5 | 4/4 |
| None | 1/5 | ||
ND, not done.
To ensure that the mice that survived infection with the sit mutant had been successfully inoculated with the mutant strain, a back challenge was performed (Table 3). Surviving mice were challenged with a lethal dose of the wild-type serotype 2a strain 21 days after the initial infection. All of the mice that had initially received the sitA strain survived a challenge with the wild type, while most of the mice (four of five) that had not previously been infected succumbed to this dose. Therefore, the mice that survived infection with the sit mutant had been successfully inoculated and had mounted an effective immune response that prevented reinfection. Similarly, the one animal that survived infection with the iucD mutant (SA240) was also protected against a subsequent challenge.
DISCUSSION
Shigella spp. have a variety of iron transport systems, but three of these, Feo, Sit, and a siderophore, are common to all of the clinical isolates examined. Differences were noted in the type of siderophore produced. Enterobactin, salmochelin, aerobactin, or combinations of these were found in different strains. This suggests that siderophore synthesis is important in some stage of the Shigella lifestyle but virulence is not dependent on the production of any particular siderophore. In contrast, studies of other pathogenic E. coli strains have shown that aerobactin production is advantageous in certain pathogens (28, 55, 59).
The Sit system, which was first described in Salmonella (63) and was shown to be required for full virulence in this pathogen (17), is primarily present in bacterial species that invade and multiply within host cells. In Salmonella, Sit has a higher affinity for manganese than for iron and is thought to be primarily a manganese transporter under physiological conditions (20). Similarly, the S. flexneri Sit system transports both iron and manganese (46) but functions well to provide iron when the bacteria are growing in the host cell cytoplasm (48). An S. flexneri mutant that lacks all iron transporters other than Sit grows normally in the intracellular environment and produces wild-type-size plaques in Henle cells in an aerobic environment. The sit mutant also grew intracellularly, presumably using the Feo and Iuc systems for iron acquisition. However, the plaques were slightly smaller, and in this work, we confirm that the sit mutant was at a competitive disadvantage by coinfecting the monolayers with the wild type and the sit mutant and directly comparing them for growth and intercellular spreading in a plaque assay. The wild type outcompeted the mutant and was recovered in a higher proportion from the infected monolayers. Further, the mutant was much less virulent in a mouse lung model of infection. In this model, the bacteria must be able to infect mice, invade lung epithelial cells, and provoke an inflammatory response. The mutant was able to infect the mice, since the survivors mounted an immune response and were resistant to challenge with the wild type, but the mutant was defective at some stage of invasion and intracellular replication. These data also indicate that a sit mutation may be appropriate to include in vaccine strains, since loss of sit causes attenuation and the sit mutant induces protective immunity.
It is unclear why the Sit system, a ferrous iron transporter, would be induced aerobically and repressed under anaerobic conditions, where ferrous iron should be more available. This regulation may reflect its dual role in iron and manganese transport, allowing increased transport of manganese, and consequently iron, when the bacteria are growing aerobically. Since the Sit system was sufficient for iron acquisition under aerobic conditions, it suggests that there is an accessible pool of ferrous iron in the presence of oxygen within the host cell cytoplasm. The cytoplasm is a reducing environment (44) which likely keeps a portion of the small pool of accessible iron in the ferrous form, even in the presence of oxygen. This is consistent with our earlier observation that the Sit system provided iron to intracellular bacteria when the cultured cells were growing aerobically but not in an anaerobic environment (3).
Both the Henle cell competition assay and the mouse virulence results indicate that the presence of Sit provides a selective advantage in the host. This is supported by the maintenance of this locus and the conservation of sit genes in virulent strains, despite the relatively high frequency of deletion. It also appears that the locus was acquired more than once in the evolution of Shigella since it is located at different sites and with different flanking sequences in various Shigella isolates. The Sit transport system is also present in enteroinvasive E. coli (48), which are derived from several ancestral lineages distinct from the Shigella strains (45). This suggests a selective advantage for the sit genes in enteroinvasive E. coli and Shigella rather than the continued presence of the genes in strains derived from a common ancestor. A number of the Sit islands contain phage-like genes, including integrases, and there are T7 promoters in the S. flexneri serotype 2a strains, suggesting phage-mediated acquisition of the islands. These T7 promoters are associated with lethality when T7 RNA polymerase is expressed within the cell.
Chen and Schneider (7) had shown that S. flexneri 2457T has an additional copy of the T7 promoter sequences in its genome, but its similarity to the T7 promoter consensus is weak, whereas the repeated sequences in the Sit island are a near-perfect match to the promoter consensus. The T7 polymerase-resistant mutants presumably still have this copy since the Sit island deletion was the only deletion detected by hybridization to microarrays representing all of the genes in the S. flexneri 2457T and E. coli K-12 genomes. The weaker match to the T7 promoter apparently makes it less deleterious to S. flexneri when T7 polymerase is expressed in the cell, and loss of the T7 promoter tandem repeats in the Sit island is sufficient for resistance to the polymerase.
The observation that RecA is required for deletion of the island suggests that the deletions are caused by homologous recombination rather than being the result of T7 polymerase expression. Thus, the presence of the tandem T7 promoters within the island in these S. flexneri strains provides a fortuitous marker for the presence of the island, allowing us to use T7 polymerase resistance to select for bacteria from which the Sit PAI has been deleted and to show that the region can be deleted at relatively high frequency.
These data indicate that S. flexneri Sit is a ferrous iron transporter that enhances the growth of the bacteria within cultured epithelial cells and the production of disease in a mouse lung model. The ability of the sit genes to promote in vivo survival is likely responsible for the retention of these genes in clinical isolates.
Acknowledgments
This work was supported by grant AI16935 from the National Institutes of Health.
We thank Jia-Wen Jessica Chang for technical assistance, Laura Runyen-Janecky and Enrique Gonzalez for construction of SM160 and pEG3, and Erin Murphy for helpful discussions.
Editor: A. J. Bäumler
Footnotes
Published ahead of print on 16 March 2009.
REFERENCES
- 1.Bagg, A., and J. B. Neilands. 1987. Ferric uptake regulation protein acts as a repressor, employing iron (II) as a cofactor to bind the operator of an iron transport operon in Escherichia coli. Biochemistry 265471-5477. [DOI] [PubMed] [Google Scholar]
- 2.Boulette, M. L. 2007. Shigella flexneri ArcA and FNR regulate iron acquisition and contribute to plaque formation under anaerobic conditions. Ph.D. dissertation. University of Texas, Austin.
- 3.Boulette, M. L., and S. M. Payne. 2007. Anaerobic regulation of Shigella flexneri virulence: ArcA regulates Fur and iron acquisition genes. J. Bacteriol. 1896957-6967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carbonetti, N. H., and P. H. Williams. 1984. A cluster of five genes specifying the aerobactin iron uptake system of plasmid ColV-K30. Infect. Immun. 467-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cartron, M. L., S. Maddocks, P. Gillingham, C. J. Craven, and S. C. Andrews. 2006. Feo—transport of ferrous iron into bacteria. Biometals 19143-157. [DOI] [PubMed] [Google Scholar]
- 6.Chang, A. C., and S. N. Cohen. 1978. Construction and characterization of amplifiable multicopy DNA cloning vehicles derived from the P15A cryptic miniplasmid. J. Bacteriol. 1341141-1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen, Z., and T. D. Schneider. 2006. Comparative analysis of tandem T7-like promoter containing regions in enterobacterial genomes reveals a novel group of genetic islands. Nucleic Acids Res. 341133-1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Colonna, B., M. Nicoletti, P. Visca, M. Casalino, P. Valenti, and F. Maimone. 1985. Composite IS1 elements encoding hydroxamate-mediated iron uptake in FIme plasmids from epidemic Salmonella spp. J. Bacteriol. 162307-316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Darling, A. E., T. J. Treangen, X. Messeguer, and N. T. Perna. 2007. Analyzing patterns of microbial evolution using the mauve genome alignment system. Methods Mol. Biol. 396135-152. [DOI] [PubMed] [Google Scholar]
- 10.Davanloo, P., A. H. Rosenberg, J. J. Dunn, and F. W. Studier. 1984. Cloning and expression of the gene for bacteriophage T7 RNA polymerase. Proc. Natl. Acad. Sci. USA 812035-2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Earhart, C. F. 2004. Iron uptake via the enterobactin system, p. 133-146. In J. H. Crosa, A. R. Mey, and S. M. Payne (ed.), Iron transport in bacteria. ASM Press, Washington, DC.
- 12.Friedman, A. M., S. R. Long, S. E. Briwn, W. J. Buikema, and F. Ausubel. 1982. Construction of a broad host range cosmid cloning vector and its uses in the genetic analysis of Rhizobium meliloti. Gene 18289-296. [DOI] [PubMed] [Google Scholar]
- 13.Hantke, K., G. Nicholson, W. Rabsch, and G. Winkelmann. 2003. Salmochelins, siderophores of Salmonella enterica and uropathogenic Escherichia coli strains, are recognized by the outer membrane receptor IroN. Proc. Natl. Acad. Sci. USA 1003677-3682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Headley, V., M. Hong, M. Galko, and S. M. Payne. 1997. Expression of aerobactin genes by Shigella flexneri during extracellular and intracellular growth. Infect. Immun. 65818-821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Headley, V. L., and S. M. Payne. 1990. Differential protein expression by Shigella flexneri in intracellular and extracellular environments. Proc. Natl. Acad. Sci. USA 874179-4183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hong, M., Y. Gleason, E. E. Wyckoff, and S. M. Payne. 1998. Identification of two Shigella flexneri chromosomal loci involved in intercellular spreading. Infect. Immun. 664700-4710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Janakiraman, A., and J. M. Slauch. 2000. The putative iron transport system SitABCD encoded on SPI1 is required for full virulence of Salmonella typhimurium. Mol. Microbiol. 351146-1155. [DOI] [PubMed] [Google Scholar]
- 18.Johnson, T. J., K. E. Siek, S. J. Johnson, and L. K. Nolan. 2006. DNA sequence of a ColV plasmid and prevalence of selected plasmid-encoded virulence genes among avian Escherichia coli strains. J. Bacteriol. 188745-758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kammler, M., C. Schon, and K. Hantke. 1993. Characterization of the ferrous iron uptake system of Escherichia coli. J. Bacteriol. 1756212-6219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kehres, D. G., A. Janakiraman, J. M. Slauch, and M. E. Maguire. 2002. SitABCD is the alkaline Mn2+ transporter of Salmonella enterica serovar Typhimurium. J. Bacteriol. 1843159-3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lawlor, K. M., P. A. Daskaleros, R. E. Robinson, and S. M. Payne. 1987. Virulence of iron transport mutants of Shigella flexneri and utilization of host iron compounds. Infect. Immun. 55594-599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lawlor, K. M., and S. M. Payne. 1984. Aerobactin genes in Shigella spp. J. Bacteriol. 160266-272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marlovits, T. C., W. Haase, C. Herrmann, S. G. Aller, and V. M. Unger. 2002. The membrane protein FeoB contains an intramolecular G protein essential for Fe(II) uptake in bacteria. Proc. Natl. Acad. Sci. USA 9916243-16248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marolda, C. L., M. A. Valvano, K. M. Lawlor, S. M. Payne, and J. H. Crosa. 1987. Flanking and internal regions of chromosomal genes mediating aerobactin iron uptake systems in enteroinvasive Escherichia coli and Shigella flexneri. J. Gen. Microbiol. 1332269-2278. [DOI] [PubMed] [Google Scholar]
- 25.Maurelli, A. T. 2007. Black holes, antivirulence genes, and gene inactivation in the evolution of bacterial pathogens. FEMS Microbiol. Lett. 2671-8. [DOI] [PubMed] [Google Scholar]
- 26.Maurelli, A. T., R. E. Fernandez, C. A. Bloch, C. K. Rode, and A. Fasano. 1998. “Black holes” and bacterial pathogenicity: a large genomic deletion that enhances the virulence of Shigella spp. and enteroinvasive Escherichia coli. Proc. Natl. Acad. Sci. USA 953943-3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McKenzie, G. J., and N. L. Craig. 2006. Fast, easy and efficient: site-specific insertion of transgenes into enterobacterial chromosomes using Tn7 without need for selection of the insertion event. BMC Microbiol. 639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mellata, M., M. Dho-Moulin, C. M. Dozois, R. Curtiss III, P. K. Brown, P. Arne, A. Bree, C. Desautels, and J. M. Fairbrother. 2003. Role of virulence factors in resistance of avian pathogenic Escherichia coli to serum and in pathogenicity. Infect. Immun. 71536-540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mey, A. R., E. E. Wyckoff, L. A. Hoover, C. R. Fisher, and S. M. Payne. 2008. Vibrio cholerae VciB promotes iron uptake via ferrous iron transporters. J. Bacteriol. 1905953-5962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mills, M., and S. M. Payne. 1995. Genetics and regulation of heme iron transport in Shigella dysenteriae and detection of an analogous system in Escherichia coli O157:H7. J. Bacteriol. 1773004-3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moss, J. E., T. J. Cardozo, A. Zychlinsky, and E. A. Groisman. 1999. The selC-associated SHI-2 pathogenicity island of Shigella flexneri. Mol. Microbiol. 3374-83. [DOI] [PubMed] [Google Scholar]
- 32.Neilands, J. B. 1984. Siderophores of bacteria and fungi. Microbiol. Sci. 19-14. [PubMed] [Google Scholar]
- 33.Oaks, E. V., M. E. Wingfield, and S. B. Formal. 1985. Plaque formation by virulent Shigella flexneri. Infect. Immun. 48124-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Payne, S. M. 1980. Synthesis and utilization of siderophores by Shigella flexneri. J. Bacteriol. 1431420-1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Payne, S. M., and R. A. Finkelstein. 1977. Detection and differentiation of iron-responsive avirulent mutants on Congo red agar. Infect. Immun. 1894-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Payne, S. M., D. W. Niesel, S. S. Peixotto, and K. M. Lawlor. 1983. Expression of hydroxamate and phenolate siderophores by Shigella flexneri. J. Bacteriol. 155949-955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Payne, S. M., E. E. Wyckoff, E. R. Murphy, A. G. Oglesby, M. L. Boulette, and N. M. Davies. 2006. Iron and pathogenesis of Shigella: iron acquisition in the intracellular environment. Biometals 19173-180. [DOI] [PubMed] [Google Scholar]
- 38.Perry, R. D., and C. L. San Clemente. 1979. Siderophore synthesis in Klebsiella pneumoniae and Shigella sonnei during iron deficiency. J. Bacteriol. 1401129-1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Philpott, D. J., J. D. Edgeworth, and P. J. Sansonetti. 2000. The pathogenesis of Shigella flexneri infection: lessons from in vitro and in vivo studies. Philos. Trans. R. Soc Lond. B Biol. Sci. 355575-586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pupo, G. M., D. K. Karaolis, R. Lan, and P. R. Reeves. 1997. Evolutionary relationships among pathogenic and nonpathogenic Escherichia coli strains inferred from multilocus enzyme electrophoresis and mdh sequence studies. Infect. Immun. 652685-2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pupo, G. M., R. Lan, and P. R. Reeves. 2000. Multiple independent origins of Shigella clones of Escherichia coli and convergent evolution of many of their characteristics. Proc. Natl. Acad. Sci. USA 9710567-10572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Purdy, G. E., and S. M. Payne. 2001. The SHI-3 iron transport island of Shigella boydii 0-1392 carries the genes for aerobactin synthesis and transport. J. Bacteriol. 1834176-4182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reeves, S. A. 2001. Iron acquisition in the intracellular environment of the host: multiple iron transport systems in Shigella dysenteriae. PhD. dissertation. University of Texas, Austin.
- 44.Rietsch, A., and J. Beckwith. 1998. The genetics of disulfide bond metabolism. Annu. Rev. Genet. 32163-184. [DOI] [PubMed] [Google Scholar]
- 45.Rolland, K., N. Lambert-Zechovsky, B. Picard, and E. Denamur. 1998. Shigella and enteroinvasive Escherichia coli strains are derived from distinct ancestral strains of E. coli. Microbiology 144(Pt. 9)2667-2672. [DOI] [PubMed] [Google Scholar]
- 46.Runyen-Janecky, L., E. Dazenski, S. Hawkins, and L. Warner. 2006. Role and regulation of the Shigella flexneri sit and MntH systems. Infect. Immun. 744666-4672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Runyen-Janecky, L. J., and S. M. Payne. 2002. Identification of chromosomal Shigella flexneri genes induced by the eukaryotic intracellular environment. Infect. Immun. 704379-4388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Runyen-Janecky, L. J., S. A. Reeves, E. G. Gonzales, and S. M. Payne. 2003. Contribution of the Shigella flexneri Sit, Iuc, and Feo iron acquisition systems to iron acquisition in vitro and in cultured cells. Infect. Immun. 711919-1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sabri, M., S. Leveille, and C. M. Dozois. 2006. A SitABCD homologue from an avian pathogenic Escherichia coli strain mediates transport of iron and manganese and resistance to hydrogen peroxide. Microbiology 152745-758. [DOI] [PubMed] [Google Scholar]
- 50.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 51.Sansonetti, P. J. 2006. The bacterial weaponry: lessons from Shigella. Ann. N. Y. Acad. Sci. 1072307-312. [DOI] [PubMed] [Google Scholar]
- 52.Schmitt, M. P., and S. M. Payne. 1991. Genetic analysis of the enterobactin gene cluster in Shigella flexneri. J. Bacteriol. 173816-825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schwyn, B., and J. B. Neilands. 1987. Universal chemical assay for the detection and determination of siderophores. Anal. Biochem. 16047-56. [DOI] [PubMed] [Google Scholar]
- 54.Torres, A. G., and S. M. Payne. 1997. Haem iron-transport system in enterohaemorrhagic Escherichia coli O157:H7. Mol. Microbiol. 23825-833. [DOI] [PubMed] [Google Scholar]
- 55.Torres, A. G., P. Redford, R. A. Welch, and S. M. Payne. 2001. TonB-dependent systems of uropathogenic Escherichia coli: aerobactin and heme transport and TonB are required for virulence in the mouse. Infect. Immun. 696179-6185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Turbyfill, K. R., R. W. Kaminski, and E. V. Oaks. 2008. Immunogenicity and efficacy of highly purified invasin complex vaccine from Shigella flexneri 2a. Vaccine 261353-1364. [DOI] [PubMed] [Google Scholar]
- 57.Vokes, S. A., S. A. Reeves, A. G. Torres, and S. M. Payne. 1999. The aerobactin iron transport system genes in Shigella flexneri are present within a pathogenicity island. Mol. Microbiol. 3363-73. [DOI] [PubMed] [Google Scholar]
- 58.Wang, R. F., and S. R. Kushner. 1991. Construction of versatile low-copy-number vectors for cloning, sequencing and gene expression in Escherichia coli. Gene 100195-199. [PubMed] [Google Scholar]
- 59.Warner, P. J., P. H. Williams, A. Bindereif, and J. B. Neilands. 1981. ColV plasmid-specific aerobactin synthesis by invasive strains of Escherichia coli. Infect. Immun. 33540-545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wei, J., M. B. Goldberg, V. Burland, M. M. Venkatesan, W. Deng, G. Fournier, G. F. Mayhew, G. Plunkett III, D. J. Rose, A. Darling, B. Mau, N. T. Perna, S. M. Payne, L. J. Runyen-Janecky, S. Zhou, D. C. Schwartz, and F. R. Blattner. 2003. Complete genome sequence and comparative genomics of Shigella flexneri serotype 2a strain 2457T. Infect. Immun. 712775-2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wyckoff, E. E., D. Duncan, A. G. Torres, M. Mills, K. Maase, and S. M. Payne. 1998. Structure of the Shigella dysenteriae haem transport locus and its phylogenetic distribution in enteric bacteria. Mol. Microbiol. 281139-1152. [DOI] [PubMed] [Google Scholar]
- 62.Wyckoff, E. E., A. R. Mey, A. Leimbach, C. F. Fisher, and S. M. Payne. 2006. Characterization of ferric and ferrous iron transport systems in Vibrio cholerae. J. Bacteriol. 1886515-6523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhou, D., W. D. Hardt, and J. E. Galan. 1999. Salmonella typhimurium encodes a putative iron transport system within the centisome 63 pathogenicity island. Infect. Immun. 671974-1981. [DOI] [PMC free article] [PubMed] [Google Scholar]