

Abstract
The well-conserved protein Hfq has emerged as the key modulator of riboregulation in bacteria. This protein is thought to function as an RNA chaperone and to facilitate base pairing between small regulatory RNA (sRNA) and mRNA targets, and many sRNAs are dependent on the Hfq protein for their regulatory functions. To address the possible role of Hfq in riboregulated circuits in Neisseria meningitidis, we generated an Hfq mutant of the MC58 strain, and the knockout mutant has pleiotropic phenotypes; it has a general growth phenotype in vitro in culture media, and it is sensitive to a wide range of stresses, including those that it may encounter in the host. Furthermore, the expression profile of a vast number of proteins is clearly altered in the mutant, and we have identified 27 proteins by proteomics. All of the phenotypes tested to date are also restored by complementation of Hfq expression in the mutant strain. Importantly, in ex vivo and in vivo models of infection the Hfq mutant is attenuated. These data indicate that Hfq plays a key role in stress response and virulence, and we propose a major role for Hfq in regulation of gene expression. Moreover, this study suggests that in meningococcus there is a large Hfq-mediated sRNA network which so far is largely unexplored.
Hfq is a well-conserved RNA binding protein which was originally identified in Escherichia coli as a host factor required for the replication of Qβ bacteriophage (14). It shares structural and functional homology with the Sm proteins in eukaryotes, which have central roles in RNA metabolism (42). It has more recently been described as a pleiotropic regulator that modulates the stability or translation of an increasing number of mRNAs (for reviews, see references 1, 6, and 62). Its role as a key mediator in many small regulatory RNA (sRNA) circuits has made it the focus of many well-studied systems. Most of our present knowledge of sRNAs has resulted from recent global search studies involving screening the genomes of certain organisms for novel sRNA genes through bioinformatic and comparative analyses and also experimental approaches (reviewed in references 2 and 64), and these studies have lead to the identification of over 80 new noncoding sRNAs in E. coli, many of which are conserved in closely related pathogens. Although the functional role of the majority of these sRNAs is unknown, those that have been characterized in detail regulate various cellular functions, including iron homeostasis, quorum sensing, virulence, metabolism, and adaptation to stresses such as envelope stress, oxidative stress, stationary phase, and other stresses (3, 9, 16, 27, 31, 44, 45, 52, 61, 63, 65).
Most sRNAs characterized to date act as posttranscriptional regulators by interacting with specific mRNA targets, modulating message stability, and/or altering accessibility to the translational machinery, and a large number of the sRNAs which act in this way have been shown to require the Hfq protein as a key mediator of the interaction with their targets (1, 15, 29, 52). This interaction occurs through imperfect stretches of base pairing, allowing recognition of multiple targets via different short, exposed segments of each trans-encoded sRNA. In most cases, these Hfq-mediated interactions have an inhibitory effect on protein expression from the target mRNA. Hfq has been proposed to be an RNA chaperone in that it binds to and often changes the secondary structures of sRNAs and/or their mRNA targets (52) in order to facilitate RNA-RNA base pairing. Recent studies revealed that Hfq accelerates strand exchange and subsequent annealing between an sRNA and its target mRNA (4). Alternatively, Hfq may increase the local concentration of the sRNA and target, thereby promoting stable duplex formation (7). Numerous sRNAs have been shown to require this protein for their own stability (32, 38, 39, 50). In addition to regulating mRNA stability or translation in an sRNA-dependent manner, Hfq has been reported to modulate the half-life of some mRNAs directly (66) or by stimulating their poly(A) adenylation (20, 37). Given the riboregulatory functions of the Hfq protein, it is not surprising that knockout mutants often have pleiotropic phenotypes. Hfq was found to be required in E. coli and Salmonella for efficient translation of rpoS mRNA encoding the general stress sigma factor σS (8, 40). In many bacteria, inactivation of the hfq gene results in sensitivity to a number of environmental stresses and alters the synthesis of many proteins, including outer membrane porins (40, 49, 60, 61). Importantly, Hfq influences the fitness and virulence of many pathogenic bacteria. Mutants lacking Hfq are often sensitive to host defense mechanisms and highly attenuated in animal models (12, 26, 34, 49, 43, 51). In contrast, in Staphylococcus aureus, an Hfq null mutant was reported to have no detectable phenotype (5), and although sRNA-mRNA interactions in S. aureus are decisive for regulation of gene expression, the authors concluded that they do not require the RNA chaperone protein Hfq.
Neisseria meningitidis is an important gram-negative bacterium that colonizes the nasopharynx of approximately 5 to 10% of the human population and, for reasons not entirely understood, in a small number of cases results in invasive meningococcal disease leading to sepsis and meningitis (46, 58). Although phylogenetic studies have shown that an hfq gene is present in over one-half of all the bacterial genomes currently sequenced (54), in many systems, such as Neisseria spp., the role of this protein has never been addressed. Furthermore, unlike genes encoding regulatory proteins, noncoding regulatory sRNAs have not been readily identified and annotated in the bacterial genome sequences that are available in the databases, and in many systems, including Neisseria spp., their existence is still largely unexplored.
In this study we address the role of Hfq as a pleiotropic riboregulator in N. meningitidis. Characterization of a knockout null mutant and a complemented derivative of this mutant showed that inactivation of the hfq gene has profound effects on a variety of phenotypes. The global protein profile of the mutant is significantly altered, and identification of some proteins whose expression levels are differentially altered in the Hfq mutant reveals possible targets for the riboregulation. The data indicate that Hfq plays a major role in the stress response and also indicate a major role for Hfq in regulation of gene expression. Moreover, this study suggests that in meningococcus there is a large Hfq-mediated sRNA network that so far is unexplored.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
The meningococal strains used in this study are all derivatives of the N. meningitidis MC58 sequenced strain (56). N. meningitidis strains were routinely cultured in GC-based (Difco) agar medium supplemented with Kellogg's supplement I (25) at 37°C in a 5% CO2-95% air atmosphere with 95% humidity. Stock preparations of strains in 10% skim milk were stored at −80°C. Each bacterial manipulation was started using an overnight culture of a frozen stock. For liquid cultures N. meningitidis strains were grown overnight on solid medium, and cells were resuspended in phosphate-buffered saline (PBS) to an optical density at 600 nm (OD600) of 1 and inoculated into broth medium at a 1:100 dilution. GC (Difco) broth supplemented with Kellogg's supplement I and 12.5 μM Fe(NO3)3 and Mueller-Hinton (Sigma, St. Louis, MO) broth supplemented with 0.2% glucose were used for liquid cultures; when required, erythromycin, chloramphenicol, and kanamycin were added to final concentrations of 5 μg/ml, 5 μg/ml, and 100 μg/ml, respectively. E. coli cultures were grown in Luria-Bertani medium, and, when required, ampicillin was added to a final concentration of 100 μg/ml.
Construction of plasmids and knockouts.
DNA manipulations were carried out routinely as described previously using standard laboratory methods (47). In order to knock out the hfq gene in the MC58 background, the pΔhfqko:Cm plasmid was constructed. Upstream and downstream flanking regions of the hfq gene were amplified by PCR with primers Hfq-1 and Hfq-2 and primers Hfq-3 and Hfq-4 (Table 1), respectively. Then in a second round of PCR the upstream and downstream fragments, which contained regions of overlap due to the design of the primers, were used for self-priming PCR amplification for five cycles, and the united fragment was amplified using external primers Hfq-1 and Hfq-4. The product was cloned into the pGEM-T (Promega) vector, and a chloramphenicol cassette from pDT2548 (67) was inserted into the BamHI site between the flanking regions, generating pΔhfqko:Cm. This plasmid was then linearized and used for transformation of the MC58 and 2996 strains to make hfq knockout mutants Δhfq and 2996Δhfq, respectively. The correct double homologous recombination event resulting in knockout of the gene was verified by PCR. For complementation of the knockout, a plasmid consisting of the ermAM erythromycin resistance genes and the Ptac promoter along with the lacI repressor for isopropyl-β-d-thiogalactopyranoside (IPTG)-inducible expression of the gene of interest, flanked by upstream and downstream regions for allelic replacement, was generated by replacing the 3.4-kb NotI-BamHI fragment carrying the promoterless lacZ gene from pSL-Fla-Ery (23) with a 1.6-kb NotI-BamHI fragment carrying the lacI repressor gene and the Ptac promoter from plasmid pPindCrgA (23), generating pFlaEry-Pind (Table 2). The hfq coding region was amplified with primers Hfq-F and Hfq-R-Ns and cloned as a 303-bp NdeI-NsiI fragment downstream of the Ptac promoter, generating pFlaPind-Hfq, so that expression of the hfq gene in the complementation construct was induced in an IPTG-inducible manner. This plasmid was used for transformation of the Δhfq mutant strain. Transformants were first isolated by selection for erythromycin resistance and then tested for the double crossover in the flanking regions by PCR, and the resultant complemented mutant was designated Δhfq_C.
TABLE 1.
Oligonucleotides used in this study
| Primer | Sequencea | Restriction enzyme site |
|---|---|---|
| Hfq-1 | attcagaattcGGTTTCCGTGCGGGTGGTAAGGC | EcoRI |
| Hfq-2 | GCTAAAGGACAAATGTTGCAggatccGCACGAAGCATGACGTGTC | BamHI |
| Hfq-3 | GACACGTGATGCTTCGTGCggatccTGCAACATTTGTCCTTTAGC | BamHI |
| Hfq-4 | attcagaagcttACGCGAAGCAGGCAGGTCTATGG | HindIII |
| Hfq-F | attcagcatATGACAGCTAAAGGACAAATGTTGCAAG | NdeI |
| Hfq-R | attcagctcgagTTATTCGGCAGGCTGCTGGACGGTTTCC | XhoI |
| Hfq-R-Ns | attcagatgcatTTATTCGGCAGGCTGCTGGACGGTTTCC | NsiI |
| Hfq-S1 | CTCAGGAGAACAACGTATTGATCG | |
| Hfq-S2 | GTTACCATCGTATTGCTGAAtTCGCCCACATC | EcoRI |
| 0747-F | GAAAGTGTGGGCATTCGAC | |
| 0747-R | TTGCTGTAACCTGCTTCCTG | |
| 16S_F | ACGGAGGGTGCGAGCGTTAATC | |
| 16S_R | CTGCCTTCGCCTTCGGTATTCCT |
Capital letters in roman type indicate N. meningitidis-derived sequences, capital letters in italics indicate E. coli-derived sequences, lowercase letters indicate sequences added for cloning purposes, and underlining indicates recognition sites.
TABLE 2.
Strains and plasmids used in this study
| Strain or plasmid | Relevant characteristics | Reference or source |
|---|---|---|
| N. meningitidis strains | ||
| MC58 | Clinical isolate, sequenced strain | 56 |
| Δhfq | Hfq null mutant of MC58, Cmr | This study |
| Δhfq_C | Complemented Hfq mutant expressing Hfq from an IPTG-inducible Ptac promoter, Cmr Eryr | This study |
| 2996 | Clinical isolate | 10 |
| 2996Δhfq | Hfq null mutant of 2996, Cmr | This study |
| E. coli strains | ||
| DH5-α | supE44 hsdR17 recA1 endA1 gyrA96 thi-1 relA1 | 21 |
| BL21(DE3) | hsdS gal (λcIts857 ind1 Sam7 nin5 lacUV5-T7 gene 1) | 53 |
| Plasmids | ||
| pGEMT | Cloning vector, Ampr | Promega |
| pΔhfqko:Cm | Construct for generating knockout of the hfq gene | This study |
| pFlaEryPind | Complementation vector for integration of the gene of interest under control of an inducible Ptac promoter into the chromosome of MC58 derivative strains between the NMB1074 and NMB1075 converging genes, Ampr Eryr | This study |
| pFlaPindHfq | Derivative of pFlaEryPind with the hfq gene cloned as an NdeI-NsiI fragment downstream of the Ptac promoter, Ampr Eryr | This study |
| pET15b | Expression vector for N-terminal His-tagged proteins | Invitrogen |
| pET15hfq | pET15b derivative for expression of recombinant Hfq protein | This study |
In vitro antimicrobial stress assays.
To investigate the sensitivities of the mutant strains to various stresses, a series of growth assays and killing assays were performed using different antimicrobial compounds. Unless otherwise stated, the Δhfq_C complemented mutant was cultured in the presence of 1 mM IPTG for induction of hfq gene expression. Membrane integrity was investigated by looking at the susceptibility to osmotic stress (30% sucrose and 5 M NaCl) and detergents (0.01 to 0.05% Tween and sodium dodecyl sulfate [SDS]). To investigate the responses of the strains to oxidative stress, paraquat (5 to 10 mM; Sigma), xanthine-xanthine oxidase (4.3 mM xanthine and 100 to 300 mU/ml xanthine oxidase; Sigma), and H2O2 (5 to 10 mM; Riedel-de Haen) killing assays were performed. For killing assays, cells from GC agar plates were harvested into GC media to obtain an OD600 of 0.05, grown to mid-log phase (OD600, 0.5 to 0.6), and then diluted to obtain a concentration of 105 to 106 CFU/ml. The killing assay was started by addition of the killing sample. Cultures were incubated at 37°C in an atmosphere containing 5% CO2 with gentle agitation, and at various time points samples were taken, plated onto Mueller-Hinton agar after serial dilution, and incubated at 37°C in an atmosphere containing 5% CO2 to determine the number of CFU. Experiments were done in triplicate and repeated on several occasions.
Ex vivo human serum assay and ex vivo whole-blood model of meningococcal bacteremia.
N. meningitidis strains were grown on GC agar overnight. Cells were harvested into Mueller-Hinton medium containing 0.25% glucose and 0.02 mM cytidine-5′-monophospho-N-acetylneuraminic acid sodium salt to obtain an OD600 of 0.05, grown to mid-log phase (OD600, 0.5 to 0.6), and then diluted to obtain a concentration of approximately 103 CFU/ml. The assay was started by addition of 100% whole human blood or human serum (240 μl) to the bacterial suspension (10 μl). Cultures were incubated at 37°C in an atmosphere containing 5% CO2 with gentle agitation, and at various time points an aliquot was removed and the number of viable bacteria was determined by plating serial dilutions onto Mueller-Hinton agar and incubating the plates overnight at 37°C in an atmosphere containing 5% CO2. Experiments were done in triplicate and repeated on several occasions. Whole venous blood, collected from healthy individuals and anticoagulated with heparin (10 U/ml), was used for whole-blood experiments. For preparation of human serum, whole blood was coagulated at 37°C for 30 min and centrifuged at 1,000 × g for 10 min at 4°C, and the supernatant was retained. Statistical analyses of the differences between the survival of the wild-type strain and the survival of mutant strains were performed using Student's t test.
RNA isolation, real-time quantitative PCR, and S1 nuclease mapping.
RNA was isolated from liquid cultures of N. meningitidis strains as previously described (11). For real-time quantitative PCR (RT-PCR), total RNA was isolated by using an RNeasy kit (Qiagen) as described by the manufacturer. Approximately 4 μg of the total RNA preparation was treated with RQ1 RNase-free DNase (Promega). RNA was then reverse transcribed using random hexamer primers and Moloney murine leukemia virus reverse transcriptase (Promega) as recommended by the manufacturer. For negative controls, all RNA samples were also incubated without reverse transcriptase. All RT-PCRs were performed in triplicate using a 25-μl mixture containing cDNA (5 μl of a 1/5 dilution), 1× brilliant SYBR green quantitative PCR master mixture (Stratagene), and approximately 5 pmol of each primer (primers 0747-F and 0747-R for the NMB0747 gene and primers 16S_F and 16S_R for the 16S rRNA normalization control [Table 1]). Amplification and detection of specific products were performed with an Mx3000 real-time PCR system (Stratagene) using the following procedure: 95°C for 10 min, followed by 40 cycles of 95°C for 30 s, 55°C for 1 min, and 72°C for 30 s and then a dissociation curve analysis. The 16S rRNA gene was used as the endogenous reference control, and relative gene expression was determined using the 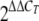 relative quantification method.
relative quantification method.
A radioactively labeled DNA probe for S1 mapping of the 5′ region of the hfq transcript from position +1 was prepared as follows. The probe was amplified from the MC58 chromosome using the Hfq-S1/Hfq-S2 primer pair, the appropriate 363-bp fragment was extracted from an agarose gel, and after purification 2 pmol of the probe was labeled at both extremities with T4 polynucleotide kinase and 4 pmol of [γ-32P]ATP. One labeled extremity was removed by digestion with EcoRI, a site for which was incorporated into the upstream primer (Hfq-S1), and the resultant 345-bp probe, labeled on the 5′ complementary strand, was purified using Chomaspin TE-100 columns (Clontech). Approximately 20 fmol of labeled probe was coprecipitated with 15 μg of total RNA and resuspended in 20 μl of hybridization buffer (80% formamide, 60 mM Tris-HCl [pH 7.5], 400 mM NaCl, 0.4 mM EDTA). The mixture was overlaid with 5 μl of paraffin oil, denatured at 100°C for 3 min, and then incubated at the melting temperature (Tm) calculated for the probe on the basis of the following formula: Tm = 81.5 + 0.5(G+C content) + 16.6(natural log of Na concentration) − 0.6(formamide concentration expressed as a percentage). After 4 to 16 h of hybridization, 180 μl of ice-cold S1 buffer (33 mM Na acetate [pH 5.2], 5 mM ZnSO4, 250 mM NaCl) and 100 U of S1 nuclease (Invitrogen) were added, and S1 nuclease digestion was carried out for 30 min at 37°C. Samples were then extracted once with phenol-chloroform, ethanol precipitated, resuspended in 5 μl sequencing loading buffer (47), and subjected to 6% urea-polyacrylamide gel electrophoresis (PAGE). Quantification of the signals from the digested probes was performed using a Phosphorimager and ImageQuant software (Molecular dynamics). A G-A sequencing reaction (33) was performed with the probe parallel to the S1 nuclease reactions to provide a molecular weight ladder for mapping the 5′ end of the transcript.
Expression and purification of the Hfq protein.
The hfq gene was amplified from the MC58 genome with the Hfq-F/Hfq-R primer pair and cloned as a 302-bp NdeI-XhoI fragment into the pET15b expression plasmid (Invitrogen), generating pET15hfq, which was subsequently transformed into E. coli strain BL21(DE3) for protein expression. Using an overnight culture of the BL21(DE3)(pET15hfq) strain, 200 ml of Luria-Bertani medium was inoculated, the culture was grown until the OD600 was 0.5, and expression of the recombinant Hfq protein containing an N-terminal histidine tag was induced by addition of 1 mM IPTG and further incubation for 3 h. The protein was purified from the harvested cells by Ni-nitrilotriacetic acid (NTA) (Qiagen) affinity chromatography under nondenaturing conditions according to the manufacturer's instructions. The purified protein preparation was then diluted to 1 μg/μl and dialyzed overnight in PBS at 4°C. To remove the His tag, the dialyzed protein was digested with thrombin (10 U/μg protein; Pharmacia/Amersham) at room temperature for 4 h, and the thrombin was then deactivated by incubation with 1 mM of phenylmethylsulfonyl fluoride at 37°C for 15 min. The digested His tag was removed by dialyzing the protein preparation twice against 1 liter of PBS at 4°C in a 6,000- to 8,000-molecular-weight-cutoff dialysis tube (Membrane Filtration Products, Inc.). The purity of the protein was estimated to be 99% by SDS-PAGE. The concentration of the protein in this preparation was determined by using the Bradford colorimetric assay (Bio-Rad), and the protein was aliquoted and stored at −80°C.
In vitro cross-linking.
For in vitro cross-linking, the dialyzed protein was diluted to obtain a concentration of 0.6 μg/μl in PBS. Disuccinimidyl suberate (DSS) was added to 10 μl of the protein solution to a final concentration of 2.5 mM, and this was followed by incubation at room temperature for 1 h. To stop the cross-linking reaction, 20 μl of protein SDS-PAGE sample buffer (47) was added, and the protein was analyzed by SDS-PAGE.
Generation of anti-Hfq antiserum and Western blot analysis.
To prepare anti-Hfq antiserum, 20 μg of purified protein was used to immunize 6-week-old female CD1 mice (Charles River Laboratories); four mice were used. The protein was given intraperitoneally, together with complete Freund's adjuvant for the first dose and incomplete Freund's adjuvant for the second (day 21) and third (day 35) booster doses. Bleed-out samples were taken on day 49 and used for Western blot analysis. For time course experiments for Hfq expression, colonies from freshly grown overnight plate cultures were suspended in PBS at an OD600 of 0.5. Each preparation was then diluted 1:10 to obtain a final starting OD600 of 0.05, identical 7-ml cultures in 14-ml tubes were grown in parallel, and the growth was monitored by determining the absorbance every hour. Every hour, one of the 7-ml cultures was pelleted in a benchtop centrifuge and resuspended to a final OD600 of 5 in PBS. For concentration-controlled IPTG induction of the Hfq protein in the complemented strain, 7-ml liquid cultures of the wild-type, Δhfq mutant, and Δhfq_C strains were grown to an OD600 of 0.5 using an inoculum with an OD600 of 0.05 and different final concentrations of IPTG, as indicated below. Samples were harvested and normalized in 1× SDS-PAGE loading buffer at a relative OD600 of 5. For Western blot analysis, 10 μg of each total protein sample in 1× SDS-PAGE loading buffer (50 mM Tris-Cl [pH 6.8], 2.5% SDS, 0.1% bromophenol blue, 10% glycerol, 5% β-mercaptoethanol, 50 mM dithiothreitol) was separated by SDS-PAGE and transferred onto a nitrocellulose filter by standard methods (47). Filters were blocked for 1 h at room temperature by agitation in blocking solution (3% skim milk, 0.1% Triton X-100 in PBS) and incubated for a further hour with a 1:1,000 dilution of the anti-Hfq protein serum in blocking solution. After washing, the filters were incubated in a 1:2,000 dilution of peroxidase-conjugated anti-mouse immunoglobulin (Dako) in blocking solution for 1 h, and the resulting signal was detected using the Supersignal West Pico chemiluminescent substrate (Pierce).
Fractionation of proteins of N. meningitidis.
Liquid cultures of meningococcus strains were grown to an OD600 of 0.5 and harvested by centrifugation at 8,000 × g for 15 min at 4°C. Secreted proteins were precipitated from each spent culture supernatant as follows. The supernatant was filtered through a 0.22-μm filter to remove bacteria, and to 22 ml of the filtrate 2.5 ml of 100% trichloroacetic acid (Sigma) was added and incubated overnight on ice. The precipitated proteins were then pelleted in a benchtop centrifuge (Beckman) at 13,000 rpm for 30 min at 4°C and washed with 500 μl of 70% ethanol, and the pellet was then air dried and resuspended in 10 μl of PBS containing complete protease inhibitor (Roche). For cytoplasmic and envelope fractions, harvested bacteria were resuspended in PBS and inactivated at 56°C for 45 min. The cells were then repelleted by centrifugation at 8,000 × g for 15 min at 4°C and resuspended in 20 mM Tris-HCl (pH 7.5) containing complete protease inhibitor. The bacteria were lysed by sonication on ice, and the cell debris was removed by centrifugation at 5,000 × g for 20 min. The supernatant was centrifuged at 50,000 × g for 75 min (29,000 rpm; Beckman Ti50) to pellet the membrane fraction. The supernatant, containing the soluble cytoplasmic fraction, was then filtered and analyzed by SDS-PAGE. The pellet containing the membrane envelope was washed with 5 ml of 20 mM bis-Tris propane (pH 6.5), 1 M NaCl, 10% glycerol and then pelleted by centrifugation at 50,000 × g for 75 min. The membrane fraction was resuspended in PBS and analyzed by SDS-PAGE.
Separation of total proteins by 2D gel electrophoresis.
N. meningitidis wild-type strain MC58 and the Δhfq mutant were grown at 37°C in GC medium until the OD600 was 0.5 to 0.6. The cells were harvested, washed once in PBS, and heat inactivated for 2 h at 65°C. Bacteria were resuspended in reswelling buffer containing 7 M urea, 2 M thiourea, 2% 3-[(cholamidopropyl)-dimethylammonio]-1-propanesulfonate (CHAPS), 2% amidosulfobetain 14, 1% dithiothreitol, 2 mM tributylphosphine, 20 mM Tris, and 2% carrier ampholyte (GE Healthcare), and 200 μg of total proteins in 0.125 ml (final volume) was absorbed overnight onto Immobiline DryStrips (7 cm; nonlinear pH 3 to 10 gradient) using an Immobiline DryStrip reswelling tray (GE Healthcare). Proteins were then separated by two-dimensional (2D) electrophoresis. The first dimension was run using an IPGphor isoelectric focusing unit (GE Healthcare) by sequentially applying 150 V for 60 min, 500 V for 35 min, 1,000 V for 30 min, 2,600 V for 10 min, 3,500 V for 15 min, 4,200 V for 15 min, and finally 5,000 V until 12,000 V·h was reached. For the second dimension, the strips were equilibrated as described previously (22), and proteins were separated on a Nu-Page 4 to 12% bis-Tris ZOOM gel (Invitrogen) using 40 mA/gel. An image of each bidimensional gel was acquired with an SI personal densitometer (Molecular Dynamics) at 16 bits and 50 μm per pixel. Images were analyzed with the software Image Master 2D Platinum 6.0 (GE Healthcare).
In-gel protein digestion and MALDI-TOF mass spectrometry analysis.
Protein spots were excised from the gels, washed with 50 mM ammonium bicarbonate-acetonitrile (50/50, vol/vol), and air dried. Dried spots were digested for 2 h at 37°C in 12 μl of 0.012-μg/μl sequencing grade modified trypsin (sequencing grade modified porcine trypsin; Promega, Madison, WI) in 5 mM ammonium bicarbonate. After digestion, 0.6 μl was loaded on a matrix-prespotted Anchorchip (PAC 384 HCCA; Bruker-Daltonics, Bremen, Germany) and air dried. Spots were washed with 0.6 μl of a solution containing 70% ethanol and 0.1% trifluoroacetic acid. Mass spectra were acquired with an ultraflex matrix-assisted laser desorption ionization—time of flight (MALDI-TOF) mass spectrometer (Bruker-Daltonics). Spectra were externally calibrated by using the combination of standards present on the PAC chip (Bruker-Daltonics). Monoisotopic peptide matching and protein searching were performed automatically using a licensed version of the MASCOT software (Matrix Sciences, London, United Kingdom) run on a local database. The MASCOT search parameters used were as follows: (i) allowed number of missed cleavages, 1; (ii) variable posttranslational modification, methionine oxidation; and (iii) peptide tolerance, ±100 ppm. Only significant hits as defined by MASCOT probability analysis were considered.
In vivo animal model.
The infant rat model was used essentially as previously described (17). The 2996 N. meningitidis strain was passaged three times in infant rats. On the day of the experiment the bacteria were grown to log phase in Mueller-Hinton medium supplemented with 0.25% glucose, washed, and resuspended at the desired concentration in PBS. Five- to 6-day-old pups from litters of outbred Wistar rats (Charles River) were challenged intraperitoneally with wild-type strain 2996 or the isogenic 2996Δhfq strain. Eighteen hours after the bacterial challenge, blood specimens were obtained by cheek puncture, and aliquots (100 μl of undiluted sera and 1:10, 1:100, and 1:1,000 dilutions) were plated onto chocolate agar for viable cell counting. The numbers of CFU per milliliter of blood were determined after overnight incubation of the plates at 37°C in an atmosphere containing 5% CO2. A statistical analysis was performed using Student's t test.
RESULTS
hfq locus of N. meningitidis.
In N. meingitidis, the hfq gene (NMB0748) is predicted to encode a protein that is similar to host factor I protein or Hfq. The encoded protein consists of 97 amino acids and shows 65% identity with the Hfq protein from E. coli. In the neisserial MC58 genome hfq is flanked upstream by a 147-bp intergenic region possibly containing its promoter and is followed downstream by a putative transcriptional terminator (Fig. 1A). This is in contrast to E. coli and other similar enterobacteria, in which hfq appears to be located in a cluster of genes which form an operon, usually transcriptionally linked to the upstream gene miaA (59).
FIG. 1.

hfq locus of N. meningitidis. (A) Diagram of the hfq locus in the genome of N. meningitidis strain MC58, showing the strategy used to construct the hfq null mutant by allelic replacement with a chloramphenicol cassette. The genes flanking the hfq gene in Neisseria are an upstream gene encoding a putative d-alanine-endopeptidase and a downstream gene encoding a conserved hypothetical protein with homology to RNA methylases. These genes are different from genes in other gram-negative and gram-positive bacteria in which a similar genetic organization has often been observed. In the sequence of the intergenic region upstream of the hfq gene underlined bold type indicates −10 and −35 hexamers and the transcriptional start site at position +1, as shown in panel B. The sequence of the putative rho-independent transcriptional terminator and its position with respect to the initiation of transcription at position +1 are also shown. (B) Mapping of the hfq promoter by an S1 nuclease protection assay. The DNA probe was radioactively labeled at one end, hybridized to 15 μg of total meningococcal RNA (lanes T1, T2, T3, T5, and T7), and digested with S1 nuclease for mapping of the 5′ end of the hfq transcript. Total RNA was prepared after 1, 2, 3, 5, and 7 h of growth in GC broth (lanes T1, T2, T3, T5, and T7, respectively). Two control samples with E. coli tRNA instead of total RNA were processed in parallel with (lane +) and without (lane −) addition of S1 nuclease. The position of the RNA-specific S1 nuclease-protected band corresponding to the 5′ end of the hfq transcript is indicated. Lane G+A contained a G-A sequence reaction mixture for the DNA probe used as a size marker (33). The nucleotide sequence of the coding strand upstream of the transcriptional start site is shown on the left.
We mapped the promoter of the hfq gene of N. meningitidis in the intergenic region directly upstream by performing S1 nuclease protection analysis. Total RNA was isolated from the MC58 strain during in vitro growth and then hybridized with the hfq-specific probe and digested with S1 nuclease. Figure 1B shows an RNA-protected band which corresponds to the 5′ end of the hfq mRNA, which maps the position 1 transcriptional initiation site to 49 nucleotides upstream of the ATG start site of the gene. Analysis of the nucleotide sequence upstream revealed −10 (TACAAT) and −35 (TGGATA) hexamers, suggesting that the hfq gene is transcribed from a σ70 promoter. The intensity of this band does not vary significantly in RNA prepared from various time points in the growth curve, suggesting that the hfq promoter is transcribed throughout the various growth phases.
Expression of the meningococcal Hfq protein and generation of an Hfq null mutant.
In order to investigate expression of the Hfq protein in the MC58 strain, we purified the Hfq protein and raised antibodies to Hfq in mice. We cloned the hfq gene into an expression vector, expressed it as a recombinant His-tagged protein in E. coli, and then purified it by Ni-NTA affinity chromatography. Figure 2A shows the results of an SDS-PAGE analysis of various fractions obtained for the expression and purification steps. The Hfq protein was highly expressed in the soluble fraction (Fig. 2A, lane 3) and gave rise to a highly pure recombinant His-Hfq protein preparation (lanes 6 to 8), and the N-terminal His tag was cleaved and removed after purification, giving rise to an untagged Hfq protein (lane 9). Using in vitro cross-linking, we investigated whether this protein oligomerized into its functional hexameric form. As Fig. 2B shows, after treatment for 1 h with the DSS cross-linker, two high-molecular-weight oligomers were visible on the gel. One of the major cross-linked forms migrated at a molecular weight of approximately 70,000, which is consistent with the hexameric form of the protein, and the other more slowly migrating form may represent two cross-linked hexamers, suggesting that the recombinant Hfq protein forms hexamers in solution. This protein preparation was used to raise antibodies against meningococcal Hfq in mice, and the resulting antiserum was tested in an immunoblot analysis with total protein samples taken from time course cultures of the MC58 strain and an Hfq null mutant strain.
FIG. 2.

(A) Expression and purification of the recombinant Hfq protein. SDS-PAGE was performed with protein extracts from uninduced (lane −) and IPTG-induced (lane +) cultures of E. coli containing the pET15bHfq expression plasmid, the cleared soluble fraction before (lane SF) and after (lane FT) binding to the Ni-NTA resin, the wash fraction (lane W), elution fractions (lanes E1 to E3), and untagged Hfq protein after thrombin digestion (lane Dig). (B) In vitro cross-linking of Hfq reveals hexameric forms in solution. For in vitro cross-linking, the untagged protein was not treated (lane −) or treated (lane +) with the cross-linking reagent DSS. The relative positions of the molecular weight markers (lane M) are indicated on the left. The positions of the oligomeric forms of the Hfq protein are indicated on the right. (C) Western blot showing the expression of the Hfq protein in wild-type strain MC58 or the Δhfq mutant over time. Total protein samples were taken at the time points shown in Fig. 3A. Five micrograms of total protein was loaded for each time point. Anti-Hfq antiserum recognizes a band at approximately 11 kDa in the wild-type strain but not in the Hfq null mutant. Antiserum against NMB2091 was also used to stain the blot as a loading control for total protein, as indicated by the asterisk.
The null mutant of MC58 was generated by replacing the hfq gene with a chloramphenicol cassette, as indicated in Fig. 1A. This mutant, when it is grown on solid medium, has an obvious growth phenotype in that it forms small pinpoint colonies after overnight incubation. Furthermore, after approximately 2 to 3 days of growth on solid medium, the centers of the colonies have a concave morphology, and, where there is confluent growth on the plate, the culture appears to be more flat and acquires a silver sheen. We grew the wild type and null mutant in liquid GC medium and took samples every hour over the time course. Consistent with the growth phenotype on plates, the null mutant exhibited a significantly altered growth curve compared with the wild type in liquid medium (Fig. 3A). In particular, it had an increased lag phase and did not reach a cell density equivalent to that of the wild type. Western blot analysis of the expression of Hfq throughout the time course (Fig. 2C) revealed that the anti-Hfq antiserum recognizes a protein band migrating to a position corresponding to approximately 11 kDa in the wild type, which is absent from the null mutant strain. Furthermore, the protein is expressed during all phases of the growth curve, which is consistent with results shown in Fig. 1B, and no significant differences in the level of expression were detected, except possibly for slight accumulation of the protein in late log and stationary phases.
FIG. 3.

(A) Growth curves for the wild type, the Δhfq mutant, and the Δhfq_C complemented mutant in GC medium supplemented with different concentrations of IPTG (in μM, as indicated after the mutant designation). Three independent sets of cultures were grown on different days, and the symbols indicate the average of the three cultures for each strain; the error bars show the standard deviations. The calculated generation times of the cultures were as follows: MC58, 48 ± 7 min; Δhfq, 78 ± 10 min; and Δhfq_C with 0, 1, 10, 100, and 1,000 μM IPTG, 80 ± 5, 80 ± 5, 70 ± 17, 55 ± 8, and 51 ± 10 min, respectively. (B) Western blotting and SDS-PAGE of cell lysates of the MC58 wild-type strain, the Δhfq mutant, and the complemented mutant, showing Hfq expression and pleiotropic effects of Hfq on protein expression in the Δhfq mutant. IPTG was added at the final concentrations indicated to the growth medium for induction of Hfq in the complemented mutant. Cells were grown in GC broth to an OD600 of 0.5, and 10 μg of total protein was added to each lane. The positions of molecular weight markers are indicated on the left. The arrows on the right indicate the positions of derepressed protein bands, and the filled circles indicate downregulated protein bands in the hfq null mutant.
Complementation of the Hfq mutant.
In order to investigate downstream polar effects due to the hfq mutation, we performed RT-PCR analysis of the NMB0747 gene using RNA prepared from the wild type and the Hfq knockout mutant. The results showed that there was a 10-fold reduction in RNA levels for NMB0747, indicating that readthrough of the putative rho-independent terminator occurs and that it is likely that hfq is coexpressed with the downstream gene. In order to determine if the phenotypes of the mutant were directly related to the lack of expression of the Hfq protein or possible polar effects, we generated a complemented mutant strain expressing a single copy of the hfq gene in trans and related this expression to possible restoration of the wild-type phenotype. In this strain, Δhfq_C (Table 2), the expression of the hfq gene was inducible by addition of IPTG, as its transcription is under the control of the Ptac promoter and the LacI repressor. This strain was grown in liquid cultures in the presence of increasing amounts of IPTG, along with the wild-type and mutant strains, and the growth curves are shown in Fig. 3A. Samples were collected at mid-log phase, and total protein extracts of each strain were used to monitor Hfq expression by Western blotting (Fig. 3B). The generation time of the mutant was significantly less (78 ± 10 min) than that of the wild type (48 ± 7 min), while the generation times of the complemented mutant were shorter in cultures with IPTG-induced increases in Hfq expression. It is worth noting that the levels of Hfq expression in the complementing strain at the time of maximal induction were approximately two- to threefold less than the wild-type levels, and this may explain the incomplete restoration of the growth phenotype. These results indicate that the growth defect of the Δhfq mutant can be restored by expression of Hfq in a dose-dependent manner.
Identification of proteins differentially expressed in the Δhfq mutant.
To obtain a preliminary picture of global changes in the expression profiles of proteins altered in the Δhfq mutant, we compared the whole-cell protein patterns in SDS-PAGE gels for culture samples obtained at mid-log phase (Fig. 3B). The lack of Hfq in the mutant results in global changes in protein expression, and many protein bands are more abundant or less abundant in the Hfq-deficient strain. Furthermore, the expression profile of the mutant is restored to a profile similar to that of the wild type after restoration of Hfq expression in the Δhfq_C strain by IPTG addition.
In order to identify some of the proteins differentially expressed due to deletion of the Hfq protein, total protein from logarithmically grown cultures of wild-type and Δhfq mutant cultures were separated by one-dimensional (1D) or 2D electrophoresis. Furthermore, cell cultures were also fractionated to obtain secreted, cytoplasmic, and envelope fractions and separated by 1D SDS-PAGE. Representative gels for triplicate samples are shown in Fig. 4, and the most altered protein spots and bands that were identified by mass spectrometry are shown in Fig. 4A and B, respectively. These spots and bands were excised and digested with trypsin, and the resulting peptides were analyzed by MALDI-TOF mass spectrometry. It is worth noting that a greater number of high-molecular-weight proteins appeared to be released into the supernatants of the hfq mutant cultures than into the supernatant of the wild-type strain cultures, although many of these proteins could not be identified. Using mass spectrometry analysis following 1D and 2D separation, we were able to identify 27 proteins that showed altered accumulation in the Δhfq mutant, and results of these analyses are shown in Table 3. Twenty of the proteins appear to be upregulated in the Δhfq mutant, and seven proteins are downregulated. The functions of the proteins whose abundance is altered can be subdivided into various groups, including energy metabolism, amino acid biosynthesis, oxidative stress responses, and outer membrane proteins (OMPs), many of which are associated with pathogenicity.
FIG. 4.

Identification of differentially abundant proteins in the Δhfq knockout mutant. (A) Total proteins of logarithmically grown wild-type strain MC58 and the Δhfq mutant were separated by 2D gel electrophoresis. Proteins were first focused on a nonlinear pH 3 to 10 gradient and then separated on a 9 to 16.5% SDS-polyacrylamide gel. Cells were grown to an OD600 of 0.5 in GC broth. (B) 1D SDS-PAGE analysis of total proteins (Total) or cytoplasmic (Cyto) or envelope (Env1 and Env2) fractions of cells of the wild type (lane +) and the Hfq mutant (lane −) grown to log phase in GC broth. Both 1D and 2D gels were stained with Coomassie brilliant blue R-250. In addition, wild-type and Hfq mutant cultures were grown to an OD600 of 0.2 in Mueller-Hinton medium containing 0.25% glucose, and 22 ml of cell-free supernatant was precipitated and loaded (Supr).
TABLE 3.
Deregulated proteins identified by proteomic analysis
| Band or spot | Designation (gene) | Protein | Predicted function | Mol wt | Relative change in Δhfq mutant (fold)a |
|---|---|---|---|---|---|
| Downregulated proteins | |||||
| C1 | NMB0758 (pnp) | Polyribonucleotide nucleotidyltransferase | RNA metabolism | 76,374 | −3.0 |
| C2 2D12 | NMB1938 (atpA) | ATP synthase F1, alpha subunit | Energy metabolism: ATP-proton motive force interconversion | 55,257 | −2.4 |
| C3 | NMB1936 (atpB) | ATP synthase F1, beta subunit | Energy metabolism: ATP-proton motive force interconversion | 50,360 | −2.1 |
| T1 C4 | NMB0763 (cysK) | Cysteine synthase | Amino acid biosynthesis: serine family | 32,800 | −2.4 |
| 2D13 | NMB0335 (dapD) | 2,3,4,5-Tetrahydropyridine-2,6-dicarboxylate N-succinyltransferase | Amino acid biosynthesis: aspartate family | 29,391 | −2.3 |
| 2D6 | NMB0138 (fusA) | Elongation factor G | Protein synthesis | 77,408 | −1.3 |
| 2D7 | NMB0618 (ppsA) | Phosphoenolpyruvate synthase | Energy metabolism: glycolysis/gluconeogenesis, carbohydrate transport, and metabolism | 87,115 | −2 |
| Upregulated proteins | |||||
| T2 E6 E17 2D1 | NMB1572 (acnB) | Aconitase hydratase 2 | Energy metabolism: tricarboxylic acid cycle | 92,657 | 2.3 |
| T3 2D2 | NMB0920 (icd) | Isocitrate dehydrogenase | Energy metabolism: tricarboxylic acid cycle | 80,113 | 1.4 |
| 2D3 | NMB0954 (gltA) | Citrate synthase | Energy metabolism: tricarboxylic acid cycle | 48,587 | 2 |
| C9 | NMB431 (pprC) | Methylcitrate synthase | Energy metabolism: propionate metabolism | 42,792 | 1.6 |
| 2D4 | NMB0435 (ackA-1) | Acetate kinase | Energy metabolism: fermentation | 43,336 | 2 |
| E2 | NMB1388 (pgi-2) | Glucose-6-phosphate isomerase | Energy metabolism: carbohydrate transport and metabolism | 62,044 | 1.3 |
| 2D10 2D11 | NMB1796 | Conserved hypothetical protein, oxidoreductase | Energy metabolism: electron transport | 20,989 | 2.9 |
| 2D8 | NMB1584 | 3-Hydroxyacid dehydrogenase | Central intermediary metabolism: other | 30,359 | 1.9 |
| E1 E16 2D5 | NMB1055 (glyA) | Serine hydroxymethyltransferase | Amino acid biosynthesis: serine family | 45,243 | 3 |
| E5 | NMB0944 (metH) | 5-Methyltetrahydropteroyltriglutamate-homocysteine methyltransferase | Amino acid biosynthesis: aspartate family | 85,024 | 1.3 |
| T5, E3 | NMB1972 (groEL) | Chaperonin, GroEL (60 kDa) | Protein fate: protein folding and stabilization | 57,387 | 1.4 |
| T7 | NMB0946 (prx) | Peroxiredoxin 2 family protein/glutaredoxin | Detoxification: oxidative stress response | 26,894 | 1.4 |
| C5 | NMB0884 (sodB) | Superoxide dismutase | Detoxification: oxidative stress response | 21,879 | 1.7 |
| C7 | NMB1590 | Conserved hypothetical protein: putative peroxidase | Unknown function: oxidative stress response | 11,526 | 1.6 |
| E4, E10 | NMB0182 (omp85) | OMP Omp85 | Outer membrane lipid biogenesis | 88,382 | 1.2b |
| E15 | NMB1053 (opc) | Class 5 OMP | Pathogenesis: adhesion | 29,973 | 1.3 |
| S10 | NMB1053 (opc) | Class 5 OMP | Pathogenesis: adhesion | 29,973 | 2.3 |
| NMB1636 | Opacity protein | 30,161 | 2.3 | ||
| S7 | NMB1541 (lbpB) | Lactoferrin binding protein B | Transport of iron cations | 81,082 | 1.8 |
| S11 | NMB0634 (fbpA) | Iron ABC transporter, periplasmic binding protein | Transport of iron cations | 35,806 | 1.5 |
| 2D9 | NMB0018 | Pilin PilE | Pathogenesis: cell motility and adhesion | 18,202 | 1.6 |
Relative abundance of protein in the Δhfq mutant compared with the wild type. The differentially regulated bands and spots were quantified using ImageQuant software, the values were normalized with respect to an unchanged internal protein control, and the relative quantities are expressed as changes compared with the wild type.
Deregulation of this band was not seen in all of the biological replicates tested.
Hfq plays a major role in stress tolerance in N. meningitidis.
As Hfq is reportedly involved in stress tolerance in many pathogens (9, 19, 44, 59) and a large number of proteins are differentially expressed in the Δhfq mutant, we tested the ability of the meningococcal Δhfq mutant to resist several environmental stresses. A series of growth assays and killing assays were performed with the wild type, the Δhfq mutant, and the Δhfq_C complemented mutant strain in the presence of different antimicrobial compounds.
The Δhfq mutant was significantly more sensitive than the wild-type strain to three membrane-perturbing detergents (the nonionic detergents Triton X-100 and Tween, as well as the anionic detergent SDS), and the results of a representative viability assay are shown in Fig. 5A. Likewise, the lack of expression of the hfq gene resulted in increased sensitivity of the null mutant strain to killing by oxidative stress, as investigated using paraquat (which generates superoxide anion radicals inside the cell), xanthine-xanthine oxidase (which generates superoxide and H2O2 outside the cell), and H2O2 assays (Fig. 5B). We also challenged the strains with 4.5 M NaCl in an osmotic stress assay. Again, the Δhfq mutant was significantly more sensitive than the strains expressing the Hfq protein (Fig. 5C). Finally, we tested whether the meningococcal mutant was more sensitive to antimicrobial peptides, such as polymyxin B or LL-37. Antimicrobial peptides are an important part of the innate immune response, which can attack the outer membrane, forming holes and thereby causing misfolding of the periplasmic proteins and envelope stress. The results shown in Fig. 5C indicate that Hfq contributes considerably to resistance to antimicrobial peptides in meningococcus. In all assays performed, the complemented mutant exhibited resistance patterns very similar to those of the wild type (Fig. 5A to C), confirming that inactivation of Hfq results in considerable sensitivity of the mutant strain to these antimicrobial compounds. The wild type, the mutant, and the complemented mutant all behaved similarly when they were incubated for the duration of the assay in GC broth, indicating that the differences in survival are not due to intrinsic growth defects (data not shown). Based on all these analyses, the Hfq protein plays an important role in the resistance of meningococcus to a wide range of stresses, many of which may be particularly physiologically relevant to the infectious cycle.
FIG. 5.

Assays of the viability of wild-type strain MC58, the Δhfq mutant, and the Δhfq_C complemented mutant. Cells were grown to an OD600 of 0.5 in GC broth (1 mM IPTG was added for expression of Hfq in the complemented mutant), diluted, and incubated with the antimicrobial compounds indicated. Samples were taken at 15, 30, and/or 60 min, and viable cell counts were determined by plating. The final concentrations of compounds used were as follows: Tween, 0.05%; SDS, 0.01%; Triton X-100, 0.1%; hydrogen peroxide, 5 mM; xanthine oxidase, 4.3 mM xanthine and 300 mU xanthine oxidase; paraquat, 5 mM; NaCl, 4.5 M; LL-37, 2 μM; and polymyxin B, 10 μg/ml.
Hfq contributes to the survival of meningococcus in ex vivo and in vivo models.
To examine how the strains responded to conditions comparable to those in the host, their survival in ex vivo whole-blood and serum assays was assessed. The human blood assay was used to assess both cellular and humoral mechanisms of killing (including the action of complement, antibody-mediated serum bactericidal activity, and opsonophagocytosis, as well as killing by neutrophils, macrophages and antimicrobial peptides), while the serum assay was used to assess killing of N. meningitidis mediated by the humoral immune response. The Hfq mutant was less able to survive in human blood and exhibited serum sensitivity (Fig. 6A and B). Once the serum was heat inactivated, the sensitivity of the Δhfq mutant was lost, and the number of CFU did not decrease over the time course of the assay (data not shown), which implies that the Δhfq mutant is sensitive to a heat-labile component of the complement pathway.
FIG. 6.

Survival of strain MC58, the Δhfq mutant, and the Δhfq_C complemented mutant in ex vivo whole blood (A) and serum (B) assays and an in vivo infant rat model (C). There was a reduced level of bacteremia with the Δhfq mutant in an infant rat infection model. Groups of animals were inoculated with two initial doses (approximately 103 and 105 CFU; specific levels of the initial inoculum were determined in each case and are indicated on the x axis), and bacterial counts for the wild type (diamonds) and the Δhfq mutant (triangles) recovered from the blood of each animal were determined after an 18-h period. The P values for comparisons of the levels of the wild type and the Δhfq mutant recovered with initial doses of 103 and 105 CFU are 0.0007 and 0.0057, respectively.
For in vivo infection studies, an isogenic hfq mutant of the adapted 2996 strain was generated for use in the infant rat model. Groups of six mice were inoculated intraperitoneally with either a high (105 CFU) or low (103 CFU) dose of the wild type or the Δhfq mutant strain. After 18 h, the animals were bled, and bacteria were counted by colony plating. Compared to the counts for the wild-type strain, the bacterial counts for the Δhfq mutant were significantly decreased (Fig. 6C). Although we cannot exclude the possibility that the growth phenotype may contribute to the reduced survival in the rat, these results suggest that Hfq plays a role in the pathogenesis of meningococcus in ex vivo human models and in the in vivo infant rat model.
DISCUSSION
The RNA-binding protein Hfq is present in many, but not all, bacteria and has emerged as a major modulator of posttranscriptional regulation in response to environmental stresses. In several pathogenic organisms, Hfq has been reported to play both major and minor roles in pathogenesis. Mutants have phenotypes ranging from severely attenuated to only mild virulence defects (12, 26, 44, 49, 51) or indeed no detectable phenotype at all (5). In this study we investigated the role of Hfq in the stress tolerance and virulence of meningococcus. We report that Hfq has a profound effect on the fitness and survival of meningococcus in the presence of a wide range of stresses (Fig. 5) and contributes to virulence in ex vivo and in vivo models of infection (Fig. 6). The stress sensitivity of the hfq mutant is consistent with the altered stress responses observed for other bacteria (9, 44, 60), suggesting that Hfq has a conserved function in meningococcus. The ability of meningococci to colonize the hostile environment of the mucosal epithelium and their ability to survive and multiply within human blood are key factors in the development of fulminant meningococcal disease. In the current models of bacteremia, the mutant is considerably attenuated (Fig. 6). The ability of this strain to survive in human blood and serum is not attributed to altered expression of known serum resistance factors of N. meningitidis, such as a capsule (24) or fHbp (28), as these factors were not altered by the mutation (data not shown). Hfq was also important for survival in the infant rat animal model over an 18-h period. Although we cannot exclude the possibility that the growth phenotype of this strain may partially explain the reduced number of bacteria recovered from the blood, this was certainly not the case in the ex vivo assays, where in media or heat-inactivated serum the three strains showed no decreases in cell numbers over the course of the assay (data not shown). Interestingly, in agreement with our findings, signature-tagged mutagenesis screening for attenuated mutants of meningococcus resulted in isolation of an Hfq mutant as 1 of 73 mutants that were attenuated in the infant rat (55).
Here we performed a proteomic analysis of cells lacking Hfq to identify proteins and processes that are directly or indirectly regulated by this protein, thus providing potential target genes for sRNA regulation. Of 27 proteins which were affected by the mutation, 20 were overproduced in the mutant, indicating that Hfq has a negative effect on expression of the majority of the proteins identified. Many of these proteins are involved in cell energetics and metabolism, amino acid biosynthesis, oxidative stress responses, and pathogenesis. Considering the pleiotropic effect of Hfq on mRNA stability and translational regulation in other bacteria, it is likely that many of the Hfq-dependent changes in protein expression are mediated by altered sRNA circuits. Of the genes encoding 27 deregulated proteins, 8 overlap with genes previously found in other systems with Hfq-altered expression levels, namely cysK, atpAB, glyA, icdA, ackA, fusA, and groEL, (19, 48, 49), whereas four genes have been reported to be under probably direct control of sRNAs in other systems, namely prpC, gltA, acnB, and sodB (31, 41).
Hfq has a negative effect on the expression of two proteins, SodB and AcnB (Fig. 4 and Table 3), whose genes have been under investigation in this laboratory as members of a group of positively Fur-regulated genes and likely candidates for sRNA regulatory control in meningococcus (11). We have recently reported evidence indicating that the Fur-positive regulation of sodB is mediated by an Hfq-dependent mechanism, as well as that there is Fur regulation of the sdhCDAB and fumB genes encoding other enzymes in the tricarboxylic acid cycle (36). The downregulation of these genes as a consequence of Fur mutation or under iron limitation conditions is abrogated by deletion of Hfq. In the case of the sdh genes, it is known that the Hfq-dependent sRNA involved is NrrF (35), and we have recently shown that Hfq increases the efficiency of duplex formation between NrrF and the sdh target in vitro (36). However, NrrF does not mediate the Hfq-dependent regulation of sodB or fumB (36) or of acnB (data not shown), and therefore, other unidentified Fur-regulated sRNAs are likely to be involved. In E. coli and Pseudomonas aeruginosa, these proteins and many others belonging to the tricarboxylic acid cycle and iron-containing proteins are regulated via the Fur-repressed sRNA analogues RyhB and PrrF, respectively (30, 41). Interestingly, we identified a number of OMPs whose accumulation was altered in the Δhfq mutant, although the major expressed OMPs PorA, PorB, and RmpB did not appear to be affected (Fig. 4). While regulation could be indirect, the fact that many OMPs are upregulated in the Δhfq mutant raises the possibility that OMP biogenesis and outer membrane composition may be extensively regulated by sRNAs in meningococcus, as has been reported for other systems (18, 61, 65).
The phenotypes associated with loss of Hfq in meningococcus may not be completely dependent on its role in directing sRNA posttranscriptional circuits but instead may be related to other Hfq functions or some additional factor or factors that are themselves targets for Hfq regulation. For instance, in E. coli Hfq is known to interact with the polynucleotide phosphorylase (PNPase) and poly(A) polymerase I (PAPI) proteins, two components of the degradasome, in an sRNA-independent manner (37). Here, PNP is identified as one of the proteins that is less abundant in the Δhfq mutant, and it is plausible that the turnover of the protein itself is altered in the absence of Hfq. Alternatively, the binding of an RNA-binding protein, such as Hfq, directly to mRNA might favor alternative mRNA secondary structures that may either promote or interfere with translational initiation or that may alter the rate of mRNA degradation, as is the case for Hfq-mediated downregulation of OmpA in E. coli (66). Many of the pleiotropic effects of Hfq mutants in other bacterial systems are related to the requirement for Hfq for efficient translation of the stationary-phase sigma factor (8, 40) or for downregulation of RpoE and the envelope stress response (12, 13, 19, 57). While N. meningitidis does not possess an RpoS-like sigma factor, a protein annotated as an extracellular function sigma factor (NMB2144) with similarity to RpoE is present. However, there are no homologues of the periplasmic protease DegS or the anti-sigma factor RseA, which are responsible for activation of RpoE in response to envelope stress.
A comparison of the initial observations for Hfq phenotypes for meningococcus and other better-studied systems shows many similarities but also some interesting differences. It appears that Hfq plays a pivotal role in virulence and stress adaptation. In the absence of a stationary-phase sigma factor and homologues of the proteins which activate the envelope stress response, the mechanisms by which Hfq could contribute to control global protein expression regulation may differ from the mechanisms in other systems. Nonetheless, due to the pleiotropic nature of the phenotypes of the Hfq mutant, it appears that Hfq plays a fundamental role in coordinating gene regulation in meningococcus, and our findings indicate that there is a large sRNA network in N. meningitidis that has yet to be uncovered. Further studies must address this possibility along with the question of how Hfq coordinates regulation of such a large number of processes.
Acknowledgments
L.F. and M.M.E.M. were the recipients of Novartis fellowships from the Ph.D. Program in Cellular, Molecular and Industrial Biology of the University of Bologna. F.O. was the recipient of a Novartis fellowship from the Ph.D. Program in Evolutionary Biology of the University of Siena.
We thank Giorgio Corsi for artwork.
Editor: J. N. Weiser
Footnotes
Published ahead of print on 17 February 2009.
REFERENCES
- 1.Aiba, H. 2007. Mechanism of RNA silencing by Hfq-binding small RNAs. Curr. Opin. Microbiol. 10134-139. [DOI] [PubMed] [Google Scholar]
- 2.Altuvia, S. 2007. Identification of bacterial small non-coding RNAs: experimental approaches. Curr. Opin. Microbiol. 10257-261. [DOI] [PubMed] [Google Scholar]
- 3.Altuvia, S., A. Zhang, L. Argaman, A. Tiwari, and G. Storz. 1998. The Escherichia coli OxyS regulatory RNA represses fhlA translation by blocking ribosome binding. EMBO J. 176069-6075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arluison, V., S. Hohng, R. Roy, O. Pellegrini, P. Reginer, and T. Ha. 2007. Spectroscopic observation of RNA chaperone activities of Hfq in post-transcriptional regulation by a small non-coding RNA. Nucleic Acids Res. 35999-1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bohn, C., C. Rigoulay, and P. Bouloc. 2007. No detectable effect of RNA-binding protein Hfq absence in Staphylococcus aureus. BMC Microbiol. 710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brennan, R. G., and T. M. Link. 2007. Hfq structure, function and ligand binding. Curr. Opin. Microbiol. 10125-133. [DOI] [PubMed] [Google Scholar]
- 7.Brescia, C. C., P. J. Mikulecky, A. L. Feig, and D. D. Sledjeski. 2003. Identification of the Hfq-binding site on DsrA RNA: Hfq binds without altering DsrA secondary structure. RNA 933-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brown, L., and T. Elliott. 1996. Efficient translation of the RpoS sigma factor in Salmonella typhimurium requires host factor I, an RNA-binding protein encoded by the hfq gene. J. Bacteriol. 1783763-3770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Christiansen, J. K., M. H. Larsen, H. Ingmer, L. Sogaard-Andersen, and B. H. Kallipolitis. 2004. The RNA-binding protein Hfq of Listeria monocytogenes: role in stress tolerance and virulence. J. Bacteriol. 1863355-3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Comanducci, M., S. Bambini, B. Brunelli, J. Adu-Bobie, B. Aricò, B. Capecchi, M. M. Giuliani, V. Masignani, L. Santini, S. Savino, D. M. Granoff, D. A. Caugant, M. Pizza, R. Rappuoli, and M. Mora. 2002. NadA, a novel vaccine candidate of Neisseria meningitidis. J. Exp. Med. 1951445-1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Delany, I., R. Grifantini, E. Bartolini, R. Rappuoli, and V. Scarlato. 2006. The effect of Neisseria meningitidis Fur mutations on global control of gene transcription. J. Bacteriol. 1882483-2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ding, Y., B. M. Davis, and M. K. Waldor. 2004. Hfq is essential for Vibrio cholerae virulence and downregulates sigma expression. Mol. Microbiol. 53345-354. [DOI] [PubMed] [Google Scholar]
- 13.Figueroa-Bossi, N., S. Lemire, D. Maloriol, R. Balbontin, J. Casadesus, and L. Bossi. 2006. Loss of Hfq activates the σE-dependent envelope stress response in Salmonella enterica. Mol. Microbiol. 62838-852. [DOI] [PubMed] [Google Scholar]
- 14.Franze de Fernandez, M. T., W. S. Hayward, and J. T. August. 1972. Bacterial proteins required for replication of phage Qβ ribonucleic acid. J. Biol. Chem. 247821-824. [PubMed] [Google Scholar]
- 15.Gottesman, S. 2005. Micros for microbes: non-coding regulatory RNAs in bacteria. Trends Genet. 21399-404. [DOI] [PubMed] [Google Scholar]
- 16.Gottesman, S., C. A. McCullen, M. Guillier, C. K. Vanderpool, N. Majdalani, J. Benhammou, K. M. Thompson, P. C. FitzGerald, N. A. Sowa, and D. J. FitzGerald. 2006. Small RNA regulators and the bacterial response to stress. Cold Spring Harbor Symp. Quant. Biol. 711-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Granoff, D. M., G. R. Moe, M. M. Giuliani, J. Abu-Bodie, L. Santini, B. Brunelli, F. Piccinetti, P. Zuno-Mitchell, S. S. Lee, P. Neri, L. Bracci, L. Lozzi, and R. Rappuoli. 2001. A novel mimetic antigen eliciting protective antibody to Neisseria meningitidis. J. Immunol. 1676487-6496. [DOI] [PubMed] [Google Scholar]
- 18.Guillier, M., S. Gottesman, and G. Storz. 2006. Modulating the outer membrane with small RNAs. Genes Dev. 202338-2348. [DOI] [PubMed] [Google Scholar]
- 19.Guisbert, E., V. A. Rhodius, N. Ahuja, E. Witkin, and C. A. Gross. 2007. Hfq modulates the σE-mediated envelope stress response and the σ32-mediated cytoplasmic stress response in Escherichia coli. J. Bacteriol. 1891963-1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hajnsdorf, E., and P. Regnier. 2000. Host factor Hfq of Escherichia coli stimulates elongation of poly(A) tails by poly(A) polymerase I. Proc. Natl. Acad. Sci. USA 971501-1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hanahan, D. 1983. Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol. 166557-580. [DOI] [PubMed] [Google Scholar]
- 22.Herbert, B. R., M. P. Molloy, A. A. Gooley, B. J. Walsh, W. G. Bryson, and K. L. Williams. 1998. Improved protein solubility in two-dimensional electrophoresis using tributyl phosphine as reducing agent. Electrophoresis 19845-851. [DOI] [PubMed] [Google Scholar]
- 23.Ieva, R., C. Alaimo, I. Delany, G. Spohn, R. Rappuoli, and V. Scarlato. 2005. CrgA, a member of the LysR family of transcriptional regulators of Neisseria meningitidis, is an inducible regulator acting as both a repressor and an activator of gene transcription. J. Bacteriol. 1873421-3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jarvis, G. A., and N. A. Vedros. 1987. Sialic acid of group B Neisseria meningitidis regulates alternative complement pathway activation. Infect. Immun. 55174-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kellogg, D. S., Jr., W. L. Peacock, Jr., W. E. Deacon, L. Brown, and C. I. Pirkle. 1963. Neisseria gonorrhoeae. I. Virulence genetically linked to clonal variation. J. Bacteriol. 851274-1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kulesus, R. R., K. Diaz-Perez, E. S. Slechta, D. S. Eto, and M. A. Mulvey. 2008. Impact of the RNA chaperone Hfq on the fitness and virulence potential of uropathogenic Escherichia coli. Infect. Immun. 763019-3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lenz, D. H., K. C. Mok, B. N. Lilley, R. V. Kulkarni, N. S. Wingreen, and B. L. Bassler. 2004. The small RNA chaperone Hfq and multiple small RNAs control quorum sensing in Vibrio harveyi and Vibrio cholerae. Cell 11869-82. [DOI] [PubMed] [Google Scholar]
- 28.Madico, G., J. A. Welsch, L. A. Lewis, A. McNaughton, D. H. Perlman, C. E. Costello, J. Ngampasutadol, U. Vogel, D. M. Granoff, and S. Ram. 2006. The meningococcal vaccine candidate GNA1870 binds the complement regulatory protein factor H and enhances serum resistance. J. Immunol. 177501-510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Majdalani, N., C. K. Vanderpool, and S. Gottesman. 2005. Bacterial small RNA regulators. Crit. Rev. Biochem. Mol. Biol. 4093-113. [DOI] [PubMed] [Google Scholar]
- 30.Massé, E., and M. Arguin. 2005. Ironing out the problem: new mechanisms of iron homeostasis. Trends Biochem. Sci. 30462-468. [DOI] [PubMed] [Google Scholar]
- 31.Massé, E., and S. Gottesman. 2002. A small RNA regulates the expression of genes involved in iron metabolism in Escherichia coli. Proc. Natl. Acad. Sci. USA 994620-4625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Massé, E., F. Escocia, and S. Gottesman. 2003. Coupled degradation of a small RNA and its mRNA targets in Escherichia coli. Genes Dev. 172374-2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maxam, A. M., and W. Gilbert. 1977. A new method for sequencing DNA. Proc. Natl. Acad. Sci. USA 74560-564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McNealy, T. L., V. Forsbach-Birk, C. Shi, and R. Marre. 2005. The Hfq homolog in Legionella pneumophila demonstrates regulation by LetA and RpoS and interacts with the global regulator CsrA. J. Bacteriol. 187127-1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mellin, J. R., S. Goswami, S. Grogan, B. Tjaden, and C. A. Genco. 2007. A novel fur- and iron-regulated small RNA, NrrF, is required for indirect fur-mediated regulation of the sdhA and sdhC genes in Neisseria meningitidis. J. Bacteriol. 1893686-3694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Metruccio, M. M. E., L. Fantappié, D. Serruto, A. Muzzi, D. Roncarati, C. Donati, V. Scarlato, and I. Delany. 2009. The Hfq-dependent small noncoding RNA NrrF directly mediates Fur-dependent positive regulation of succinate dehydrogenase in Neisseria meningitidis. J. Bacteriol. 1911330-1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mohanty, B. K., V. F. Maples, and S. R. Kushner. 2004. The Sm-like protein Hfq regulates polyadenylation dependent mRNA decay in Escherichia coli. Mol. Microbiol. 54905-920. [DOI] [PubMed] [Google Scholar]
- 38.Moll, I., T. Afonyushkin, O. Vytvytska, V. R. Kaberdin, and U. Bläsi. 2003. Coincident Hfq binding and RNase E cleavage sites on mRNA and small regulatory RNAs. RNA 91308-1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Møller, T., T. Franch, P. Højrup, D. R. Keene, H. P. Bächinger, R. G. Brennan, and P. Valentin-Hansen. 2002. Hfq: a bacterial Sm-like protein that mediates RNA-RNA interaction. Mol. Cell 923-30. [DOI] [PubMed] [Google Scholar]
- 40.Muffler, A., D. D. Traulsen, D. Fischer, R. Lange, and R. Hengge-Aronis. 1997. The RNA-binding protein HF-I plays a global regulatory role which is largely, but not exclusively, due to its role in expression of the σS subunit of RNA polymerase in Escherichia coli. J. Bacteriol. 179297-300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oglesby, A. G., J. M. Farrow III, J. H. Lee, A. P. Tomaras, E. P. Greenberg, E. C. Pesci, and M. L. Vasil. 2008. The influence of iron on Pseudomonas aeruginosa physiology: a regulatory link between iron and quorum sensing. J. Biol. Chem. 28315558-15567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pannone, B. K., and S. L. Wolin. 2000. Sm-like proteins wRING the neck of mRNA. Curr. Biol. 10R478-481. [DOI] [PubMed] [Google Scholar]
- 43.Pichon, C., and B. Felden. 2005. Small RNA genes expressed from Staphylococcus aureus genomic and pathogenicity islands with specific expression among pathogenic strains. Proc. Natl. Acad. Sci. USA 10214249-14254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Robertson, G. T., and R. M. Roop, Jr. 1999. The Brucella abortus host factor I (HF-I) protein contributes to stress resistance during stationary phase and is a major determinant of virulence in mice. Mol. Microbiol. 34690-700. [DOI] [PubMed] [Google Scholar]
- 45.Romby, P., F. Vandenesch, and E. G. Wagner. 2006. The role of RNAs in the regulation of virulence-gene expression. Curr. Opin. Microbiol. 9229-236. [DOI] [PubMed] [Google Scholar]
- 46.Rosenstein, N. E., B. A. Perkins, D. S. Stephens, T. Popovic, and J. M. Hughes. 2001. Meningococcal disease. N. Engl. J. Med. 3441378-1388. [DOI] [PubMed] [Google Scholar]
- 47.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 48.Sittka, A., S. Lucchini, K. Papenfort, C. M. Sharma, K. Rolle, T. T. Binnewies, J. C. Hinton, and J. Vogel. 2008. Deep sequencing analysis of small noncoding RNA and mRNA targets of the global post-transcriptional regulator, Hfq. PLoS Genet. 4e1000163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sittka, A., V. Pfeiffer, K. Tedin, and J. Vogel. 2007. The RNA chaperone Hfq is essential for the virulence of Salmonella typhimurium. Mol. Microbiol. 63193-217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sonnleitner, E., M. Schuster, T. Sorger-Domenigg, E. P. Greenberg, and U. Bläsi. 2006. Hfq-dependent alterations of the transcriptome profile and effects on quorum sensing in Pseudomonas aeruginosa. Mol. Microbiol. 591542-1558. [DOI] [PubMed] [Google Scholar]
- 51.Sonnleitner, E., S. Hagens, F. Rosenau, S. Wilhelm, A. Habel, K. E. Jager, and U. Bläsi. 2003. Reduced virulence of a hfq mutant of Pseudomonas aeruginosa O1. Microb. Pathog. 35217-228. [DOI] [PubMed] [Google Scholar]
- 52.Storz, G., J. A. Opdyke, A., and Zhang. 2004. Controlling mRNA stability and translation with small, noncoding RNAs. Curr. Opin. Microbiol. 7140-144. [DOI] [PubMed] [Google Scholar]
- 53.Studier, F. W., and B. A. Moffatt. 1986. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J. Mol. Biol. 189113-130. [DOI] [PubMed] [Google Scholar]
- 54.Sun, X., I. Zhulin, and R. M. Wartell. 2002. Predicted structure and phyletic distribution of the RNA-binding protein Hfq. Nucleic Acids Res. 303662-3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sun, Y. H., S. Bakshi, R. Chalmers, and C. M. Tang. 2000. Functional genomics of Neisseria meningitidis pathogenesis. Nat. Med. 61269-1273. [DOI] [PubMed] [Google Scholar]
- 56.Tettelin, H., N. J. Saunders, J. Heidelberg, A. C. Jeffries, K. E. Nelson, J. A. Eisen, K. A. Ketchum, D. W. Hood, J. F. Peden, R. J. Dodson, W. C. Nelson, M. L. Gwinn, R. DeBoy, J. D. Peterson, E. K. Hickey, D. H. Haft, S. L. Salzberg, O. White, R. D. Fleischmann, B. A. Dougherty, T. Mason, A. Ciecko, D. S. Parksey, E. Blair, H. Cittone, E. B. Clark, M. D. Cotton, T. R. Utterback, H. Khouri, H. Qin, J. Vamathevan, J. Gill, V. Scarlato, V. Masignani, M. Pizza, G. Grandi, L. Sun, H. O. Smith, C. M. Fraser, E. R. Moxon, R. Rappuoli, and J. C. Venter. 2000. Complete genome sequence of Neisseria meningitidis serogroup B strain MC58. Science 2871809-1815. [DOI] [PubMed] [Google Scholar]
- 57.Thompson, K. M., V. A. Rhodius, and S. Gottesman. 2007. σE regulates and is regulated by a small RNA in Escherichia coli. J. Bacteriol. 1894243-4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tinsley, C., and X. Nassif. 2001. Meningococcal pathogenesis: at the boundary between the pre- and post-genomic eras. Curr. Opin. Microbiol. 447-52. [DOI] [PubMed] [Google Scholar]
- 59.Tsui, H. C., and M. E. Winkler. 1994. Transcriptional patterns of the mutLmiaA superoperon of Escherichia coli K-12 suggest a model for posttranscriptional regulation. Biochimie 761168-1177. [DOI] [PubMed] [Google Scholar]
- 60.Tsui, H. C., H. C. Leung, and M. E. Winkler. 1994. Characterization of broadly pleiotropic phenotypes caused by an hfq insertion mutation in Escherichia coli K-12. Mol. Microbiol. 1335-49. [DOI] [PubMed] [Google Scholar]
- 61.Valentin-Hansen, P., J. Johansen, and A. A. Rasmussen. 2007. Small RNAs controlling outer membrane porins. Curr. Opin. Microbiol. 10152-155. [DOI] [PubMed] [Google Scholar]
- 62.Valentin-Hansen, P., M. Eriksen, and C. Udesen. 2004. The bacterial Sm like protein Hfq: a key player in RNA transactions. Mol. Microbiol. 511525-1533. [DOI] [PubMed] [Google Scholar]
- 63.Vanderpool, C. K. 2007. Physiological consequences of small RNA-mediated regulation of glucose-phosphate stress. Curr. Opin. Microbiol. 10146-151. [DOI] [PubMed] [Google Scholar]
- 64.Vogel, J., and C. M. Sharma. 2005. How to find small non-coding RNAs in bacteria. Biol. Chem. 3861219-1238. [DOI] [PubMed] [Google Scholar]
- 65.Vogel, J., and K. Papenfort. 2006. Small non-coding RNAs and the bacterial outer membrane. Curr. Opin. Microbiol. 9605-611. [DOI] [PubMed] [Google Scholar]
- 66.Vytvytska, O., I. Moll, V. R. Kaberdin, A. von Gabain, and U. Blasi. 2000. Hfq (HF1) stimulates ompA mRNA decay by interfering with ribosome binding. Genes Dev. 141109-1118. [PMC free article] [PubMed] [Google Scholar]
- 67.Wang, Y., and D. E. Taylor. 1990. Chloramphenicol resistance in Campylobacter coli: nucleotide sequence, expression, and cloning vector construction. Gene 9423-28. [DOI] [PubMed] [Google Scholar]