

Abstract
Pseudomonas aeruginosa is an opportunistic pathogen that causes chronic infections in individuals suffering from the genetic disorder cystic fibrosis. In P. aeruginosa, the transcriptional regulator AlgR controls a variety of virulence factors, including alginate production, twitching motility, biofilm formation, quorum sensing, and hydrogen cyanide (HCN) production. In this study, the regulation of HCN production was examined. Strains lacking AlgR or the putative AlgR sensor AlgZ produced significantly less HCN than did a nonmucoid isogenic parent. In contrast, algR and algZ mutants showed increased HCN production in an alginate-producing (mucoid) background. HCN production was optimal in a 5% O2 environment. In addition, cyanide production was elevated in bacteria grown on an agar surface compared to bacteria grown in planktonic culture. A conserved AlgR phosphorylation site (aspartate at amino acid position 54), which is required for surface-dependent twitching motility but not alginate production, was found to be critical for cyanide production. Nuclease protection mapping of the hcnA promoter identified a new transcriptional start site required for HCN production. A subset of clinical isolates that lack this start site produced small amounts of cyanide. Taken together, these data show that the P. aeruginosa hcnA promoter contains three transcriptional start sites and that HCN production is regulated by AlgZ and AlgR and is maximal under microaerobic conditions when the organism is surface attached.
Pseudomonas aeruginosa is a gram-negative opportunistic pathogen that can cause acute infections in patients suffering from cancer (6), human immunodeficiency virus/AIDS (27, 51), and severe burns (9). P. aeruginosa can also cause chronic infections in patients affected with the genetic disorder cystic fibrosis (CF) (8, 31). Mutation of the gene that encodes the CF transmembrane conductance regulator, a chloride ion channel found in epithelial cells, results in a dehydrated layer of mucus in the airways of the lungs. The impaired mucociliary clearance entraps bacteria and allows for the establishment of a chronic infection (65, 85). Pulmonary colonization by P. aeruginosa is the leading cause of morbidity and mortality of CF patients (8).
During chronic CF infection, P. aeruginosa strains acquire mutations that cause them to undergo a phenotypic switch from a nonmucoid to a mucoid phenotype. The mucoid phenotype results from the production of copious amounts of the capsular exopolysaccharide alginate (23). The presence of a mucoid strain in the CF lung is associated with a worsening clinical prognosis (22, 24). Production of alginate aids the organism in the evasion of phagocytosis and in resistance to killing by neutrophils and macrophages (11, 64, 74), quenching of oxygen intermediates (46, 76), and antibiotic resistance (1). Alginate biosynthesis is regulated by multiple factors (34, 69) including the transcriptional regulator AlgR, a response regulator belonging to the bacterial two-component signaling system superfamily (21, 35). AlgR is required for virulence in an acute septicemia model (48) and regulates a variety of other virulence factors, including twitching motility (80, 81), hydrogen cyanide (HCN) production (13), and biofilm formation and quorum sensing (58).
The effects of cyanide on various cell types have been extensively tested, and depending on cell type and level of oxidative stress, cyanide causes either apoptosis or necrosis (66, 67). P. aeruginosa was one of the first bacteria shown to undergo cyanogenesis (18). Growing evidence suggests that HCN production may be an important virulence factor. The presence of cyanide in burn wounds was directly associated with P. aeruginosa infections (33). More recently, the ability of P. aeruginosa to paralyze and kill Caenorhabditis elegans in an in vitro model was shown to be dependent on hcnC (30). HCN production has also proven to be clinically relevant in the CF lung as (i) P. aeruginosa CF isolates produced higher levels of HCN than did laboratory strains (13), (ii) cyanide has been detected in the sputum of P. aeruginosa-infected CF and bronchiectasis patients (12, 26, 40, 71), and (iii) detection of HCN in sputum and bronchoalveolar lavage fluid directly correlated with a worsening prognosis (12). The reported HCN concentrations in the sputum of infected patients ranged from 1.6 to 3.25 times the lethal blood concentration (71).
HCN is a secondary metabolite of P. aeruginosa produced from glycine in a poorly understood oxidative reaction catalyzed by HCN synthase (15, 82). HCN is not produced by P. aeruginosa under anaerobic conditions (15) but is expressed under low oxygen tensions (14). Optimal production requires the anaerobic regulator ANR and the quorum sensing regulators LasR and RhlR, which bind the hcnA promoter at high cell densities during late exponential growth (62). There are two reported transcriptional start sites for hcnA in P. aeruginosa (62) and one in Pseudomonas fluorescens (45). P. aeruginosa T1 is regulated by the rhl and las quorum sensing systems, while T2 relies on a synergistic effect of LasR, RhlR, and ANR. GacA and RsmA function as posttranscriptional regulators of HCN production through juxtaposition of the hcnA ribosomal binding site and indirectly by positively and negatively regulating amounts of N-acylhomoserine, respectively (43, 61, 70). Additionally, AlgR directly regulates HCN production in P. aeruginosa through control of T1 and T2 of the hcnA promoter. Moreover, AlgR has recently been shown to repress HCN production in nonmucoid strains and to activate production in mucoid strains, demonstrating AlgR's ability to switch from a repressor to an activator (13).
The purpose of this study was to examine the role of AlgZ, the putative sensor for AlgR in P. aeruginosa HCN production. Two independent research groups identified the open reading frame (PA5262) that encodes AlgZ almost simultaneously (80, 89). The first group identified this gene while screening a transposon library for mutants that lost twitching motility and termed the gene fimS (80). The second group identified this same open reading frame while examining alginate production and called it AlgZ (89). The algZ or fimS gene is located directly upstream of algR and shares homology with other two-component sensors. PA3385 has also been referred to as algZ in the literature (2-4, 86) but has since been renamed amrZ (3). In addition to identifying a previously unreported hcnA transcriptional start site, the present study expands the role of AlgZ and AlgR by providing evidence that this putative sensor/regulator pair control HCN production through a surface attachment-specific mechanism that requires the conserved AlgR aspartate at position 54. These data suggest that AlgZ functions as a kinase in regulating HCN production in P. aeruginosa.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
The strains and plasmids used in this study are listed in Table 1. Escherichia coli was grown on Luria-Bertani (LB) agar (Difco). P. aeruginosa was grown on Pseudomonas isolation agar (PIA; Difco). Both were incubated at 37°C overnight. LB agar was supplemented with kanamycin (25 μg/ml) or ampicillin (50 μg/ml), and PIA was supplemented with tetracycline (300 μg/ml), streptomycin (10 μg/ml), or gentamicin (10 μg/ml) for selection, plasmid maintenance, or genetic manipulation.
TABLE 1.
Strains used in this study
| Strain or plasmid | Description | Mucoid phenotypea | Reference or source |
|---|---|---|---|
| P. aeruginosa strains | |||
| PAO1 | Wild type | NM | 38 |
| PAZ-1 | ΔalgZ | NM | This study |
| PAO6886 | algZ::Tcr | NM | 89 |
| PSL317 | ΔalgR | NM | 48 |
| PSL317Z | algZ::Tcr ΔalgR | NM | This study |
| PAO568 | mucA2 | M | 29 |
| PDO300 | mucA22 | M | 54 |
| PAZ568 | mucA2 ΔalgZ | M | This study |
| PAO6884 | mucA2 algZ::Tcr | M | 89 |
| PAR568 | mucA2 ΔalgR | NM | 13 |
| PAO6885 | mucA2 algZ::GmralgR::Tcr | NM | 89 |
| TUMC92 | Clinical isolate | NM | 13 |
| TUMC92Z | ΔalgZ | NM | This study |
| TUMC92R | ΔalgR | NM | 13 |
| FRD-1 | mucA | M | 32 |
| FRD-1Z | mucA ΔalgZ | M | This study |
| FRD-1R | mucA algR::Smr | NM | 50 |
| WFPAPAO1 | Wild type | NM | 50 |
| WFPA8 | algR(D54N) | NM | 50 |
| WFPA13 | algR(D85N) | NM | 50 |
| WFPA16 | algR(D54N D85N) | NM | 50 |
| WFPA8A | mucA algR(D54N) | NM | This study |
| WFPA13A | mucA algR(D85N) | M | This study |
| WFPA16A | mucA algR(D54N D85N) | M | This study |
| WFPA8Z | mucA algR(D54N) | NM | This study |
| WFPA13Z | mucA algR(D85N) | M | This study |
| WFPA16Z | mucA algR(D54N D85N) | M | This study |
| WFPA8AZ | mucA ΔalgZ algR(D54N) | M | This study |
| WFPA13AZ | mucA ΔalgZ algR(D85N) | M | This study |
| WFPA16AZ | mucA ΔalgZ algR(D54N D85N) | M | This study |
| PAO1 vfr | Δvfr | NM | This study |
| PAK | Hyperpiliated | N | 39 |
| PAK mucA | mucA22 | M | 87 |
| PAK algR | algR::Gmr | NM | 87 |
| PAK mucA algR | mucA22 algR::Gmr | NM | 87 |
| PA14 | Wild type | NM | 68 |
| PA103 | Wild type | NM | 47 |
| 19660 | Clinical isolate | NM | 83 |
| X13273 | Clinical isolate | NM | 83 |
| S54485 | Clinical isolate | NM | 83 |
| CF27 | Clinical isolate | M | 83 |
| PAK pscC | ΔpscC | NM | 84 |
| PAK HCN KI | ΔpscC; PAO1 hcnA promoter | NM | This study |
| Plasmids | |||
| pCR2.1 | TA cloning vector; Apr Kanr | Invitrogen | |
| pCVD442 | Gene replacement vector; TcrsacB+ | 26 | |
| pZKO442 | algZ in-frame deletion vector; Apr | This study | |
| pUCP18 | Expression vector; Tcr | 78 | |
| pUCP18Z | algZ expression vector for complementation | This study | |
| pRK2013 | Helper plasmid for conjugation, tra functions | 28 | |
| pEXGmGW | Gateway converted pEX18Gm | 83 | |
| phcnAKI | pEXGmGW with hcnA PAO1 promoter | This study |
M, mucoid; NM, nonmucoid.
DNA manipulations.
The oligonucleotides used in this study are listed in Table 2. The plasmid pZKO442, a derivative of pCVD442 (25), was used to create a 907-bp in-frame deletion of the algZ gene in multiple strains. The deletion plasmid was constructed by a two-step crossover PCR (37) with oligonucleotide primers algZkoXbaF, algZkoR′2, algZkoF′2, and algZkoSstR using the PAO1 chromosome as the template DNA. Oligonucleotides algZkoXbaF and algZkoSstR were used to engineer XbaI and SstI restriction enzyme sites to the 5′ and 3′ ends of the final PCR product, respectively. The resulting 1,743-bp fragment was cloned into pCVD442 using the restriction enzymes XbaI and SstI. The plasmid pUCP18Z was used to complement the algZ deletion strains. To construct this plasmid, a 2,326-bp fragment was amplified by PCR with oligonucleotides algZko1 and algZko4 to obtain the algZ open reading frame and promoter region from the PAO1 chromosome. This fragment was cloned into pCR2.1 (Invitrogen) and subcloned into the XbaI and KpnI restriction sites of pUCP18.
TABLE 2.
Oligonucleotides used in this study
| Name | Oligonucleotide sequence | Gene | Locationa |
|---|---|---|---|
| algZkoXbaF′ | CCCTCTAGACCCAGCAACTGGCAGGTGATCTCG | algZ | −889 to −866 (+) |
| algZkoR′2 | ACGCCTGTCGCGTGCCTTTCGACGCTTGAATCGGATAGGCATCGACAGAG | algZ | −8 to +17 (−) |
| algZkoF′2 | CGTCGAAAGGCACGCGACAGGCGT | algZ | +925 to +948 (+) |
| algZkoSstR′ | CCCGAGCTCGAGGCCTTTCAGGTAGAGCTGGA | algZ | +1736 to +1758 (−) |
| algZKO1F | TGTCTTCCTGGTTGTCCTTGTTGTA | algZ | −1212 to −1188 (+) |
| S1algRhindIII | CCCAAGCTTCCAGAGGTTCGTCATCGACAATCAG | algZ | +1090 to +1114 (−) |
| algZKO4R | CGAGTGGCGATGCGGATT | algZ | +1950 to +1967 (−) |
| hcnAprimetext | GTGTTGACGACGTTCAAGAAGGTGCAT | hcnA | −495 to +24 (−) |
| hcnAS1BglI | GCAAAGCCGGCTCACCCGTGGAGG | hcnA | −3166 to −3137 |
| PhcnA KI 5′ | TACAAAAAAGCAGGCTCCGCTTCCGACCGCCCCAGGCGGCTGA | hcnA | −1622 to −1600 (+) |
| PhcnA KI 3′ | TACAAGAAAGCTGGGTCCGCAGTTTTTCCAGCATCCGGGTCTA | hcnA | +533 to +508 (−) |
| ArgHF | ATATATGAGCTCGGACCTGTCCGACCTGTTCC | argH | −482 to −426 (+) |
| HemCR | ATATATGAGCTCGGCTGGCGTAGGTGTTCGAG | hemC | +1197 to +1177 (−) |
Location numbering is relative to the translational start site of gene. (+), coding strand; (−), antisense strand.
DNA manipulations.
In-frame deletions of algZ were created in laboratory and clinical P. aeruginosa strains by introducing the algZ deletion plasmid (pZKO442 [ΔalgZ]) through triparental conjugation (42) and selection of carbenicillin-sensitive and sucrose-resistant double recombinants using the helper plasmid pRK2013 (28). Deletion of algZ was confirmed by PCR and Southern blotting with a digoxigenin-labeled probe (Roche) using oligonucleotides algZko1 and algZko4 (data not shown). Western blot analysis with a monoclonal anti-AlgR antibody, 3H9, confirmed that there were no polar effects on AlgR expression in algZ deletion strains. In-frame deletion of the algR gene was confirmed by PCR with oligonucleotides ArgHF and HemCR and Western blot analysis with anti-AlgR antibody (20). The PAO1 ΔalgR algZ::Tcr mutant was created by electroporating fragmented chromosomal DNA from PAO6886 (algZ::Tcr) into PSL317 and selection of tetracycline-resistant recombinants, as described by Choi et al. (17). Deletion of algR and insertional inactivation of algZ was confirmed by Southern blot analysis using digoxigenin-labeled probes (Roche) created with primers ArgHF and HemCR or algZko1 and algZko4, respectively (data not shown).
The entire hcnA promoter region from PAO1 was amplified from genomic DNA with primers PhcnA KI 5′ and PhcnA KI 3′ (Table 2). The resulting PCR fragment (700 bp) was engineered with AttB1 and AttB2 sequences at the 5′ and 3′ ends, respectively, and cloned into pEXGmGW (84) via cloning to create the hcnA knock-in (KI) vector phcnAKI. This 700-bp PAO1 hcnA promoter fragment was introduced into an attenuated PAK strain (PAKΔpscC), which lacks a functioning type III secretion system, as previously described (84). Positive hcnA KI mutants were identified by PCR and confirmed by sequencing.
S1 nuclease protection assays.
RNA for S1 nuclease protection assays was isolated from PAO1, PSL317 (ΔalgR), and PAZ (ΔalgZ) grown in LB to stationary phase using CsCl as previously described (52). An S1 nuclease protection assay was performed as previously described using 100 μg of RNA with the following modifications (51). The 519-bp region of the hcnA promoter ranging from −495 to +24 (numbering relative to the translational start site) was cloned into M13mp18. Single-stranded phage DNA was isolated and used as a template for the uniformly labeled ([α-32P]dCTP; NEN DuPont) single-stranded DNA probe. Two probes were constructed using either of the oligonucleotides hcnAprimetext and hcnAS1BglI. Probes constructed with hcnAprimetext were digested with BglI, and probes constructed with hcnAS1BglI were cut with BglI. Digested probes were purified on a 5% polyacrylamide gel. Total RNA (100 μg) was hybridized to each probe at 67°C for 1 h. Reactions were then digested with S1 nuclease for 30 min and then precipitated. S1 reaction mixtures were electrophoresed through an 8% acrylamide-urea gel adjacent to a sequencing ladder generated using the same oligonucleotide as the probe.
HCN production assay of P. aeruginosa strains.
HCN was measured using a modification of the protocols described by Zlosnik and Williams (91) and Gallagher and Manoil (30). Laboratory and clinical P. aeruginosa isolates were grown on PIA plates aerobically for 16 h at 37°C. The plates were then inverted and individually sealed with lids containing 1 ml of 4 M NaOH. The inverted plates were then placed inside a trigas incubator under microaerobic conditions (5% O2, 5% CO2, and 90% N2) at 30°C for 4 h. After a 4-hour incubation at 30°C and after 20 h total, the NaOH/NaCN was collected and measured directly using a cyanide micro-ion-selective electrode (Lazar Research Laboratories) connected to an Accument Basic AB15 pH meter (Fisher Scientific) set to read mVs. Values recorded from known concentrations of a KCN standard ranging from 1 × 10−5 to 1 M were used to generate a line of regression on a semilog graph. Unknown values were calculated using the slope and y intercept of this line. For the time course, cells were incubated with the NaOH for 4 hours prior to the collection time indicated. Bacterial cells were collected from the plates, total protein was determined by the Bradford assay (Bio-Rad) as previously described (13), and HCN production was reported as μmol HCN/mg protein.
Pseudomonas isolation liquid medium (2% peptone, 0.14% MgCl2, 1% K2SO4, 0.0025% Irgasan) broth cultures were grown in a sidearm flask with an attached enclosed chamber with shaking at 150 rpm under temperature and oxygen conditions identical to those of the plate-grown cultures. Four hours prior to the collection time point 1 ml of 4 M NaOH was added to the sidearm collection chambers to act as a cyanide trap. The micro-ion-selective electrode was used to measure the cyanide concentration in the NaOH/NaCN collected in the attached chamber and broth directly, and the concentrations were added together to obtain the total amount of cyanide produced in the broth culture. The cells were harvested by centrifugation, and total protein was determined as described above. Six biological replicates were performed for each strain. One-way analysis of variance (ANOVA) and the Tukey-Kramer multiple test were used for statistical analysis.
Optimization of oxygen concentration for P. aeruginosa HCN production.
PAO1 was grown on PIA plates aerobically for 16 h at 37°C. The plates were then inverted, individually sealed with lids containing 1 ml of 4 M NaOH, and incubated for 4 additional hours at 30°C in a trigas incubator under various oxygen conditions (1%, 3%, 5%, 7%, 10%, and 15% O2). The NaOH/NaCN and total protein were collected and measured as described above.
RESULTS
P. aeruginosa HCN production is surface attachment specific.
HCN is produced by P. aeruginosa during the transition from logarithmic to stationary growth phase at low oxygen tensions and high cell densities due to regulation of the hcnA promoter by the synergistic effects of the anaerobic regulator ANR and the quorum sensing regulators LasR and RhlR (14, 16, 62). Cyanide production is also under the control of the transcriptional regulator AlgR (13). RhlA and RhlI have recently been shown to be controlled by AlgR through an attachment-specific mechanism (58). A recent report showed increased cyanide production from P. aeruginosa grown in a static glass bead biofilm system (72). In order to determine if HCN production is increased during surface growth, HCN production by PAO1 was measured following growth of cells in broth and on agar media. Planktonic cultures of P. aeruginosa PAO1 produced a maximal amount of 0.94 μmol HCN/mg protein during the transition from mid-log to stationary phase (8 h) consistent with previous findings (14, 16) (Fig. 1A). Cultures grown on agar plates produced various amounts of HCN over the time course with an increase at 20 h to 1.5 μmol HCN/mg protein (Fig. 1B). These results show that surface-grown P. aeruginosa in stationary phase produces more HCN than do planktonically grown cells.
FIG. 1.

PAO1 HCN production in liquid and plate media and under different oxygen tensions. (A) HCN production (solid squares and solid line) by PAO1 in liquid Pseudomonas isolation broth and bacterial CFU/ml (solid triangles and dotted line) over time. (B) HCN production (solid squares and solid line) by PAO1 grown on PIA plates and bacterial CFU/ml (solid triangles and dotted line) over time. (C) HCN production under various oxygen tensions. Statistical significance was determined by one-way ANOVA and the Tukey-Kramer test. ***, P < 0.001.
Optimal oxygen tension for P. aeruginosa HCN production.
HCN is produced at low oxygen tensions during planktonic growth (14, 16), but the optimal oxygen concentration for surface-attached cells is unknown. To determine the range of oxygen concentrations that permit HCN production, P. aeruginosa was grown at various oxygen levels and HCN production was measured. PAO1 produced the maximum amount of HCN in a 5% oxygen environment (Fig. 1C). Consistent with previous studies, HCN production was greatly reduced as oxygen tensions neared anaerobic or aerobic conditions (14, 16).
Identification of previously unidentified hcnA transcriptional start site.
AlgR directly regulates HCN production in P. aeruginosa by controlling expression of hcnA from the T1 and T2 transcriptional start sites (13). The effects of algZ deletion on the transcription of the hcnA promoter were examined under microaerobic growth conditions by an S1 nuclease protection assay. This assay showed that AlgZ, like AlgR, is required for repression of hcnA transcription in a nonmucoid background (Fig. 2A). Interestingly, the assay also revealed an additional transcriptional start site (designated T3) upstream of the two previously identified sites (13, 62). The intensity of this band of protection indicates that the amount of transcript produced from the T3 site is equal to or greater than that of the T1 and T2 transcripts. The T3 start site is a G residue located −221 bp from the translational initiation codon.
FIG. 2.

The hcnA promoter is controlled by AlgZ and AlgR. (A) S1 nuclease protection assay of the hcnA promoter. Previously mapped T1 and T2 transcriptional start sites (indicated by respective labels and arrows) of the nonmucoid strain PAO1 and ΔalgR and ΔalgZ derivatives. The open triangle with the adjacent black line denotes the beginning of the common deletion region found upstream of the hcnA promoter. (B) S1 nuclease protection assay identifying T3 transcriptional start site. The arrow indicates the band of protection indicating the +1 start site. An asterisk denotes the first nucleotide in the antisense probe. (C) Sequence of the hcnA promoter region. The shaded sequence denotes the exoY deletion region. Binding sites are underlined for AlgR (AlgR Binding Site), ANR (ANR Box), RhlR and LasR (lux α and lux β), and ribosome (RBS). +1, HcnA translational start site; boldface nucleotides, hcnA open reading frame.
A naturally occurring deletion upstream of hcnA results in low cyanide production.
Previously, Wolfgang et al. identified a region upstream of hcnA in PAO1 (termed the exoY deletion region) that is absent in several other laboratory and clinical strains (83). The hcnA T3 transcriptional start site and previously identified AlgR and Lux β binding sites are all within this deleted region (Fig. 2B) (13, 62). Measurement of HCN production on the laboratory strain PAK, which features this chromosomal deletion, showed low levels of cyanide production (∼0.3 μmol/mg protein) in comparison to PAO1 (∼1.4 μmol/mg protein) (Fig. 3A) when measured under microaerobic conditions. Mutations in PAK algR, mucA2, or both did not affect the amount of cyanide produced by these isogenic strains (Fig. 3A). Sequence analysis of the hcnA promoter region in strains PA14 (HCN production = 0.4 μmol/mg protein) and PA103 (HCN production = 0.1 μmol/μg protein) revealed that they did not contain the hcnA T3 promoter either (data not shown). Insertion of the PAO1 hcnA promoter onto the PAK chromosome by homologous recombination restored HCN production to PAO1 wild-type levels in the PAK background (Fig. 3A). Moreover, cyanide measurements of blood isolate X13273, urinary tract infection isolate S54485, eye isolate 19660, and clinical isolate CF27, all previously shown to contain the same deletion region (83), revealed that these strains also produce very little HCN (Fig. 3B).
FIG. 3.
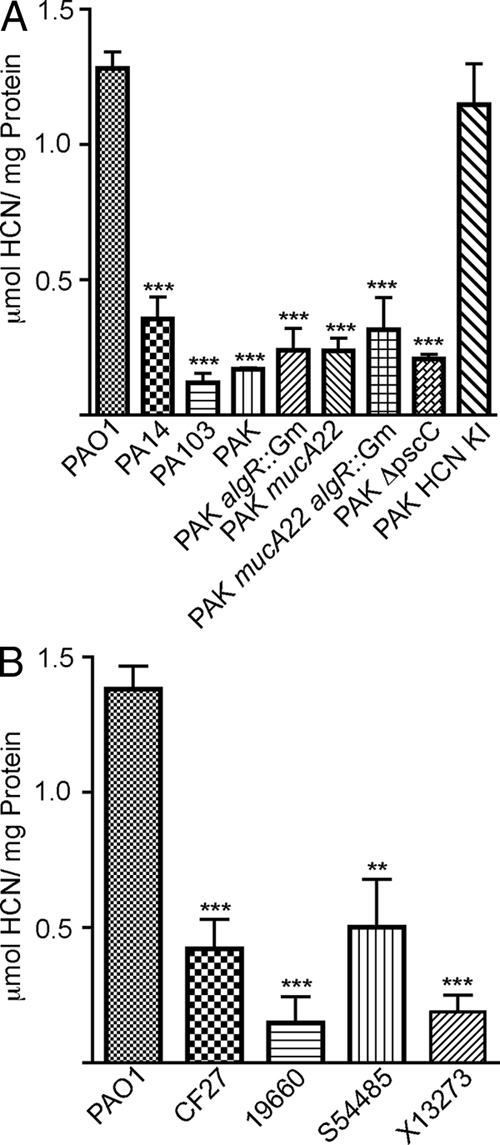
Strains with the exoY chromosomal deletion region upstream of hcnA produced decreased amounts of cyanide. (A) HCN production from nonmucoid wild-type PAO1 and laboratory strains with the exoY deletion region (PA14, PA103, and PAK), the PAK algR deletion (PAK algR::Gm), the PAK mucA22 mutation, the PAK mucA22 algR deletion, the PAK pscC deletion (PAK ΔpscC), and the PAK pscC deletion with the PAO1 hcnA promoter (PAK HCN KI). (B) HCN produced from PAO1, CF isolate CF27, eye isolate 19660, urinary tract infection isolate S54485, and blood isolate X13273. Statistical significance was determined by one-way ANOVA and the Tukey-Kramer test. **, P < 0.01, and ***, P < 0.001, compared to the isogenic parent.
AlgZ activates and represses HCN production in P. aeruginosa.
Previous analysis of HCN production in P. aeruginosa strains indicated that mucoid strains produce approximately sevenfold more HCN than do their nonmucoid parental strains. In-frame algR deletions in nonmucoid strains produced ∼3-fold more HCN and ∼1.5-fold less HCN in a mucoid background, showing that AlgR switches from a repressor to an activator of the hcnA promoter depending upon mucoid status (13). Both AlgR and AlgZ are involved in alginate production (19, 89) and required for twitching motility (5, 80, 81). An S1 nuclease protection assay indicated that AlgZ also plays a role in HCN regulation (Fig. 2A). Cyanide production levels from in-frame algZ deletion and insertional algZ mutants in both nonmucoid and mucoid backgrounds were measured under microaerobic conditions on agar plates. Mutations in algR, algZ, or both resulted in a ∼3-fold increase in HCN production in nonmucoid laboratory strains (Fig. 4A) compared to the wild type. In mucoid laboratory strains, mutations in algR, algZ, or both resulted in an ∼3.5-fold decrease in HCN production (Fig. 4B). In order to determine if the results observed with laboratory strains and their isogenic mutants are consistent with those for clinical isolates, HCN production was measured in mucoid and nonmucoid CF isolates and their isogenic algR and algZ mutants. Consistent with the results for the laboratory strains, mutations in algR or algZ resulted in an ∼1.5-fold increase in HCN production in nonmucoid clinical isolates (Table 3). In mucoid clinical isolates, mutations in algR or algZ resulted in a decrease in HCN production (Table 3). Additionally, complementation of the algZ mutant with the wild-type allele restored HCN production to wild-type levels in all strains (Fig. 4A and B). These results provide evidence that AlgZ works through AlgR to regulate HCN production.
FIG. 4.

AlgZ and AlgR control HCN production. HCN production for nonmucoid (A) and mucoid (B) algZ and algR mutants was measured as for Fig. 3. (A) PAO1, PSL317 (ΔalgR), PAZ (ΔalgZ), PAO6886 (algZ::Tc), PAO6886 harboring pUCP18Z (algZ::Tc + algZ), and PSL317Z (ΔalgR algZ::Tc). (B) PAO568, PAR568 (ΔalgR mucA2), PAZ568 (ΔalgZ mucA2), PAO6884 (algZ::Tc mucA2), PAO6884 harboring plasmid pUCP18Z (algZ::Tc mucA2 + algZ), and PAO6885 (algR::Gm algZ::Tc mucA2). Statistical significance was determined as for Fig. 3.
TABLE 3.
Effects of algR and algZ mutations in clinical isolates on HCN production
| Strain | Description or genotype | HCN production (mean μmol/μg protein ± SD) |
|---|---|---|
| TUMC92 | Clinical | 5.64 ± 0.64 |
| TUMC92R | ΔalgR | 9.30 ± 0.32 |
| TUMC92Z | ΔalgZ | 8.02 ± 0.69 |
| FRD-1 | Clinical | 7.60 ± 0.62 |
| FRD-1R | ΔalgR | 2.38 ± 0.47 |
| FRD-1Z | ΔalgZ | 2.89 ± 0.08 |
AlgZ and AlgR regulation of HCN production is surface attachment specific.
AlgZ and AlgR are both required for fimbrial biogenesis (80, 81) and affect alginate production (55, 56, 88). Both of these phenotypes are associated with biofilm formation and surface colonization (60, 78). Moreover, AlgR control of the rhlI and rhlA promoters has recently been shown to be surface attachment specific (58). In order to determine if AlgZ and AlgR regulation of HCN production was dependent on surface growth, cyanide assays were performed on nonmucoid and mucoid strains both grown in broth or on agar plates in stationary phase, 8 h for broth-grown strains and 20 h for plate-grown strains under microaerobic conditions (see Materials and Methods) (Fig. 1A and B). In agreement with our previous data, strains grown in broth produced approximately half (Table 4) the amount of HCN observed when the same strains were grown on plates (Fig. 4). In broth, neither algR nor algZ mutation had an effect on HCN production in either background (Table 4) whereas the same strains grown on agar plates resulted in threefold changes in HCN production (Fig. 4). Taken together, these results indicate that AlgZ/AlgR-dependent cyanide production was regulated through a surface-attachment-specific mechanism.
TABLE 4.
Effects of algR and algZ mutations in broth-grown strains on HCN production
| Strain | Genotype | HCN production (mean μmol/μg protein ± SD) |
|---|---|---|
| PAO1 | Wild type | 0.63 ± 0.05 |
| PSL317 | PAO1 ΔalgR | 0.55 ± 0.01 |
| PAZ | PAO1 ΔalgZ | 0.55 ± 0.01 |
| PAO568 | mucA2 | 0.55 ± 0.01 |
| PAR568 | PAO568 ΔalgR | 0.57 ± 0.01 |
| PAZ568 | PAO568 ΔalgZ | 0.63 ± 0.01 |
AlgR D54 is critical for HCN production.
The aspartate at position 54 of AlgR is highly conserved among two-component regulators and is able to accept phosphate from E. coli CheA in vitro (20, 50). An additional aspartate at position 85 is conserved in the AlgR homologues Staphylococcus aureus LytR and E. coli YehT but is not capable of accepting phosphorylation. AlgR D85 has been implicated in controlling the amount of pilin subunits exported (81). To determine the role of these conserved aspartates in HCN production, site-directed AlgR mutants containing D54N, D85N, or D54N D85N chromosomal alleles (81) were tested for their ability to produce HCN under microaerophilic conditions on PIA. These strains were made mucoid by mutating the mucA gene, and HCN production was measured from all isolates. Though the parental strain of these mutants, a PAO1 strain obtained from the Wozniak laboratory, produced less cyanide than did our PAO1 strain, the same trends for HCN production were observed for an algR mutant and an algZ mutant as with our PAO1 strain (data not shown). The AlgR D54N and AlgR D54N D85N mutants produced increased HCN levels in nonmucoid strains (∼2-fold [Fig. 5A]) while producing lower levels in mucoid (mucA::Tcr) strains (∼3-fold [Fig. 5B]). Similar to the results obtained from assays of twitching motility (81) and alginate production (50), the D85N mutant had no effect on HCN production. Deletion of algZ resulted in the same effects on HCN production as those found with the AlgR D54N mutant (Fig. 5). These data show that repression of HCN production in nonmucoid strains and activation in mucoid strains are both dependent on AlgZ and AlgR D54. Taken together, these data strongly suggest that phosphorylation of AlgR D54 is required for HCN regulation and that AlgZ is required for phosphorylation of AlgR. However, these data also indicate that phosphorylation of AlgR is not the mechanism responsible for its ability to switch from a repressor in nonmucoid P. aeruginosa to an activator in mucoid isolates for HCN production.
FIG. 5.
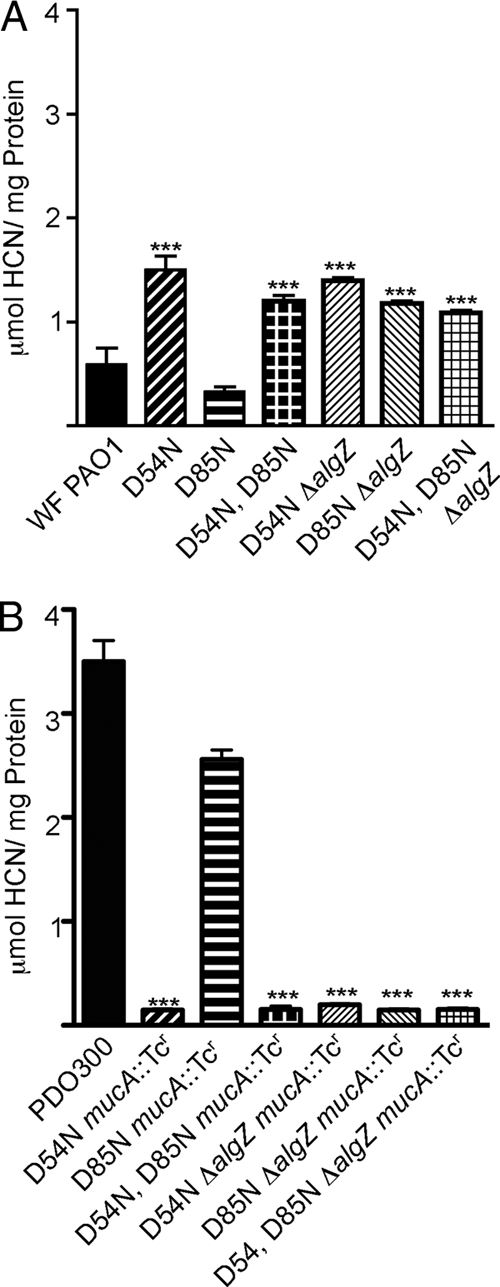
AlgR D54 is critical for HCN. (A) HCN production was measured from WFPAO1, WFPAO1 with chromosomal algR mutation D54N (AlgR D54N), WFPAO1 with chromosomal algR mutation D85N (AlgR D85N), WFPAO1 with double chromosomal algR mutations D54N and D85N (AlgR D54N D85N), WFPAO1 containing chromosomal algR mutation D54N and an algZ deletion (D54N ΔalgZ), and WFPAO1 containing chromosomal mutations in algR D54N and D85N and an algZ deletion (D54N D85N ΔalgZ). (B) HCN production from mucoid derivatives (mucA::Tc) of the same site-directed algR and algZ mutants as those in panel A. Statistical significance was determined as described for Fig. 3.
DISCUSSION
This study shows that mutations in algZ or algR affect HCN production in P. aeruginosa and that these effects are partially due to the phosphorylation state of AlgR. In addition, results shown here demonstrate that the hcnA promoter contains three transcriptional start sites with the most upstream T3 promoter being required for maximal activity. The hcnA promoter is controlled at transcriptional and posttranscriptional levels by many different regulators including ANR (62, 90), LasR and RhlR (62), GacA (7, 44, 61, 70), RsmA (10, 36, 44), and AlgR (13, 49). Two hcnA promoters have been previously reported for P. aeruginosa, termed T1 and T2 (62). T1 is regulated by quorum sensing alone, while T2 relies on a synergistic effect of LasR, RhlR, and ANR (62). In P. fluorescens, there are three RsmA/RsmE recognition elements that function as posttranscriptional regulators of the hcnA promoter by directly binding to the 5′ leader region (44). The chromosome of P. aeruginosa also contains rsmA, and deletion of this gene increased HCN production, suggesting that RsmA may function similarly in P. aeruginosa (63, 70). GacA also indirectly controls HCN production by positively regulating the rhlI promoter and hence amounts of N-butanoyl-homoserine-lactone (63, 70). The effects of algZ and algR deletion on hcnA transcription could be partially indirect, as AlgR represses rhlI transcription when P. aeruginosa is attached (58). However, AlgR has been shown to bind directly to the hcnA promoter in vitro, indicating that it controls HCN production directly (13). The most plausible explanation is that AlgR is interacting with other regulators (RhlR, ANR, or others) to control hcnA. Further studies are under way to determine the effects of AlgR, ANR, RsmA, LasR, and RhlR interactions on the hcnA promoter.
S1 nuclease protection assays (Fig. 2) established that there is one additional transcriptional start site upstream of the two previously reported. The hcnA′-′lacZ translational fusion used in a previous study that contained the T3 transcriptional start site showed higher β-galactosidase activity than did those fusions that lacked this promoter (62). The new start site is located in the exoY deletion region and is found in several laboratory and clinical strains (83). Results from Fig. 3 indicate that T3 may be at least as important as T1 and T2, as the absence of this 5′ region of the hcnA promoter resulted in extremely low levels of HCN production. These data suggest that the T3 promoter is responsible for a considerable amount of hcnA expression in P. aeruginosa. Interestingly, with the exception of PAK and CF27, most of the strains examined with this deletion region are exoU positive and exhibit increased type III secretion activity compared to PAO1, suggesting that only one of these virulence factors may be featured in a given strain (83).
It has been proposed that AlgZ acts as the sensor to AlgR in a two-component regulatory system (80, 89). The gene encoding AlgZ is directly upstream of algR, and both proteins share homology to two other sensor/regulator pairs, S. aureus lytS and lytR and E. coli yehU and yehT. In fact, AlgR, LytR, and AgrA compose a unique branch of the two-component system family, called the LytTR family (59). In this study we provide additional evidence that AlgZ acts as the AlgR sensor. Deletion of algZ resulted in the same phenotype as did an algR deletion and the site-directed algR(D54N) strain with respect to HCN production, strongly indicating that AlgZ acts through AlgR to control this promoter.
The AlgR response regulator responds to different environmental conditions depending upon the promoter examined. For instance, AlgR responds to nitrate concentrations on the algD promoter (57) and to surface contact or perhaps lowered oxygen tensions in control of the rhlI and rhlA promoters (58). HCN is not produced by P. aeruginosa under aerobic or anaerobic conditions (15) but is expressed under low oxygen tensions that range from 0.1 μM to 180 μM dissolved oxygen in liquid medium (14). This broad range of HCN production was not observed here (Fig. 1); however, the conditions used in this study were differing concentrations of atmospheric oxygen and not dissolved oxygen. The results presented here show that HCN production was maximal at 5% (1.5 μM O2) and is within the range previously reported. Part of the oxygen dependence can be explained by the requirement for the anaerobic regulator ANR for hcnA transcription (62, 90). However, it is not clear how or if AlgR and ANR interact directly. The current data support a model where AlgR is repressing hcnA and ANR is activating hcnA, in a nonmucoid background. In the mucoid background, the data indicate that there may be some synergy between these two transcriptional regulators to activate hcnA, as both are required for maximal hcnA expression.
As with AlgZ and AlgR control of the rhlI and rhlA promoters (58), changes in HCN production were not observed in liquid cultures of P. aeruginosa in strains containing mutations in algR or algZ. These results together suggest that AlgR may respond to either a microaerobic environment or surface attachment. Recently, Ryall et al. have shown that bacteria of the Burkholderia cepacia complex are cyanogenic under biofilm conditions (a glass bead system and agar plate) but not in liquid cultures (73). The results shown in Fig. 1A are in agreement with those studies. Cultures grown on agar have been described as open-air biofilms (41) and share many characteristics, including surface attachment, high cell densities, and coordinated cellular behavioral patterns (41, 75). Type IV pili could directly mediate either interaction and simultaneously act as a sensor for attachment and have been shown to affect gene transcription of lipC (53). Alternatively, surface growth could have a nonspecific effect. For example, surface growth could result in high local concentrations of signaling molecules (N-acylhomoserine lactones) or alterations in the local available O2 concentration, all things known to affect HCN production (14, 62, 63). In order to address this possibility, HCN was measured in a pilA strain that resulted in a slightly lower but not statistically significant amount of HCN compared to the amount produced by PAO1 (data not shown). Further studies are being performed to elucidate this potentially important mechanism.
P. aeruginosa has been proposed to grow in a biofilm in the CF lung, and this environment also contains the low oxygen tension and high cell density required for HCN production (77, 85). A steep oxygen gradient is present within secondary bronchi, reported as more than 70 times lower than the partial pressure of oxygen (pO2) found in the primary bronchi (85). Recently, reports have shown that HCN is detectable in the sputum and bronchoalveolar lavage fluid (12, 40, 71) of CF patients and is associated with impaired lung function. These reports are consistent with our previous data showing that 86% of CF isolates produced HCN (13) and with the data of others who have measured volatile compounds from CF sputum samples and detected HCN in vitro (12). Altogether, these data indicate that HCN may be an important virulence determinant for P. aeruginosa in CF.
Previous studies show that AlgZ and AlgR are both activators of twitching motility, with AlgZ acting in an AlgR-dependent manner. It has also been shown that AlgZ is a repressor of alginate production in the mucoid strain PAO568 (88), while AlgR acts as an activator (19, 55, 56). The conserved AlgR aspartate at position 54 does not play a role in alginate production in P. aeruginosa (50); however, it is required for twitching motility (81). The different phenotypes observed in alginate and twitching motility with respect to AlgR D54N suggest both a phosphorylation-dependent and a phosphorylation-independent mechanism for AlgR regulation. Analysis of HCN production by P. aeruginosa strains demonstrated that AlgR switches from a repressor to an activator of the hcnA promoter depending upon mucoid status (13). This study demonstrates that AlgR control of HCN production requires AlgR D54, indicating that some regulation occurs through the phosphorylation-dependent mechanism. In contrast, the AlgR switch from repressor to activator is still observed in the AlgR D54N site-directed mutants in comparisons of the nonmucoid to the mucoid background. These data provide evidence that phosphorylation is not the mechanism by which AlgR switches from a repressor to an activator in the mucoid background. Furthermore, these data suggest that AlgZ acts as a kinase in the microaerobic environment necessary for HCN production. Further investigation into the protein-protein interactions and posttranslational modification of AlgR is required to explain the repressor/activator switch.
Acknowledgments
We thank Daniel Wozniak for strains WFPAO1, WFPA8, WFPA13, and WFPA16.
This work was supported by National Institutes of Health grant RO1-AI050812 (M.J.S.), training grant 5 T32 AI052066-06 (W.L.C.), and Cystic Fibrosis Foundation Research grant WOLFGA06P0 (M.C.W.).
Footnotes
Published ahead of print on 6 March 2009.
REFERENCES
- 1.Anwar, P. W., J. L. Strap, and J. W. Costerton. 1992. Establishment of aging biofilms: possible mechanism of bacterial resistance to antimicrobial therapy. Antimicrob. Agents Chemother. 361347-1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baynham, P. J., A. L. Brown, L. L. Hall, and D. J. Wozniak. 1999. Pseudomonas aeruginosa AlgZ, a ribbon-helix-helix DNA-binding protein, is essential for alginate synthesis and algD transcriptional activation. Mol. Microbiol. 331069-1080. [DOI] [PubMed] [Google Scholar]
- 3.Baynham, P. J., D. M. Ramsey, B. V. Gvozdyev, E. M. Cordonnier, and D. J. Wozniak. 2006. The Pseudomonas aeruginosa ribbon-helix-helix DNA-binding protein AlgZ (AmrZ) controls twitching motility and biogenesis of type IV pili. J. Bacteriol. 188132-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baynham, P. J., and D. J. Wozniak. 1996. Identification and characterization of AlgZ, an AlgT-dependent DNA-binding protein required for Pseudomonas aeruginosa algD transcription. Mol. Microbiol. 2297-108. [DOI] [PubMed] [Google Scholar]
- 5.Belete, B., H. Lu, and D. J. Wozniak. 2008. Pseudomonas aeruginosa AlgR regulates type IV pilus biosynthesis by activating transcription of the fimU-pilVWXY1Y2E operon. J. Bacteriol. 1902023-2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bergen, G. A., and J. H. Shelhamer. 1996. Pulmonary infiltrates in the cancer patient. New approaches to an old problem. Infect. Dis. Clin. N. Am. 10297-325. [DOI] [PubMed] [Google Scholar]
- 7.Blumer, C., S. Heeb, G. Pessi, and D. Haas. 1999. Global GacA-steered control of cyanide and exoprotease production in Pseudomonas fluorescens involves specific ribosome binding sites. Proc. Natl. Acad. Sci. USA 9614073-14078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boat, T. F., M. J. Welsh, and A. L. Beaudet. 1989. Cystic fibrosis, p. 2649-2680. In C. R. Scriver, A. L. Beaudet, W. S. Sly, and D. Valle (ed.), The metabolic basis of inherited disease, 6th ed., vol. II. McGraw-Hill, New York, NY. [Google Scholar]
- 9.Bodey, G. P., R. Bolivar, V. Fainstein, and L. Jadeja. 1983. Infections caused by Pseudomonas aeruginosa. Rev. Infect. Dis. 5279-313. [DOI] [PubMed] [Google Scholar]
- 10.Burrowes, E., C. Baysse, C. Adams, and F. O'Gara. 2006. Influence of the regulatory protein RsmA on cellular functions in Pseudomonas aeruginosa PAO1, as revealed by transcriptome analysis. Microbiology 152405-418. [DOI] [PubMed] [Google Scholar]
- 11.Cabral, D. A., B. A. Loh, and D. P. Speert. 1987. Mucoid Pseudomonas aeruginosa resists nonopsonic phagocytosis by human neutrophils and macrophages. Pediatr. Res. 22429-431. [DOI] [PubMed] [Google Scholar]
- 12.Carroll, W., W. Lenney, T. Wang, P. Spanel, A. Alcock, and D. Smith. 2005. Detection of volatile compounds emitted by Pseudomonas aeruginosa using selected ion flow tube mass spectrometry. Pediatr. Pulmonol. 39452-456. [DOI] [PubMed] [Google Scholar]
- 13.Carterson, A. J., L. A. Morici, D. W. Jackson, A. Frisk, S. E. Lizewski, R. Jupiter, K. Simpson, D. A. Kunz, S. H. Davis, J. R. Schurr, D. J. Hassett, and M. J. Schurr. 2004. The transcriptional regulator AlgR controls cyanide production in Pseudomonas aeruginosa. J. Bacteriol. 1866837-6844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Castric, P. A. 1983. Hydrogen cyanide production by Pseudomonas aeruginosa at reduced oxygen levels. Can. J. Microbiol. 291344-1349. [DOI] [PubMed] [Google Scholar]
- 15.Castric, P. A. 1975. Hydrogen cyanide, a secondary metabolite of Pseudomonas aeruginosa. Can. J. Microbiol. 21613-618. [DOI] [PubMed] [Google Scholar]
- 16.Castric, P. A. 1994. Influence of oxygen on the Pseudomonas aeruginosa hydrogen cyanide synthase. Curr. Microbiol. 2919-21. [Google Scholar]
- 17.Choi, K. H., A. Kumar, and H. P. Schweizer. 2006. A 10-min method for preparation of highly electrocompetent Pseudomonas aeruginosa cells: application for DNA fragment transfer between chromosomes and plasmid transformation. J. Microbiol. Methods 64391-397. [DOI] [PubMed] [Google Scholar]
- 18.Clawson, B. J., and C. C. Young. 1913. Preliminary report on the production of hydrocyanic acid bacteria. J. Biol. Chem. 15419-422. [Google Scholar]
- 19.Deretic, V., R. Dikshit, W. M. Konyecsni, A. M. Chakrabarty, and T. K. Misra. 1989. The algR gene, which regulates mucoidy in Pseudomonas aeruginosa, belongs to a class of environmentally responsive genes. J. Bacteriol. 1711278-1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deretic, V., J. H. Leveau, C. D. Mohr, and N. S. Hibler. 1992. In vitro phosphorylation of AlgR, a regulator of mucoidy in Pseudomonas aeruginosa, by a histidine protein kinase and effects of small phospho-donor molecules. Mol. Microbiol. 62761-2767. [DOI] [PubMed] [Google Scholar]
- 21.Deretic, V., M. J. Schurr, and H. Yu. 1995. Pseudomonas aeruginosa, mucoidy and chronic infection phenotype in cystic fibrosis. Trends Microbiol. 3351-356. [DOI] [PubMed] [Google Scholar]
- 22.Di Sant'Agnese, P. A., and D. H. Andersen. 1946. Celiac syndrome: chemotherapy in infections of the respiratory tract associated with cystic fibrosis of the pancreas; observations with penicillin and drugs of the sulphonamide groups, with special reference to penicillin aerosol. Am. J. Dis. Child. 7217-61. [PubMed] [Google Scholar]
- 23.Doggett, R. G. 1969. Incidence of mucoid Pseudomonas aeruginosa from clinical sources. Appl. Microbiol. 18936-937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Doggett, R. G., G. M. Harrison, R. N. Stillwell, and E. S. Wallis. 1966. An atypical Pseudomonas aeruginosa associated with cystic fibrosis of the pancreas. J. Pediatr. 68215-221. [Google Scholar]
- 25.Donnenberg, M. S., and J. B. Kaper. 1991. Construction of an eae deletion mutant of enteropathogenic Escherichia coli by using a positive selection suicide vector. Infect. Immun. 594310-4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Enderby, B., D. Smith, W. Carroll, and W. Lenney. 2009. Hydrogen cyanide as a biomarker for Pseudomonas aeruginosa in the breath of children with cystic fibrosis. Pediatr. Pulmonol. 44142-147. [DOI] [PubMed] [Google Scholar]
- 27.Fichtenbaum, C. J., K. F. Woeltje, and W. G. Powderly. 1994. Serious Pseudomonas aeruginosa infections in patients infected with human immunodeficiency virus: a case-control study. Clin. Infect. Dis. 19417-422. [DOI] [PubMed] [Google Scholar]
- 28.Figurski, D. H., and D. R. Helinski. 1979. Replication of an origin-containing derivative of plasmid RK2 dependent on a plasmid function provided in trans. Proc. Natl. Acad. Sci. USA 761648-1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fyfe, J. A. M., and J. R. W. Govan. 1984. Chromosomal loci associated with antibiotic hypersensitivity in pulmonary isolates of Pseudomonas aeruginosa. J. Gen. Microbiol. 130825-834. [DOI] [PubMed] [Google Scholar]
- 30.Gallagher, L. A., and C. Manoil. 2001. Pseudomonas aeruginosa PAO1 kills Caenorhabditis elegans by cyanide poisoning. J. Bacteriol. 1836207-6214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gilligan, P. H. 1991. Microbiology of airway disease in patients with cystic fibrosis. Clin. Microbiol. Rev. 435-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goldberg, J. B., and D. E. Ohman. 1984. Cloning and expression in Pseudomonas aeruginosa of a gene involved in the production of alginate. J. Bacteriol. 1581115-1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goldfarb, W. B., and H. Margraf. 1967. Cyanide production by Pseudomonas aeruginosa. Ann. Surg. 165104-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Govan, J. R., and V. Deretic. 1996. Microbial pathogenesis in cystic fibrosis: mucoid Pseudomonas aeruginosa and Burkholderia cepacia. Microbiol. Rev. 60539-574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Govan, J. R., D. W. Martin, and V. P. Deretic. 1992. Mucoid Pseudomonas aeruginosa and cystic fibrosis: the role of mutations in muc loci. FEMS Microbiol. Lett. 79323-329. [DOI] [PubMed] [Google Scholar]
- 36.Heeb, S., C. Blumer, and D. Haas. 2002. Regulatory RNA as mediator in GacA/RsmA-dependent global control of exoproduct formation in Pseudomonas fluorescens CHA0. J. Bacteriol. 1841046-1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Higuchi, R. 1989. Using PCR to engineer DNA, p. 61-70. In H. A. Erlich (ed.), PCR technology: principles and applications for DNA amplification. Stockton Press, New York, NY.
- 38.Holloway, B. W. 1955. Genetic recombination in Pseudomonas aeruginosa. J. Gen. Microbiol. 13572-581. [DOI] [PubMed] [Google Scholar]
- 39.Holloway, B. W., V. Krishnapillai, and A. F. Morgan. 1979. Chromosomal genetics of Pseudomonas. Microbiol. Rev. 4373-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Julak, J., E. Stranska, V. Rosova, H. Geppert, P. Spanel, and D. Smith. 2006. Bronchoalveolar lavage examined by solid phase microextraction, gas chromatography-mass spectrometry and selected ion flow tube mass spectrometry. J. Microbiol. Methods 6576-86. [DOI] [PubMed] [Google Scholar]
- 41.Kolter, R., and E. P. Greenberg. 2006. Microbial sciences: the superficial life of microbes. Nature 441300-302. [DOI] [PubMed] [Google Scholar]
- 42.Konyecsni, W. M., and V. Deretic. 1988. Broad-host-range plasmid and M13 bacteriophage-derived vectors for promoter analysis in Escherichia coli and Pseudomonas aeruginosa. Gene 74375-386. [DOI] [PubMed] [Google Scholar]
- 43.Lapouge, K., M. Schubert, F. H. Allain, and D. Haas. 2008. Gac/Rsm signal transduction pathway of gamma-proteobacteria: from RNA recognition to regulation of social behaviour. Mol. Microbiol. 67241-253. [DOI] [PubMed] [Google Scholar]
- 44.Lapouge, K., E. Sineva, M. Lindell, K. Starke, C. S. Baker, P. Babitzke, and D. Haas. 2007. Mechanism of hcnA mRNA recognition in the Gac/Rsm signal transduction pathway of Pseudomonas fluorescens. Mol. Microbiol. 66341-356. [DOI] [PubMed] [Google Scholar]
- 45.Laville, J., C. Blumer, C. Von Schroetter, V. Gaia, G. Defago, C. Keel, and D. Haas. 1998. Characterization of the hcnABC gene cluster encoding hydrogen cyanide synthase and anaerobic regulation by ANR in the strictly aerobic biocontrol agent Pseudomonas fluorescens CHA0. J. Bacteriol. 1803187-3196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Learn, D. B., E. P. Brestel, and S. Seetharama. 1987. Hypochlorite scavenging by Pseudomonas aeruginosa alginate. Infect. Immun. 551813-1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu, P. V. 1973. Exotoxins of Pseudomonas aeruginosa. I. Factors that influence the production of exotoxin A. J. Infect. Dis. 128506-513. [DOI] [PubMed] [Google Scholar]
- 48.Lizewski, S. E., D. S. Lundberg, and M. J. Schurr. 2002. The transcriptional regulator AlgR is essential for Pseudomonas aeruginosa pathogenesis. Infect. Immun. 706083-6093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lizewski, S. E., J. R. Schurr, D. W. Jackson, A. Frisk, A. J. Carterson, and M. J. Schurr. 2004. Identification of AlgR-regulated genes in Pseudomonas aeruginosa by use of microarray analysis. J. Bacteriol. 1865672-5684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ma, S., U. Selvaraj, D. E. Ohman, R. Quarless, D. J. Hassett, and D. J. Wozniak. 1998. Phosphorylation-independent activity of the response regulators AlgB and AlgR in promoting alginate biosynthesis in mucoid Pseudomonas aeruginosa. J. Bacteriol. 180956-968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Manfredi, R., A. Nanetti, M. Ferri, and F. Chiodo. 2000. Pseudomonas spp. complications in patients with HIV disease: an eight-year clinical and microbiological survey. Eur. J. Epidemiol. 16111-118. [DOI] [PubMed] [Google Scholar]
- 52.Martin, D. W., M. J. Schurr, H. Yu, and V. Deretic. 1994. Analysis of promoters controlled by the putative sigma factor AlgU regulating conversion to mucoidy in Pseudomonas aeruginosa: relationship to sigma E and stress response. J. Bacteriol. 1766688-6696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Martinez, A., P. Ostrovsky, and D. N. Nunn. 1999. LipC, a second lipase of Pseudomonas aeruginosa, is LipB and Xcp dependent and is transcriptionally regulated by pilus biogenesis components. Mol. Microbiol. 34317-326. [DOI] [PubMed] [Google Scholar]
- 54.Mathee, K., O. Ciofu, C. Sternberg, P. W. Lindum, J. I. Campbell, P. Jensen, A. H. Johnsen, M. Givskov, D. E. Ohman, S. Molin, N. Hoiby, and A. Kharazmi. 1999. Mucoid conversion of Pseudomonas aeruginosa by hydrogen peroxide: a mechanism for virulence activation in the cystic fibrosis lung. Microbiology 1451349-57. [DOI] [PubMed] [Google Scholar]
- 55.Mohr, C. D., N. S. Hibler, and V. Deretic. 1991. AlgR, a response regulator controlling mucoidy in Pseudomonas aeruginosa, binds to the FUS sites of the algD promoter located unusually far upstream from the mRNA start site. J. Bacteriol. 1735136-5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mohr, C. D., J. H. Leveau, D. P. Krieg, N. S. Hibler, and V. Deretic. 1992. AlgR-binding sites within the algD promoter make up a set of inverted repeats separated by a large intervening segment of DNA. J. Bacteriol. 1746624-6633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mohr, C. D., D. W. Martin, W. M. Konyecsni, J. R. Govan, S. Lory, and V. Deretic. 1990. Role of the far-upstream sites of the algD promoter and the algR and rpoN genes in environmental modulation of mucoidy in Pseudomonas aeruginosa. J. Bacteriol. 1726576-6580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Morici, L. A., A. J. Carterson, V. E. Wagner, A. Frisk, J. R. Schurr, K. H. Zu Bentrup, D. J. Hassett, B. H. Iglewski, K. Sauer, and M. J. Schurr. 2007. Pseudomonas aeruginosa AlgR represses the Rhl quorum-sensing system in a biofilm-specific manner. J. Bacteriol. 1897752-7764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nikolskaya, A. N., and M. Y. Galperin. 2002. A novel type of conserved DNA-binding domain in the transcriptional regulators of the AlgR/AgrA/LytR family. Nucleic Acids Res. 302453-2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.O'Toole, G. A., and R. Kolter. 1998. Flagellar and twitching motility are necessary for Pseudomonas aeruginosa biofilm development. Mol. Microbiol. 30295-304. [DOI] [PubMed] [Google Scholar]
- 61.Pessi, G., and D. Haas. 2001. Dual control of hydrogen cyanide biosynthesis by the global activator GacA in Pseudomonas aeruginosa PAO1. FEMS Microbiol. Lett. 20073-78. [DOI] [PubMed] [Google Scholar]
- 62.Pessi, G., and D. Haas. 2000. Transcriptional control of the hydrogen cyanide biosynthetic genes hcnABC by the anaerobic regulator ANR and the quorum-sensing regulators LasR and RhlR in Pseudomonas aeruginosa. J. Bacteriol. 1826940-6949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pessi, G., F. Williams, Z. Hindle, K. Heurlier, M. T. Holden, M. Camara, D. Haas, and P. Williams. 2001. The global posttranscriptional regulator RsmA modulates production of virulence determinants and N-acylhomoserine lactones in Pseudomonas aeruginosa. J. Bacteriol. 1836676-6683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pier, G. B., G. J. Small, and H. B. Warren. 1990. Protection against mucoid Pseudomonas aeruginosa in rodent models of endobronchial infections. Science 249537-540. [DOI] [PubMed] [Google Scholar]
- 65.Pilewski, J. M., and R. A. Frizzell. 1999. Role of CFTR in airway disease. Physiol. Rev. 79S215-S255. [DOI] [PubMed] [Google Scholar]
- 66.Prabhakaran, K., L. Li, J. L. Borowitz, and G. E. Isom. 2004. Caspase inhibition switches the mode of cell death induced by cyanide by enhancing reactive oxygen species generation and PARP-1 activation. Toxicol. Appl. Pharmacol. 195194-202. [DOI] [PubMed] [Google Scholar]
- 67.Prabhakaran, K., L. Li, J. L. Borowitz, and G. E. Isom. 2002. Cyanide induces different modes of death in cortical and mesencephalon cells. J. Pharmacol. Exp. Ther. 303510-519. [DOI] [PubMed] [Google Scholar]
- 68.Rahme, L. G., E. J. Stevens, S. F. Wolfort, J. Shao, R. G. Tompkins, and F. M. Ausubel. 1995. Common virulence factors for bacterial pathogenicity in plants and animals. Science 2681899-1902. [DOI] [PubMed] [Google Scholar]
- 69.Ramsey, D. M., and D. J. Wozniak. 2005. Understanding the control of Pseudomonas aeruginosa alginate synthesis and the prospects for management of chronic infections in cystic fibrosis. Mol. Microbiol. 56309-322. [DOI] [PubMed] [Google Scholar]
- 70.Reimmann, C., M. Beyeler, A. Latifi, H. Winteler, M. Foglino, A. Lazdunski, and D. Haas. 1997. The global activator GacA of Pseudomonas aeruginosa PAO positively controls the production of the autoinducer N-butyryl-homoserine lactone and the formation of the virulence factors pyocyanin, cyanide, and lipase. Mol. Microbiol. 24309-319. [DOI] [PubMed] [Google Scholar]
- 71.Ryall, B., J. C. Davies, R. Wilson, A. Shoemark, and H. D. Williams. 2008. Pseudomonas aeruginosa, cyanide accumulation and lung function in cystic fibrosis and non-cystic fibrosis bronchiectasis patients. Eur. Respir. J. 32740-747. [DOI] [PubMed] [Google Scholar]
- 72.Reference deleted.
- 73.Ryall, B., X. Lee, J. E. Zlosnik, S. Hoshinoa, and H. D. Williams. 2008. Bacteria of the Burkholderia cepacia complex are cyanogenic under biofilm and colonial growth conditions. BMC Microbiol. 8108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schwarzmann, S., and J. R. Boring. 1971. Antiphagocytic effect of slime from a mucoid strain of P. aeruginosa. Infect. Immun. 3762-767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shapiro, J. A. 1998. Thinking about bacterial populations as multicellular organisms. Annu. Rev. Microbiol. 5281-104. [DOI] [PubMed] [Google Scholar]
- 76.Simpson, J. A., S. E. Smith, and R. T. Dean. 1989. Scavenging by alginate of free radicals released by macrophages. Free Radic. Biol. Med. 6347-353. [DOI] [PubMed] [Google Scholar]
- 77.Singh, P. K., A. L. Schaefer, M. R. Parsek, T. O. Moninger, M. J. Welsh, and E. P. Greenberg. 2000. Quorum-sensing signals indicate that cystic fibrosis lungs are infected with bacterial biofilms. Nature 407762-764. [DOI] [PubMed] [Google Scholar]
- 78.Stapper, A. P., G. Narasimhan, D. E. Ohman, J. Barakat, M. Hentzer, S. Molin, A. Kharazmi, N. Hoiby, and K. Mathee. 2004. Alginate production affects Pseudomonas aeruginosa biofilm development and architecture, but is not essential for biofilm formation. J. Med. Microbiol. 53679-690. [DOI] [PubMed] [Google Scholar]
- 79.West, S. E., H. P. Schweizer, C. Dall, A. K. Sample, and L. J. Runyen-Janecky. 1994. Construction of improved Escherichia-Pseudomonas shuttle vectors derived from pUC18/19 and sequence of the region required for their replication in Pseudomonas aeruginosa. Gene 14881-86. [DOI] [PubMed] [Google Scholar]
- 80.Whitchurch, C. B., R. A. Alm, and J. S. Mattick. 1996. The alginate regulator AlgR and an associated sensor FimS are required for twitching motility in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 939839-9843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Whitchurch, C. B., T. E. Erova, J. A. Emery, J. L. Sargent, J. M. Harris, A. B. Semmler, M. D. Young, J. S. Mattick, and D. J. Wozniak. 2002. Phosphorylation of the Pseudomonas aeruginosa response regulator AlgR is essential for type IV fimbria-mediated twitching motility. J. Bacteriol. 1844544-4554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wissing, F. 1974. Cyanide formation from oxidation of glycine by a Pseudomonas species. J. Bacteriol. 1171289-1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wolfgang, M. C., B. R. Kulasekara, X. Liang, D. Boyd, K. Wu, Q. Yang, C. G. Miyada, and S. Lory. 2003. Conservation of genome content and virulence determinants among clinical and environmental isolates of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 1008484-8489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wolfgang, M. C., V. T. Lee, M. E. Gilmore, and S. Lory. 2003. Coordinate regulation of bacterial virulence genes by a novel adenylate cyclase-dependent signaling pathway. Dev. Cell 4253-263. [DOI] [PubMed] [Google Scholar]
- 85.Worlitzsch, D., R. Tarran, M. Ulrich, U. Schwab, A. Cekici, K. C. Meyer, P. Birrer, G. Bellon, J. Berger, T. Weiss, K. Botzenhart, J. R. Yankaskas, S. Randell, R. C. Boucher, and G. Doring. 2002. Effects of reduced mucus oxygen concentration in airway Pseudomonas infections of cystic fibrosis patients. J. Clin. Investig. 109317-325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wozniak, D. J., A. B. Sprinkle, and P. J. Baynham. 2003. Control of Pseudomonas aeruginosa algZ expression by the alternative sigma factor AlgT. J. Bacteriol. 1857297-7300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wu, W., H. Badrane, S. Arora, H. V. Baker, and S. Jin. 2004. MucA-mediated coordination of type III secretion and alginate synthesis in Pseudomonas aeruginosa. J. Bacteriol. 1867575-7585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yu, H., J. C. Boucher, N. S. Hibler, and V. Deretic. 1996. Virulence properties of Pseudomonas aeruginosa lacking the extreme-stress sigma factor AlgU (σE). Infect. Immun. 642774-2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yu, H., M. Mudd, J. C. Boucher, M. J. Schurr, and V. Deretic. 1997. Identification of the algZ gene upstream of the response regulator AlgR and its participation in control of alginate production in Pseudomonas aeruginosa. J. Bacteriol. 179187-193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zimmermann, A., C. Reimmann, M. Galimand, and D. Haas. 1991. Anaerobic growth and cyanide synthesis of Pseudomonas aeruginosa depend on anr, a regulatory gene homologous with fnr of Escherichia coli. Mol. Microbiol. 51483-1490. [DOI] [PubMed] [Google Scholar]
- 91.Zlosnik, J. E., and H. D. Williams. 2004. Methods for assaying cyanide in bacterial culture supernatant. Lett. Appl. Microbiol. 38360-365. [DOI] [PubMed] [Google Scholar]