

Abstract
We characterized the nanLET operon in Bacteroides fragilis, whose products are required for the utilization of the sialic acid N-acetyl neuraminic acid (NANA) as a carbon and energy source. The first gene of the operon is nanL, which codes for an aldolase that cleaves NANA into N-acetyl mannosamine (manNAc) and pyruvate. The next gene, nanE, codes for a manNAc/N-acetylglucosamine (NAG) epimerase, which, intriguingly, possesses more similarity to eukaryotic renin binding proteins than to other bacterial NanE epimerase proteins. Unphosphorylated manNAc is the substrate of NanE, while ATP is a cofactor in the epimerase reaction. The third gene of the operon is nanT, which shows similarity to the major transporter facilitator superfamily and is most likely to be a NANA transporter. Deletion of any of these genes eliminates the ability of B. fragilis to grow on NANA. Although B. fragilis does not normally grow with manNAc as the sole carbon source, we isolated a B. fragilis mutant strain that can grow on this substrate, likely due to a mutation in a NAG transporter; both manNAc transport and NAG transport are affected in this strain. Deletion of the nanE epimerase gene or the rokA hexokinase gene, whose product phosphorylates NAG, in the manNAc-enabled strain abolishes growth on manNAc. Thus, B. fragilis possesses a new pathway of NANA utilization, which we show is also found in other Bacteroides species.
Many bacteria have the ability to release sialic acids from complex glycoproteins and oligosaccharides present in the media or on cell surfaces at sites of colonization or infection. To use the released sialic acids as a rich source of carbon and nitrogen for growth, bacteria must have the ability to transport these compounds into the cell and convert the nine carbon sugars into intermediates that enter the central glycolytic pathways. The utilization of N-acetyl neuraminic acid (NANA), one of the sialic acids, has been well studied in Escherichia coli (36, 37), Haemophilus spp. (1, 35), and Clostridium spp. (38), to name a few.
In many microorganisms, the genes for NANA utilization are arranged in an operon that may be regulated by a repressor protein, termed NanR. A comprehensive review of the organization and composition of several prokaryotic operons involved in NANA utilization has been published (36). Many of these operons share common components, including a transport gene for NANA (nanT), a gene encoding an aldolase (nanA) that splits NANA into pyruvate and N-acetyl mannosamine (manNAc), a gene encoding a kinase activity (nanK) that phosphorylates manNAc to form manNAc 6-P and, finally, an epimerase gene (nanE) whose product converts manNAc 6-P to N-acetylglucosamine 6-P (NAG 6-P). NAG 6-P then enters the common pathway of aminosugar utilization (21). For a schematic of the NANA utilization pathway in E. coli, the current paradigm of prokaryotic NANA utilization, see Fig. 7A.
FIG. 7.

Comparison of the NANA and amino sugar utilization pathways of E. coli (A) and B. fragilis (B). (A) Pathway adapted from Vimr et al. (36). GlcN, d-glucosamine. (B) Enzymes in boldface have been assayed by our laboratory. The NagA reaction has not been experimentally verified by us; however, there is an annotated sequence of such a gene in the B. fragilis 638R partial genome sequence. X designates the transporter, probably a NAG transporter, that contains the enabling mutation present in the strain ADB77M.
Bacteroides fragilis possesses a neuraminidase activity, which can liberate free NANA from complex glycoproteins and oligosaccharides. Godoy et al. (11) have established a role for this activity in the ability of B. fragilis to proliferate in two in vivo model systems of infection. In the present study, we establish the NANA catabolic pathway in B. fragilis, define a three-gene nanLET, NANA utilization operon (Fig. 1), and demonstrate the regulation of this operon by the B. fragilis NanR protein. Moreover, we demonstrate a new pathway for manNAc utilization in B. fragilis.
FIG. 1.

Schematic of the nanLET/nanR operon. The nanR gene product is a ROK family repressor protein. The nanL gene product is a NANA lyase (aldolase). The nanE gene product possesses similarity to the mammalian RnBP, also known to be an N-acetylglucosamine 2-epimerase. The nanT gene product is a transport protein of the major facilitator superfamily. The hyp gene encodes a hypothetical protein. Relevant deletion constructs and the schematics of the deletions are listed.
MATERIALS AND METHODS
Reagents.
All chemicals used in the present study were obtained from Sigma Chemical Co. (St. Louis, MO) or Fisher Scientific Co. (Agawam, MA) unless otherwise specified. Oligonucleotide primers were synthesized by IDT (Coralville, IA). NANA was purchased from Nacalai Tesque (Kyoto, Japan). The radiolabeled amino sugar [3H]manNAc (specific activity, 20 Ci/mmol) was purchased from American Radiolabeled Chemical Co. (ARC Co., St. Louis, MO). [14C]manNAc (specific activity, 15 Ci/mmol) and [14C]NANA (specific activity, 55 mCi/mmol) were generous gifts from Eric Vimr (University of Illinois, Champaign-Urbana).
Bacterial strains, plasmids, and growth conditions.
Bacterial strains and plasmids used in the present study are described in Table 1. B. fragilis cells were grown in brain heart infusion broth (BHIS) supplemented with 0.5% yeast extract and 15 μg of hematin/ml (33) in an anaerobic chamber (Coy Laboratory Products) at 37°C with an atmosphere of 85% N2, 10% H2, and 5% CO2 (Airgas East). Strains deficient in glucose utilization were grown on BHIS with the addition of 0.5% (wt/vol) galactose. Thymine (200 μg/ml) was added for growth of thyA mutant strains. E. coli strain DH5α (39) was used for cloning, and HB101/RK231 (12) was used for mobilization of plasmids from DH5α to B. fragilis, as previously described (33). E. coli cells were grown in Luria broth (Difco) at 37°C. Ampicillin (100 μg/ml), tetracycline (2 μg/ml for B. fragilis, 10 μg/ml for E. coli), chloramphenicol (25 μg/ml), rifampin (50 μg/ml), gentamicin (50 μg/ml), kanamycin (50 μg/ml), trimethoprim (80 μg/ml), and erythromycin (8 μg/ml) were used as indicated.
TABLE 1.
Bacterial strains and plasmids
| Strain or plasmid | Relevant genotype or descriptiona | Source or referenceb |
|---|---|---|
| Strains | ||
| B. fragilis | ||
| ADB77 | TM4000, ΔthyA; Tpr | 3 |
| CJB100 | ADB77:ΔrokA1 | 6 |
| ADB77M | ADB77:manNAc growth enabled+ | This study |
| MD100 | ADB77:ΔnanL1 | This study |
| RC201 | ADB77:ΔnanE1 | This study |
| RC140 | ADB77:ΔnanT1 | This study |
| RC122 | ADB77:ΔnanR1 | This study |
| ADB77MΔL1 | ADB77M:ΔnanL1 | This study |
| ADB77MΔE | ADB77M:ΔnanE1 | This study |
| ADB77MΔT | ADB77M:ΔnanT1 | This study |
| ADB77MΔrok | ADB77M:ΔrokA1 | This study |
| RC122T | RC122:ΔnanT | This study |
| ADB77::pRAG210 | ADB77, disruption of the nanL open reading frame with pRAG210 | This study |
| B. thetaiotaomicron | ||
| VPI 5482 | ΔnanL nanE+ ΔnanT rokA+ | VPI |
| TAL21586 | Clinical B. thetaiotaomicron isolate | TAL |
| E. coli | ||
| DH5α | λ nonlysogen; Cms Amps | 39 |
| HB101 | rpsL20; Tets Amps | 33 |
| BL21 | F−hsdSB (rB− mB−) gal dcm rne131 | Invitrogen |
| Plasmids | ||
| RK231 | RP4 derivative, Tetr Tra+ Amps | 12 |
| pYT102 | p15A ori; Cmr; RP4 oriT, B. fragilis suicide vector containing B. fragilis thyA+; Tetr | 32 |
| pLITMUS29 | pUC19 ori; M13 ori; Ampr; lacZα | NEB |
| pKDel12 | pYT102 ΔrokA | 6 |
| pMBD1 | pLITMUS29 containing the 3′ fragment of ΔnanL1 | This study |
| pMBD2 | pLITMUS29 containing the 5′ fragment of ΔnanL1 | This study |
| pMBD4 | pLITMUS29 ΔnanL1 | This study |
| pMBD5 | pYT102 ΔnanL1 | This study |
| pRC150 | pYT102 ΔnanT1 | This study |
| pRC200 | pYT102 ΔnanE1 | This study |
| pRC120 | pYT102 ΔnanR1 | This study |
| pRAG210 | pYT102 containing a 852-bp internal fragment of the nanL gene | This study |
| pET24 | pBR322 ori lacI; Kanr | Invitrogen |
| pRC4-3 | pET24 containing nanEHis6 | This study |
Cm, chloramphenicol; Tet, tetracycline; Amp, ampicillin; Tp, trimethoprim; Kan, kanamycin; s, sensitive; r, resistant.
VPI, Virginia Polytechnic Institute, Blacksburg, VA; TAL, Tufts Anaerobic Laboratory, Boston, MA; NEB, New England Biolabs, Beverly, MA.
For B. fragilis growth experiments, overnight cultures in BHIS broth containing thymine, were harvested by centrifugation, washed twice with modified phosphate-buffered saline (MPBS), resuspended in MPBS and used to inoculate SAMM broth, a defined minimal medium (32) to a starting A600 of ∼0.05. Growth was monitored by monitoring the absorbance at 600 nm. All minimal medium cultures containing NANA as the main carbon source were supplemented with 0.02% glucose or galactose to allow for adaptation to growth on NANA.
Sugar accumulation studies. (i) [3H]manNAc accumulation.
Cells were grown anaerobically at 37°C in SAMM broth containing thymine and 0.5% manNAc or containing manNAc and 0.5% xylose and then harvested at an A600 of 0.5, washed twice with MPBS, and resuspended in fresh SAMM broth with thymine and 0.5% xylose. [3H]manNAc was added to a final concentration of 4 mM (specific activity, 0.25 μCi/mmol), followed by incubation of the cells anaerobically at 37°C for 15 min with agitation. A 1-ml aliquot was harvested on nitrocellulose filters (0.22-μm pore size; Millipore, Inc., Bedford, MA) and washed with 3 ml of fresh SAMM broth with no additions. Cell-associated radioactivity was determined with a Beckman model LS5000TD scintillation counter. To determine the protein concentration, a separate aliquot was removed before the addition of labeled substrate, sonicated for 45 s using a Branson model 250 sonifier, and then used with Bio-Rad (Hercules, CA) protein assay reagent according to the manufacturer's instructions. The total accumulation of [3H]manNAc by each strain was expressed as nmol/mg of whole-cell protein.
(ii) [3H]NAG accumulation.
[3H]NAG accumulation was measured as described above, except that cells were grown in SAMM broth containing thymine and 0.5% NAG and then incubated with 4 mM [3H]NAG (specific activity, 0.25 μCi/mmol). The inhibition of [3H]NAG accumulation by unlabeled manNAc and the inhibition of [3H]manNAc accumulation by unlabeled NAG were performed as described above, except a range of inhibitor concentrations (4 to 40 mM) were examined. The kinetics of inhibition were determined by analysis of Lineweaver-Burke plots or Dixon plots (10).
(iii) [14C]NANA accumulation.
Cells grown in SAMM broth with the addition of thymine and 0.5% xylose were prepared as described above. [14C]NANA was added to a final concentration of 10 μM (specific activity, 10 μCi/mmol), and the cells were incubated anaerobically at 37°C for up to 30 min with agitation. At several time points, 0.2-ml aliquots of cells were harvested on nitrocellulose filters, and the cell-associated radioactivity was determined. The total accumulation of [14C]NANA by each strain was expressed as nmol/mg of whole-cell protein.
Cloning, DNA sequencing and analysis, and strain manipulation.
Details of all cloning and strain manipulations can be found in the supplemental material.
Purification of an oligohistidine-tagged NanE protein.
A description of the purification procedures can be found in the supplemental material.
Enzyme assays.
A description of the NANA lyase assay as used in the present study can be found in the supplemental material (7).
The NanE activity in B. fragilis cell extracts (50 to 100 μg of total protein per reaction) was detected by chromatographic separation of the substrate manNAc from reaction products. Assays were performed with 25 mM unlabeled manNAc in 0.05 M Tris (pH 8.0) and 10 mM MgCl2 in the presence or absence of ATP (30 mM final concentration) in a final volume of 15 μl. Assay mixtures were incubated at 37°C for 1 h and then heat inactivated at 70°C for 20 min. A 10-μl aliquot of the reaction mixture was spotted onto 1% sodium borate-treated Whatman 3MM filter paper and allowed to dry. Descending paper chromatography was then performed as described previously (26), using butanol-pyridine-H2O (6:4:3) as the solvent. Chromatograms were baked at 95°C for 5 min, developed using Ehrlich's reagent, and then baked at 50°C for 10 min or until colored spots appeared. Standards of NAG6-P, NAG, and manNAc were run in parallel.
The activity of the purified NanE6HIS was measured as described above. To each reaction, a total of 5 μg of NanEHis6 was added to 5 mM unlabeled manNAc and 0.1 μCi of [14C]manNAc (final specific activity, 0.8 mCi/mmol) in 0.05 M Tris-10 mM MgCl2 (pH 8.0), in the presence or absence of ATP (30 mM final concentration), or the nonhydrolyzable adenosine 5′-O-3-thiotriphosphate (ATP-γ-S) (20 mM final concentration). Descending paper chromatography was performed as described above. After drying, the chromatograms were exposed on a Kodak MD146-931 phosphor screen for 20 to 30 min and scanned on a Storm 850 PhosphorImager. Spot intensities were quantified by using ImageQuant 1.2 imaging software (Molecular Dynamics).
To measure kinetic parameters of the NanE enzymatic reaction, a coupled assay that included purified RokA kinase, pyruvate kinase, and lactate dehydrogenase was used. The reaction mixture contained ATP (2 mM), NADH (0.6 mM), phosphoenolpyruvate (5 mM), 1 μl of pyruvate kinase (from rabbit muscle [EC 2.7.1.40], 1,351 U of activity/ml), 5 μl of lactate dehydrogenase (12,500 U/ml), and 1 μl of purified RokA protein (0.75 μg [6]) in an assay buffer consisting of 0.1 M Tris-HCl (pH 7.0) and 0.01 M MgCl2. Purified NanEHis6 protein and manNAc (final concentrations ranged from 33.5 μM to 13.4 mM from a 0.67 M stock solution) were then added, and the reaction was monitored by measuring the decrease in the A340 as NAD+ was formed from NADH at 25°C. All auxiliary enzymes were present in excess. The Km and Vmax of NanE were determined by analysis of a double reciprocal (Lineweaver-Burk) plot.
Phylogenetic analyses.
Sequences of the nanL, nanA, and its homologs and of nanE and its homologs were aligned by using the CLUSTAL W method. Phylogenetic trees were calculated by using MEGA 3.1 or MEGA 4.0 (16) using the neighbor-joining algorithm. The confidence limits were estimated by using 500 bootstrapping replicates. Phylogenetic trees were also calculated by using the minimum evolution algorithm, and the confidence limits were estimated by using 500 bootstrapping replicates.
RESULTS
Identification of an operon encoding genes required for NANA utilization.
The action of B. fragilis NanH on sialic acid-containing oligosaccharides and glycoproteins produces free NANA which B. fragilis can use as the main carbon and energy source in minimal medium (Fig. 2A). We used a genomics approach to find additional genes required for NANA catabolism. We hypothesized that these genes would be regulated by the presence of NANA in the medium and might share upstream regulatory sequences. Thus, we used the DNA sequences upstream of the nanH gene (25) to search the B. fragilis NCTC 9343 and the unfinished B. fragilis 638R genome databases for matches. Indeed, a matching region was found just upstream of a gene cluster whose protein products showed similarities to proteins involved in NANA catabolism in other organisms. The B. fragilis gene cluster contains a gene encoding a NANA lyase homolog (nanL), followed by nanE, encoding a homolog of the mammalian renin binding protein (RnBP); a nanT homolog, based on its similarity to a transporter of the major facilitator superfamily; and a 444-bp open reading frame that encodes a hypothetical protein.
FIG. 2.

Growth phenotypes of nanR and nanLET gene knockout strains, and manNAc-utilizing strains. (A) Strains were grown in SAMM broth containing thymine, 0.5% NANA, and 0.02% glucose as described in Materials and Methods. Strains: ADB77 (wild type, ▪) and RC122 (ΔnanR, ▴), ΔL1 (ΔnanL, ▾), RC201 (ΔnanE, ⧫), RC140 (ΔnanT, •). (B) Growth on manNAc. Strains were inoculated in SAMM with thymine and 0.5% manNAc as described in Materials and Methods. Strains: ADB77 (▪), ADB77M (▴), ADB77MΔL1 (▾), ADB77MΔT (•), ADB77MΔE (⧫). (C) Growth of manNAc-utilizing strain and derivatives on NANA. Symbols represent the same strains as in panel B. Strains were inoculated into SAMM with thymine, 0.5% NANA, and 0.02% glucose as described in Materials and Methods. All curves shown here are representative of three separate growth experiments.
B. fragilis NanR is a negative regulator of NANA utilization gene expression.
A gene encoding a predicted regulatory protein, with similarities to many ROK family regulators (Fig. 1), is located 297 bp upstream of and divergent to this nanLET cluster. We refer to this gene as nanR (R. Caughlan et al., unpublished data). An in-frame deletion of the nanR gene was used to examine whether NanR plays a role in NANA utilization, as well as in the regulation of the nanLET operon. Since the nanR deletion strain RC122 grows in minimal medium with NANA as the principal carbon source (Fig. 2A), the B. fragilis NanR protein, like the E. coli NanR protein (15), is not a positive regulator of the NANA utilization genes. Extracts of RC122 grown with different carbon sources were assayed for NANA lyase expression by coupling the removal of pyruvate produced during the lyase reaction to a lactate dehydrogenase reaction in the presence of NADH. Figure 3 shows that RC122 extracts from any of the growth conditions tested exhibited constitutive levels of lyase activity; compare this to extracts from wild-type cells that show elevated activity only if the strain is grown in the presence of NANA. This result establishes that NanR is a negative regulator of the nanLET operon.
FIG. 3.

NANA lyase (aldolase) activities of B. fragilis strains. All strains were grown in SAMM broth with thymine and 0.5% of the indicated sugar. Extracts were prepared from cells and NANA lyase (aldolase) activity was measured as indicated in the supplemental material. ADB77 (wild type) cells were grown in xylose (X), glucose (G), and NANA (N). ADB77M cells were grown in glucose, NANA, and manNAc (M). RC122 (ΔnanR) cells were grown in xylose, glucose, and NANA.
Deletions of nanL, nanE, or nanT affect the growth of B. fragilis on NANA as a main carbon source.
To establish the role of each of the nanLET genes in NANA utilization, we created strains bearing in-frame deletions of each gene as described in the supplemental material and tested these strains for growth with NANA as the main carbon and energy source. The nanL in-frame deletion strain MD100, the nanE in-frame deletion strain RC200, and the nanT in-frame deletion strain RC140 all grew poorly, if at all, on NANA (Fig. 2A). Thus, NanL, NanE, and NanT are directly involved in NANA utilization in B. fragilis. Enzymatic and functional assays were then performed to establish the specific role of each gene in NANA utilization.
NanL is an N-acetyl neuraminate lyase.
B. fragilis wild-type strain ADB77 was grown in minimal medium containing xylose, glucose, or NANA. As shown in Fig. 3, extracts of NANA-grown cells possess 15 times more NANA lyase activity than extracts of cells grown in the presence of glucose or xylose. Interestingly, the lyase activity in extracts of cells grown in glucose is similar to the activity of extracts from cells grown in xylose, suggesting that there is no “glucose effect” on the expression of the nanL gene. Extracts prepared from the nanL deletion strain MD100 were devoid of lyase activity, as expected, under all growth conditions tested (data not shown).
NanE is a N-acetylglucosamine 2-epimerase.
The nanE gene product is annotated in the NCTC genome project as having homology to the porcine RnBP, a polypeptide first characterized by its ability to tightly bind and inactivate the peptidase renin (20, 34). RnBPs from human and porcine kidney have been shown to possess N-acetylglucosamine 2-epimerase activity (18, 28, 30). We tested for this activity in extracts of the wild-type ADB77 and RC201 (ΔnanE) using manNAc and ATP as described in Materials and Methods. If B. fragilis NanE utilizes manNAc6-P as does the NanE from E. coli (22), we would expect this assay to yield NAG6-P. However, the NanE reaction might produce nonphosphorylated NAG since this is the product of the RnBP reaction, i.e., manNAc ↔ NAG. Indeed, B. fragilis extracts containing NanE catalyzed the formation of NAG from manNAc (Fig. 4A) and only formed NAG6-P if the extracts also contained the RokA kinase, which can phosphorylate NAG (6). In ΔrokA extracts, only NAG could be detected; there was no production of NAG6-P (data not shown). That there is no activity in B. fragilis capable of phosphorylating manNAc to produce manNAc6-P can be deduced by the failure to detect this product when extracts of the ΔnanE strain are incubated with manNAc and ATP (Fig. 4A). These observations, along with the inability of rokA mutants to utilize NANA (6), lend support to the conclusions that B. fragilis lacks any NanK activity that would phosphorylate manNAc to form manNAc6-P (as seen in E. coli) and that the RokA protein phosphorylates the NAG which is produced by NanE acting on manNAc, which is produced by NanL action on NANA.
FIG. 4.

NanE enzymatic activity. (A) manNAc is converted to NAG in an ATP-dependent manner. Extracts of B. fragilis cells with (ADB77, lanes 2 and 4) or without (RC201, lanes 1 and 3) an intact nanE gene were incubated with manNAc with or without ATP as described in Materials and Methods. Lane 5 is manNAc incubated in buffer without added extract. Arrows indicate the positions of NAG, manNAc, and NAG6-P standards, as determined in separate experiments. (B) manNAc→NAG steady-state ratio is 1:3. Purified NanEHis6 was incubated with ATP and [14C]manNAc (specific activity, 20 μCi/mmol) for the times indicated. (C) ATP, but not ATP, hydrolysis is required for manNAc epimerase activity. Purified NanEHis6 was incubated with or without ATP or ATP-γ-P and [14C]manNAc (specific activity, 20 μCi/mmol) as indicated. Each reaction in panel C was incubated for 18 h at 37°C. In panels B and C, the positions of manNac and NAG were determined in separate experiments.
Further characterization of NanE kinetics was performed with an oligohistidine-tagged version of the protein, NanEHis6 that had been expressed in E. coli and purified by Ni2+ affinity chromatography. The activity of purified NanEHis6 was tested in the coupled assay using manNAc, purified RokA kinase (6), pyruvate kinase, and lactate dehydrogenase. The Km for manNAc was determined to be 4.63 mM, which is slightly lower than the Km of the human, rat, and porcine NAG 2-epimerases (13.2, 7.6, and 13.7 mM, respectively) as reported by Takahashi et al. (27, 30). The Vmax of the manNAc→NAG conversion was determined to be 9.61 μmol/min/mg and, as shown in Fig. 4B, the manNAc→NAG conversion reached steady state after 5 h at a manNAc/NAG ratio of 1:3. For the NAG→manNAc reaction, after 18 h the conversion of NAG to manNAc was poor, and the NAG/manNAc ratio was only 10:1, indicating that the favored reaction is to convert manNAc to NAG. To determine whether ATP hydrolysis was necessary for NanE activity, NanEHis6 was incubated with manNAc and ATP-γ-S, a nonhydrolyzable ATP analog (Fig. 4C). We could still detect epimerase activity in the assays with ATP-γ-S, indicating that NanE activity does not require ATP hydrolysis. This result is similar to the finding of Takahashi et al. (27, 29), which showed that the eukaryotic N-acetylglucosamine 2-epimerases required ATP as a cofactor and not as an energy source.
The nanT deletion mutant is deficient in NANA accumulation.
Martinez et al. (17) identified a NANA transporter protein in E. coli. However, the putative B. fragilis NanT protein (412 amino acids) is shorter in length than the E. coli NanT protein (496 amino acids) and is only 21% identical to the E. coli NanT. Having previously shown that the deletion of nanT abolishes NANA utilization in B. fragilis, we tested whether or not the deletion affected NANA accumulation. We incubated strains RC122 (ΔnanR) and RC122T (ΔnanRΔnanT) with [14C]NANA as described in Materials and Methods. The nanR deletion strains were used since they express the NANA utilization genes constitutively (see Fig. 3 for results with NanL activity) and would express the nanLET gene cluster even in the absence of NANA. Thus, as shown in Fig. 5, the strain that is lacking the NanR repressor (RC122) accumulated 10 times more NANA over the course of a 30-min time period than the ΔnanR ΔnanT double-deletion strain (RC122T). We also examined NANA accumulation in strain ADB77::pRAG210, which has a large insertion in the nanL gene and is unable to grow on NANA (Fig. 5). Strain ADB77::pRAG210 accumulated four- to fivefold less labeled NANA than the wild-type strain after 15 min, suggesting that the insertion in nanL exerts a polar effect on the expression of nanT and, thus, the nanLET gene cluster is expressed as an operon.
FIG. 5.
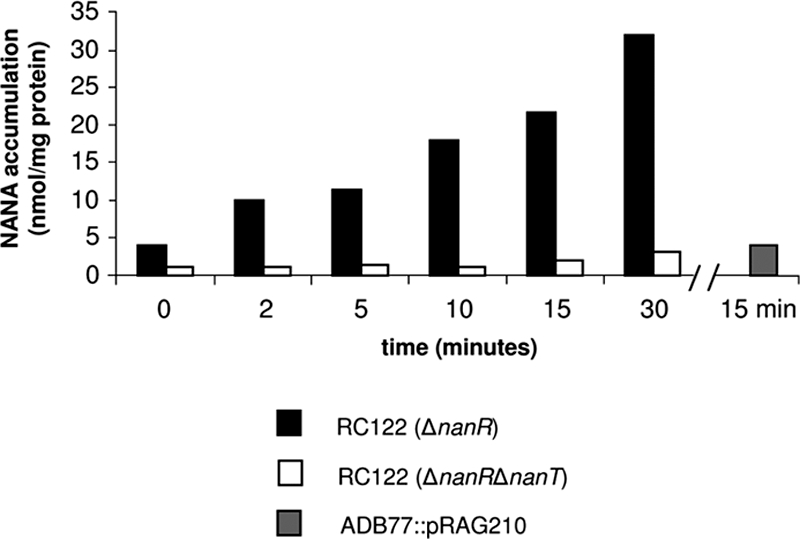
NANA accumulation in RC122, RC122T, and the nanL disruption strain ADB77::pRAG210. Cells were incubated with labeled sugar (specific activity, 10 μCi/mmol) for a time course of 30 min as described in Materials and Methods. Aliquots were harvested at the times indicated on the x axis. The [14C]NANA specific activity was 0.1 μCi/mmol. The experiment with ADB77::pRAG210 cells was incubated for 15 min as described in Materials and Methods.
Isolation of a manNAc utilizing strain of B. fragilis.
Wild-type B. fragilis strains do not grow on manNAc as the main carbon and energy source (Fig. 2B). However, since wild-type B. fragilis grows on NANA through a pathway that generates intracellular manNAc, it could be that the block in the utilization of external manNAc results from an inability to transport this substrate into the cytoplasm. We therefore attempted to isolate a mutant strain of B. fragilis that could now utilize extracellular manNAc. We used the method of Plumbridge and Vimr (21), which allowed them to isolate mutants of E. coli K-12 that grew better than the wild-type strain on manNAc as a carbon source. For details of the method used to isolate this strain, see the supplemental material. One such manNac-utilizing strain of B. fragilis was designated ADB77M. It grows on manNAc as the main carbon and energy source with a doubling time of 3 h; the parental strain failed to grow on this medium (Fig. 2B). Strain ADB77M was tested for NANA lyase activity. Assays of extracts from cells of ADB77 and ADB77M grown in glucose showed the same low level of expression, while extracts from the same cells grown in NANA showed the fully induced level of activity (Fig. 3). However, extracts of ADB77M grown on manNAc as the main carbon source showed high levels of lyase activity, similar to what was observed in the fully induced, NANA-grown wild-type cells. This suggests that either manNAc is capable of inducing expression of the genes in the nanLET operon (at least nanL) or that manNAc is altered in the cell to create the true inducer of nanL (or nanLET) expression.
ADB77M is able to accumulate manNAc.
We postulate that the enabling mutation in strain ADB77M alters a transporter, or the expression of a transporter, that imports manNAc into the cell. To test this hypothesis, we performed [3H]manNAc accumulation experiments in strain ADB77M and compared these results with the wild-type strain, ADB77. As shown in Fig. 6, the enabled mutant strain ADB77M accumulates three times more manNAc in a 15-min time period than does wild-type ADB77. This might result from changes in the NanT transporter, however, the nanT deletion derivative of ADB77M, ADB77MΔT, still grows on manNAc as the main carbon source (Fig. 2B), eliminating a role for NanT in manNAc uptake. In contrast, strain ADB77M exhibits increased uptake of NAG compared to the wild-type ADB77 (Fig. 6). Although the exact genetic alteration(s) in ADB77M, the manNAc-utilizing strain, are not known, we suggest that alteration of an existing NAG transporter may be responsible for manNAc uptake in this strain.
FIG. 6.

manNAc and NAG uptake in ADB77 (wild type) and ADB77M. Cells were incubated with labeled sugar (specific activity, 2.5 μCi/mmol) as described in Materials and Methods. Accumulation experiments were performed in triplicate.
This hypothesis is also supported by a determination of the affinities of wild-type ADB77 and the mutant ADB77M strains for manNAc in an accumulation assay. Each strain was incubated with various concentrations of manNAc or NAG, and the accumulation of amino sugar over time was analyzed by double reciprocal plots. Interestingly, the affinities for NAG in the wild-type ADB77 and the mutant ADB77M are similar with Kms of 30.95 and 47 mM, respectively. However, there is a marked difference between the two strains in the affinity for manNAc. The Km of manNAc in mutant strain ADB77M is 55.4 mM, similar to the Km for NAG. However, the Km for manNAc in wild-type ADB77 is 60 times greater than that of ADB77M at 337.75 mM. Using different manNAc concentrations, we measured the [NAG] required to inhibit manNAc uptake. We found that the Ki,NAG of the wild-type strain ADB77 was approximately 122 mM, whereas the Ki,NAG of the enabled mutant ADB77M was 61 mM. We also measured accumulation of radiolabeled NAG in the presence of unlabeled manNAc, and determined a Ki,manNAc for wild-type ADB77 of 225 mM, and a Ki,manNAc for the enabled mutant ADB77M was 60 mM.
Role of nanLET operon expression in growth of ADB77M on manNAc.
To test whether the nanLET operon plays a role in manNAc utilization in the enabled mutant ADB77M, we constructed a series of strains with deletions in each of the nanLET genes in the ADB77M background (see the supplemental material) and tested their ability to grow with manNAc as carbon source. The nanL derivative of ADB77M (ADB77MΔL1, Table 1) grows on manNAc, as expected, although it cannot grow on NANA (Fig. 2B and 2C). These data suggest that the NanL gene product is not necessary for utilization of manNAc. A similar result was found with the nanT deletion derivative of ADB77M (data not shown). However, a nanE deletion derivative of ADB77M (ADB77MΔE, Table 1) is defective for growth on either manNAc or NANA as the main carbon and energy source (Fig. 2B and C) underscoring its importance in manNAc utilization.
Role of the RokA kinase in NANA and manNAc utilization.
We have previously shown that the B. fragilis rokA deletion strain, CJB100, cannot utilize NANA as the main carbon and energy source. We have also shown that the purified RokA protein does not phosphorylate NANA or manNAc in vitro (6). To examine whether a mutation in the rokA gene in the ADB77M background would similarly affect manNAc utilization, we constructed the ADB77MΔrok strain, with an internal deletion within the rokA gene. Indeed, this strain no longer grows with manNAc as a carbon source (data not shown). Taken together, this evidence establishes that the RokA kinase is required for NANA and manNAc utilization in B. fragilis. However, given previous results, it is unlikely that RokA phosphorylates manNAc but instead phosphorylates NAG, the product of NanE action on manNAc in the B. fragilis cell.
DISCUSSION
Comparison of NANA utilization pathways in B. fragilis and E. coli.
The NANA utilization pathway in B. fragilis differs significantly from the previously described pathway in E. coli (22) and other bacteria (35, 36). Figure 7 presents a comparison of these pathways. Differences occur only after NANA is cleaved by the lyase, NanL or NanA, to form manNAc and pyruvate. In E. coli and many other bacteria, including well-studied pathogens, manNAc is immediately phosphorylated by a NanK kinase to produce manNAc 6-phosphate. The E. coli NanE epimerase then converts manNAc 6-P to NAG 6-P, which then enters the amino-sugar utilization pathway (22). In contrast, in B. fragilis the NanE epimerase enzyme converts unphosphorylated manNAc to NAG, using ATP as a cofactor. In this respect the B. fragilis NanE catalytic function is similar to the mammalian RnBP/NAG 2-epimerases. In B. fragilis, NAG is now phosphorylated by the RokA kinase to yield NAG 6-P, which is further metabolized. RokA-deficient mutants of B. fragilis are unable to utilize NANA (or manNAc when the rokA deletion is introduced into the manNAc enabled strain ADB77M). This implies that B. fragilis does not contain a specific manNAc kinase (NanK) or a hexokinase other than RokA that can phosphorylate manNAc.
Phylogenetic comparisons between the nanE gene products from B. fragilis, other Bacteroides species, and other bacterial NAG 2-epimerases involved in NANA utilization suggests that they may have independent origins. A phylogenetic tree was constructed from a multiple-sequence alignment of eukaryotic RnBP/NAG 2-epimerases and bacterial NanE homologs. Clearly, the manNAc 6-P 2-epimerases, such as those from E. coli, C. perfringens, and P. multocida, form their own distinct clade, as do all RnBPs including that of B. fragilis (Fig. 8). Indeed, the B. fragilis NanE is closely related to the mammalian RnBPs/NAG 2-epimerases, and it groups into a larger clade that includes all RnBPs, clearly excluding E. coli NanE and the other manNAc 6-P 2-epimerases (Fig. 8). This suggests two distinct origins of the NanE epimerase groups. Surprisingly, NanE homologs from Bacteroides thetaiotaomicron and the Bacteroidetes group member Flavobacteriales strain ALC-1 do not group as closely as might be expected with the enzymes of other Bacteroidetes, such as Tannerella forsythia, B. fragilis, Bacteroides ovatus, Parabacteroides distasonis, and several other intestinal Bacteroidetes (Fig. 8). This could represent different degrees of divergence of the nanE sequence since its acquisition by each of these organisms.
FIG. 8.

Phylogenetic tree of NanE proteins and RnBPs, including B. fragilis NanE (boxed). The tree was constructed by using the neighbor-joining method and was visualized with MEGA 3.1. hyp prot, hypothetical protein.
Sequence alignment of B. fragilis NanE and eukaryotic RnBP's indicates marked differences, as well as similarities, between the B. fragilis NanE and the RnBPs (see Fig. S2 in the supplemental material). Takahashi, et al. describe several cysteine residues that are important for the function of the human epimerase enzyme (known as C1 to C10) (31). Only one of these cysteines appears to be conserved between B. fragilis NanE and RnBPs (C1, see Fig. S2 in the supplemental material). Obviously, the absence of the other cysteine residues in B. fragilis NanE does not affect the epimerase activity. Furthermore, Itoh et al. (14) have produced a crystal structure of the porcine kidney NAG 2-epimerase, which identified active site residues in the protein, many of which are aromatic side chains such as tryptophan and phenylalanine. These residues are conserved in the other mammalian RnBPs/NAG 2-epimerases and strikingly, most of these residues are conserved in B. fragilis NanE (see Fig. S2 in the supplemental material). In a study by Itoh et al. (14), the NAG 2-epimerase crystals were saturated with NAG, and conserved residues that might play a role in sugar contacts were determined. These same residues are present not only in B. fragilis NanE but also in the epimerases of T. forsythia and B. thetaiotaomicron (see Fig. S2 in the supplemental material). The genome sequences of several intestinal Bacteroidetes, such as B. vulgatus (40), B. caccae, B. ovatus, Parabacteroides distasonis, and Parabacteroides contain nanE homologs, with all of the aforementioned conserved residues (data not shown). There are several other enzymes in B. fragilis that show greater similarity to “eukaryotic” enzymes than to bacterial enzymes. The B. fragilis aconitase shares striking similarity to the eukaryotic mitochondrial aconitase (3). This has been interpreted to suggest that a Bacteroides species may have been the symbiont that eventually became the mitochondrion in eukaryotic cells.
In contrast to the similarities of B. fragilis NanE and the eukaryotic RnBP, there does not appear to be a close eukaryotic homolog of B. fragilis NanL. A phylogenetic tree was constructed using a multiple-sequence alignment of NANA lyase proteins (see Fig. S1 in the supplemental material), including NanL homologs from other Bacteroidetes, the E. coli NanA protein and its homologs, and putative NANA lyase proteins from other commensal or pathogenic bacteria. NanL proteins from the Bacteroidetes form a distinct clade, with the exception of the putative NanL of F. bacterium. This clade is clearly distinct from the E. coli/Salmonella/Shigella clade of NanA proteins, suggesting either two separate origins of NANA lyase or a large divergence between NanL homologs and NanA homologs. Notably absent from the phylogenetic tree shown in Fig. S1 in the supplemental material is a NanL homolog from B. thetaiotaomicron (see below).
As was demonstrated by the phylogenetic tree in Fig. 8, B. fragilis and E. coli NanE proteins are not similar to each other on the primary sequence level. The same conclusion can also be drawn when comparing B. fragilis NanL to E. coli NanA (see Fig. S1 in the supplemental material). The degree of difference, however, between NanE proteins is much greater than the degree of difference between NanL and NanA. NanL is 34% identical (56% positive) at the primary sequence level compared to NanA, while E. coli and B. fragilis NanE proteins share no significant similarity. Furthermore, B. fragilis NanE (377 amino acids) is significantly larger than E. coli NanE (229 amino acids). In contrast, the NanL (302 amino acids) and NanA (297 amino acids) proteins are similar in size. This suggests two very different origins for the B. fragilis NanL and NanE proteins.
Sialic acid utilization systems in other Bacteroidetes.
Sialic acids are available in large quantities in the human colon (23, 24). It is not surprising that B. fragilis can use NANA and other sialic acid-containing substrates for growth. Indeed, B. fragilis has been shown to express a variety of glycoside hydrolases (4), including neuraminidase (8, 11, 19), that can convert mucins into usable carbon sources. The ability to metabolize NANA gives B. fragilis a competitive advantage in the niche of the colonic mucosa and also at sites of infection, since it is capable of cleaving and utilizing sialic acid-rich components of intestinal mucins and epithelial cell glycoproteins. Based on the availability of sialic acids in the colon, we might expect other colonic residents, including other Bacteroides species, to possess a NANA utilization system similar to that in B. fragilis. Surprisingly, E. coli strains lack neuraminidase activity, although neuraminidase activity has been detected in many other colonic Bacteroides species (23). Recently, genome sequences of several colonic Bacteroides have become available. Many of these genomes, such as that of Bacteroides vulgatus and Parabacteroides distasonis, contain nanLET gene clusters with close sequence similarity to B. fragilis nanLET. These genomes also contain a nanH (neuraminidase) homolog and a rokA homolog. The genomes of two Bacteroidetes, Bacteroides uniformis and Porphyromonas gingivalis, appear to contain neuraminidase homologs but do not contain nanLET genes. The genome of one Bacteroides species, Bacteroides capillosus, does not appear to have a neuraminidase gene or any NANA utilization genes. B. capillosus also does not appear to have a rokA homolog in its genome, making it a unique Bacteroides species among the species whose genome sequences are available.
Analysis of the published B. thetaiotaomicron VPI 5482 genome reveals the presence of a putative neuraminidase gene but not a NANA lyase (nanL) gene. This suggests that B. thetaiotaomicron should not be able to use NANA for growth. Furthermore, we found that B. thetaiotaomicron strains VPI 5482 and TAL21586 do not grow in minimal medium supplemented with NANA (data not shown). Interestingly, the B. thetaiotaomicron VPI 5482 genome seems to possess three separate manNAc 2-epimerase genes, whose products are similar to the B. fragilis NanE. The role of these epimerase candidates in B. thetaiotaomicron metabolism is as yet unknown.
Neuraminidase activity has also been detected in oral Bacteroidetes (19), including, but not limited to, P. gingivalis, T. forsythia, and Prevotella sp. The sialidase gene of T. forsythia, siaHI, has been isolated and characterized (13). This sialidase does not share sequence homology with any other bacterial sialidase/neuraminidase, including B. fragilis NanH or the neuraminidase candidates from B. thetaiotaomicron or P. gingivalis. A search of the T. forsythia genome database (http://www.oralgen.lanl.gov/oralgen/bacteria/tfor/) reveals the presence of a nanLET-like gene cluster, suggesting that T. forsythia should be capable of utilizing NANA as a carbon and energy source. The gene arrangement and sequences of the putative protein products of the nanLET-like cluster in T. forsythia are similar to the products of the B. fragilis nanLET operon, although there is no nanR-like gene present upstream of and divergent to the nanLET cluster. A search of the P. gingivalis and Prevotella intermedia sequences reveals possible neuraminidase genes but no other genes whose protein products could be involved in NANA utilization.
There is considerable interest in the question of when sialic acids first appeared in living systems (see references 2, 5, 9, and 36 for recent discussions). Until recently, it had been accepted that sialic acids were absent until the appearance of the deuterostomes, perhaps 500 million years ago. With the discovery of sialic acid biosynthesis capabilities in several bacteria, especially human pathogens, it is now considered possible that sialic acids appeared billions of years before the split. Complicating the issue is the likelihood that lateral gene transfer between bacteria and eukaryotes has played an important role in the current genetic makeup of sialic acid-synthesizing and sialic acid-utilizing organisms (5). No matter what the actual explanation turns out to be, the NANA utilization system of B. fragilis represents another solution to sialic acid metabolism. B. fragilis and all other bacteria that metabolize NANA cleave NANA into manNAc and pyruvate. In B. fragilis and presumably in other Bacteroides strains that contain a similar NanE protein, manNAc is epimerized to NAG, which is then phosphorylated by the RokA hexokinase to form NAG 6-P, an intermediate in the common pathway for aminosugar utilization. In E. coli and other related bacteria, another pathway-specific enzyme is required to convert manNAc to manNAc 6-P, specifically the kinase, NanK. manNAc6-P is then epimerized to NAG6-P by an enzyme specific for the phosphorylated sugar derivatives. It is not clear which pathway came first in time and, indeed, the subject becomes even more complicated with the realization that the B. fragilis pathway contains two proteins, RokA (6) and NanE, with close similarity to eukaryotic proteins. If it can be shown that sialic acid biosynthesis and degradation systems were present in bacteria long before the appearance of eukaryotes capable of sialic acid biosynthesis, it may be possible to conclude that the bacteria were the ultimate source of the enzymes required for these pathways. Without such proof, it remains impossible to decide the question.
Supplementary Material
Acknowledgments
This paper is dedicated to the memory of Francis P. Tally, who introduced the senior author to the world of B. fragilis and to the importance of sialic acids.
This study was supported by Public Health Service grant AI19497 from the National Institute of Allergy and Infectious Disease, National Institutes of Health.
We are grateful to Eric Vimr for gifts of substrates and for his advice and continued interest in this project.
Footnotes
Published ahead of print on 20 March 2009.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Allen, S., A. Zaleski, J. W. Johnston, B. W. Gibson, and M. A. Apicella. 2005. Novel sialic acid transporter of Haemophilus influenzae. Infect. Immun. 735291-5300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Angata, T., and A. Varki. 2002. Chemical diversity in the sialic acids and related alpha-keto acids: an evolutionary perspective. Chem. Rev. 102439-469. [DOI] [PubMed] [Google Scholar]
- 3.Baughn, A. D., and M. H. Malamy. 2002. A mitochondrial-like aconitase in the bacterium Bacteroides fragilis: implications for the evolution of the mitochondrial Krebs cycle. Proc. Natl. Acad. Sci. USA 994662-4667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berg, J. O., L. Lindqvist, and C. E. Nord. 1980. Purification of glycoside hydrolases from Bacteroides fragilis. Appl. Environ. Microbiol. 4040-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bravo, I. G., S. Garcia-Vallve, A. Romeu, and A. Reglero. 2004. Prokaryotic origin of cytidylyltransferases and alpha-ketoacid synthases. Trends Microbiol. 12120-128. [DOI] [PubMed] [Google Scholar]
- 6.Brigham, C. J., and M. H. Malamy. 2005. Characterization of the RokA and HexA broad-substrate-specificity hexokinases from Bacteroides fragilis and their role in hexose and N-acetylglucosamine utilization. J. Bacteriol. 187890-901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reference deleted.
- 8.Corfield, A. P., S. A. Wagner, J. R. Clamp, M. S. Kriaris, and L. C. Hoskins. 1992. Mucin degradation in the human colon: production of sialidase, sialate O-acetylesterase, N-acetylneuraminate lyase, arylesterase, and glycosulfatase activities by strains of fecal bacteria. Infect. Immun. 603971-3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Koning, A. P., F. S. Brinkman, S. J. Jones, and P. J. Keeling. 2000. Lateral gene transfer and metabolic adaptation in the human parasite Trichomonas vaginalis. Mol. Biol. Evol. 171769-1773. [DOI] [PubMed] [Google Scholar]
- 10.Dixon, M., and E. C. Webb. 1964. Enzymes, 2nd ed. Academic Press, Inc., New York, NY.
- 11.Godoy, V. G., M. M. Dallas, T. A. Russo, and M. H. Malamy. 1993. A role for Bacteroides fragilis neuraminidase in bacterial growth in two model systems. Infect. Immun. 614415-4426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guiney, D. G., P. Hasegawa, and C. E. Davis. 1984. Plasmid transfer from Escherichia coli to Bacteroides fragilis: differential expression of antibiotic resistance phenotypes. Proc. Natl. Acad. Sci. USA 817203-7206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ishikura, H., S. Arakawa, T. Nakajima, N. Tsuchida, and I. Ishikawa. 2003. Cloning of the Tannerella forsythensis (Bacteroides forsythus) siaHI gene and purification of the sialidase enzyme. J. Med. Microbiol. 521101-1107. [DOI] [PubMed] [Google Scholar]
- 14.Itoh, T., B. Mikami, I. Maru, Y. Ohta, W. Hashimoto, and K. Murata. 2000. Crystal structure of N-acyl-d-glucosamine 2-epimerase from porcine kidney at 2.0 Å resolution. J. Mol. Biol. 303733-744. [DOI] [PubMed] [Google Scholar]
- 15.Kalivoda, K. A., S. M. Steenbergen, E. R. Vimr, and J. Plumbridge. 2003. Regulation of sialic acid catabolism by the DNA binding protein NanR in Escherichia coli. J. Bacteriol. 1854806-4815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kumar, S., K. Tamura, and M. Nei. 2004. MEGA3: integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief Bioinform. 5150-163. [DOI] [PubMed] [Google Scholar]
- 17.Martinez, J., S. Steenbergen, and E. Vimr. 1995. Derived structure of the putative sialic acid transporter from Escherichia coli predicts a novel sugar permease domain. J. Bacteriol. 1776005-6010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maru, I., Y. Ohta, K. Murata, and Y. Tsukada. 1996. Molecular cloning and identification of N-acyl-d-glucosamine 2-epimerase from porcine kidney as a renin-binding protein. J. Biol. Chem. 27116294-16299. [DOI] [PubMed] [Google Scholar]
- 19.Moncla, B. J., P. Braham, and S. L. Hillier. 1990. Sialidase (neuraminidase) activity among gram-negative anaerobic and capnophilic bacteria. J. Clin. Microbiol. 28422-425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murakami, K., S. Hirose, S. Chino, N. Ueno, and H. Miyazaki. 1982. Properties of renin-binding protein. Clin. Exp. Hypertens. A 42073-2081. [DOI] [PubMed] [Google Scholar]
- 21.Plumbridge, J., and E. Vimr. 1999. Convergent pathways for utilization of the amino sugars N-acetylglucosamine, N-acetylmannosamine, and N-acetylneuraminic acid by Escherichia coli. J. Bacteriol. 18147-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ringenberg, M. A., S. M. Steenbergen, and E. R. Vimr. 2003. The first committed step in the biosynthesis of sialic acid by Escherichia coli K1 does not involve a phosphorylated N-acetylmannosamine intermediate. Mol. Microbiol. 50961-975. [DOI] [PubMed] [Google Scholar]
- 23.Robbe, C., C. Capon, B. Coddeville, and J. C. Michalski. 2004. Structural diversity and specific distribution of O-glycans in normal human mucins along the intestinal tract. Biochem. J. 384307-316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Robbe, C., C. Capon, E. Maes, M. Rousset, A. Zweibaum, J. P. Zanetta, and J. C. Michalski. 2003. Evidence of regio-specific glycosylation in human intestinal mucins: presence of an acidic gradient along the intestinal tract. J. Biol. Chem. 27846337-46348. [DOI] [PubMed] [Google Scholar]
- 25.Russo, T. A., J. S. Thompson, V. G. Godoy, and M. H. Malamy. 1990. Cloning and expression of the Bacteroides fragilis TAL2480 neuraminidase gene, nanH, in Escherichia coli. J. Bacteriol. 1722594-2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Spivak, C. T., and S. Roseman. 1959. Preparation of N-Acetyl-d-mannosamine (2-acetimido-2-deoxy-d-mannose) and d-mannosamine hydrochloride (2-Amino-2-deoxy-d-mannose). J. Am. Chem. Soc. 812403-2404. [Google Scholar]
- 27.Takahashi, S., K. Hori, K. Takahashi, H. Ogasawara, M. Tomatsu, and K. Saito. 2001. Effects of nucleotides on N-acetyl-d-glucosamine 2-epimerases (renin-binding proteins): comparative biochemical studies. J. Biochem. 130815-821. [DOI] [PubMed] [Google Scholar]
- 28.Takahashi, S., M. Kumagai, S. Shindo, K. Saito, and Y. Kawamura. 2000. Renin inhibits N-acetyl-d-glucosamine 2-epimerase (renin-binding protein). J. Biochem. 128951-956. [DOI] [PubMed] [Google Scholar]
- 29.Takahashi, S., H. Ogasawara, K. Takahashi, K. Hori, K. Saito, and K. Mori. 2002. Identification of a domain conferring nucleotide binding to the N-acetyl-d-glucosamine 2-epimerase (renin binding protein). J. Biochem. 131605-610. [DOI] [PubMed] [Google Scholar]
- 30.Takahashi, S., K. Takahashi, T. Kaneko, H. Ogasawara, S. Shindo, and M. Kobayashi. 1999. Human renin-binding protein is the enzyme N-acetyl-d-glucosamine 2-epimerase. J. Biochem. 125348-353. [DOI] [PubMed] [Google Scholar]
- 31.Takahashi, S., K. Takahashi, T. Kaneko, H. Ogasawara, S. Shindo, K. Saito, and Y. Kawamura. 2001. Identification of functionally important cysteine residues of the human renin-binding protein as the enzyme N-acetyl-d-glucosamine 2-epimerase. J. Biochem. 129529-535. [DOI] [PubMed] [Google Scholar]
- 32.Tang, Y. P., and M. H. Malamy. 2000. Isolation of Bacteroides fragilis mutants with in vivo growth defects by using Tn4400′, a modified Tn4400 transposition system, and a new screening method. Infect. Immun. 68415-419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thompson, J. S., and M. H. Malamy. 1990. Sequencing the gene for an imipenem-cefoxitin-hydrolyzing enzyme (CfiA) from Bacteroides fragilis TAL2480 reveals strong similarity between CfiA and Bacillus cereus beta-lactamase II. J. Bacteriol. 1722584-2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ueno, N., H. Miyazaki, S. Hirose, and K. Murakami. 1981. A 56,000-dalton renin-binding protein in hog kidney is an endogenous renin inhibitor. J. Biol. Chem. 25612023-12027. [PubMed] [Google Scholar]
- 35.Vimr, E., C. Lichtensteiger, and S. Steenbergen. 2000. Sialic acid metabolism's dual function in Haemophilus influenzae. Mol. Microbiol. 361113-1123. [DOI] [PubMed] [Google Scholar]
- 36.Vimr, E. R., K. A. Kalivoda, E. L. Deszo, and S. M. Steenbergen. 2004. Diversity of microbial sialic acid metabolism. Microbiol. Mol. Biol. Rev. 68132-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vimr, E. R., and F. A. Troy. 1985. Identification of an inducible catabolic system for sialic acids (nan) in Escherichia coli. J. Bacteriol. 164845-853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Walters, D. M., V. L. Stirewalt, and S. B. Melville. 1999. Cloning, sequence, and transcriptional regulation of the operon encoding a putative N-acetylmannosamine-6-phosphate epimerase (nanE) and sialic acid lyase (nanA) in Clostridium perfringens. J. Bacteriol. 1814526-4532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Woodcock, D. M., P. J. Crowther, J. Doherty, S. Jefferson, E. DeCruz, M. Noyer-Weidner, S. S. Smith, M. Z. Michael, and M. W. Graham. 1989. Quantitative evaluation of Escherichia coli host strains for tolerance to cytosine methylation in plasmid and phage recombinants. Nucleic Acids Res. 173469-3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu, J., M. A. Mahowald, R. E. Ley, C. A. Lozupone, M. Hamady, E. C. Martens, B. Henrissat, P. M. Coutinho, P. Minx, P. Latreille, H. Cordum, A. Van Brunt, K. Kim, R. S. Fulton, L. A. Fulton, S. W. Clifton, R. K. Wilson, R. D. Knight, and J. I. Gordon. 2007. Evolution of symbiotic bacteria in the distal human intestine. PLoS Biol. 5e156. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.