

Abstract
Pseudomonas aeruginosa is recognized for its ability to colonize diverse habitats, ranging from soil to immunocompromised people. The formation of surface-associated communities called biofilms is one factor thought to enhance colonization and persistence in these diverse environments. Another factor is the ability of P. aeruginosa to diversify genetically, generating phenotypically distinct subpopulations. One manifestation of diversification is the appearance of colony morphology variants on solid medium. Both laboratory biofilm growth and chronic cystic fibrosis (CF) airway infections produce rugose small-colony variants (RSCVs) characterized by wrinkled, small colonies and an elevated capacity to form biofilms. Previous reports vary on the characteristics attributable to RSCVs. Here we report a detailed comparison of clonally related wild-type and RSCV strains isolated from both CF sputum and laboratory biofilm cultures. The clinical RSCV had many characteristics in common with biofilm RSCVs. Transcriptional profiling and Biolog phenotypic analysis revealed that RSCVs display increased expression of the pel and psl polysaccharide gene clusters, decreased expression of motility functions, and a defect in growth on some amino acid and tricarboxylic acid cycle intermediates as sole carbon sources. RSCVs also elicited a reduced chemokine response from polarized airway epithelium cells compared to wild-type strains. A common feature of all RSCVs analyzed in this study is increased levels of the intracellular signaling molecule cyclic di-GMP (c-di-GMP). To assess the global transcriptional effects of elevated c-di-GMP levels, we engineered an RSCV strain that had elevated c-di-GMP levels but did not autoaggregate. Our results showed that about 50 genes are differentially expressed in response to elevated intracellular c-di-GMP levels. Among these genes are the pel and psl genes, which are upregulated, and flagellum and pilus genes, which are downregulated. RSCV traits such as increased exopolysaccharide production leading to antibiotic tolerance, altered metabolism, and reduced immunogenicity may contribute to increased persistence in biofilms and in the airways of CF lungs.
Pseudomonas aeruginosa is responsible for chronic infections in the airways of cystic fibrosis (CF) patients (13). During the course of chronic infection, P. aeruginosa forms biofilms, which are thought to promote persistence by protecting the bacterium from antibiotics and host clearance. P. aeruginosa also undergoes phenotypic and genotypic diversification. A manifestation of this diversification is the appearance of colony morphology variants among CF sputum sample isolates. One clear example of this phenomenon, which has been termed “dissociative” behavior (42), is the appearance of mucoid colonies. Mucoidy is characterized by overproduction of the exopolysaccharide (EPS) alginate, a polymer of 1,4-β-linked mannuronic acid and its epimer, guluronic acid (13). The appearance of mucoid colonies is thought to correlate with a downturn in the patient's prognosis and the onset of chronic colonization (12, 30). Besides mucoid colonies, rugose small-colony variants (RSCVs) have been isolated from CF sputum. RSCVs are selected for during the course of chronic infection, which suggests that they may play a role in pathogenesis (36). RSCVs autoaggregate and are hyperadherent (10, 15), and recent studies have linked their appearance to prolonged antibiotic treatment (10, 14).
P. aeruginosa biofilms grown in the laboratory also produce colony morphology variants, including RSCVs (5, 9, 21). Like CF sputum-derived RCSVs, laboratory-derived RSCVs autoaggregate in liquid culture and hyperadhere to surfaces. We have characterized biofilm-derived RSCVs and found that at least one EPS biosynthetic locus, the psl gene cluster, contributes to the autoaggregation and hyperadherence phenotypes (21). The psl locus (PA2231 to PA2245) is responsible for the production of a mannose-rich EPS (11, 19, 25). Cyclic di-GMP (c-di-GMP) signaling has also been implicated in the RSCV phenotype (17, 26). For example, mutations in wspF, which result in constitutive activation of the diguanylate cyclase, WspR, result in elevated intracellular c-di-GMP levels. A wspF mutation is capable of converting a wild-type smooth strain into an RSCV, and subsequent depletion of c-di-GMP in a wspF mutant strain can convert the morphology back to a wild-type morphology (7, 17).
The relationship between biofilm-derived and CF-derived RSCVs is unclear. Although these variants share some common phenotypes, such as hyperadherence and autoaggregation in liquid culture, some interesting phenotypic differences have been reported. For example, von Gotz et al. performed a transcriptional analysis of a clinical RSCV and found that type III secretion genes were induced in RSCVs (41). These investigators also reported that the RSCV cells were hypermotile and more cytotoxic to macrophages. The motility of these cells is in stark contrast to the reduced motility reported for biofilm and other clinical RSCVs (17, 21). It is difficult to know with certainty whether the motility phenotypes of clinical isolates were RSCV specific because the phenotypes of the wild-type parents of the clinical RSCV isolates were unknown.
Here, we report isolation and characterization of clonally related wild-type and RSCV strains from the same CF sputum sample. Our results show that the transcriptional profiles of the clinical and laboratory RSCVs are very similar. We demonstrated that the pel and psl EPS loci contributed to the clinical RSCV phenotype. Also, all clinical and laboratory RSCVs examined in this study require the intracellular signaling molecule c-di-GMP for the RSCV phenotype. Due to the importance of c-di-GMP for this phenotype, we determined the transcriptional profile of an RSCV strain (with an elevated c-di-GMP level) and compared it to that of a psl pel mutant derivative that has an elevated intracellular c-di-GMP level but does not autoaggregate. We identified two subsets of differentially expressed genes. One subset appears to be differentially expressed as a consequence of liquid culture autoaggregation. The second, smaller subset is differentially expressed in response to elevated intracellular c-di-GMP levels. One set of regulated functions suggests that there is increased expression of factors involved in dampening the host immune response. Compared with the progenitor wild-type strains, both clinical and biofilm RSCVs elicited a reduced chemokine response from polarized airway epithelium. Our study suggests that RSCVs represent an adaptation that allows P. aeruginosa to persist in CF biofilm communities.
MATERIALS AND METHODS
Strains and growth media.
All relevant P. aeruginosa strains and plasmids are described in Table 1. Three separate samples from a longitudinal CF collection (6, 7), samples 009 37 to 009 39, produced two distinct colony morphologies on agar plates. The two colony morphotypes were separated and designated CF37wt to CF39wt for colonies resembling PAO1 colonies and CF37s to CF39s for colonies similar to colonies of laboratory-derived RSCV strains. Cultures were routinely grown on LB agar or in LB broth. Antibiotic selection was performed with 1.0 μg/ml carbenicillin and 1.5 μg/ml of gentamicin for clinical strains CF37 to CF39 and with 300 μg/ml carbenicillin and 100 μg/ml gentamicin for all other P. aeruginosa strains. Plasmids were maintained in Escherichia coli using 15 μg/ml gentamicin and 100 μg/ml ampicillin.
TABLE 1.
Bacterial strains and plasmids
| Strain(s) or plasmid | Relevant genotype or phenotype | Reference |
|---|---|---|
| P. aeruginosa strains | ||
| PAO1 | Wild type | |
| MJK8 | Biofilm-derived rugose strain (isogenic to PAO1) | 21 |
| MJK8ΔcupA3 | cupA3 polar mutant of MJK8 | This study |
| MJK8ΔcupA-FLP | cupA3 nonpolar mutant of MJK8 | This study |
| MJK8Δpslpel | pslBCD pelA mutant of MJK8 | This study |
| MJK8Δ1169 | PA1169 mutant of MJK8 | This study |
| CF37wt to CF39wt | Clinical longitudinal isolates with wild-type phenotype | 6, 21 |
| CF38s and CF39s | Clinical longitudinal isolates with rugose phenotype | 6, 21 |
| CF39sΔpslBCD | pslBCD mutant of CF39s | This study |
| CF39sΔpelA | pelA mutant of CF39s | This study |
| CF39sΔpslBCDΔpelA | pslBCD pelA mutant of CF39s | This study |
| WF801 | PAO1ΔpslBCD expressing PBAD-psl | 1 |
| WF800 | pslBCD mutant of PAO1 expressing PBAD-psl | 23 |
| Plasmids | ||
| pFLP2 | Source of FLP recombinase, Ampr | 18 |
| pEX18.Ap | Suicide cloning vector, Ampr | 18 |
| pSP858 | Source of gentamicin resistance cassette, Ampr Gentr | 18 |
| pSP5 | WspF expresson vector, Ampr | This study |
| pSP6 | PA2133 expression, Ampr | This study |
| pUCP18 | E. coli-P. aeruginosa expression vector, Ampr | 34 |
| pMPSL-KO1 | pslBCD allelic replacement vector | 21 |
| pMPELA | pelA allelic replacement vector | This study |
| pMCUPA3 | cupA3 allelic replacement vector | This study |
| pM1169 | PA1169 allelic replacement vector | This study |
To determine doubling times, the appropriate strains were inoculated into separate 250-ml baffled Erlenmeyer flasks containing 50 ml LB broth and a stir bar. The flasks were incubated at 37°C with shaking, and samples were removed periodically over a 3-h period. Each sample was divided into aliquots to measure growth in two different ways: optical density at 600 nm (OD600) and protein content. The OD600 sample aliquots were vortexed for 10 s, and the OD600 was measured in duplicate. The cells from the protein content sample aliquot were pelleted, washed, and resuspended in 100 mM Tris-HCl (pH 7.2). Three aliquots of the resuspended cells were placed in a microtiter plate, and the cells were lysed with a microplate horn sonicator (20 kHz; XL-2020; Misonix, Inc., Farmingdale, NY). The cells were sonicated for 1 min at an output of 7.5, followed by a 1-min rest period; this cycle was repeated a total of 12 times. The lysed cells were subjected to a standard microplate protein assay (Coomassie Plus protein assay reagent kit; Pierce Biotechnology, Inc., Rockford, IL).
Mutant construction.
PCR was used to confirm the presence of psl and pel DNA in the clinical RSCV backgrounds. Mutants were generated by allelic exchange (18). psl mutants were constructed using a previously described allelic replacement construct, pMPSL-KO1(21). A pelA allelic replacement construct (pMPELA) was generated by first amplifying a 5-kb DNA fragment encompassing PA3064 and cloning it into suicide vector pEX18.Ap. A blunt-ended SacI fragment from pPS858 was then inserted into the blunt-ended SmaI and XbaI sites. The resulting plasmid, pMPELA, was mated into P. aeruginosa strains, and mutants were selected on Pseudomonas isolation agar containing gentamicin. Double-recombinant mutants were selected on LB agar plates containing 5% sucrose and confirmed by PCR. A PA1169 mutant strain was generated by amplifying a 3.4-kb segment of genomic DNA encompassing PA1169 and cloning it into pEX18.Ap via SacI and HindIII sites incorporated into the primers. Internal ClaI and BamHI sites were used to replace most of PA1169 with the blunt-ended SacI fragment from pPS858, resulting in plasmid pM1169. Mutant strains were generated as described above. To create a cupA3 mutant, primers cupA3-1 and cupA3-2 were used to amplify a 3.6-kb sequence including cupA3, which was cloned into pEX18.Ap using SacI and HindIII. The resistance cassette from pPS858 was inserted via blunt-ended EcoRI and SalI sites found in the cupA3 gene, creating plasmid pMCUPA3. Following allelic replacement of the cupA3 gene, the resistance cassette was excised using pFLP2, and colonies were selected for sucrose resistance and carbenicillin sensitivity (18).
Microarray analysis.
Cultures were grown in 250-ml baffled flasks in LB broth supplemented with 100 mM morpholinepropanesulfonic acid (MOPS) (pH 7). Stir bars were added to reduce clumping, and cultures were shaken at 37°C. Log-phase cells at an OD 600 of 0.25 were harvested in RNA Protect (Qiagen, Inc., Valencia, CA). RNA and cDNA were prepared as described by Schuster et al. (33). Samples were hybridized to Affymetrix P. aeruginosa GeneChips at the University of Iowa DNA Facility. Replicates from biologically distinct experiments were analyzed for each strain. Data were analyzed using GeneSpring software and the online program Cyber-T (http://visitor.ics.uci.edu/genex/cybert/index.html).
Reverse transcription (RT)-quantitative PCR (qPCR).
Cultures were grown as described above and harvested during logarithmic growth. RNA was isolated using reagents from a RiboPure bacterial kit (Ambion, Austin, TX). Cells were pelleted and resuspended in 700 μl RNAwiz. The cell suspension was vortexed for 20 s, sonicated for 5 min, and passed through a 19.5-gauge syringe needle 10 times. Cell suspensions from duplicate cultures were transferred to a single 2-ml RNase-free microcentrifuge tube with 500 μl of 100-μm zirconia beads and bead beaten for 10 min. The remainder of the RNA purification procedure was performed according to the manufacturer's instructions. RNA was eluted with two 50-μl aliquots of elution solution. Residual DNA was removed by DNase I treatment as described in the RiboPure bacterial kit instruction manual. DNase-treated RNA was ethanol precipitated, and the pellet was resuspended in RiboPure bacterial elution solution.
Duplicate cDNA synthesis reactions were performed with random hexamer primers and avian myeloblastosis virus reverse transcriptase (Roche, Germany). Four micrograms of RNA was mixed with 2 μl of primer (10 μM) and nuclease-free water (Ambion, Austin, TX) to obtain a final volume of 10 μl. The RNA mixture was denatured at 70°C for 5 min and placed on ice. A 4-μl aliquot of 5× first-strand buffer, 2 μl of a deoxynucleoside triphosphate mixture (10 mM of each nucleoside; New England Biolabs, Beverly, MA), 2 μl of water, and 2 μl of avian myeloblastosis virus reverse transcriptase were added to each reaction mixture on ice. Each tube was incubated at 42°C for 1.5 h in a Primus 25 personal cycler with a heated lid (MWG Biotech, High Point, NC). At the end of the first-strand synthesis, the reaction mixtures were placed on ice. Negative controls for RT-qPCR were prepared by omitting the reverse transcriptase during the cDNA synthesis reactions, and these controls were used to verify that significant amounts of contaminating genomic DNA were not present in the total RNA preparations. Duplicate cDNA reaction mixtures were pooled and used to perform triplicate real-time qPCRs. qPCRs were performed with an Applied Biosystems 7900HT real-time PCR system (Applied Biosystems, Foster City, CA) using DyNAmo HS SYBR green qPCR kit reagents (Finnzymes, Finland) and following the manufacturer's instructions. Primers are listed in Table 1. The final primer concentration was 0.3 μM for all reactions. The following thermocycler program was used: 95°C for 15 min, followed by 40 cycles of 94°C for 10 s, 60°C for 30 s, and 72°C for 30 s. Fivefold serial dilutions of cDNA prepared from a mixture containing equal aliquots of cDNA from the strains were used to construct a relative standard curve. The cycle number at which the fluorescence crossed a selected threshold value during linear amplification was correlated to a relative quantity. Real-time PCR product dissociation curves were used to verify the specificity of the amplified product. Relative quantities of PA3064 and PA1169 were normalized to relative quantities of PA4946 and PA1615.
wspF and PA2133 complementation.
To express wspF in RSCV backgrounds, plasmid pSP5 was generated by cloning wspF plus 18 bp upstream of the start codon into the EcoRI and XbaI sites of pUCP18. To express an EAL domain protein in RSCV backgrounds, plasmid pSP6 was generated by cloning PA2133 plus 22 bp upstream of the start codon into the EcoRI and XbaI sites of pUCP18. Constructs were electroporated into P. aeruginosa strains.
2D TLC.
Procedures for two-dimensional (2D) thin-layer chromatography (TLC) were adapted from procedures described previously (3, 38). Bacteria were grown overnight in MOPS minimal medium with 30 mM succinate and 0.15 mM KH2PO4 and then subcultured in the same medium to an OD600 of 0.05. After one or two doublings, 0.1 mCi of [32P]orthophosphate (Perkin-Elmer) was added to 1 ml of culture and incubation was continued overnight (12 h), after which 10 μl of 1 M cold formic acid was added to 100 μl of the labeled cell culture.
Polyethyleneimine-cellulose plates (Selecto Scientific) were washed in 0.5 M LiCl and then in distilled H2O and allowed to air dry. Radioactivity in formic acid extracts was measured using a scintillation counter. Equal amounts of total radioactivity from each extract were spotted on a TLC plate. Plates were soaked in methanol for 5 min, air dried, and developed in 0.2 M NH4HCO3 (pH 7.8) in the first dimension. The plates were then soaked in methanol for 15 min, dried, and developed in the second dimension with 1.5 M KH2PO4 (pH 3.65). Plates were soaked again in methanol for 15 min, air dried, and exposed to a phosphorimager screen overnight. The data were collected with a Storm 860 phosphorimager and analyzed using ImageQuant software (Molecular Dynamics).
Phenotype MicroArray analysis.
Phenotype MicroArray analysis (Biolog, Hayward, CA) was performed in duplicate according to the manufacturer's instructions. P. aeruginosa strains were streaked on plates containing LB agar diluted 1:5 (2 g tryptone per liter, 1 g yeast extract per liter, 2 g NaCl per liter, 15 g agar per liter). Colonies were scraped from the plates and suspended in IF-0 inoculating fluid to obtain 85% transmittance. Cells were diluted 1:200 in IF-0 minimal medium containing Biolog redox dye mixture A, and 100-μl aliquots were added to carbon source plates (PM1 and PM2). For the remaining metabolic plates (PM3 to PM8), inocula were supplemented with 20 mM sodium succinate and 2 μM ferric citrate. The plates were incubated at 37°C in an OmniLog plate reader, and growth was measured kinetically by determining the colorimetric reduction of a tetrazolium dye. Data were analyzed with the Biolog Kinetic and Parametric software (version 1.20.02). The keys for determining the test conditions for each well in Fig. 8 and Fig. S6 in the supplemental material are available at http://www.biolog.com/pmMicrobialCells.html.
FIG. 8.
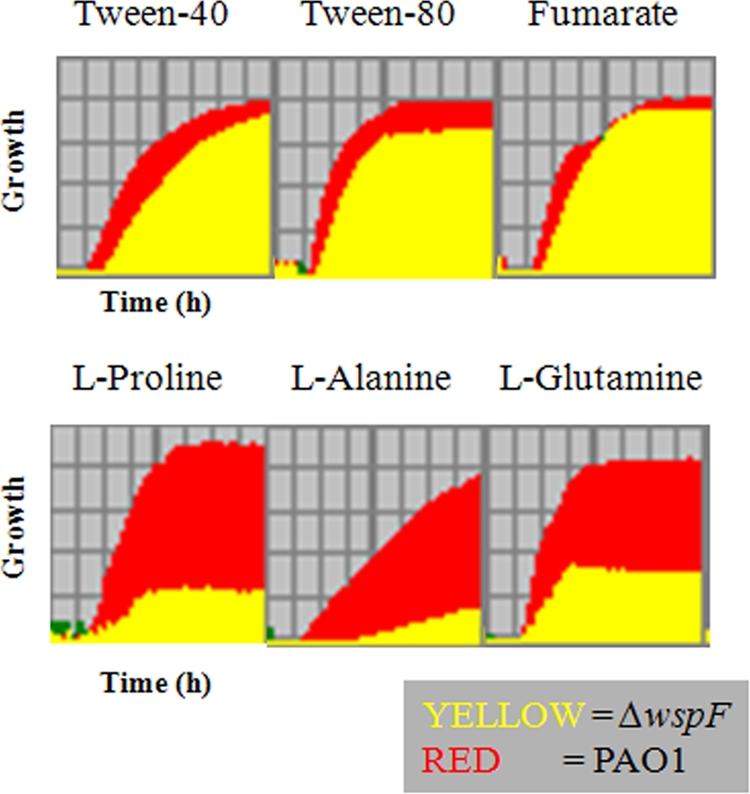
Biolog growth profiles for PAO1 and PAO1ΔwspF on selected carbon sources. Growth (y axis) over time (x axis) is shown. The growth period was 24 h.
Polarized epithelium chemokine signaling assays. (i) Preparation of bacterial cultures.
P. aeruginosa cultures grown for 16 h in LB broth were vortexed for 30 s to disperse aggregates. Cells were harvested, washed with phosphate-buffered saline (PBS), and diluted in PBS to obtain a final concentration of 106 CFU per 50 μl.
(ii) Airway epithelial cell culture.
Calu-3 cells (ATCC HTB-55) were prepared and grown at the air-liquid interface with medium containing Ultroser-G (Biosepra, France) as previously described (20, 37). Penicillin, streptomycin, gentamicin, and fluconazole used for initial establishment of epithelial cultures were removed by repeated apical and basolateral washing with 1:1 Dulbecco's modified Eagle's medium—Ham's F-12 medium (Gibco, Carlsbad, CA) and antibiotic-free medium for 4 to 5 days prior to apical stimulation with bacteria. Additionally, to decrease the stimulatory effects of serum products, Ultroser-G was omitted from the medium starting 3 days prior to interleukin-8 (IL-8) or NF-κB experiments.
(iii) Airway epithelial NF-κB signaling.
Airway epithelial cultures were initially prepared as described above. Cell cultures were basolaterally infected with a replication-incompetent adenoviral vector (multiplicity of infection, 10) containing NF-κB response elements driving a luciferase reporter as previously described (32, 37). One day after transduction, epithelia were apically inoculated with 1 × 106 CFU of bacteria suspended in 50 μl of PBS using the following bacteria (the numbers of replicate independent experiments are indicated in parentheses): PAO1 (10), MJK8 (10), MJK8PA1169 (4), MJK8ΔpslBCDpelA (6), CF39wt (4), CF39s (4), and the PA1169 mutant strain (4). As a negative control, epithelia were incubated with 50 μl of PBS apically and 500 μl of medium basolaterally, and as a positive control, apical PBS and basolateral medium contained 100 ng/ml recombinant human IL-1β (Sigma, St. Louis, MO). Four hours following stimulation with PBS, IL-1β, or bacteria, cells were lysed, and luciferase activity was measured (luciferase assay system; Promega Madison, WI) according to the manufacturer's recommendations using a Monolight 3010 luminometer (Pharmogen, San Diego, CA).
(iv) Basolateral chemokines and cytokine abundance.
Airway epithelial cultures were prepared and grown in antibiotic- and serum-free conditions as described above. Cultures were inoculated with bacteria as described above for NF-κB signaling. We measured IL-8 protein concentrations in basolateral medium using IL-8 (human IL-8 OptEIA set; BD Biosciences, San Jose, CA) enzyme-linked immunosorbent assay kits according to the manufacturer's instructions. All enzyme-linked immunosorbent assays were performed in duplicate to ensure reproducibility. With one exception, experiments were performed with matching cultures for NF-κB experiments but without adenoviral reporter vector infection. The numbers of replicate-independent experiments for the bacterial strains were as follows: PAO1, nine; MJK8, nine; MJK8PA1169, five; MJK8ΔpslBCDpelA, four; CF39wt, five; CF39s, five; and PA1169, five.
RESULTS
Isolation of RSCV and wild-type colony variants from the same CF clinical sample.
Three separate samples designated CF37 to CF39 from a longitudinal CF P. aeruginosa collection produced two distinct colony morphologies. Strains with the two colony morphotypes were designated CF37wt to CF39wt (strains with smooth, wild-type colonies) and CF37s to CF39s (strains with RSCV colonies). The RSCV isolates formed small, rough colonies on Congo red agar plates and autoaggregated in liquid cultures, in which most of the biomass was in a tight pellicle that was difficult to disperse (Fig. 1A). These characteristics are similar to those described for both clinical and laboratory-derived RSCV strains (9, 14, 15, 21). To determine if the RSCV and wild-type clinical colony types were clonally related, we performed restriction fragment length polymorphism analysis. The patterns on a pulsed-field gel electrophoresis (PFGE) gel were identical for all six isolates, suggesting that they are related (Fig. 1B). As a control, we also compared PAO1 and the PAO1-derived biofilm RSCV strain MJK8 and found that they had identical restriction patterns that were distinct from those of the clinical isolates (data not shown). We used two isolates, CF39wt and CF39s, for subsequent experiments.
FIG. 1.

Characterization of clinical isolate CF39s. (A) CF39s has a rugose colony morphology (top) and autoaggregates in liquid culture (bottom) compared to CF39wt. (B) Restriction fragment length analysis of clinical isolates CF37s to CF39s and CF37wt to CF39wt by PFGE, showing identical banding patterns for RCSV and wild-type isolates.
Complementation analysis suggests that there are two classes of RSCVs.
First, we sought to determine if the genetic basis of the clinical RSCV and the genetic basis of our laboratory RSCV were similar. One well-characterized genetic route to the RSCV phenotype is a mutation in the wspF gene (7). MJK8, our laboratory-derived RSCV, was complemented by supplying wspF in trans on a plasmid. Further analysis of this strain revealed an in-frame deletion in the wspA gene (see Fig. S1 in the supplemental material). Interestingly, a wspF-bearing plasmid failed to complement the clinical RSCV CF39s (Fig. 2; see Fig. S2 in the supplemental material). This observation led us to examine whether most biofilm-derived RSCVs are due to wspF mutations. To test this possibility, we independently isolated 50 RSCVs from separate biofilms formed by PAO1 grown for 4 days in drip biofilm reactors (5). We provided each of these RSCV isolates with wspF in trans and found that this gene complemented 34 of them (68%), as shown by the formation of smooth colonies (Fig. 2, class A). We sequenced the wspF alleles of several of these isolates and found only point and frameshift mutations in the gene that presumably disrupted its function (data not shown). A large fraction of the RSCV isolates (∼32%; class B) were not complemented by wspF and appeared to harbor mutations in other loci. To confirm that biofilms can produce wsp-independent RSCVs, we utilized a PAO1 ΔwspR strain. This strain cannot produce RSCVs via mutation in wspF, since the output for the Wsp system depends on WspR (17). PAO1 ΔwspR biofilms produced RSCVs at approximately the same frequency as PAO1 biofilms (data not shown).
FIG. 2.

Two classes of RSCVs have been isolated based on complementation patterns. Class A RSCVs, but not class B, RSCVs are complemented by supplying wspF in trans (pWspF). Both classes of RSCVs are suppressed by depleting c-di-GMP in the cells by expressing the phosphodiesterase encoded by PA2133 (pPA2133). The images show colony morphology, while the graphs show the adherence in a microtiter dish binding assay.
RSCVs have elevated levels of intracellular c-di-GMP.
Elevated intracellular c-di-GMP levels have been linked to RSCV-like phenotypes in several bacterial species, including P. aeruginosa (8). Previous work suggested that a wspF mutation leads to increased c-di-GMP levels and an autoaggregative phenotype in PAO1 by stimulating the activity of the diguanylate cyclase WspR.
Depletion of c-di-GMP pools by overexpression of the c-di-GMP phosphodiesterase encoded by PA2133 suppressed the RSCV phenotype in a wspF derivative of PAO1 (17). Consistent with this, when we overexpressed PA2133 in our collection of biofilm-derived RSCV isolates, we found that the RSCV phenotype was suppressed in every strain tested, including representatives of the class B isolates, such as CF39s (Fig. 2; see Fig. S2 in the supplemental material).
We next tested whether our clinical and laboratory-derived RSCVs, CF39s and MJK8, produce elevated intracellular c-di-GMP levels. A 2D TLC assay was used to determine the relative cellular c-di-GMP levels, and the results showed that CF39s and MJK8 had significantly higher levels of intracellular c-di-GMP than the corresponding wild-type strains, CF39wt and PAO1 (Fig. 3).
FIG. 3.

Our clinical and laboratory RSCVs both display elevated c-di-GMP levels: 2D TLC of cells labeled with 32PPi. Cells were extracted and hydrolyzed following labeling, and the labeled nucleotide fraction was chromatgraphed as previously described (17). Both wild-type strains (PAO1 and CF39wt) produced little measurable c-di-GMP, while the two RSCVs (MJK8 and CF39s) produced a dark, radiolabeled spot that migrated with c-di-GMP (indicated by an arrow).
We also tested biofilm formation by the RSCV strains harboring PA2133- and wspF-expressing vectors. As anticipated, MJK8 hyperadherence was lost when either PA2133 or wspF was expressed, while only PA2133 expression reduced the biofilm level in CF39s (Fig. 2). These data reinforce the importance of elevated c-di-GMP levels in RSCV isolates of both laboratory and clinical origin, regardless of the underlying mutation that confers the RSCV phenotype.
Transcriptional profiling suggests that clinical and laboratory RSCVs have similar patterns of gene expression.
To further characterize our clinical and laboratory-derived RSCVs, we compared the transcriptional profiles of CF39s and CF39wt cells grown in liquid culture. The cultivation conditions were identical to those used in a previous transcriptional profiling study, which compared PAO1 and our laboratory biofilm-derived RSCV, MJK8 (21).
We found that the transcriptional profiles of clinical (Fig. 4 and Table 2) and laboratory-derived (21) isolates were very similar (see Table S1 in the supplemental material for a list of differentially expressed genes). A total of 472 genes were upregulated in CF39s compared to CF39wt, while 256 genes were downregulated (using a twofold cutoff and a P value of <0.01). Many quorum-sensing-controlled genes (e.g., the hcn locus) and genes involved in denitrifying metabolism were significantly upregulated in CF39s compared to CF39wt. The EPS loci pel and psl were also upregulated in CF39s (17, 21). In CF39s, expression of the psl locus was increased 1.5- to 2.4-fold, while expression of the pel locus was increased 4.3- to 15.7-fold. Interestingly one of the most highly upregulated genes in CF39s was PA1169 (loxA), which encodes a lipoxygenase that is active with arachidonic acid, a substrate for inflammatory signaling in humans (40). This gene was also highly expressed in MJK8 compared to PAO1. We confirmed this result by RT-qPCR (data not shown).
FIG. 4.

Selected genes that are differentially expressed in the RSCV background: genes that were either upregulated (red and orange arrows) or downregulated (light and dark green arrows). Stripes indicate that there was a marginal difference in expression.
TABLE 2.
Differentially expressed genes in CF39s and CF39wt
| PA no. | Gene | Change (fold) | Functiona |
|---|---|---|---|
| PA0083 | 2.48 | T6SS HSI-I conserved hypothetical protein | |
| PA0084 | 2.94 | T6SS HSI-I conserved hypothetical protein | |
| PA0085 | hcp1 | 2.70 | T6SS HSI-I Hcp1 |
| PA0088 | 2.13 | T6SS HSI-I hypothetical protein | |
| PA0090 | clpV1 | 2.29 | T6SS HSI-I probable ClpV1 |
| PA0091 | vgrG1a | 2.02 | T6SS HSI-I VgrG1a |
| PA0095 | vgrG1b | 2.28 | T6SS HSI-I VgrG1b |
| PA0285 | −2.04 | Putative diguanylate cyclase/phosphodiesterase (GGDEF/EAL domains) | |
| PA0861 | 2.37 | Putative diguanylate cyclase/phosphodiesterase (GGDEF/EAL domains) | |
| PA1079 | flgD | −2.26 | Flagellar basal body rod modification protein FlgD |
| PA1081 | flgF | −2.23 | Flagellar basal body rod protein FlgF |
| PA1097 | fleQ | −2.00 | Transcriptional regulator FleQ |
| PA1105 | fliJ | −2.50 | Flagellar protein FliJ |
| PA1168 | 225.21 | Hypothetical protein | |
| PA1169 | 261.52 | Probable lipoxygenase | |
| PA1431 | rsaL | 2.61 | Regulatory protein RsaL |
| PA1656 | 3.14 | T6SS HSI-II hypothetical protein | |
| PA1657 | 5.28 | T6SS HSI-II conserved hypothetical protein | |
| PA1658 | 5.86 | T6SS HSI-II conserved hypothetical protein | |
| PA1659 | 5.62 | T6SS HSI-II hypothetical protein | |
| PA1660 | 4.74 | T6SS HSI-II hypothetical protein | |
| PA1661 | 2.22 | T6SS HSI-II hypothetical protein | |
| PA1662 | clpV2 | 2.90 | T6SS HSI-II probable ClpV2 |
| PA1663 | 2.15 | T6SS HSI-II probable transcriptional regulator | |
| PA1664 | 2.59 | T6SS HSI-II hypothetical protein | |
| PA1667 | 3.45 | T6SS HSI-II hypothetical protein | |
| PA1668 | 2.83 | T6SS HSI-II hypothetical protein | |
| PA1669 | icmF2 | 2.23 | T6SS HSI-II IcmF2 |
| PA1871 | lasA | 8.84 | LasA protease precursor |
| PA2193 | hcnA | 33.91 | Hydrogen cyanide synthase HcnA |
| PA2194 | hcnB | 25.20 | Hydrogen cyanide synthase HcnB |
| PA2195 | hcnC | 24.31 | Hydrogen cyanide synthase HcnC |
| PA2231 | pslA | 2.04 | PslA |
| PA2232 | pslB | 2.00 | Probable phosphomannose isomerase/GDP-mannose pyrophosphorylase |
| PA2233 | pslC | 2.40 | Probable glycosyl transferase |
| PA2235 | pslE | 2.42 | Hypothetical protein |
| PA2239 | pslI | 2.31 | Probable transferase |
| PA2361 | icmF3 | 2.62 | T6SS HSI-III IcmF3 |
| PA2364 | 2.11 | T6SS HSI-III hypothetical protein | |
| PA2365 | 3.24 | T6SS HSI-III conserved hypothetical protein | |
| PA2366 | 9.28 | T6SS HSI-III conserved hypothetical protein | |
| PA2367 | hcp3 | 2.26 | T6SS HSI-III Hcp3 |
| PA2368 | 2.59 | T6SS HSI-III hypothetical protein | |
| PA2371 | clpV3 | 2.21 | T6SS HSI-III ClpV3 |
| PA2372 | 6.69 | T6SS HSI-III hypothetical protein | |
| PA2440 | 8.45 | Hypothetical protein | |
| PA2441 | 12.80 | Hypothetical protein | |
| PA2567 | 6.29 | Putative diguanylate cyclase/phosphodiesterase (GGDEF/EAL domains) | |
| PA2652 | −2.34 | Probable chemotaxis transducer | |
| PA2653 | −6.19 | Probable transporter | |
| PA3058 | pelG | 4.35 | PelG |
| PA3059 | pelF | 5.73 | PelF |
| PA3060 | pelE | 7.39 | PelE |
| PA3061 | pelD | 11.47 | PelD |
| PA3062 | pelC | 15.74 | PelC |
| PA3063 | pelB | 5.08 | PelB |
| PA3064 | pelA | 5.06 | PelA |
| PA3361 | lecB | 8.08 | Fucose-binding lectin PA-IIL |
| PA3476 | rhlI | 2.11 | Autoinducer synthesis protein RhlI |
| PA3477 | rhlR | 2.39 | Transcriptional regulator RhlR |
| PA3478 | rhlB | 4.73 | Rhamnosyltransferase chain B |
| PA3479 | rhlA | 11.00 | Rhamnosyltransferase chain A |
| PA3704 | wspE | −2.00 | Probable chemotaxis sensor/effector fusion protein |
| PA3705 | wspD | −2.87 | Hypothetical protein |
| PA3706 | wspC | −2.27 | Probable protein methyltransferase |
| PA3707 | wspB | −2.00 | Hypothetical protein |
| PA3708 | wspA | −2.28 | Probable chemotaxis transducer |
| PA3724 | lasB | 10.46 | Elastase LasB |
| PA4307 | pctC | −8.17 | Chemotactic transducer PctC |
| PA4624 | 6.22 | Hypothetical protein | |
| PA4625 | 7.60 | Hypothetical protein | |
| PA4843 | −3.04 | Putative diguanylate cyclase (GGDEF domain) |
T6SS, type VI secretion system.
Pel and Psl EPS each contribute to the RSCV phenotype.
Using our array data, we sought to determine genetic elements that contribute to the RSCV phenotype. Transcriptional profile data revealed that pel and psl gene expression was elevated. We also demonstrated previously that the psl EPS gene cluster contributes to the RSCV phenotype of MJK8 (21). Therefore, we constructed mutants of strain CF39s with pel and psl mutations to determine if these genes contributed to the RSCV phenotype of a clinical strain. A pslBCD mutation reduced autoaggregation in shaken liquid cultures, resulting in turbid cultures with a thick pellicle on the tube wall (Fig. 5A). A pelA mutation had little effect on the liquid culture phenotype, but the pellicle was disrupted more easily than it was in the parent strain. Neither pslBCD nor pelA individual mutations altered the colony morphology of the RSCV isolate. However, a CF39s pslBCD pelA double mutant strain had a smooth colony morphology and exhibited no autoaggregation in liquid culture (Fig. 5A). Identical phenotypes were observed for single and double mutations in MJK8 (see Fig. S3 in the supplemental material), confirming that the pel and psl gene clusters contributed to the RSCV phenotype of MJK8.
FIG. 5.

Characterization of CF39s and pel and psl mutant derivatives. (A) Parent strain CF39s autoaggregates in liquid culture. Aggregation is eliminated by introduction of a pslBCD mutation, while introduction of a pelA mutation has no effect on the phenotype. A pslBCD pelA double mutant forms no pellicle in the glass tube and does not autoaggregate. (B) Adherence of clinical isolate CF39s and EPS mutant derivatives of this isolate in a microtiter dish biofilm assay. CF39s is hyperadherent compared to the related strain CF39wt. Introduction of a pslBCD or pelA mutation into CF39s decreases adherence by at least one-half. Adherence is almost eliminated in the EPS double mutant of CF39s.
We previously found that in MJK8 there was a Psl-dependent increase in biofilm formation (21). To determine if there was a corresponding increase in the clinical RSCV, we tested relative biofilm formation using the microtiter dish adherence assay. The clinical RSCV produced over 20 times more biofilm biomass than the corresponding wild-type strain (Fig. 5B). The clinical wild-type and RSCV strains had nearly identical doubling times under these cultivation conditions (data not shown). As they do in MJK8, the EPS loci psl and pel contribute to biofilm formation in CF39s. Both pslBCD and pelA single mutations resulted in less biofilm biomass (approximately one-third less) than the parent strain CF39s. A pslBCD pelA double-mutant strain had a marked defect in adherence, forming less biofilm than even the wild-type isolate (Fig. 5B).
The data in Fig. 5 suggest that high-level expression of either the Pel or Psl EPS is sufficient to produce the RSCV phenotype. To test this hypothesis further, we used a PAO1 strain engineered to conditionally express the psl operon (23). In this strain, the psl promoter was replaced with the arabinose-inducible PBAD promoter so that the strain produced the Pel transcript in an arabinose dose-dependent manner. We found that overexpression of the psl transcript resulted in phenotypes on solid medium and in liquid culture similar to those of RSCVs (Fig. 6A and B), indicating that psl overexpression is sufficient to produce the RSCV phenotype.
FIG. 6.

Overexpression of the psl gene cluster produces RSCV phenotypes in solid and liquid cultures. An arabinose-inducible PBAD promoter was placed in front of the psl gene cluster in PAO1 to form strain WF801. (A) Strain WF801 forms RSCVs on solid medium that become progressively more rugose when the strain is grown in the presence of increasing concentrations of arabinose. (B) Strain WF801 autoaggregates in liquid culture, and the degree of autoaggregation increases with increasing amounts of arabinose. This strain was compared to wild-type strain PAO1 and its isogenic pslBCD mutant, strain WFPA800.
The cupA locus does not contribute to the RSCV phenotype.
The cupA operon consists of six genes that specify production of a fimbria belonging to the chaperone usher pathway. The cupA locus has been shown to be involved in biofilm formation in P. aeruginosa wild-type strains (39). In PAO1, the cupA locus is most highly expressed under oxygen-limiting conditions (2). CupA fimbriae were also found to be expressed at high levels in an RSCV-like variant using Western analysis (26). In two separate studies, transposon insertions into the cupA operon suppressed RSCV formation, suggesting that CupA fimbriae may play an important role in the RSCV phenotype (7, 26). Although cupA expression was not elevated in RSCV transcriptional profiles, CupA fimbriae still could play an integral role in different RSCV phenotypes.
To test whether the cupA locus contributes to the RSCV phenotype in our strains, we created a cupA3 deletion with a 1.6-kb cassette that could be excised with FLP recombinase (18). Prior to excision of the antibiotic cassette, the MJK8ΔcupA3 insertion mutant strain produced smooth colonies and did not autoaggregate in liquid culture. These observations seemed to confirm previous reports. However, when the resistance cassette was excised from the MJK8ΔcupA3, leaving an unmarked cupA3 deletion, the RSCV phenotype was restored (Fig. 7). We hypothesized that the cupA3 insertion cassette had a polar effect that caused elevated expression of the downstream genes in the operon, including PA2133. As noted above, this gene encodes a c-di-GMP phosphodiesterase. Thus, induced expression of this gene could lead to lower levels of intracellular c-di-GMP and reversal of the RSCV phenotype. To test this possibility, we measured the c-di-GMP levels in the mutant strains. Indeed, the c-di-GMP levels were significantly reduced in the cupA3 insertion mutant strain, whereas the c-di-GMP levels in the unmarked deletion strain were high and similar to those in the parent strain, MJK8 (see Fig. S4 in the supplemental material).
FIG. 7.

The cupA operon does not contribute to RSCV phenotypes. (A) Diagram of the cupA operon. A downstream gene, PA2133, contains an EAL domain with c-di-GMP-degrading activity. (B) A MJK8 cupA3 insertion mutant containing a resistance cassette from pSP858, MJK8ΔcupA3, does not autoaggregates in liquid culture and does not form a pellicle. When the resistance cassette is excised with FLP recombinase (MJK8ΔcupA3-FLP), autoaggregation and pellicle formation are restored. We also deleted the cupA1 gene in MJK8 and PAO1, which resulted in no observable phenotype. We mutated cupA3 by replacing the gene with a resistance cassette from pSP858, which restored wild-type colony morphology to MJK8 and eliminated autoaggregation in liquid culture. Additionally, in a culture of this mutant, no pellicle was produced. These phenotypes are similar to those of the parent strains harboring an EAL overexpression construct, so we hypothesized that the mutation may have a polar effect on the downstream EAL domain gene, PA2133. When the resistance cassette is excised from cupA3 with FLP recombinase, colony morphology and autoaggregation are restored to the parental MJK8 phenotypes.
Global transcriptional response to elevated c-di-GMP levels.
The data in Fig. 2 indicate that c-di-GMP is a key feature of the RSCV phenotype in P. aeruginosa. This intracellular signal is known to have regulatory effects at many levels. It has allosteric effects on alginate and Pel EPS biosynthesis (22, 27). In addition, the transcription factor FleQ regulates expression of psl, pel, and other genes in response to c-di-GMP (16). Our initial transcriptional profile comparisons of RSCVs and the corresponding wild-type strains were complicated by the fact that RSCV cells aggregate in liquid culture. We hypothesized that in the RSCVs, expression of certain genes is affected simply by the physical and chemical environment caused by autoaggregation, while other genes may be differentially expressed in direct response to elevated c-di-GMP levels. To examine this possibility, we took advantage of pel and psl mutations in the RSCV background. We first demonstrated that an MJK8 pelA pslBCD double-mutant strain had elevated levels of c-di-GMP, while it did not autoaggregate (see Fig. S5 in the supplemental material). Since MJK8 may harbor chromosomal mutations in addition to the deletion in wspA, we then constructed a clean wspF pelA pslBCD triple mutant of strain PAO1. As expected, this strain had elevated levels of c-di-GMP, while it did not autoaggregate in liquid culture (data not shown). We then compared the transcription profile of the wspF pelA pslBCD strain that produced high levels of c-di-GMP with the transcription profile of a PAO1 pelA pslBCD control strain that produced wild-type levels of c-di-GMP.
We found significantly fewer differentially expressed genes than we found when we compared an RSCV and a wild-type strain. A total of 48 genes were differentially expressed at least twofold; 31 genes were downregulated, and 15 genes were upregulated (Table 3). As expected, the first two genes in the psl cluster, pslA and pslB, were slightly upregulated (the rest of the psl cluster was not differentially expressed due to polar effects of the pslBCD mutation). Some interesting genes include the FleQ-regulated PA2440 and PA2441 genes and PA4624 and PA4625 genes, which were induced in the high-level c-di-GMP background (upregulated 5.4- and 5.7-fold and 11.7- and 9.7-fold, respectively). These genes encode proteins with homology to polysaccharide modification proteins and adhesins, respectively. In addition, genes not under transcriptional control of FleQ, such as PA0169 to PA0172, were upregulated. PA0169 encodes a protein harboring a GGDEF motif, which has been shown to catalyze c-di-GMP synthesis. Several of the downregulated genes encode functions related to motility, including several flagellar and chemotaxis genes, as well as genes involved in type IV pilus-mediated twitching motility.
TABLE 3.
Genes differentially expressed in an elevated c-di-GMP background
| PA no. | Gene | Change (fold) | Function |
|---|---|---|---|
| PA0020 | −1.6 | Hypothetical protein | |
| PA0026 | plcB | −1.9 | Phospholipase C |
| PA0408 | pilG | −1.6 | Response regulator, twitching motility |
| PA0409 | pilH | −1.9 | Response regulator, twitching motility |
| PA0652 | vfr | −1.5 | Regulator of virulence |
| PA1077 | flgB | −2.1 | Flagellar basal body rod protein |
| PA1078 | flgC | −2.3 | Flagellar basal body rod protein |
| PA1079 | flgD | −1.8 | Flagellar basal body rod modification |
| PA1080 | flgE | −1.9 | Flagellar hook protein |
| PA1081 | flgF | −2.4 | Flagellar basal body rod protein |
| PA1082 | flgG | −2.0 | Flagellar basal body rod protein |
| PA1083 | flgH | −2.0 | Flagellar L-ring protein precursor |
| PA1086 | flgK | −1.7 | Flagellar hook-associated protein |
| PA1087 | flgL | −1.9 | Flagellar hook-associated protein type 3 |
| PA1089 | −2.0 | Hypothetical protein | |
| PA1093 | −2.1 | Hypothetical protein | |
| PA1094 | fliD | −2.0 | Flagellar capping protein |
| PA1095 | −1.8 | Hypothetical protein | |
| PA1096 | −2.0 | Hypothetical protein | |
| PA1098 | fleS | −1.8 | Two-component sensor, flagellum regulation |
| PA1099 | fleR | −2.2 | Two-component regulator, flagellum regulation |
| PA1100 | fliE | −1.7 | Flagellar hook-basal body complex |
| PA1101 | fliF | −1.8 | Flagellum M-ring outer membrane protein |
| PA1441 | −2.0 | Hypothetical protein | |
| PA1452 | flhA | −1.8 | Flagellar biosynthesis protein |
| PA1456 | cheY | −1.6 | Chemotaxis response regulator |
| PA1545 | −2.2 | Hypothetical protein | |
| PA1705 | pcrG | −2.0 | Regulator in type III secretion |
| PA1718 | pscE | −1.8 | Type III export protein PscE |
| PA2654 | −2.4 | Methyl-accepting chemotaxis transducer | |
| PA2761 | −3.1 | Hypothetical protein | |
| PA3351 | flgM | −1.5 | Anti-sigma factor, flagellum regulation |
| PA3526 | motY | −1.9 | Sodium-driven flagellum motor protein |
| PA3713 | spdH | −2.6 | Spermidine dehydrogenase |
| PA3762 | −2.4 | Hypothetical protein | |
| PA4221 | fptA | −2.9 | Fe(III)-pyochelin outer membrane receptor |
| PA4296 | pprB | −1.9 | Two-component response regulator |
| PA4307 | pctC | −7.2 | Methyl-accepting chemotaxis transducer |
| PA4309 | pctA | −2.1 | Methyl-accepting chemotaxis transducer |
| PA4310 | pctB | −1.7 | Methyl-accepting chemotaxis transducer |
| PA4523 | −5.1 | Hypothetical protein | |
| PA4616 | −3.3 | Probable C4-dicarboxylate-binding protein | |
| PA4633 | −2.3 | Methyl-accepting chemotaxis transducer | |
| PA4704 | −2.2 | Hypothetical protein | |
| PA5040 | pilQ | −1.8 | Type 4 fimbrial biogenesis protein |
| PA5041 | pilP | −1.9 | Type 4 fimbrial biogenesis protein |
| PA5042 | pilO | −2.3 | Type 4 fimbrial biogenesis protein |
| PA5043 | pilN | −2.0 | Type 4 fimbrial biogenesis protein |
| PA5044 | pilM | −2.0 | Type 4 fimbrial biogenesis protein |
| PA5072 | −2.2 | Methyl-accepting chemotaxis transducer | |
| PA5122 | −1.8 | Hypothetical protein | |
| PA5139 | −1.7 | Hypothetical protein | |
| PA5404 | −2.3 | Hypothetical protein | |
| PA0169 | 5.1 | Putative diguanylate cyclase (GGDEF protein) | |
| PA0170 | 4.4 | Hypothetical protein | |
| PA0171 | 3.9 | Hypothetical protein | |
| PA0172 | 3.6 | Hypothetical protein | |
| PA2231 | pslA | 2.4 | Polysaccharide biosynthesis protein |
| PA2232 | pslB | 4.7 | Polysaccharide biosynthesis protein |
| PA2406 | 4.3 | Hypothetical protein | |
| PA2407 | 4.6 | Probable adhesion protein | |
| PA2440 | 5.4 | Putative polysaccharide deacteylase | |
| PA2441 | 5.7 | ||
| PA2560 | 2.0 | Hypothetical protein | |
| PA3340 | 2.2 | Hypothetical protein | |
| PA3819 | 1.7 | Hypothetical protein | |
| PA3901 | fecA | 1.7 | Fe(III) dicitrate transport protein |
| PA4312 | 2.1 | Hypothetical protein | |
| PA4624 | 9.7 | Hypothetical protein | |
| PA4625 | 11.7 | Putative hemagglutinin-adhesin | |
| PA5212 | 1.6 | Hypothetical protein | |
| PA5526 | 1.8 | Hypothetical protein |
Thus, our transcriptional profiling data indicate that the differences for a majority of the differentially expressed genes in RSCV strains are due to liquid culture autoaggregation rather than to responses to elevated intracellular c-di-GMP levels per se.
The RSCV phenotype has a major impact on P. aeruginosa physiology.
The array data indicate that one of the most pronounced effects of autoaggregation is the effect on P. aeruginosa metabolism. Several of the differentially regulated genes in the RSCV background encode functions related to metabolism, for example, genes involved in nitrate respiration, which were upregulated.
To better characterize the impact of the RSCV phenotype on general metabolism, we performed Biolog phenotypic profiling with an RSCV strain, PAO1ΔwspF, and compared this strain to wild-type strain PAO1 and a PAO1 wspF pslBCD pelA mutant. In general, we found that RSCVs did not grow as well on amino acids as sole carbon sources as the wild-type strain (Fig. 8; see Fig. S6 in the supplemental material). However, RSCVs did grow as well as the wild-type strain on hydrophobic carbon sources, such as Tween 40 and Tween 80, and on some tricarboxylic acid cycle intermediates, including fumarate and α-ketoglutarate. The PAO1 and PAO1ΔwspF strains appeared to utilize various nitrogen sources equally well, with one exception, dipeptides. In addition, these two strains were able to utilize different phosphorus sources equally well, but the PAO1ΔwspF strain was defective in utilization of cysteine and cysteine-related compounds as sulfur sources. To determine whether the PAO1ΔwspF phenotypes are due to elevated c-di-GMP levels or autoaggregation, we profiled a PAO1 wspF pslBCD pelA mutant. The pattern of this mutant was almost identical to that of PAO1, indicating that autoaggregation was probably responsible for the differences observed in the PAO1ΔwspF mutant Biolog profiles. (see Fig. S6 in the supplemental material).
Clinical and laboratory RSCVs elicit a reduced chemokine response from polarized airway epithelium cells.
Several features of RSCVs suggest that they may be less immunostimulatory in the host. These features include reduced flagellar synthesis and motility and an increase in expression of potential immunomodulatory functions (such as loxA and PA4625). Previous research demonstrated that loxA encodes a protein that has lipoxygenase activity using arachidonic acid as a substrate. Products of LoxA activity could decrease inflammatory signaling in the human host. Therefore, we hypothesized that this gene might have an anti-inflammatory role in RSCV cells, causing them to dampen the host inflammatory response (40).
We sought to determine the eukaryotic immune responses of human airway epithelia to both laboratory and clinical RSCVs compared to related wild-type strains. Since the loxA gene was highly expressed in RSCV strains, we included CF39s and MJK8 loxA mutant strains. Infection with 106 bacteria elicited IL-8 and NF-κB responses from polarized human lung epithelial cells 4 h after infection. RSCV strains were thoroughly resuspended prior to inoculation to reduce indirect effects due to aggregation. In both assays, RSCVs elicited a dampened immune response compared to the wild-type isolates elicited. We also observed decreased immune responses with the loxA mutant strains, which elicited similarly reduced IL-8 and NF-κB levels compared to those elicited by the parent RSCV strains (Fig. 9), indicating that PA1169 is likely not responsible for the reduced IL-8 or NF-κB responses observed in RSCV isolates.
FIG. 9.

Production of the cytokine IL-8 and NF-κB on human polarized airway epithelia. Equal numbers of the strains of bacteria indicated were layered onto airway epithelia, and cytokine levels and NFκB signaling were determined after 4 h. Compared to the PAO1 and CF39wt wild-type strains, strains MJK8, MJK8ΔloxA, and CF39s elicited reduced IL-8 (A) and NF-κB (B) responses. The MJK8ΔpslBCDpelA double mutant showed responses similar to those of wild-type strain PAO1. *, P < 0.05; **, P < 0.01.
DISCUSSION
Previous studies have reported isolation of P. aeruginosa RSCVs, and there is evidence that these variants play a role in disease pathogenesis. In this study, we report isolation and characterization of clonally related RSCV and wild-type isolates from a CF sputum sample. We demonstrated that our clinical RSCV displayed many of the same characteristics as laboratory biofilm RSCV isolates. One such characteristic is the contribution of the Pel and Psl EPS to the phenotype. Another similarity is the elevated c-di-GMP levels in the clinical RSCV compared to the clinical wild-type strain. Overexpression of a phosphodiesterase that degrades intracellular c-di-GMP eliminated the autoaggregation and hyper-biofilm-formation phenotypes in all the RSCV strains tested. This suggests that c-di-GMP plays a central role in modulating the transition between wild-type and RSCV phenotypes.
Our transcriptome and Biolog profiling data suggest that the clinical and laboratory-derived RSCVs that we examined are physiologically similar. The autoaggregation of RSCV cells in liquid culture affects the expression of a large subset of genes, including some genes that impact metabolism. For example, RSCV strains were characterized by increased expression of denitrification genes. The expression of these genes likely occurred in response to anaerobic pockets that developed in aggregates of cells as they consumed the available oxygen. Biolog phenotyping data suggest that autoaggregation has other effects on metabolism. RSCV cells were deficient in growth on several carbon sources (Fig. 8). This also appeared to be a consequence of autoaggregation, since the wspF pelA pslBDCD triple mutant (which exhibited no autoaggregation but had elevated c-di-GMP levels) had wild-type carbon utilization patterns. This suggests that in laboratory biofilms and CF airways, the carbon sources that RSCVs can utilize may be limited. Interestingly, we found that the type VI secretion locus HSI-I was upregulated in the clinical RSCV. Antibodies to Hcp, the secreted effector, have been used to detect Hcp in sputum from a CF patient infected for a long time; thus, type VI secretion may be another hallmark of chronic infections (28).
The differential expression of a second, smaller subset of genes in RSCVs is a consequence of elevated cyclic-di-GMP levels. It appears that motility and EPS production are major functions that are transcriptionally controlled by c-di-GMP. Some of the differentially expressed genes are known to be controlled by FleQ, a cyclic-di-GMP-responsive transcriptional repressor (16). However, some genes whose expression is impacted by elevated cyclic-di-GMP levels (e.g., PA0169 to PA0172) are not regulated by FleQ. This suggests that there may be other cyclic-di-GMP-sensitive transcriptional regulators.
Data presented here and elsewhere show that the pel and psl EPS biosynthetic loci are key contributors to the RSCV phenotype. Single mutations in either of these gene clusters have only a partial effect on RSCV phenotypes. Only pel psl double mutations fully convert RSCVs to wild-type strains. It is not clear whether these EPS types are functionally redundant or if they have unique roles in RSCVs. Interestingly, in contrast to previous reports, the cupA operon did not appear to contribute to the RSCV phenotype (26). Insertion-deletion mutations in cupA3 appeared to suppress the RSCV phenotype; however, excision of the antibiotic cassette insertion resulted in reversion back to the RSCV phenotype. One potential explanation for this is that the antibiotic resistance cassette induces expression of the last gene in the operon, PA2133 encoding the c-di-GMP-degrading phophodiesterase. Our analysis of intracellular c-di-GMP levels supports this; the c-di-GMP levels were depleted in the insertion mutant, while excision of the antibiotic resistance cassette restored c-di-GMP levels (see Fig. S4 in the supplemental material). Meissner et al. reported upregulation of CupA fimbriae in RSCV backgrounds. We have evidence that liquid culture autoaggregation creates anaerobic pockets in the aggregates. Since microaerobic conditions induce cupA expression, we think that reduced oxygen tension in RSCVs might have contributed to the cupA expression in the study of Meissner et al. (2, 26).
The complementation analysis suggests that there are two classes of variants based on wspF complementation. Even though these two classes appear to have many phenotypic similarities, we do not know whether there are any important phenotypic differences. Although the levels of intracellular c-di-GMP are high for both classes, the quantities may differ. This may affect Pel and Psl expression levels, which in turn have the potential to impact the degree of autoaggregation. RSCVs characterized by higher cyclic-di-GMP levels may have differences in key phenotypes, such as antimicrobial resistance. A challenge for the future is to identify the mutations that confer the RSCV phenotype for class B strains. Once this has been achieved, functional comparisons can be made among isogenic RSCV strains. von Gotz et al. reported that an RSCV-like strain isolated from a CF patient is highly motile and extremely cytotoxic and expresses high levels of type III secretion genes (41). In our transcriptional analysis, we found that the RSCVs did not express elevated levels of type III secretion genes. The differences between the results illustrate the fact that not all RSCV strains are exactly alike (15, 21).
What are the selective forces in the CF lung that are responsible for selecting for RSCV formation? Are these selective forces the same as those that produce RSCVs in laboratory-cultured biofilms? Since chronic CF infections involve biofilms, some selective pressure present in both laboratory biofilms and CF biofilms might amplify the RSCV phenotype (35). Previous studies have shown that RSCVs exhibit elevated tolerance to hydrogen peroxide (5). Recent work by Boles and Singh also identified oxidative stress as a selective pressure for diversification in laboratory biofilms (4). One stimulus for the generation of mucoid variants in the lung is thought to be reactive oxygen species (24). Perhaps oxidative stress present in both lab and CF biofilms is a key selective pressure for RSCVs as well.
RSCV strains appear to grow relatively poorly on certain carbon and sulfur sources, including amino acids. Amino acids are suggested to be a major constituent of airway secretions, and they presumably support the growth of P. aeruginosa and other colonizing species (29). RSCVs may not be able to compete with wild-type strains (or other bacterial species) for amino acids, which may explain why they are never the predominant organisms in P. aeruginosa strains isolated from a given patient. The heightened resistance of RSCVs to antimicrobials may allow them to successfully compete for growth substrates in microniches subjected to elevated antimicrobial stress, where wild-type P. aeruginosa strains and other bacterial species would be impaired. Alternatively, RSCVs may be better adapted to use other growth substrates with which they do not have such a severe growth handicap, such as fatty acid and hydrophobic substrates. For example, RSCVs grew as well as wild-type strains on Tween substrates. The CF airways contain host-derived fatty acids and lipids. In addition, hydrophobic substrates are also present in older laboratory biofilms as cells begin to lyse. Consideration of all these points leads to the hypothesis that RSCVs may occupy a nutritional niche in these biofilm environments.
The CF host mounts a tremendous immune response to chronic infection (13). This response probably exhibits temporal and spatial variability in the airways. Given their highly autoaggregative nature, RSCVs probably exist in the airways as distinct aggregates. The local host immune response surrounding these aggregates may be dampened due to reduced expression of flagella and an increase in expression of RSCV-induced immunomodulatory functions. Thus, RSCVs may represent a nidus of persistence that can contribute to reseeding of the airways after a course of antibiotic treatment. Precisely how the RSCV strains, of both clinical and biofilm origin, elicit a reduced inflammatory response is unknown. One observation from the array data is that flagellar genes are downregulated in RSCV strains. Since flagellar expression is known to cause Toll-like receptor 5-mediated inflammation in the host, this might be one factor contributing to the response in our assays (31). Collectively, the hyper-biofilm-formation, increased antibiotic tolerance, and reduced inflammation traits of RSCVs suggest they are a subpopulation geared toward persistence in biofilms and the CF airway environment.
Supplementary Material
Acknowledgments
We thank Gary Doern for performing the PFGE assays. We thank Jon Penterman and Pradeep Singh for providing unpublished data.
We acknowledge NSF (grant MCB0822405), CFF (grant PARSEK05G0), and NIH (grant R01 AI61396 and grant GM56665 to C.S.H.).
Footnotes
Published ahead of print on 27 March 2009.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Allesen-Holm, M., K. B. Barken, L. Yang, M. Klausen, J. S. Webb, S. Kjelleberg, S. Molin, M. Givskov, and T. Tolker-Nielsen. 2006. A characterization of DNA release in Pseudomonas aeruginosa cultures and biofilms. Mol. Microbiol. 591114-1128. [DOI] [PubMed] [Google Scholar]
- 2.Alvarez-Ortega, C., and C. S. Harwood. 2007. Responses of Pseudomonas aeruginosa to low oxygen indicate that growth in the cystic fibrosis lung is by aerobic respiration. Mol. Microbiol. 65153-165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bochner, B. R., and B. N. Ames. 1982. Complete analysis of cellular nucleotides by two-dimensional thin layer chromatography. J. Biol. Chem. 2579759-9769. [PubMed] [Google Scholar]
- 4.Boles, B. R., and P. K. Singh. 2008. Endogenous oxidative stress produces diversity and adaptability in biofilm communities. Proc. Natl. Acad. Sci. USA 10512503-12508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boles, B. R., M. Thoendel, and P. K. Singh. 2004. Self-generated diversity produces “insurance effects” in biofilm communities. Proc. Natl. Acad. Sci. USA 10116630-16635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burns, J. L., R. L. Gibson, S. McNamara, D. Yim, J. Emerson, M. Rosenfeld, P. Hiatt, K. McCcoy, R. Castile, A. L. Smith, and B. W. Ramsey. 2001. Longitudinal assessment of Pseudomonas aeruginosa in young children with cystic fibrosis. J. Infect. Dis. 183444-452. [DOI] [PubMed] [Google Scholar]
- 7.D'Argenio, D. A., M. W. Calfee, P. B. Rainey, and E. C. Pesci. 2002. Autolysis and autoaggregation in Pseudomonas aeruginosa colony morphology mutants. J. Bacteriol. 1846481-6489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.D'Argenio, D. A., and S. I. Miller. 2004. Cyclic di-GMP as a bacterial second messenger. Microbiology 1502497-2502. [DOI] [PubMed] [Google Scholar]
- 9.Deziel, E., Y. Comeau, and R. Villemur. 2001. Initiation of biofilm formation by Pseudomonas aeruginosa 57RP correlates with emergence of hyperpiliated and highly adherent phenotypic variants deficient in swimming, swarming, and twitching motilities. J. Bacteriol. 1831195-1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Drenkard, E., and F. M. Ausubel. 2002. Pseudomonas biofilm formation and antibiotic resistance are linked to phenotypic variation. Nature 416740-743. [DOI] [PubMed] [Google Scholar]
- 11.Friedman, L., and R. Kolter. 2004. Two genetic loci produce distinct carbohydrate-rich structural components of the Pseudomonas aerguinosa biofilm matrix. J. Bacteriol. 1864457-4465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gibson, R. L., J. L. Burns, and B. W. Ramsey. 2003. Pathophysiology and management of pulmonary infections in cystic fibrosis. Am. J. Respir. Crit. Care Med. 168918-951. [DOI] [PubMed] [Google Scholar]
- 13.Govan, J. R. W., and V. Deretic. 1996. Microbial pathogenesis in cystic fibrosis: mucoid Pseudomonas aeruginosa and Burkholderia cepacia. Microbiol. Rev. 60539-574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haussler, S., B. Tummler, H. Weissbrodt, M. Rohde, and I. Steinmetz. 1999. Small-colony variants of Pseudomonas aeruginosa in cystic fibrosis. Clin. Infect. Dis. 29621-625. [DOI] [PubMed] [Google Scholar]
- 15.Haussler, S., I. Ziegler, A. Lottel, F. von Gotz, M. Rohde, D. Wehmhohner, S. Saravanamuthu, B. Tummler, and I. Steinmetz. 2003. Highly adherent small-colony variants of Pseudomonas aeruginosa in cystic fibrosis lung infection. J. Med. Microbiol. 52295-301. [DOI] [PubMed] [Google Scholar]
- 16.Hickman, J. W., and C. S. Harwood. 2008. Identification of FleQ from Pseudomonas aeruginosa as a c-di-GMP-responsive transcription factor. Mol. Microbiol. 69376-389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hickman, J. W., D. F. Tifrea, and C. S. Harwood. 2005. A chemosensory system that regulates biofilm formation through modulation of cyclic diguanylate levels. Proc. Natl. Acad. Sci. USA 10214422-14427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoang, T. T., R. R. Karkhoff-Schweizer, A. J. Kutchma, and H. Schweizer. 1998. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: applications for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 21277-86. [DOI] [PubMed] [Google Scholar]
- 19.Jackson, K. D., M. Starkey, S. Kremer, M. R. Parsek, and D. J. Wozniak. 2004. Identification of psl, a locus encoding a potential exopolysaccharide that is essential for Pseudomonas aeruginosa PAO1 biofilm formation. J. Bacteriol. 1864466-4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karp, P. H., T. O. Moninger, S. P. Weber, T. S. Nesselhauf, J. L. Launspach, J. Zabner, and M. J. Welsh. 2002. An in vitro model of differentiated human airway epithelia. Methods for establishing primary cultures. Methods Mol. Biol. 188115-137. [DOI] [PubMed] [Google Scholar]
- 21.Kirisits, M. J., L. Prost, M. Starkey, and M. R. Parsek. 2005. Characterization of colony morphology variants isolated from Pseudomonas aeruginosa biofilms. Appl. Environ. Microbiol. 714809-4821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee, V., J. M. Matewish, J. L. Kessler M. Hyodo, Y. Hayakawa, and S. Lory. 2007. A cyclic-di-GMP receptor required for bacterial exopolysaccharide production. Mol. Microbiol. 651474-1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma, L., K. D. Jackson, R. M. Landry, M. R. Parsek, and D. J. Wozniak. 2006. Analysis of Pseudomonas aeruginosa conditional Psl variants reveals roles for the Psl polysaccharide in adhesion and maintaining biofilm structure postattachment, J. Bacteriol. 1888213-8221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mathee, K., O. Ciofu, C. Sternberg, P. W. Lindum, J. I. Campbell, P. Jensen, A. H. Johnsen, M. Givskov, D. E. Ohman, S. Molin, N. Hoiby, and A. Kharazmi. 1999. Mucoid conversion of Pseudomonas aeruginosa by hydrogen peroxide: a mechanism for virulence activation in the cystic fibrosis lung. Microbiology 1451349-1357. [DOI] [PubMed] [Google Scholar]
- 25.Matsukawa, M., and E. P. Greenberg. 2004. Putative exopolysaccharide synthesis genes influence Pseudomonas aeruginosa biofilm development. J. Bacteriol. 1864449-4456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meissner, A., V. Wild, R. Simm, M. Rohde, C. Erck, F. Bredenbruch, M. Morr, U. Römling, and S. Häussler. 2007. Pseudomonas aeruginosa cupA-encoded fimbriae expression is regulated by a GGDEF and EAL domain-dependent modulation of the intracellular level of cyclic diguanylate. Environ. Microbiol. 92475-2485. [DOI] [PubMed] [Google Scholar]
- 27.Merighi, M., V. T. Lee, M. Hyodo, Y. Hayakawa, and S. Lory. 2007. The second messenger bis-(3′-5′)-cyclic-GMP and its PilZ domain-containing receptor Alg44 are required for alginate biosynthesis in Pseudomonas aeruginosa. Mol. Microbiol. 65876-895. [DOI] [PubMed] [Google Scholar]
- 28.Mougous, J. D., M. E. Cuff, S. Raunser, A. Shen, M. Zhou, C. A. Gifford, A. L. Goodman, G. Joachimiak, C. L. Ordonez, S. Lory, T. Walz, A. Joachimiak, and J. J. Mekalanos. 2006. A virulence locus of Pseudomonas aeruginosa encodes a protein secretion apparatus. Science 3121526-1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Palmer, K. L., L. M. Mashburn, P. K. Singh, and M. Whiteley. 2005. Cystic fibrosis sputum supports growth and cues key aspects of Pseudomonas aeruginosa physiology. J. Bacteriol. 1875267-5277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pedersen, S. S., N. Hoiby, F. Espersen, and C. Koch. 1992. Role of alginate in infection with mucoid Pseudomonas aeruginosa in cystic fibrosis. Thorax 476-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Prince, A. 2006. Flagellar activation of epithelial signaling. Am. J. Respir. Cell Mol. Biol. 34548-551. [DOI] [PubMed] [Google Scholar]
- 32.Sanlioglu, S., C. M. Williams, L. Samavati, N. S. Butler, G. Wang, P. B. McCray, Jr., T. C. Ritchie, G. W. Hunninghake, E. Zandi, and J. F. Engelhardt. 2001. Lipopolysaccharide induces Rac1-dependent reactive oxygen species formation and coordinates tumor necrosis factor-alpha secretion through IKK regulation of NF-kappa B. J. Biol. Chem. 27630188-30198. [DOI] [PubMed] [Google Scholar]
- 33.Schuster, M., C. P. Lostroh, T. Ogi, and E. P. Greenberg. 2003. Identification, timing, and signal specificity of Pseudomonas aeruginosa quorum-controlled genes: a transcriptome analysis. Proc. Natl. Acad. Sci. USA 1852066-2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schweizer, H. P. 1991. Improved broad-host-range lac-based plasmid vectors for the isolation and characterization of protein fusions in Pseudomonas aeruginosa. Gene 10387-92. [DOI] [PubMed] [Google Scholar]
- 35.Singh, P. K., A. L. Schaefer, M. R. Parsek, T. O. Moninger, M. J. Welsh, and E. P. Greenberg. 2000. Quorum-sensing signals indicate that cystic fibrosis lungs are infected with bacterial biofilms. Nature 407762-764. [DOI] [PubMed] [Google Scholar]
- 36.Smith, E. E., D. G. Buckley, Z. Wu, C. Saenphimmachak, L. R. Hoffman, D. A. D'Argenio, S. I. Miller, B. W. Ramsey, D. P. Speert, S. M. Moskowitz, J. L. Burns, R. Kaul, and M. V. Olson. 2006. Genetic adaptation by Pseudomonas aeruginosa to the airways of cystic fibrosis patients. Proc. Natl. Acad. Sci. USA 1038487-8492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Starner, T. D., N. Zhang, G. Kim, M. A. Apicella, and P. B. McCray, Jr. 2006. Haemophilus influenzae forms biofilms on airway epithelia: implications in cystic fibrosis. Am. J. Respir. Cell Mol. Biol. 174213-220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tischler, A. D., and A. Camilli. 2004. Cyclic diguanylate (c-di-GMP) regulates Vibrio cholerae biofilm formation. Mol. Microbiol. 53857-869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vallet, I., J. W. Olson, S. Lory, A. E. Lazdunski, and A. Filloux. 2001. The chaperone/usher pathways of Pseudomonas aeruginosa: identification of fimbrial gene clusters (cup) and their involvement in biofilm formation. Microbiology 986911-6916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vance, R. E., S. Hong, K. Gronert, C. N. Serhan, and J. J. Mekalanos. 2004. The opportunistic pathogen Pseudomonas aeruginosa carries a secretable arachidonate 15-lipoxygenase. Proc. Natl. Acad. Sci. USA 1012135-2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.von Gotz, F., S. Haussler, D. Jordan, S. S. Saravanamuthu, D. Wehmhoner, A. Strussmann, J. Lauber, I. Attree, J. Buer, B. Tummler, and I. Steinmetz. 2004. Expression analysis of a highly adherent and cytotoxic small-colony variant of Pseudomonas aeruginosa isolated from a lung of a patient with cystic fibrosis. J. Bacteriol. 1863837-3847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zierdt, C. H., and P. J. Schmidt. 1964. Dissociation in Pseudomonas aeruginosa. J. Bacteriol. 871003-1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.