

Abstract
Each Sindbis virus (SINV) surface glycoprotein has two sites for N-linked glycosylation (E1 positions 139 and 245 [E1-139 and E1-245] and E2 positions 196 and 318 [E2-196 and E2-318]). Studies of SINV strain TE12 mutants with each site eliminated identified the locations of carbohydrates by cryo-electron microscopy (S. V. Pletnev et al., Cell 105:127-136, 2001). In the current study, the effects of altered glycosylation on virion infectivity, growth in cells of vertebrates and invertebrates, heparin binding, virulence in mice, and replication in mosquitoes were assessed. Particle-to-PFU ratios for E1-139 and E2-196 mutant strains were similar to that for TE12, but this ratio for the E1-245 mutant was 100-fold lower than that for TE12. Elimination of either E2 glycosylation site increased virus binding to heparin and increased replication in BHK cells. Elimination of either E1 glycosylation site had no effect on heparin binding but resulted in an approximately 10-fold decrease in virus yield from BHK cells compared to the TE12 amount. No differences in pE2 processing were detected. E2-196 and E2-318 mutants were more virulent in mice after intracerebral inoculation, while E1-139 and E1-245 mutants were less virulent. The E1-245 mutant showed impaired replication in C7/10 mosquito cells and in Culex quinquefasciatus after intrathoracic inoculation. We conclude that the increased replication and virulence of E2-196 and E2-318 mutants are primarily due to increased efficiency of binding to heparan sulfate on mammalian cells. Lack of glycosylation at E1-139 or E1-245 impairs replication in vertebrate cells, while E1-245 also severely affects replication in invertebrate cells.
Sindbis virus (SINV), the prototype alphavirus in the family Togaviridae, is an arthropod-borne virus that cycles between mosquitoes and wild birds and causes summertime outbreaks of rashes and arthritis in humans (17, 34). In mice, SINV causes encephalomyelitis and serves as a useful model for the study of alphavirus infections of the central nervous system (CNS). The virion is enveloped and has icosahedral symmetry, with a single linear strand of positive-sense RNA surrounded by a capsid and two transmembrane glycoproteins, E1 and E2, that heterodimerize and then form trimeric spikes on the surface. Each of the viral glycoproteins has two N-linked glycosylation sites at E1 positions 139 and 245 (E1-139 and E1-245) and at E2 positions 196 and 318 (E2-196 and E2-318) that are used (4, 42). Some glycosylation is required for proper folding and transport of the glycoproteins, as total ablation of glycosylation by treatment with tunicamycin results in failure of E1 and E2 to be transported through the Golgi apparatus to the cell surface (31, 32).
The type of carbohydrate, high-mannose carbohydrate or complex oligosaccharide, at a glycosylation site varies with the type and growth state of the cell infected (3, 18, 23, 28, 29, 35, 51). In insect cells, all sites have high-mannose sugars (22) due to the absence of N-acetylglucosaminyl-, galactosyl-, and sialyltransferases (5). In vertebrate cells, the distribution of complex and high-mannose carbohydrates is largely determined by the accessibility of the site to processing enzymes in the Golgi apparatus (23).
E1 has an extracellular domain that contains the internal fusion peptide. The carbohydrate at E1-139 consists of complex oligosaccharides in vertebrate cells (35), while the carbohydrate at E1-245 has a mixture of complex and high-mannose oligosaccharides (24, 35). These glycosylation patterns are consistent with E1-139 being located away from the center of the trimer spikes and more accessible to processing enzymes and E1-245 being located near the base of the spikes and less accessible, as shown by cryo-electron microscopy difference maps (41). Studies of the related alphaviruses Ross River virus and Semliki Forest virus indicate that the heterohexameric spike complexes surround an apparently hollow area at the center of the spikes (8, 12, 21) that in SINV is occupied by the E1-245 carbohydrate (41).
E2 is the receptor binding protein, and it interacts with the capsid protein during virion morphogenesis and has an extracellular domain that covers much of E1 to constitute the majority of the exposed protein in the virion spikes (39, 52, 52). The carbohydrate at E2-196 consists mostly of complex oligosaccharides, while the carbohydrate at E2-318 is of the high-mannose type (24, 35). Studies using antibodies and antibody escape mutants (9, 37, 44, 50), cryo-electron microscopy (41, 49), and mass spectrometry (40) to determine surface conformation and exposed peptides indicated that the carbohydrate at E2-196 is exposed on the surface of the spike, near the top. The carbohydrate at E2-318 is located at the bottom of the spike complex, on top of the membrane.
Previous studies have shown that deleting or inserting SINV glycosylation sites affects virus assembly, binding to host cells, and antibody reactivity (9, 10, 32). Because the presence or absence of glycosylation affects various aspects of the SINV life cycle, small changes in E1 or E2 structure can affect neurovirulence, and effects can be cell type specific, we analyzed the effects of deleting each of the glycosylation sites on virion infectivity, heparin binding, virus replication in vertebrate and invertebrate cells, glycoprotein processing and trafficking, neurovirulence in mice, and infection of mosquitoes.
MATERIALS AND METHODS
Viruses and cells.
BHK-21 cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin G, and 100 μg/ml streptomycin sulfate at 37°C in a 5% CO2 atmosphere. C7/10 mosquito cells (46) were grown at 28°C in 5% CO2 in minimal essential medium supplemented with 10% FBS, 5% tryptose phosphate broth, penicillin, and streptomycin.
Recombinant strains of SINV TE12 (33) mutated to change the asparagine at E1-139, E1-245, E2-196, or E2-318 to glutamine were previously described (41) and were acquired as cDNAs (courtesy of Richard Kuhn, Purdue University, and Dennis Brown, North Carolina State University). Full-length RNA was transcribed from the cDNAs by using the mMessage mMachine in vitro transcription kit for SP6 promoters (Ambion) and transfected into BHK-21 cells by using Lipofectin. Supernatant fluids were collected, and viral titers were determined by plaque assay on BHK cells.
35S-labeled virus was prepared by growing virus in cysteine/methionine-free DMEM supplemented with 3.3 μCi/ml Translabel 35S (MP Biomedicals, Irvine, CA). Virus was purified by polyethylene glycol precipitation followed by sedimentation on a potassium tartrate gradient as previously described (6). Virus was resuspended in phosphate-buffered saline (PBS; pH 7.2) containing 0.5% bovine serum albumin and stored at −70°C until used.
Particle-to-PFU ratio.
The ratio of virus particles to PFU was determined by electron microscopy. Direct counts from electron micrographs were done by mixing potassium tartrate-banded virus previously assayed for infectious virus with a known concentration of 100-nm polystyrene beads (Molecular Probes). This mixture was applied to a glow-discharged 400-mesh Parlodion-coated copper electron microscopy grid and allowed to dry. Grids were stained with uranyl acetate and examined at ×15,000 magnification. Ten randomly chosen fields were photographed and digitized for each grid. Images were examined under various conditions of brightness and contrast at a final magnification of ×95,000 to ensure that all virus particles and polystyrene beads were counted. The average numbers of virus particles and polystyrene beads for the 10 electron micrographs were used to calculate the virus particle concentration as follows: X = concentration of beads × no. of virus particles/no. of beads.
Heparin-Sepharose chromatography.
The heparin binding properties of the viruses were determined as previously described (6). Briefly, 35S-labeled virus was loaded onto a HiTrap heparin high-performance 1-ml column (Amersham Pharmacia) in 100 μl of column binding buffer (100 mM NaCl, 5 mM phosphate, 0.5% bovine serum albumin, pH 7.5) and eluted with a 100 mM-to-500 mM NaCl gradient. One-milliliter fractions were collected and analyzed for 35S cpm and for [NaCl] by measuring conductivity. Each virus was tested three or four, times and the concentrations of NaCl at which there was a peak in virus elution, as determined by radioactivity, were averaged.
Virus replication in tissue culture.
BHK or C7/10 cells were infected at a multiplicity of infection (MOI) of 8 to 10. Virus was suspended in DMEM and 2% FBS and added to cells for 1 h at 37°C. Cells were washed, and medium was replaced. At each time point, 50 μl of supernatant fluid was harvested and replaced with an equal volume for each of three wells. Titers of the samples were determined by plaque assay on BHK cells.
Pulse-chase analysis of glycoproteins.
Five hours after infection (MOI = 5), BHK-21 cells were incubated with Met- and Cys-free DMEM for 1 h followed by the addition of 50 μCi/ml Translabel 35S for 20 min. Cells were washed with PBS and incubated with chase medium (DMEM-10% FBS plus 15 mg/liter l-Met). At various times after the pulse, cells were washed with cold PBS and lysed with radioimmunoprecipitation assay buffer (1% NP-40, 0.1% sodium dodecyl sulfate, 0.5% Na deoxycholate, 50 mM Tris-HCl [pH 8.0], 150 mM NaCl). Samples were stored at −80°C and analyzed simultaneously. SINV structural proteins were immunoprecipitated with a polyclonal rabbit antibody (25) and protein A-conjugated beads (Thermo Scientific, Waltham, MA). Protein processing was visualized by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and autoradiography.
Infection of mice.
Groups of 19 to 23 CD-1 or C57BL/6 mice (Charles River, Wilmington, MA) were infected intracerebrally with 1,000 PFU virus in 20 μl Hanks balanced salt solution (HBSS). Control mice were inoculated with the same volume of HBSS. CD-1 mice were infected at 2 days, 1 week, 2 weeks, 3 weeks, or 10 weeks of age. C57BL/6 mice were infected at 3 weeks of age. Mice were observed daily for morbidity and mortality.
To assess virus replication, brains and spinal cords were collected from three 17-day-old CD-1 mice at 3, 5, and 7 days after infection with virus. Tissues were weighed, homogenized, and assayed for plaque formation on BHK cells. All animal experiments were performed in accordance with protocols approved by the Johns Hopkins Animal Care and Use Committee.
Infection of mosquitoes.
Culex quinquefasciatus were generously supplied as pupae by Robin Todd (Insect Control Research, Baltimore, MD). Pupae were allowed to eclose in the insectary at Johns Hopkins. Adults were kept at 26°C, 80% relative humidity, and a 14-h/10-h light-dark cycle and allowed to feed on 15% sucrose ad libitum. At 3 to 5 days of age, 40 adult female mosquitoes were injected in the membranous area anterior to the mesepisternum with 100 PFU of virus in a volume of 0.4 μl HBSS (43). After infection, mosquitoes were held at 24°C, 80% relative humidity, and a 12-h/12-h light-dark cycle. Three mosquitoes were collected daily for up to 8 days after infection and frozen at −70°C. For virus titrations, mosquitoes were homogenized in 1 ml PBS, and the homogenates were clarified and assayed for infectious virus by plaque formation on BHK cells. Experiments were repeated twice, and data were averaged.
Statistical analysis.
Mouse mortality data were subjected to Kaplan-Meier survival analysis using the log rank test. One-way analysis of variance (ANOVA) with Dunnett's correction was used to compare heparin binding data. Two-way ANOVA was used to identify differences between replication curves by using the Bonferroni correction (GraphPad Prism, version 4).
RESULTS
Virion infectivity.
To determine whether the virus particles produced by BHK cells were similarly infectious, the particle-to-PFU ratios for the viruses were assessed by using a combination of electron microscopy to count virus particles and assays for infectious virus. TE12 and E1-139 and E2-196 mutant strains had similar particle-to-PFU ratios (73, 65, and 21, respectively), while the E1-245 mutant was approximately 100-fold less infectious (with a particle-to-PFU ratio of 9,710). We were not successful in determining a ratio for the E2-318 mutant, because the virions could not be visualized on the micrographs.
Heparin binding.
Many viruses, including most laboratory strains of SINV, use the abundantly expressed negatively charged glycosaminoglycan heparan sulfate (HS) for initial interaction with host cells (6, 11, 14, 30, 48, 57). To assess the effects of deletion of individual glycosylation sites on SINV interaction with heparin, the NaCl concentration required to elute each of the viruses from a heparin-Sepharose column was determined (Fig. 1; Table 1). Viruses grown in BHK cells with mutations at either site in E1 bound heparin equivalently to the parent TE12 virus. However, viruses lacking either E2 glycosylation site bound heparin more strongly than the parent virus, with the greatest effect for the E2-196 mutant. All elution profiles had sharply defined peaks except for the E2-318 mutant, suggesting heterogeneity in this virion population (Fig. 1E and F).
FIG. 1.

Representative heparin-Sepharose NaCl elution profiles. 35S-labeled viruses were grown in BHK or C7/10 cells, gradient purified, and applied to heparin-Sepharose columns. Elution was performed with a 100 mM-to-500 mM NaCl gradient. Fractions were collected and analyzed for 35S cpm and [NaCl] to determine the NaCl concentration at which there was peak virus elution.
TABLE 1.
Average NaCl concentrations at which the parent and mutant TE12 viruses elute from heparin-Sepharosea
| Source cell | Virus | Avg [NaCl] ± SEM (mM) | Pb |
|---|---|---|---|
| BHK | TE12 | 318 ± 1.5 | |
| E1-139 mutant | 320 ± 1.9 | NS | |
| E1-245 mutant | 315 ± 2.5 | NS | |
| E2-196 mutant | 369 ± 5.0 | <0.01 | |
| E2-318 mutant | 343 ± 1.9 | <0.01 | |
| C7/10 | TE12 | 340 ± 2.3 | <0.01 |
| E2-196 mutant | 341 ± 3.3 | <0.01 | |
| E2-318 mutant | 333 ± 2.7 | <0.01 |
Viruses (TE12 parent strain and TE12 strains with asparagine-to-glutamine mutations at single glycosylation sites) were grown in either BHK cells or C7/10 mosquito cells.
P values refer to the comparison of the elution concentration to that of TE12 grown in BHK cells (ANOVA with Dunnett's correction). NS, not significant.
Because HS and sialic acid (often the terminal monosaccharide in mammalian cell-derived N-linked carbohydrates) are both negatively charged, the absence of E2 glycosylation may decrease charge repulsion in the interaction with HS. To assess this possibility, TE12 and E2-196 and E2-318 mutants were grown in C7/10 mosquito cells to produce virus without sialic acid. TE12 grown in C7/10 cells bound to heparin more strongly than TE12 grown in BHK cells, suggesting that charge repulsion does play a role in HS binding. E2-196 (P = 0.01) and E2-318 (P = 0.037) mutants grown in C7/10 cells bound heparin less strongly than either grown in BHK cells, indicating that factors in addition to the negative charge of sialic acid are involved.
Virus growth in BHK cells.
To identify the effects of mutations on virus replication, virus growth in BHK cells was assessed (Fig. 2). Both of the E1 glycosylation-deficient mutants grew less well than TE12. The E1-139 mutant reached lower titers (P value of <0.001 at 24 h), while the E1-245 mutant both reached lower peak titers and grew more slowly than TE12 (P value of <0.001 between 8 and 24 h). In contrast, both of the E2 glycosylation-deficient mutants grew better than the parent virus (for the E2-196 mutant, the P value was <0.001 at 10 and 20 h; for the E2-318 mutant, the P value was <0.001 at 6, 8, and 10 h).
FIG. 2.
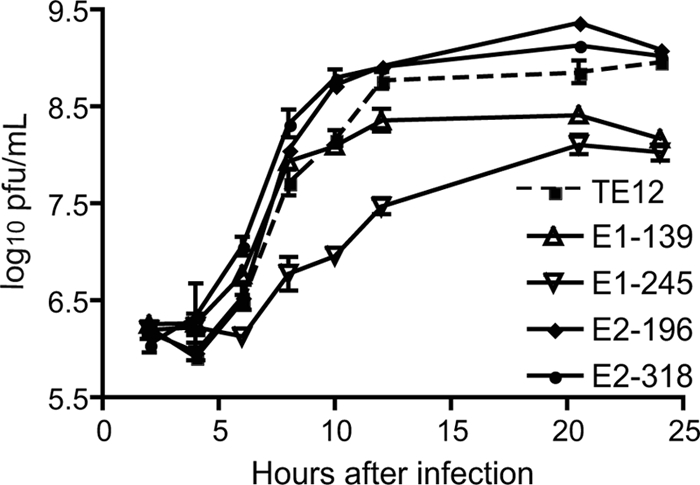
Effects of elimination of individual glycosylation sites on virus replication in BHK cells. BHK cells were infected at an MOI of 8 with TE12 or an E1-139, E1-245, E2-196, or E2-318 mutant and monitored for production of infectious virus. Viruses with loss of E1 glycosylation at either site replicated less well than the parent virus, while viruses with loss of E2 glycosylation at either site replicated as well as or better than the parent virus.
To determine if glycosylation site mutations affected structural protein maturation from the precursor polyprotein to individual proteins, we monitored protein processing (Fig. 3). E1 and pE2 are transported together from the endoplasmic reticulum to the plasma membrane, and pE2 is processed to E2 shortly before virus release. Viral glycoproteins were synthesized to similar degrees in TE12- and E1-139, E1-245, and E2-196 mutant-infected cells but were undetectable in E2-318 mutant-infected cells (data not shown). No differences were detected in processing of precursor polyprotein pE3E2E1 to pE2 or pE2 to E2.
FIG. 3.

Synthesis and processing of pE2. BHK cells were infected (MOI = 5) with TE12 or an E1-139, E1-245, or E2-196 mutant. Six hours after infection, cells were labeled with 35S for 20 min (m) and then chased for the indicated times. Structural proteins from 35S-labeled cell lysates were immunoprecipitated with rabbit antibody to SINV and visualized by autoradiography. C, capsid.
Virulence in mice.
Because mice show an age-dependent susceptibility to SINV-induced encephalomyelitis (16, 27, 53), virulence for CD-1 mice was assessed across a broad age range (Fig. 4). All viruses caused fatal disease in mice infected at 2 days (Fig. 4A) and 1 week (Fig. 4B) of age. However, death due to the E1-245 mutant occurred later for 2-day-old mice (P = 0.0107), and deaths due to the E1-245 mutant and the E1-139 mutant were later than those due to TE12 or the E2 mutant viruses for 1-week-old mice (P = 0.019). Most 2-week-old mice infected with the E1 mutants survived, but most of the mice infected with TE12 or the E2 mutants did not survive (P < 0.0001) (Fig. 4C). Thus, deletion of either glycosylation site in E1 reduced virulence in mice.
FIG. 4.

Effects of elimination of individual glycosylation sites on neurovirulence in mice. Groups of 19 to 23 CD-1 mice at 2 days (A), 1 week (B), 2 weeks (C), or 3 weeks (D) of age or C57BL/6 (B6) mice at 3 weeks of age (E) were inoculated intracerebrally with 1,000 PFU of TE12 or an E1-139, E1-245, E2-196, or E2-318 mutant and followed for mortality. Deaths due to the E1-245 mutant were later than deaths due to TE12 among 2-day-old (P = 0.0107) and 1-week-old (P = 0.019) mice. Both E1-139 and E1-245 mutants were less virulent in 2-week-old mice (P < 0.0001) than TE12. The E2-196 and E2-318 mutants were more virulent in 3-week-old CD-1 (P = 0.0251) and B6 (P = 0.0026) mice.
All 3-week-old animals survived infection with TE12, but deaths occurred among the mice infected with the E2-196 (35% mortality) and E2-318 (30% mortality) mutants (Fig. 4D) (P = 0.0251). Very few 10-week-old mice died after infection with any of the viruses (data not shown). Because CD-1 mice are outbred, we also assessed virulence of these viruses in C57BL/6 mice known to be particularly susceptible to SINV encephalomyelitis (Fig. 4E) (54, 55). In 3-week-old C57BL/6 mice, both E2 mutant viruses were more virulent than TE12, with 43% mortality for mice infected with the E2-196 mutant and 77% mortality for mice infected with the E2-318 mutant, in comparison to 14% mortality for mice infected with TE12 (P = 0.0026).
Virus replication in the CNS.
Because virulence was most clearly differentiated in mice 2 or 3 weeks of age (Fig. 4), virus replication in the CNS in 17-day-old CD-1 mice was assessed (Fig. 5). The E1-139 and E1-245 mutants generally replicated less well and were cleared more rapidly from both the brain (Fig. 5, left panel) and the spinal cord (Fig. 5, right panel) than TE12 or the E2 glycosylation mutants. Clearance of virus from the spinal cord was slower for the E2 mutants than for TE12, and none of the E2-196 mutant-infected mice survived to day 7.
FIG. 5.

Virus replication in the brain and spinal cord. CD-1 mice, 17 days old, were infected intracerebrally with 1,000 PFU of TE12 or an E1-139, E1-245, E2-196, or E2-318 mutant. Brains and spinal cords were taken from three mice from each group 3, 5, and 7 days after infection and assayed for infectious virus by plaque formation on BHK cells. The E1-139 and E1-245 mutants generally replicated less well and were cleared more rapidly than TE12. Clearance of virus from the spinal cord was slower for the E2 mutants than for TE12. Horizontal lines indicate the limits of virus detection.
Virus growth in C7/10 cells.
To determine the consequences of deletion of glycosylation sites on replication of SINV in invertebrate cells, C7/10 mosquito cells were infected, and virus production was monitored (Fig. 6A and B). Replication of the E1-139 mutant was more rapid than TE12 at early times after infection, while growth of the E2 mutants was slower. The E1-245 mutant replicated substantially less well than the other viruses.
FIG. 6.

Virus replication in C7/10 mosquito cells and in Culex quinquefasciatus. Cells were infected at an MOI of 10 with TE12 or an E1-139 or E1-245 mutant (A) or TE12 or an E2-196 or E2-318 mutant (B) and monitored for production of infectious virus by plaque formation in BHK cells. Mosquitoes were inoculated intrathoracically with 100 PFU of TE12 or an E1-139 or E1-245 mutant (C) or TE12 or an E2-196 or E2-318 mutant (D) and monitored daily for virus replication.
Virus growth in mosquitoes.
To determine the effect of mutations on virus replication in mosquitoes, adult Culex quinquefasciatus mosquitoes were infected by intrathoracic inoculation, and virus replication was monitored (Fig. 6C and D). For all viruses except the E1-245 mutant, levels increased steadily over the first 3 to 4 days after inoculation and plateaued at similar levels. Levels of the E1-245 mutant were lower at all times examined.
DISCUSSION
As with other enveloped RNA viruses (2, 15, 47), the effects of loss of glycosylation had various effects on the biologic properties of SINV depending on the specific site mutated and the cell in which the virus replicated. Some glycosylation at sites on E1 or E2 is essential for SINV replication because total inhibition of glycosylation with tunicamycin completely prevents virus production (31, 32). However, effects are more subtle if only single sites are affected, and mutation of each of the four glycosylation sites in E1 and E2 of SINV had distinct biologic effects. Each protein has one site (E1-139 and E2-196) with a complex carbohydrate that is relatively exposed on the virion surface and deletion of these sites did not affect virion infectivity. Deletion of either of the glycosylation sites in E2 improved the affinity with which virions bound to heparin, improved replication in BHK cells, and increased virulence for mice but had little effect on replication in C7/10 mosquito cells or in mosquitoes. Each protein also has one site (E1-245 and E2-318) with a mostly high-mannose carbohydrate that is close to the membrane and important for virion function (41). The deletion of E1-245 decreased virion infectivity 100-fold, and the E2-318 mutant was not stable under the conditions used for analysis. The deletion of the glycosylation sites in E1 did not affect heparin binding but decreased replication in BHK cells and virulence for mice, particularly for the E1-245 mutant, for which replication was impaired in C7/10 cells as well. Previous observations that altered alphavirus glycosylation does not affect the processing of pE2 (13, 36, 38) were confirmed.
Interaction with the cellular glycosaminoglycan HS is an initial electrostatic binding event for the E2 glycoproteins of many strains of SINV, including TE12, the parent strain for these studies (6, 7, 26). Efficient HS binding is charge dependent (56) and associated with basic amino acids at four regions in the E2 linear sequence: positions 62 to 76, 114, 157 to 159, and 230 (7, 30, 30). Elimination of the carbohydrate moiety at E2-196 may make the HS binding site for SINV more accessible. However, complex carbohydrates synthesized in vertebrate cells have negatively charged terminal sialic acid residues that could contribute to alterations in HS binding due to elimination of charge repulsion. This was confirmed by demonstration of a reduction in heparin binding for TE12 grown in mosquito cells that lack the ability to modify complex carbohydrates with sialic acid. Furthermore, the position of the E2-318 carbohydrate at the base of the spike (41, 58) is not in the region of predicted HS binding by E2.
Improved replication of E2-196 and E2-318 mutants in BHK cells is consistent with improved HS binding and the ability of positively charged proteins to inhibit cellular infection with alphaviruses that bind HS (56). In contrast, replication in C7/10 cells was slowed slightly, and there was no difference in replication in mosquitoes, perhaps due to lack of importance for HS in binding to invertebrate cells. The ability to bind HS is strongly selected in vitro, as it greatly improves the efficiency with which mammalian cells are infected (6, 30) and can even expand the host range of alphaviruses (20). Improved efficiency of infection of both vertebrate and invertebrate cells with elimination of glycosylation sites has also been observed for West Nile virus, but the mechanism was not identified (19).
The importance of HS binding for virulence is more complicated. Binding to HS leads to more rapid hepatic clearance from circulation (7). Prolonged viremia in vivo selects against HS binding by replacement of positively charged amino acids in E2 with uncharged or acidic amino acids (7). However, once the virus is in the CNS, binding to HS appears to improve the ability of SINV to replicate in neurons (1, 45). In the current study, virus was inoculated intracerebrally, and the E2 mutants with improved HS binding replicated better and caused higher mortality in 2- to 3-week-old mice, further suggesting that binding to HS increases neurovirulence.
Compared to the case for the E2 mutants, the effects of deletion were more varied for viruses without glycosylation at E1-139 or E1-245 in vertebrate cells, and neurovirulence was decreased, even in young mice. The E1-245 mutant was particularly impaired, with decreased virion infectivity and decreased replication in invertebrate cells and mosquitoes as well as vertebrate cells. The E1-245 site is near the base and close to the threefold axis of the spike (58), and it may participate in regulation of the conformational change in the spike required for fusion and entry (41). The mechanisms by which elimination of E1 glycosylation sites affect replication in a cell type-specific way are not clear and will require further investigation.
Acknowledgments
This work was funded by a research grant (R01 NS18596) and a training grant (T32 AI007417) from the National Institutes of Health.
We thank Ronald Schnaar, Andrew Byrnes, and Carolyn Machamer for many helpful discussions, Marcia Lyons and Debra Hauer for their expert technical assistance, and Robin Todd for supplying mosquito pupae.
Footnotes
Published ahead of print on 18 March 2009.
REFERENCES
- 1.Bear, J. S., A. P. Byrnes, and D. E. Griffin. 2006. Heparin-binding and patterns of virulence for two recombinant strains of Sindbis virus. Virology 347183-190. [DOI] [PubMed] [Google Scholar]
- 2.Beasley, D. W., M. C. Whiteman, S. Zhang, C. Y. Huang, B. S. Schneider, D. R. Smith, G. D. Gromowski, S. Higgs, R. M. Kinney, and A. D. Barrett. 2005. Envelope protein glycosylation status influences mouse neuroinvasion phenotype of genetic lineage 1 West Nile virus strains. J. Virol. 798339-8347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burge, B. W., and J. H. Strauss, Jr. 1970. Glycopeptides of the membrane glycoprotein of Sindbis virus. J. Mol. Biol. 47449-466. [DOI] [PubMed] [Google Scholar]
- 4.Burke, D., and K. Keegstra. 1979. Carbohydrate structure of Sindbis virus glycoprotein E2 from virus grown in hamster and chicken cells. J. Virol. 29546-554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Butters, T. D., R. C. Hughes, and P. Vischer. 1981. Steps in the biosynthesis of mosquito cell membrane glycoproteins and the effects of tunicamycin. Biochim. Biophys. Acta 640672-686. [DOI] [PubMed] [Google Scholar]
- 6.Byrnes, A. P., and D. E. Griffin. 1998. Binding of Sindbis virus to cell-surface heparan sulfate. J. Virol. 727349-7356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Byrnes, A. P., and D. E. Griffin. 2000. Large-plaque mutants of Sindbis virus show reduced binding to heparan sulfate, heightened viremia and slower clearance from the circulation. J. Virol. 74644-651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheng, R. H., R. J. Kuhn, N. H. Olson, M. G. Rossmann, H.-K. Choi, R. J. Smith, and T. S. Baker. 1995. Nucleocapsid and glycoprotein organization in an enveloped virus. Cell 80621-630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davis, N. L., D. F. Pence, W. J. Meyer, A. L. Schmaljohn, and R. E. Johnston. 1987. Alternative forms of a strain specific neutralizing antigenic site on the Sindbis virus E2 glycoprotein. Virology 161101-108. [DOI] [PubMed] [Google Scholar]
- 10.Durbin, R. K., and V. Stollar. 1986. Sequence analysis of the E2 gene of a hyperglycosylated, host restricted mutant of Sindbis virus and estimation of mutation rate from frequency of revertants. Virology 154135-143. [DOI] [PubMed] [Google Scholar]
- 11.Fry, E. E., S. M. Lea, T. Jackson, J. W. Newman, F. M. Ellard, W. E. Blakemore, R. Abu-Ghazaleh, A. Samuel, A. M. King, and D. I. Stuart. 1999. The structure and function of a foot-and-mouth disease virus-oligosaccharide receptor complex. EMBO J. 18543-554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fuller, S. D., J. A. Berriman, S. J. Butcher, and B. E. Gowen. 1995. Low pH induces swiveling of the glycoprotein heterodimers in the Semliki Forest virus spike complex. Cell 81715-725. [DOI] [PubMed] [Google Scholar]
- 13.Garoff, H., and R. T. Schwarz. 1978. Glycosylation is not necessary for membrane insertion and cleavage of Semliki Forest virus membrane proteins. Nature 274487-490. [DOI] [PubMed] [Google Scholar]
- 14.Giroglou, T., L. Florin, F. Schafer, R. E. Streeck, and M. Sapp. 2001. Human papillomavirus infection requires cell surface heparan sulfate. J. Virol. 751565-1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goffard, A., N. Callens, B. Bartosch, C. Wychowski, F. L. Cosset, C. Montpellier, and J. Dubuisson. 2005. Role of N-linked glycans in the functions of hepatitis C virus envelope glycoproteins. J. Virol. 798400-8409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Griffin, D. E. 1976. Role of the immune response in age-dependent resistance of mice to encephalitis due to Sindbis virus. J. Infect. Dis. 133456-464. [DOI] [PubMed] [Google Scholar]
- 17.Griffin, D. E. 2007. Alphaviruses, p. 1023-1067. In D. L. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus (ed.), Fields virology. Lippincott Williams & Wilkins, Philadelphia, PA.
- 18.Hakimi, J., and P. H. Atkinson. 1980. Growth-dependent alterations in oligomannosyl glycopeptides expressed in Sindbis virus glycoproteins. Biochemistry 195619-5624. [DOI] [PubMed] [Google Scholar]
- 19.Hanna, S. L., T. C. Pierson, M. D. Sanchez, A. A. Ahmed, M. M. Murtadha, and R. W. Doms. 2005. N-linked glycosylation of West Nile virus envelope proteins influences particle assembly and infectivity. J. Virol. 7913262-13274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heil, M. L., A. Albee, J. H. Strauss, and R. J. Kuhn. 2001. An amino acid substitution in the coding region of the E2 glycoprotein adapts Ross River virus to utilize heparan sulfate as an attachment moiety. J. Virol. 756303-6309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Helenius, A. 1995. Alphavirus and flavivirus glycoproteins: structures and functions. Cell 81651-653. [DOI] [PubMed] [Google Scholar]
- 22.Hsieh, P., and P. W. Robbins. 1984. Regulation of asparagine-linked oligosaccharide processing. Oligosaccharide processing in Aedes albopictus mosquito cells. J. Biol. Chem. 2592375-2382. [PubMed] [Google Scholar]
- 23.Hsieh, P., M. R. Rosner, and P. W. Robbins. 1983. Host-dependent variation of asparagine-linked oligosaccharides at individual glycosylation sites of Sindbis virus glycoproteins. J. Biol. Chem. 2582548-2554. [PubMed] [Google Scholar]
- 24.Hubbard, S. C. 1988. Regulation of glycosylation. The influence of protein structure on N-linked oligosaccharide processing. J. Biol. Chem. 26319303-19317. [PubMed] [Google Scholar]
- 25.Jackson, A. C., T. R. Moench, and D. E. Griffin. 1987. The pathogenesis of spinal cord involvement in the encephalomyelitis of mice caused by neuroadapted Sindbis virus infection. Lab. Investig. 56418-423. [PubMed] [Google Scholar]
- 26.Jan, J.-T., A. P. Byrnes, and D. E. Griffin. 1999. Characterization of a Chinese hamster ovary cell line developed by retroviral insertional mutagenesis that is resistant to Sindbis virus infection. J. Virol. 734919-4924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnson, R. T., H. F. McFarland, and S. E. Levy. 1972. Age-dependent resistance to viral encephalitis: studies of infections due to Sindbis virus in mice. J. Infect. Dis. 125257-262. [DOI] [PubMed] [Google Scholar]
- 28.Keegstra, K., and D. Burke. 1977. Comparison of the carbohydrate of Sindbis virus glycoprotein with the carbohydrate of host glycoproteins. J. Supramol. Struct. 7371-379. [DOI] [PubMed] [Google Scholar]
- 29.Keegstra, K., B. Sefton, and D. Burke. 1975. Sindbis virus glycoproteins: effect of the host cell on the oligosaccharides. J. Virol. 16613-620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Klimstra, W. B., K. D. Ryman, and R. E. Johnston. 1998. Adaptation of Sindbis virus to BHK cells selects for use of heparan sulfate as an attachment receptor. J. Virol. 727357-7366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leavitt, R., S. Schlesinger, and S. Kornfeld. 1977. Impaired intracellular migration and altered solubility of nonglycosylated glycoproteins of vesicular stomatitis virus and Sindbis virus. J. Biol. Chem. 2529018-9023. [PubMed] [Google Scholar]
- 32.Leavitt, R., S. Schlesinger, and S. Kornfeld. 1977. Tunicamycin inhibits glycosylation and multiplication of Sindbis and vesicular stomatitis virus. J. Virol. 21375-385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lustig, S., A. C. Jackson, C. S. Hahn, D. E. Griffin, E. G. Strauss, and J. H. Strauss. 1988. The molecular basis of Sindbis virus neurovirulence in mice. J. Virol. 622329-2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mackenzie, J. S., M. D. Lindsay, R. J. Coelen, A. K. Broom, R. A. Hall, and D. W. Smith. 1994. Arboviruses causing human disease in the Australasian zoogeographic region. Arch. Virol. 136447-467. [DOI] [PubMed] [Google Scholar]
- 35.Mayne, J. T., J. R. Bell, E. G. Strauss, and J. H. Strauss. 1985. Pattern of glycosylation of Sindbis virus envelope proteins synthesized in hamster and chicken cells. Virology 142121-133. [DOI] [PubMed] [Google Scholar]
- 36.McDowell, W., P. A. Romero, R. Datema, and R. T. Schwarz. 1987. Glucose trimming and mannose trimming affect different phases of the maturation of Sindbis virus in infected BHK cells. Virology 16137-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meyer, W. J., and R. E. Johnston. 1993. Structural rearrangement of infecting Sindbis virions at the cell surface: mapping of newly accessible epitopes. J. Virol. 675117-5125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Naim, H. Y., and H. Koblet. 1988. Investigation of the role of glycans for the biological activity of Semliki Forest virus grown in Aedes albopictus cells using inhibitors of asparagine-linked oligosaccharides trimming. Arch. Virol. 10273-89. [DOI] [PubMed] [Google Scholar]
- 39.Owen, K. E., and R. J. Kuhn. 1997. Alphavirus budding is dependent on the interaction between the nucleocapsid and hydrophobic amino acids on the cytoplasmic domain of the E2 envelope glycoprotein. Virology 230187-196. [DOI] [PubMed] [Google Scholar]
- 40.Phinney, B. S., K. Blackburn, and D. T. Brown. 2000. The surface conformation of Sindbis virus glycoproteins E1 and E2 at neutral and low pH, as determined by mass spectrometry-based mapping. J. Virol. 745667-5678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pletnev, S. V., W. Zhang, S. Mukhopadhyay, B. R. Fisher, R. Hernandez, D. T. Brown, T. S. Baker, M. G. Rossmann, and R. J. Kuhn. 2001. Locations of carbohydrate sites on alphavirus glycoproteins show that E1 forms an icosahedral scaffold. Cell 105127-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rice, C. M., and J. H. Strauss. 1981. Nucleotide sequence of the 26S mRNA of Sindbis virus and deduced sequence of the encoded virus structural proteins. Proc. Natl. Acad. Sci. USA 782062-2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rosen, L., and D. Gubler. 1974. The use of mosquitoes to detect and propagate dengue viruses. Am. J. Trop. Med. Hyg. 231153-1160. [DOI] [PubMed] [Google Scholar]
- 44.Russell, D. L., J. M. Dalrymple, and R. E. Johnston. 1989. Sindbis virus mutations which coordinately affect glycoprotein processing, penetration, and virulence in mice. J. Virol. 631619-1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ryman, K. D., C. L. Gardner, C. W. Burke, K. C. Meier, J. M. Thompson, and W. B. Klimstra. 2007. Heparan sulfate binding can contribute to the neurovirulence of neuroadapted and nonneuroadapted Sindbis viruses. J. Virol. 813563-3573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sarver, N., and V. Stollar. 1977. Sindbis virus-induced cytopathic effect in clones of Aedes albopictus (Singh) cells. Virology 80390-400. [DOI] [PubMed] [Google Scholar]
- 47.Shi, X., K. Brauburger, and R. M. Elliott. 2005. Role of N-linked glycans on bunyamwera virus glycoproteins in intracellular trafficking, protein folding, and virus infectivity. J. Virol. 7913725-13734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Smit, J. M., B.-L. Waarts, K. Kimata, W. B. Klimstra, R. Bittman, and J. Wilschut. 2002. Adaptation of alphaviruses to heparan sulfate: interaction of Sindbis and Semliki Forest viruses with liposomes containing lipid-conjugated heparin. J. Virol. 7610128-10137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Smith, T. J., R. H. Cheng, N. H. Olson, P. Peterson, E. Chase, R. J. Kuhn, and T. S. Baker. 1995. Putative receptor binding sites on alphaviruses as visualized by cryoelectron microscopy. Proc. Natl. Acad. Sci. USA 9210648-10652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Strauss, E. G., D. S. Stec, A. L. Schmaljohn, and J. H. Strauss. 1991. Identification of antigenically important domains in the glycoproteins of Sindbis virus by analysis of antibody escape variants. J. Virol. 654654-4664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Strauss, J. H., Jr., B. W. Burge, and J. E. Darnell. 1970. Carbohydrate content of the membrane protein of Sindbis virus. J. Mol. Biol. 47437-448. [DOI] [PubMed] [Google Scholar]
- 52.Strauss, J. H., and E. G. Strauss. 1994. The alphaviruses: gene expression, replication and evolution. Microbiol. Rev. 58491-562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Taylor, R. M., H. S. Hurlbut, T. H. Work, J. R. Kingsbury, and T. E. Frothingham. 1955. Sindbis virus: a newly recognized arthropod-transmitted virus. Am. J. Trop. Med. Hyg. 4844-846. [DOI] [PubMed] [Google Scholar]
- 54.Thach, D. C., T. Kimura, and D. E. Griffin. 2000. Differences between C57BL/6 and BALB/cBy mice in mortality and virus replication after intranasal infection with neuroadapted Sindbis virus. J. Virol. 746156-6161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thach, D. C., S. R. Kleeberger, P. C. Tucker, and D. E. Griffin. 2001. Genetic control of neuroadapted Sindbis virus replication in female mice maps to chromosome 2 and associates with paralysis and mortality. J. Virol. 758674-8680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Waarts, B. L., O. J. Aneke, J. M. Smit, K. Kimata, R. Bittman, D. K. Meijer, and J. Wilschut. 2005. Antiviral activity of human lactoferrin: inhibition of alphavirus interaction with heparan sulfate. Virology 333284-292. [DOI] [PubMed] [Google Scholar]
- 57.WuDunn, D., and P. G. Spear. 1989. Initial interaction of herpes simplex virus with cells is binding to heparan sulfate. J. Virol. 6352-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang, W., S. Mukhopadhyay, S. V. Pletnev, T. S. Baker, R. J. Kuhn, and M. G. Rossmann. 2002. Placement of the structural proteins in Sindbis virus. J. Virol. 7611645-11658. [DOI] [PMC free article] [PubMed] [Google Scholar]