

Abstract
Human cytomegalovirus (HCMV) exists indefinitely in infected individuals by a yet poorly characterized latent infection in hematopoietic cells. We previously demonstrated a requirement for the putative UL138 open reading frame (ORF) in promoting a latent infection in CD34+ hematopoietic progenitor cells (HPCs) infected in vitro. In our present study, we have identified two coterminal transcripts of 2.7 and 3.6 kb and a 21-kilodalton (kDa) protein (pUL138) that are derived from the UL138 locus with early-late gene kinetics during productive infection. The UL138 transcripts and protein are detected in both fibroblasts and HPCs. A recombinant virus, FIX-UL138STOP, that synthesizes the UL138 transcripts but not the protein exhibited a partial loss-of-latency phenotype in HPCs, similar to the phenotype observed for the UL138-null recombinant virus. This finding suggests that the UL138 protein is required for latency, but it does not exclude the possibility that the UL138 transcripts or other ORFs also contribute to latency. The mechanisms by which pUL138 contributes to latency remain unknown. While the 86- and 72-kDa immediate-early proteins were not detected in HPCs infected with HCMV in vitro, pUL138 did not function directly to suppress expression from the major immediate-early promoter in reporter assays. Interestingly, pUL138 localizes to the Golgi apparatus in infected cells but is not incorporated into virus particles. The localization of pUL138 to the Golgi apparatus suggests that pUL138 contributes to HCMV latency by a novel mechanism. pUL138 is the first HCMV protein demonstrated to promote an infection with the hallmarks of latency in CD34+ HPCs.
Human cytomegalovirus (HCMV) is an ancient herpesvirus that persists in 60 to 99% of the human population worldwide through a latent infection that is asymptomatic in healthy individuals (35). Reactivation of HCMV from latency can result in life-threatening pathology in immunocompromised individuals, including stem cell and solid organ transplant recipients, AIDS patients, and cancer patients undergoing intensive chemotherapy (8, 13, 35). In addition to overt pathologies associated with reactivation from latency, the impact of viral coexistence on our biology is ill-defined but encompasses a wide range of possibilities, including conferring protection from other microbial infections (5), contributing to the development of vascular disease (22, 53), or gradually exhausting the host's immune defenses (40, 55, 64, 65). Viral persistence or latency is a poorly understood phenomenon in virology, yet it is critical to understanding how viruses assimilate into and affect our biology and cause disease.
HCMV latency is best characterized in hematopoietic cells of the myeloid lineage. While the primary cellular reservoir for latent HCMV is unknown, latency has been studied experimentally in a wide variety of primary progenitor cells, including CD34+ and CD34+/CD38− cells (15-17, 29, 70), and myeloid-lineage cells, including granulocyte-macrophage progenitor cells (GM-Ps) (19, 25, 26) and CD34+-derived dendritic cells (42). Indeed, latent HCMV genomes have been detected in CD34+ cells (32, 66), GM-Ps (19, 26, 46), and monocytes (34, 48, 58, 59) from latently infected, healthy individuals. HCMV latency and reactivation from latency are intimately associated with hematopoietic cell differentiation (47-49, 59). Because HCMV genomes are detected in cells as primitive as CD34+ cells in the hierarchy of hematopoietic differentiation (32, 45, 66), we have chosen primary human CD34+ hematopoietic progenitor cells (HPCs) as the basis for our studies.
Little is known regarding the contribution of viral determinants to latency. Several HCMV transcripts and proteins have been detected in hematopoietic cells infected in vitro or derived from healthy, seropositive individuals. First, transcripts originating from the major immediate-early (IE) region via alternative start sites and the proteins they encoded were detected following infection of GM-Ps in vitro and in latently infected individuals (26, 27). Second, a variant of the viral interleukin-10 (IL-10) homologue encoded by UL111.5A was detected in GM-Ps infected in vitro and in monocytes from seropositive individuals (24). Third, a transcript antisense to the UL81-82 genes was detected in monocytes from healthy seropositive individuals (6). Despite the expression of these factors in hematopoietic cells infected endogenously or in vitro, a role has yet to be demonstrated for the latency-associated IE transcripts or the UL81-82 and UL111.5A gene products in HCMV latency. Further, the open reading frame 94 (ORF94) protein encoded by the latency-associated IE transcripts was dispensable for establishing and maintaining a latent infection in vitro (68). We previously identified sequences including the putative ULb′ genes, UL136 to -142, that are required for an infection with the hallmarks of latency in HPCs infected in vitro (16). Specifically, recombinant viruses lacking the UL138 ORF exhibited a loss-of-latency phenotype and replicated productively in HPCs infected in vitro. Further, UL138 transcripts were detected in CD34+ cells and monocytes from healthy seropositive individuals (16).
UL138 is a previously uncharacterized putative HCMV ORF. Our present study represents an initial characterization of the UL138 gene products and investigated their role in the latent infection. In productively infected fibroblasts, UL138 is carried on two large, polyadenylated, coterminal transcripts of 2.7 and 3.6 kb that are colinear with the viral genome. These transcripts give rise to a 21-kDa protein (pUL138) corresponding to the predicted coding sequence of UL138. pUL138 was required for an infection with the hallmarks of latency in vitro. However, our data suggest that the UL138 transcripts or other ULb′ ORFs between UL133 and UL142 also contribute to the in vitro latency phenotype. pUL138 localizes to the Golgi apparatus as an integral type I membrane protein where the majority of protein resides on the cytoplasmic face of the Golgi membranes. Given the localization of pUL138 in the Golgi apparatus, pUL138 may contribute to latency by mediating protein trafficking or the cellular stress response. pUL138 is the first virus-encoded protein demonstrated to function in HCMV latency in HPCs infected in vitro. Further characterization of pUL138 in HPCs and the mechanisms by which pUL138 functions in latency is the focus of our ongoing research.
MATERIALS AND METHODS
Cells.
Human lung fibroblasts (MRC5) (ATCC, Manassas, VA) were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 10 mM HEPES, 1 mM sodium pyruvate, 2 mM l-glutamine, 0.1 mM nonessential amino acids, 100 U/ml penicillin, and 100 μg/ml streptomycin. Human astrocytoma cells (U373) (ATCC) were cultured in DMEM supplemented with 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin. 293 cells (ATCC) were cultured in DMEM supplemented with 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin. ARPE-19 cells (ATCC) were cultured in a 1:1 mixture of DMEM and Ham's F-12 medium supplemented with 1.2 g/liter sodium bicarbonate, 2.5 mM l-glutamine, 15 mM HEPES, 0.5 mM sodium pyruvate, and 10% FBS. The M2-10B4 murine stromal cell line expressing human IL-3 and granulocyte colony-stimulating factor and the S1/S1 murine stromal cell line expressing human IL-3 and stem cell factor were generously provided by Stem Cell Technologies Ltd. on behalf of D. Hogge (Terry Fox Laboratory, University of British Columbia, Vancouver, British Columbia) and were cultured as recommended (33). All cells were maintained at 37°C with 5% CO2.
For most experiments, bone marrow cells were obtained from waste produced during bone marrow harvest from healthy donors at the University Medical Center at the University of Arizona via a protocol approved by the Institutional Review Board. Alternatively, fresh bone marrow from cadavers was purchased from the National Disease Research Interchange (Philadelphia, PA). CD34+ cells were isolated as described previously (17).
Viruses.
The FIXwt virus strain has been cloned as a bacterial artificial chromosome (BAC) and engineered to express the green fluorescent protein (GFP) (D. Yu and T. Shenk, unpublished results). Virus stocks were propagated by electroporation of infectious BAC DNA into MRC5 cells and purified by density gradient centrifugation through a 20% d-sorbitol cushion at 20,000 rpm in an SW28 rotor (Beckman Coulter, Fullerton, CA) for 80 min at 22°C. Virions were resuspended in Iscove's modified DMEM containing 2% bovine serum albumin (BSA) and stored at −80°C. Infectious virus yields were determined by 50% tissue culture infective dose (TCID50) on MRC5 fibroblasts.
Recombinant viruses were constructed in Escherichia coli by linear recombination in a two-step positive/negative selection that leaves no trace of the engineering process (67, 69). For FIX-UL138STOP, PCR fragments carrying galK with homologous flanking sequences to the targeted region of the HCMV genome in the FIXwt strain were synthesized using forward (5′-CCATGGACGATCTGCCGCTGAACGTCGGGTTACCCATCATCGGCGTGCCTGTTGACAATTA ATCATC-3′) and reverse (5′-TCGTGCCAATGGTAAGCTAGATAGCAGAGAATGGCCACGATCAGCACGAGTCAGCACTGTCCTGCTCCTT-3′) primers and the pGalK plasmid (50 ng) (67). The primer sequences specific to galK are underlined, and the remaining portion of the primer is homologous to HCMV sequences flanking the targeted region. galK PCR products were digested with DpnI, gel purified, and electroporated into recombinogenic SW102 E. coli containing the FIXwt BAC. galK-positive recombinants were selected for growth on M63 minimal plates and screened on MacConkey agar plates, both supplemented with 0.2% galactose and 15 μg/ml chloramphenicol. In the second step, 100-bp complementary oligonucleotides containing UL138 with a stop codon substituted for codon 15 (ATG → TAA) (5′-CCATGGACGATCTGCCGCTGAACGTCGGGTTACCCATCATCGGCGTGTAACTCGTGCTGATC GTGGCCATTCTCTGCTATCTAGCTTACCATTGGCACGA-3′) were annealed and electroporated into recombinogenic SW102 E. coli containing the FIX-galK+ BAC. galK recombinants were selected on M63 minimal plates containing 0.2% 2-deoxygalactose and 15 μg/ml chloramphenicol. Recombinants were further screened by BAC digestion, PCR, and sequencing. Infectious BAC DNA was isolated and electroporated into MRC5 cells to produce virus stocks.
The recombinant FIX-UL138myc virus was constructed similarly by amplifying galK using forward (5′-TGTACAAAAGAGAGAGACTGGGACGTAGATCCGGACAGAGGACGGTCACCCCTGTTGACAATTAATCATCGGC-3′) and reverse (5′-GTCAAAACGACATTACCGCGATCCGCTCCCCTCTTTTTTCTTTTTCTCATTCAGCACTGTCCTGCTCC-3′) primers, and the PCR product was substituted for UL138 in the FIXwt BAC genome in SW102 E. coli. The primer sequences specific to galK are underlined, and other sequences are specific to sequences flanking UL138. galK was then replaced with a PCR product encoding UL138 with an in-frame, C-terminal Myc epitope tag. This PCR product was synthesized using the FIXwt BAC as a template and forward (5′-TCATCCGCGACTCTACGAC-3′) and reverse (5′-GTCAAAACGACATTACCGCGATCCGCTCCCCTCTTTTTTCTTTTTCTCATtcaCAGATCCTCTTCTGA GATGAGTTTTTGTTCCGTGTATTCTTGATGATAATGTACC-3′) primers, where the Myc epitope sequences are underlined, the stop codon is lower case, and HCMV sequences are in uppercase. Recombinant clones were selected as described previously (67) and screened by restriction digestion, PCR, and sequencing.
The replication of recombinant viruses was determined by infecting MRC5 cells at a multiplicity of infection (MOI) of 0.01. Cells and medium were collected over a 20-day time course and sonicated. Virus titers in each lysate were determined by TCID50 on MRC5 cells.
Northern blotting.
Total RNA was isolated from cells infected at an MOI of 2 using TRIzol LS reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. Where indicated, polyadenylated RNA was further purified using the NucleoTrap mRNA mini kit (Macherey-Nagel, Bethlehem, PA). To analyze transcript stability, 4 μg/ml actinomycin D was added to infected cells at 16 h postinfection (hpi) and RNA isolated over a time course. RNA was glyoxylated (4), subjected to 1% agarose gel electrophoresis, and transferred to a Nytran nylon membrane using a Turboblotter (Whatman, Keene, NH). RNA was cross-linked to the membrane using a UV Stratalinker (Stratagene, La Jolla, CA). Antisense riboprobes recognizing ULb′ coding sequences were generated from PCR products (primers are listed in Table 1) cloned into the pGEM-T Easy vector (Promega, Madison, WI). Vectors were linearized and transcribed in vitro to generate [α-32P]CTP-radiolabeled probes using the SP6/T7 riboprobe combination system (Promega). Probes were hybridized to blots in 4× SSC (0.6 M NaCl, 0.06 M sodium citrate [pH 7.0]), 5× Denhardt's solution (0.01% [wt/vol] Ficoll 400, 0.01% [wt/vol] polyvinylpyrrolidone, 0.01% wt/vol BSA), 1% sodium dodecyl sulfate, 50% formamide, and 10 μg/ml denatured salmon sperm DNA at 60°C overnight. Membranes were washed (4), and hybridization was detected by exposure to autoradiographic film or by phosphorilluminescence.
TABLE 1.
Primers for Northern analysis templates
| ORF | Primer | Sequence |
|---|---|---|
| UL133 | Forward | 5′-ATG GGT TGC GAC GTG CAC GAT C-3′ |
| Reverse | 5′-TTA CGT TCC GGT CTG ATG CTG C-3′ | |
| UL135 | Forward | 5′-ATG TCC GTA CAC CGG CCC TTC-3′ |
| Reverse | 5′-TCA GGT CAT CTG CAT CGA CTC G-3′ | |
| UL136 | Forward | 5′-ATG TCA GTC AAG GGC GTG GAG-3′ |
| Reverse | 5′-TTA CGT AGC GGG AGA TAC GGC G-3′ | |
| UL138 | Forward | 5′ATG GAC GAT CTG CCG CTG AAC G-3′ |
| Reverse | 5′-TCA CGT GTA TTC TTG ATG ATA ATG-3′ | |
| UL138b | Forward | 5′-AAA AAG AGG GGA GCG GAT CG-3′ |
| Reverse | 5′-ATC ACC GCC ACC ATT ACC AC-3′ | |
| UL139 | Forward | 5′-ATG CTG TGG ATA TTA ATT TTA TTT GC-3′ |
| Reverse | 5′-TCA CCG AGG CGG AGG TGG AAA T-3′ |
RACE.
Total RNA was isolated from infected MRC5 fibroblasts at 18 hpi, digested with DNase, and purified using the Nucleospin RNA-II kit (Macherey-Nagel). The 5′ ends of the UL138 transcripts were mapped using the SMART rapid amplification of cDNA ends (RACE) cDNA amplification kit IIa (Clontech, Mountain View, CA). A poly(dT)25VN primer was annealed to 1 μg of total RNA and reverse transcribed with 200 U Superscript II (Invitrogen) with 7% dimethyl sulfoxide for 90 min at 42°C. The 5′ end was amplified with primers to the SMART-IIa oligonucleotide and a gene-specific primer (GSP) to the UL138 ORF (5′-CAACAGCGGATCAGCCAGCGGTAGCTCAAA-3′) using Advantage 2 polymerase (Clontech) with 7% DMSO, 1 M betaine, and 400 nM primer. Cycling conditions were 94°C for 30 s; 30 cycles of 94.5°C for 30 s, 59°C for 30 s, and 72°C for 4.5 min; and then a final extension at 72°C for 10 min. The 3′ ends of the transcripts were mapped using the 3′ RACE system, version 2.0 (Invitrogen). Total RNA (1 μg) was annealed with a 3′ adapter primer and reverse transcribed using 200 U Superscript III for 60 min at 55 °C, followed by a 15-min enzyme inactivation at 75°C and a 30-min digestion with 2 U of RNase H. The 3′ end of the transcript was amplified with the GSP to the 5′ end of the UL138 ORF (5′-ATGGACGATCTGCCGCTGAA-3′) and the adapter primer mixture using Advantage 2 polymerase and the following conditions: 94°C for 30 s, followed by 30 cycles of 94°C for 30 s and 65°C for 30 s, and then 72°C for 3.5 min. All RACE products were gel purified, cloned into the pGEM-T Easy vector, and sequenced.
Plasmids.
Wild-type UL138 or UL138 containing a premature stop codon (TAA) at codon 15 (ATG) was PCR amplified from BAC DNA using the UL138 forward primer (5′-GGGGAATTCCAGAGGACGGTCACCATGGAC-3′) and the UL138 reverse primer (5′-GCGCTCTAGAGCCGTGTATTCTTGATGATAATG-3′). Synthesized fragments were digested and cloned in frame into the EcoRI and XbaI sites of pEF1/Myc-His (Invitrogen), generating carboxy-terminally Myc-tagged UL138 expression vectors, pEF1-UL138WT and pEF1-UL138STOP. pGL3-MIEP1400 was created by PCR amplifying the entire HCMV major IE promoter (MIEP), including the modulator and enhancer region (31), and cloning it into the KpnI and HindIII sites of the pGL3-Basic firefly luciferase plasmid (Promega). The simian virus 40 (SV40)-driven Renilla luciferase plasmid pRL-SV40 was a generous gift of T. Bowden (University of Arizona). The pSVH-1 IE1/2 expression plasmid was a generous gift from J. Nelson (Oregon Health Sciences University) (52).
Immunoblotting.
Protein lysates (5 to 10 μg of protein per lane) were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to 0.22-μm nitrocellulose (Hybond-ECL; Amersham Biosciences, Piscataway, NJ) or 0.45-μm polyvinylidene difluoride (Immobilon-FL, Millipore, Billerica, MA) membranes. Membranes were blocked in Tris-buffered saline (TBS) (25 mM Tris [pH 8.0], 137 mM NaCl, 3 mM KCl, 1.5 mM MgCl2, pH 8.0) plus 5% nonfat dry milk and 2.5 mg/ml BSA overnight at 4°C. Blots were incubated in primary antibody in TBS supplemented with 2.5 μg/ml BSA and 0.05% Tween 20 (TBS-BT) for 1 h at room temperature. Primary antibodies are listed in Table 2. Blots were washed three times in TBS-BT for 15 min each and then incubated with either goat anti-mouse or goat anti-rabbit immunoglobulin G (IgG) secondary antibodies conjugated to horseradish peroxidase (Invitrogen) in TBS-BT plus 5% normal goat serum. Membranes were developed with the ECL Plus Western blotting detection system (Amersham Biosciences, Piscataway, NJ) according to the manufacturer's instructions and exposed to autoradiography film. Alternatively, protein blots were visualized using goat anti-mouse IgG(H+L) DyLight 680-conjugated and goat anti-rabbit IgG(H+L) DyLight 800-conjugated secondary antibodies (Pierce, Rockford, IL) in conjunction with the Odyssey infrared imaging system (Li-Cor, Lincoln, NE).
TABLE 2.
Primary antibodies used for immunofluorescence and immunoblotting
| Antigena | Antibody | Source | Typeb | Dilution
|
|
|---|---|---|---|---|---|
| Immunofluorescencec | Immunoblottingd | ||||
| UL138 | 5484, custom | Open Biosystems | R | 1:250 | 1:1,000 |
| Myc epitope | Clone 9E10 | Millipore | M | 1:1500 | 1:5,000 |
| Myc epitope | 71D10 | Cell Signaling | R | 1:200 | NDe |
| MHC-I, HLA-B/C | HC10 | Giftf | M | ND | 1:50 |
| IE1/2 | 3H4 | Giftg | M | ND | 1:100 |
| pp71 | 2H10-9 | Giftg | M | ND | 1:100 |
| α-Tubulin | DM 1A | Sigma | M | ND | 1:12,000 |
| β-Actin | A2103 | Sigma | M | ND | 1:1,000 |
| Golgi GM130 | Clone 35 | BD Transduction Laboratories | M | 1:100 | ND |
| Golgi G58K | Clone 58K-9 | Gene Tex | M | 1:125 | ND |
| GS 27 | Clone 25 | BD Transduction Laboratories | M | 1:100 | ND |
| PDI | RL 77 | Affinity Bioreagents | M | 1:50 | ND |
| EEA1 | 14/EEA1 | BD Transduction Laboratories | M | 1:250 | ND |
GM, Golgi matrix; GS, Golgi SNARE.
R, rabbit; M, mouse monoclonal.
Dilution in PBS supplemented with BSA and Tween 20; incubated for 60 to 90 min at room temperature or at 4°C overnight in the case of the rabbit anti-Myc antibody.
Dilution in TBS supplemented with BSA and Tween 20; incubated for 60 to 90 min at room temperature.
ND, not done.
Provided by Lonnie Lybarger (51).
Generous gift from Tom Shenk, Princeton University.
Latency and reactivation.
Freshly isolated CD34-enriched bone marrow cells were infected at an MOI of 2 PFU per cell for 20 h in Iscove's modified DMEM supplemented with 10% BIT9500 serum substitute (Stem Cell Technologies, Vancouver, BC, Canada), 2 mM l-glutamine, 20 ng/ml low-density lipoprotein (Sigma, St. Louis, MO), and 50 μM 2-mercaptoethanol. Following infection, cells were washed in citrate buffer (40 mM Na citrate, 10 mM KCl, 135 mM NaCl, pH 3.0) for 1 min to inactivate unabsorbed virus. Infected cells were labeled with CD34+-specific monoclonal antibodies conjugated to phycoerythrin (BD Biosciences Immunocytometry Systems, San Jose, CA). Pure populations of infected (GFP-positive) CD34+ HPCs were isolated by fluorescence-activated cell sorting (FACSAria; BD Biosciences Immunocytometry Systems) and cultured in long-term bone marrow cultures on irradiated (2,000 rads, 60Co gamma-irradiator) (Atomic Energy of Canada, Mississauga, ON, Canada; University of Arizona, Tucson, AZ) stromal cell monolayers as described previously (16, 33). Briefly, irradiated recombinant M2-10B4 murine fibroblasts engineered to express recombinant human IL-3 and granulocyte colony-stimulating factor and S1/S1 murine fibroblasts engineered to express recombinant human stem cell factor and IL-3 (a gift from D. Hogge, Terry Fox Laboratory, BC, Canada; provided through StemCell Technologies) were mixed at a 1:1 ratio and seeded onto collagen-coated dishes at 1.5 × 105 cells per ml in human long-term culture medium (alpha modification of Eagle's minimum essential medium supplemented with 12.5% horse serum, 12.5% fetal bovine serum, 0.2 mM i-inositol, 20 μM folic acid, 0.1 mM 2-mercaptoethanol, 2 mM l-glutamine, and 1 μM hydrocortisone) (33). Infected CD34+ cells were cultured above irradiated feeder monolayers in collagen-coated transwells (Costar, Corning, NY). To measure latency and reactivation at 11 to 14 days postinfection, 10,000 infected HPCs were placed in each of 12 to 24 wells of a 96-well tissue culture plate containing MRC5 fibroblasts. To differentiate virus made as a result of reactivation from virus preexisting in long-term cultures, an equal number of infected HPCs were mechanically disrupted and plated on fibroblasts. The fraction of wells containing GFP-expressing fibroblasts was scored 20 days later (16).
Luciferase.
U373 cells were seeded onto 24-well plates at 1 × 105 cells per well 2 days prior to transfection. Cells were transfected using Lipofectamine 2000 (Invitrogen) in Opti-MEM I (Invitrogen). Briefly, U373 cells were incubated with 4 μg of Lipofectamine 2000; 10 ng pGL3-MIEP1400 firefly reporter plasmid; 20 ng pRL-SV40 Renilla reporter plasmid; and 0.1, 0.5, 1.0, or 3.0 μg pEF1-UL138WT or pEF1-UL138STOP effector plasmid compensated with pEF1-EMPTY effector plasmid to maintain 3 μg total effector DNA per transfection. At 6 h posttransfection, the medium was replaced with normal growth medium. Luciferase activity was assayed at 48 h posttransfection using the dual-luciferase reporter assay system (Promega) and a Berthold FB12 luminometer (Berthold Detection Systems, Oak Ridge, TN) according to the manufacturers’ instructions. Relative luciferase activity was calculated by normalizing the firefly luciferase activity to the Renilla luciferase activity.
293 cells were seeded onto 12-well plates at 1 × 105 cells per well and transfected with 1.5 μg Lipofectamine 2000 per well. 293 cell transfections included 10 ng pGL3-MIEP1400 firefly reporter plasmid; 20 ng pRL-SV40 Renilla reporter plasmid; 100 ng pSVH-1 IE1/2 expression plasmid; and 0.1, 0.5, 1.0, or 1.5 μg pEF1-UL138WT or pEF1-UL138STOP effector plasmid compensated with pEF1-EMPTY effector plasmid to maintain 1.5 μg total effector DNA per transfection. Transfections were performed and assayed as discussed above.
qRT-PCR.
RNA was isolated and DNase digested using the NucleoSpin RNA II kit (Macherey-Nagel) according to manufacturer's instructions. cDNA was generated using oligo(dT) primers and Superscript II reverse transcriptase (Invitrogen) according to the manufacturer's instructions. Quantitative real-time reverse transcriptase (qRT-PCR) was performed using D-Lux primers (Invitrogen) specific for UL122 (5′-cgggACAGGAAGACATCAAGCCcG-3′ and 5′-TTGTTGCGGTACTGGATGGTAAA-3′) and UL138 (5′-cgggCGTCGATCTGTTGAAACCcG-3′ and 5′-CATGGCTACGGTGGTGAACTG-3′), where lowercase letters indicate nonhomologous sequences or non-HCMV sequences. Linear fragments for the generation of UL122 and UL138 standard curves were synthesized using primer sets specific for the targeted HCMV genomic regions: for UL122 exon 5, 5′-GACAACCCACTCTTGCAGCG-3′ and 5′-ATTGCGCACCTTCTCGTTGT-3′, and for UL138, 5′-ATGGACGATCTGCCGCTGAA-3′ and 5′-TCACGTGTATTCTTGATGATAA-3′. Real-time PCRs were run on an ABI 7300 real-time PCR system (Applied Biosystems, Foster City, CA) using Platinum PCR SuperMix-UDG with ROX (Invitrogen). Reaction conditions were as follows: 50°C for 2 min; 95°C for 10 min; 60 cycles of 95°C for 15 s and 58°C for 30 s, melting curve analysis consisting of 95°C for 15 s and 60°C for 30 s, and a final step at 95°C for 15 s. β-Actin expression was measured using certified Lux primers (Invitrogen) and used as a control for template loading in the real-time reactions. Real-time data were analyzed using Applied Biosystems 7300 sequence detection software v1.3.1 (Applied Biosystems).
Immunofluorescence.
For transient expression of pUL138, MRC5 fibroblasts were transfected using Amaxa nucleofection according to the manufacturer's instructions (Lonza, Gaithersburg, MD). Transfected cells were processed for immunofluorescence at 72 h posttransfection. For virus infection, MRC5 fibroblasts were seeded at 5 × 104 cells per well onto glass coverslips in 24-well plates and either mock infected or infected with either FIX-UL138myc or FIXsubUL138 at an MOI of 2. Cells were washed in phosphate-buffered saline (PBS) (136.8 mM NaCl, 9.6 mM Na2HPO4, 2.68 mM KCl, 1.47 mM KH2PO4), fixed in 2% paraformaldehyde in PBS at room temperature for 20 min, and permeabilized with 0.1% Triton X-100 in PBS at room temperature for 15 min. Cells were blocked in PBS containing 2.5 mg/ml BSA and 1% normal goat serum for 30 min at 4°C. The primary antibodies used are listed in Table 2. Secondary antibodies conjugated to fluorescent molecules included Alexa Fluor 647-conjugated goat anti-mouse and Alexa Fluor 546-conjugated goat anti-rabbit IgG(H+L) (Invitrogen). Secondary antibodies were applied in PBS plus 0.05% Tween 20 for 1 h at room temperature. Cells were washed with PBS plus 0.05% Tween 20 and incubated in 1 μg/ml DAPI (4′,6′-diamidino-2-phenylindole) in PBS for 5 min at room temperature. Coverslips were mounted on slides with Aqua Polymount (Polysciences, Inc, Warrington, PA) and visualized using a Zeiss 510 Meta confocal microscope (Carl Zeiss Microimaging, Inc. Thornwood, NY). Images were recolored artificially.
Microsomes.
A confluent 10-cm dish of ARPE-19 cells was transfected with 30 μg of Lipofectamine 2000 and 30 μg of pEF1-UL138WT. Alternatively, MRCS cells were infected with FIXwt or FIXsubUL138 or were mock infected. Crude membrane fractions were prepared by resuspending cells in buffer A (250 mM sucrose, 50 mM triethanolamine, 1 mM EDTA, 6 mM magnesium acetate, 50 mM potassium acetate, 1 mM dithiothreitol) at a density of 2 × 107 cells per ml and sonicated three times for 10 s. Lysates were centrifuged at 3000 × g for 5 min at 4°C. Supernatants and pellets were separated. Supernatants were centrifuged at 12,000 × g for 10 min at 4°C. Supernatants and pellets were again separated and supernatants centrifuged at 25,000 × g for 30 min. Pellets were resuspended in buffer A. Pellets and supernatants were analyzed by immunoblotting. The orientation of pUL138 in the 25K light membrane fraction was determined by digesting intact microsomes with 0.5 ng/μl proteinase K for 30 min at 37°C in the presence or absence of 9 mM phenylmethylsulfonyl fluoride and immunoblotting.
Microsomal membranes were salt stripped by resuspension in 100 mM Na2CO3, incubated on ice for 1 h, and pelleted at 50,000 rpm in a TLA-100.3 rotor (Beckman Coulter) at 4°C for 45 min. Stripped membranes were resuspended in buffer A and analyzed by immunoblotting.
Purification of extracellular virions.
Extracellular virions were isolated from cell-free tissue culture supernatants from FIXwt-infected MRC5 cells by density gradient centrifugation through a 20% d-sorbitol cushion for 80 min at 20,000 rpm in an SW28 rotor (Beckman Coulter) at 22°C. Virions were further purified by banding on glycerol-tartrate gradients as described previously (56). Briefly, virions were resuspended in 0.1 ml TN (50 mM Tris [pH 7.4], 100 mM NaCl) and layered on top of a linear 9-ml glycerol-tartrate gradient formed in TN. Gradients were centrifuged at 34,000 rpm for 90 min at 22°C in an SW41 Ti rotor (Beckman Coulter) and then further centrifuged at 34,000 rpm for 18 h at 4°C. Virions were visualized using incandescent light, collected, diluted in 10 ml TN, and centrifuged at 34,000 rpm for 80 min at 22°C in an SW41 Ti rotor (Beckman Coulter). Pelleted particles were resuspended in 0.1 ml TN and analyzed by immunoblotting.
RESULTS
UL138 is encoded on two large transcripts during productive infection.
Using our experimental model for HCMV infection in HPCs, we have identified sequences in the ULb′ region of the genome, specifically the putative UL138 ORF, that are required for a quiescent infection with the hallmarks of latency in vitro (16). To begin to characterize the gene products derived from the UL138 locus, we analyzed transcription through this region in productively infected MRC5 fibroblasts by Northern blotting. Total RNA was analyzed by Northern blotting using a radiolabeled antisense ribonucleotide probe to the entire UL138 coding sequence. Transcripts were detected at 2.7 and 3.6 kb (Fig. 1A) as early as 6 hpi and accumulated throughout the time course of infection. To characterize the viral kinetics of UL138 transcription, cells were treated with cycloheximide (CHX) or phosphonoacetic acid (PAA), inhibitors of translation and viral DNA replication, respectively. UL138 transcript levels were somewhat reduced in the presence of PAA and undetectable in the presence of CHX. These results suggest that UL138 expression in fibroblasts is dependent on the synthesis of the IE proteins. In contrast, UL138 expression is enhanced by, but not dependent on, viral DNA synthesis. Taken together, these results suggest that UL138 is expressed with early-late gene kinetics during productive infection in fibroblasts.
FIG. 1.

Analysis of UL138 transcripts. (A) Total RNA isolated from FIXwt-infected (MOI = 2) MRC5 cells over a time course was analyzed by Northern blotting using a radiolabeled antisense UL138-specific probe. To determine the kinetics of UL138 expression, cells were treated with CHX or PAA and RNA was isolated at the times indicated. (B) Poly(A)+ purified RNA isolated from infected MRC5 cells at 24 hpi was analyzed by Northern blotting using radiolabeled antisense probes specific to the ULb′ ORFs indicated or a sense probe specific to UL138. The positions of the 3- and 4-kb RNA markers are shown to the left of each blot. (C) Schematic representation of 5′ and 3′ RACE results. GSPs for 5′ (gsp2) and 3′ (gsp1) RACE are shown by open arrows. The results shown in each panel are representative of at least three experiments.
The size of the UL138 transcripts was surprising given that the UL138 ORF is 510 nucleotides in length. To map the transcripts by Northern blotting, we used radiolabeled antisense RNA probes to three ORFs upstream of UL138 (UL133, UL135, and UL136) and two ORFs downstream (newORF11 [36], herein referred to as UL138b, and UL139) (Fig. 1B). Our analysis indicates that the poly(A)+ purified UL138 transcripts expressed in fibroblasts likely share a common 3′ end, including sequences to UL138b. The 5′ ends of the 2.7- and 3.6-kb transcripts include ORFs as far upstream as UL135 and UL133, respectively. The antisense ribonucleotide probes to the ORFs upstream and downstream of UL138 did not identify any other transcripts in addition to the 2.7- and 3.6-kb transcripts. Further, no transcripts were detected using a sense probe to the UL138 coding sequence, indicating that transcription originates from a single strand in this region of the genome.
We precisely mapped the 5′ and 3′ termini of each transcript by RACE using overlapping primers to UL138 (Fig. 1C). cDNA derived from mock- or FIXwt-infected fibroblasts at 24 hpi was cloned and sequenced. From these analyses, the 5′ end of the 3.6-kb transcript is at nucleotide position 190476, 8 nucleotides upstream of the predicted start of UL133. The 5′ end of the 2.7-kb transcript is at nucleotide position 189551, 24 nucleotides downstream of the predicted start of UL135. In multiple experiments, we detected a single 3′ RACE product, indicating that both the 3.6- and 2.7-kb transcripts terminate at a polyadenylation site immediately downstream of UL138b (nucleotide 186833). Using the 5′ and 3′ ends, we cloned and sequenced the UL138 cDNAs. Both transcripts are colinear with FIX genomic sequences, suggesting that these transcripts are not spliced.
UL138 is transcribed with differential kinetics in fibroblasts and CD34+ HPCs.
Using qRT-PCR, we analyzed the expression kinetics of polyadenylated UL138 transcripts in fibroblasts and CD34+ HPCs infected in vitro with FIXwt. Transcripts were quantitated using a standard curve derived from the PCR product for UL122 and UL138. β-Actin expression levels were used as a control for template loading. In productively infected MRC5 cells, UL138 was expressed starting at early times postinfection, consistent with our RNA analyses in Fig. 1A. UL138 transcripts accumulated as the infection progressed to levels exceeding those of UL122 transcripts encoding the IE2 86-kDa protein (Fig. 2A).
FIG. 2.

Kinetics of UL138 expression. Total RNA was isolated from MRC5 cells (A) or CD34+ HPCs (B) over a time course following infection with FIXwt at an MOI of 2. cDNAs were detected by qRT-PCR using UL138- and UL122-specific D-Lux primers and quantified by comparison to standard curves generated from PCR-amplified gene sequences. Reactions using RNA derived from mock-infected cells or infected cells where the reverse transcriptase was omitted (RT−) from the cDNA synthesis served as negative controls. The black and gray bars represent data for UL138 and UL122, respectively. The insets show real time ΔRn curves for β-actin at each time point to demonstrate that template loading into each reaction mixture was equivalent. Bars represent the averages from two experiments each performed in triplicate for panel A and the averages from two experiments for panel B. Standard deviations are shown for the replicates in panel A. The brackets in panel B represent the range of the data points in the two experiments performed.
The trend of UL138 expression differed in HPCs infected with FIXwt (Fig. 2B). UL138 transcripts were detected at maximal levels immediately following infection but steadily decreased during the time course of infection. UL122 transcripts were detected at lower levels during early points of the time course than UL138. We have detected both UL122 and UL123 in infected HPCs previously using microarrays (16, 17). It is not known if UL122 and UL138 are expressed in the same cell or if UL122 and UL138 are expressed in mutually exclusive subpopulations of infected cells. However, we have not detected IE1 72-kDa or IE2 86-kDa proteins in HPCs by either immunoblotting (see Fig. 4C) or immunofluorescence (data not shown). Over the time course, both UL122 and UL138 transcripts decreased to levels that were undetectable by 15 days postinfection. These data are consistent with the previously observed loss of HCMV gene expression in HPCs infected in vitro (15). The loss of detectable UL138 expression may reflect a global silencing of HCMV transcription that is proposed to occur during the establishment of latency (23, 37, 43). Alternatively, the loss of UL138 transcripts could also reflect a loss of viral genomes during cellular proliferation, a limitation of in vitro culture of CD34+ HPCs (15). Nevertheless, UL138 was detected in CD34+ cells as well as CD14+ monocytes derived from healthy seropositive individuals (16).
FIG. 4.

pUL138 expression. (A) Protein lysates derived from mock-infected MRC5 cells or cells infected with FIXwt, FIXsubUL138, or FIX-UL138STOP at an MOI of 2 were analyzed by immunoblotting using rabbit anti-UL138 antiserum (5484), identifying a 21-kDa protein corresponding to UL138. (B) Immunoblot analysis of the kinetics of pUL138 expression compared to the IE1 and IE2 proteins. Protein lysates were derived from mock-infected MRC5 cells or cells infected with FIXwt at an MOI of 2 over a time course and analyzed using rabbit anti-UL138 (5484) and mouse anti-IE1/2 (3H4) antibodies. α-Tubulin levels were used as a control for loading; the identity of the 43-kDa band in the mock infection is unknown but is specific to the α-tubulin antibody. (C) Protein lysates derived from MRC5 cells infected with FIXwt (positive control [+C], far left lane), mock-infected CD34+ HPCs, or CD34+ HPCs infected with FIXwt were analyzed by immunoblotting with rabbit anti-UL138 (5484) and anti-IE1/2 (3H4) antibodies. β-Actin served as a loading control.
UL138 encodes a 21-kDa protein during productive infection.
To determine if UL138 encodes a protein, we generated a rabbit polyclonal antiserum against a 19-amino-acid peptide (MAVTAPLTDVDLLKPVTGS) near the carboxyl terminus of the UL138 coding sequence (Fig. 3A). The antiserum was affinity purified using the immunizing peptide. In addition, we engineered several recombinant viruses (Fig. 3B). FIX-UL138STOP was engineered by substituting a stop codon (TAA) at codon 15 (ATG) of the UL138 coding sequence (Fig. 3B, FIX-UL138STOP). To aid our investigation into the protein-coding capacity of UL138, we also constructed a recombinant virus that expresses UL138 with an in-frame, C-terminal Myc epitope tag. Similar to the UL138-null viruses lacking the entire UL138 coding sequence (Fig. 3B, FIXsubUL138 and FIX(ur)sub2) (16), FIX-UL138STOP and FIX-UL138myc replicated with wild-type kinetics (Fig. 3C).
FIG. 3.

UL138 amino acid sequence and recombinant UL138 viruses. (A) Amino acid sequence of pUL138. The putative transmembrane domain is shaded, and the immunizing peptide used to create the rabbit anti-UL138 antibody is underlined. (B) Schematic of the recombinant viruses FIXsubUL138, FIX-UL138STOP, and FIX-UL138myc. (C) Multistep growth curve analysis of FIX recombinant viruses. MRC5 cells were infected at 0.01 TCID50 per cell and virus titers measured by TCID50 over the time course indicated. Values represent the averages from two independent experiments.
Lysates derived from MRC5 cells infected with FIXwt, FIXsubUL138, or FIX-UL138STOP were analyzed by immunoblotting using the rabbit anti-UL138 antibody. In FIXwt-infected cells, we detected a protein of approximately 21 kDa corresponding to the 170-amino-acid-coding sequence of UL138 (Fig. 4A). This protein was not detected in mock-infected cells or cells infected with either FIXsubUL138 or FIX-UL138STOP. The UL138 protein is detected by 6 hpi, coincident with IE1 and IE2 protein synthesis, and continues to accumulate during the productive infection in MRC5 cells (Fig. 4B). We next analyzed lysates derived from infected (GFP-positive) CD34+ HPCs by immunoblotting to determine if pUL138 was synthesized during a nonproductive infection resembling latency. Interestingly, the 21-kDa protein was detected at 2 (Fig. 4C), 4, and 8 (data not shown) days postinfection despite the small number of transcripts detected by qRT-PCR in HPCs (Fig. 2B). The IE proteins were not detected in infected HPCs.
To confirm that the 2.7- and 3.6-kb transcripts encoding pUL138 are stably synthesized in cells infected with FIX-UL138STOP, we analyzed RNA derived from MRC5 cells infected with FIXwt or FIX-UL138STOP and treated with actinomycin D at 16 hpi. FIX-UL138STOP synthesized both UL138 transcripts as in the FIXwt infection. Over the 36-hour time course, UL138 transcripts synthesized in both FIXwt- and FIX-UL138STOP-infected cells were stable and did not exhibit significantly different rates of decay that could abrogate protein production (Fig. 5). These results indicate that the UL138 transcripts synthesized during FIX-UL138STOP infection, while stable, do not serve as templates for pUL138 synthesis.
FIG. 5.

The UL138 transcripts synthesized in cells infected with FIX-UL138STOP are stable. Cells infected with FIXwt (FIX) or FIX-UL138STOP (STOP) at an MOI of 2 were treated with 4 μg/ml actinomycin D at 16 hpi. Total RNA was isolated over a time course and analyzed by Northern blotting using a radiolabeled antisense probe to UL138. 18S and 28S rRNAs for each blot are stained with methylene blue and served as a loading control (lower two panels).
The UL138 protein is required for HCMV latency.
Virus-encoded RNAs and proteins have been identified and demonstrated to function in the latent programs of alpha- and gammaherpesviruses. Our previous studies demonstrated that sequences carrying UL138 were required for HCMV to establish and/or maintain latency in vitro (16). To determine if pUL138 is required for latency, we analyzed infectious-center formation in pure populations of CD34+ HPCs infected with FIXwt, FIX(ur)sub2 (lacking UL136 to UL142) (16), FIXsubUL138, or FIX-UL138STOP. After 12 days in long-term culture, 10,000 infected CD34+ cells or an equivalent cell lysate was transferred to each of 12 to 24 wells of a 96-well dish seeded with fibroblasts. The cell lysate is a critical control for distinguishing virus preformed during the culture period prior to reactivation from virus formed as a result of reactivation. The fractions of wells containing GFP-positive fibroblasts for both the reactivation and preformed virus control experiments were scored 20 days later. FIXwt exhibited a typical latency phenotype where the fraction of wells scoring GFP positive was fivefold greater than that of the cell lysate (P = 0.004) (Fig. 6). In contrast, FIX(ur)sub2, lacking 5 kb of the ULb′ region including UL138, exhibits a complete loss-of-latency phenotype (P = 0.21), with equivalent fractions of GFP-positive wells for the reactivation and preformed virus control. FIXsubUL138 and FIX-UL138STOP exhibited partial loss-of-latency phenotypes. The differences between the fractions of GFP-positive wells prior to and following reactivation were 1.4- and 1.7-fold for FIXsubUL138 and FIX-UL138STOP, respectively. Student's t test values for FIXsubUL138 and FIXsubUL138STOP were 0.03 and 0.09, respectively. The partial loss-of-latency phenotype exhibited in FIX-UL138STOP infection indicates a requirement for pUL138 in the latent infection in vitro. However, the incomplete nature of the loss-of-latency phenotype exhibited by FIXsubUL138 and FIX-UL138STOP compared to the complete loss of latency exhibited by FIX(ur)sub2 (16) suggests that the UL138 transcripts or other ORFs in this region may also contribute to the latent infection of HCMV in vitro.
FIG. 6.
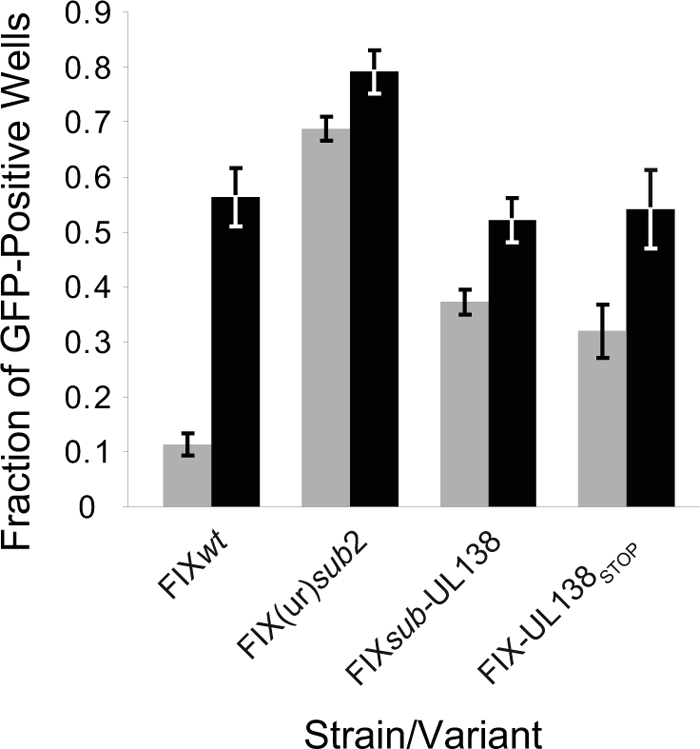
The UL138 protein is required for HCMV latency. Pure populations of HPCs infected with FIXwt, FIX(ur)sub2, FIXsubUL138, or FIX-UL138STOP at an MOI of 2 were isolated by fluorescence-activated cell sorting. Following 12 days in long-term culture, 10,000 infected CD34+ HPCs or an equivalent cell lysate was seeded into each of 12 to 24 wells of a 96-well dish containing MRC5 cells in a cytokine-rich medium. Wells were scored at 20 to 24 days postinfection for GFP-positive fibroblasts. The fraction of wells scoring positive for virus production in the reactivation experiments (black bars) is compared to the preformed virus detected in cell lysates (gray bars) in five or six independent experiments for FIXwt, FIX(ur)sub2, and FIX-UL138STOP and three independent experiments for FIXsubUL138. The standard errors of the means are shown. The P values for the comparisons of the fraction of GFP-positive wells in the reactivation to the preformed virus are 0.004, 0.21, 0.03, and 0.09 for FIXwt, FIX(ur)sub2, FIXsubUL138, and FIX-UL138STOP, respectively.
pUL138 does not downregulate IE gene expression.
pUL138 might contribute to latency by downregulating gene expression from the MIEP to suppress viral replication. Indeed, recombinant virus lacking UL138 replicates to greater levels in infected HPCs (16), indicating greater IE gene expression. To determine if UL138 directly affects MIEP activity, the coding sequence for UL138, including its Kozak sequence, was cloned in frame into the pEF1/Myc-His expression vector, generating a promoter-driven carboxy-terminally Myc-tagged UL138 construct, pEF1-UL138WT. A similar vector, pEF1-UL138STOP, was constructed by cloning the UL138 coding sequence containing a stop codon (Fig. 3B) to prevent protein expression. We performed dual-luciferase assays to analyze the activity of the MIEP in the presence or absence of pUL138. U373 cells were transfected with increasing amounts of pEF1-UL138WT or pEF1-UL138STOP and a constant amount of the reporter vectors, pGL3-MIEP1400 and pRL-SV40. pGL3-MIEP1400 expresses the firefly luciferase gene from the complete MIEP, including enhancer and modulator regions, and pRL-SV40 expresses Renilla luciferase from the SV40 early promoter. pEF1-UL138STOP serves as a negative control and produced results similar to those with a pEF1-EMPTY vector control (data not shown). At 48 h posttransfection, firefly luciferase activity was measured and normalized to Renilla luciferase activity to control for transfection efficiency. UL138 expression did not affect the expression of luciferase from the MIEP regardless of dose (Fig. 7A). Expression of pUL138 was confirmed by immunoblotting (data not shown). This experiment was repeated with other cell lines, including HeLa, ARPE-19, and 293 cells, with similar results (data not shown). Additionally, in experiments using only the enhancer region of the MIEP to drive luciferase expression, pUL138 also failed to repress MIEP activity (data not shown). These results provide evidence that pUL138 does not function by downregulating expression from the MIEP in the cells tested.
FIG. 7.

pUL138 does not suppress IE gene expression. (A) U373 cells were transfected with increasing amounts (0.1, 0.5, 1.0, and 3.0 μg) of pEF1-UL138WT, carrying UL138, or pEF1-UL138STOP, carrying UL138 containing a stop codon substituted at codon 15 as a negative control. Cells were also transfected with 10 ng of pGL3-MIEP1400 expressing the firefly luciferase gene from the MIEP and 20 ng pRL-SV40 expressing Renilla luciferase from the SV40 promoter as a control for transfection efficiency. (B) 293 cells were transfected with increasing amounts (0.1, 0.5, 1.0, and 1.5 μg) of pEF1-UL138WT or pEF1-UL138STOP as described above. Cells were also transfected with 100 ng of pSVH-1, encoding both IE1 and IE2 driven from the MIEP, in addition to pGL3-MIEP1400 (10 ng) and pRL-SV40 (20 ng). In both panels A and B, the total amount of DNA transfected was kept constant over the titrations using pEF1-EMPTY. Luciferase activity was measured at 48 to 72 h following transfection. (C) Protein lysates derived from FIXwt- or FIXsubUL138-infected (MOI = 1) MRC5 fibroblasts were isolated over a time course following infection and analyzed by immunoblotting using rabbit anti-UL138 and mouse anti-IE1/2 (3H4), with mouse anti-α-tubulin as a loading control.
We next wanted to explore the possibility that pUL138 might modulate activity of the MIEP through interaction with the IE proteins (IE1 and IE2) in an inhibitory feedback mechanism to control IE gene expression. To determine if pUL138 requires IE1 and/or IE2 in order to alter MIEP expression, we expanded our dual-luciferase assays to include an expression plasmid, pSVH-1, encoding both the IE1 72-kDa and IE2 86-kDa proteins. We transfected 293 cells with increasing amounts of pEF1-UL138WT or pEF1-UL138STOP and constant amounts of pGL3-MIEP1400, pRL-SV40, and pSVH-1. Expression of IE1, IE2, and pUL138 in these experiments was confirmed by immunoblotting (data not shown). Firefly and Renilla luciferase activities were quantified as described for Fig. 7A. In the presence of the IE proteins, pUL138 failed to diminish transcriptional activation from the MIEP and, if anything, slightly induced activation in a dose-dependent manner (Fig. 7B). Dual-luciferase assays were also performed using expression vectors encoding either the IE1 72-kDa or IE2 86-kDa protein alone, with similar results (data not shown). These results indicate that pUL138 does not repress expression from the MIEP even in the presence of IE1 and/or IE2 in the cells tested. Immunoblot analysis of lysates further revealed that pUL138 expression did not reduce the levels of IE1 or IE2 proteins in transfected cells (data not shown).
Finally, we analyzed IE1 72-kDa and IE2 86-kDa protein levels during infection in the presence or absence of pUL138. MRC5 cells were infected with FIXwt or FIXsubUL138 at an MOI of 1, and protein lysates were collected over a time course following infection and analyzed by immunoblotting. Consistent with reporter assays (Fig. 7A and B), UL138 expression did not suppress IE1 or IE2 expression in the context of a productive infection (Fig. 7C). Indeed, the expression of the IE1 72-kDa and IE2 86-kDa proteins was delayed, and the IE2 86-kDa protein was expressed to lower levels in the FIXsubUL138 infection than in the FIXwt infection. Delayed and diminished levels of IE1 72-kDa and IE2 86-kDa protein expression were observed in multiple experiments using independent virus preparations. Taken together, these data indicate that UL138 does not downregulate expression from the MIEP. However, our results do not address the possibility that UL138 alters IE1/2 function, nor do they rule out the possibility that pUL138 affects the MIEP in a context-dependent manner.
pUL138 localizes to the Golgi apparatus.
To determine the subcellular localization of pUL138, MRC5 fibroblasts were nucleofected with pEF1-UL138WT or empty vector and analyzed by indirect immunofluorescence using a rabbit anti-Myc antibody. pUL138 localized to a discrete perinuclear compartment resembling the Golgi apparatus (Fig. 8A). Further analysis demonstrated that pUL138 colocalized with or was juxtaposed to the trans-Golgi/trans-Golgi network marker G58K, the medial-trans-Golgi marker GS27, and the cis-medial-Golgi marker GM130 during transient expression. The localization of pUL138 in the secretory pathway was specific to the Golgi apparatus, as it did not colocalize with a marker for the endoplasmic reticulum (protein disulfide isomerase [PDI]) or early endosomes (early endosomal antigen 1 [EEA1]). pUL138 localization to the Golgi apparatus is preserved in multiple cell types, including APRE-19, HeLa, and 293 cells transiently expressing pUL138 (data not shown). We observe identical localization of the wild-type (non-epitope-tagged) pUL138 in infection using our polyclonal antiserum to pUL138, indicating that the Myc epitope tag has not altered pUL138 localization. Localization of pUL138 to the Golgi apparatus is consistent with the presence of multiple Golgi localization motifs in the protein sequence, including three tyrosine sorting motifs (YXXΦ) and a single acidic cluster dileucine motif (DXXLL), both of which have been shown to target herpesvirus proteins to the Golgi apparatus (2, 20, 28, 30).
FIG. 8.

pUL138 localizes to the Golgi apparatus. (A) MRC5 cells were nucleofected with the pEF1-UL138WT expression vector or empty vector (pEF1-EMPTY) as a negative control. pUL138 localization was visualized by indirect double-label immunofluorescence using a rabbit antibody (71D10) specific to the Myc epitope. Cell structures were labeled using mouse monoclonal antibodies to the Golgi markers G58K, GM130, and GS27; to the endoplasmic reticulum marker PDI; and to the early endosomal marker EEA1. In cells transfected with pEF1-EMPTY, the Golgi apparatus was marked using the GM130 Golgi marker. (B) MRC5 cells were mock infected or infected with FIX-UL138myc or FIXsubUL138 at an MOI of 2. pUL138 was localized to the Golgi apparatus by indirect immunofluorescence over a time course using the rabbit anti-Myc antibody (71D10) and the mouse anti-GM130 antibody. Cell nuclei are indicated by the DNA stain DAPI. Localization was visualized by confocal microscopy.
The localization of pUL138 to the Golgi apparatus does not require other proteins specific to virus infection, since Golgi localization occurs in the absence of viral infection. To determine if pUL138 localizes to the Golgi apparatus during viral infection, MRC5 cells were mock infected or infected with FIX-UL138myc or FIXsubUL138 at an MOI of 2. Cells were fixed and stained by indirect immunofluorescence at 12, 24, 36, and 48 hpi using rabbit anti-Myc and the GM130 Golgi marker. pUL138 was localized to the Golgi cisternae and juxtaposed with or colocalized to GM130 by 12 hpi (Fig. 8B) in a pattern similar to that observed in transient-expression studies. The localization of pUL138 to the Golgi apparatus suggests that pUL138 may function by modulating cellular or viral processes mediated through the Golgi apparatus, such as viral egress, intracellular protein trafficking, apoptosis, and the cellular stress response. Further studies are under way to localize pUL138 in HPCs. While pUL138 is associated with membranes in HPCs, we have not yet demonstrated exclusive association of pUL138 with Golgi membranes (data not shown).
pUL138 is a type 1 integral membrane protein.
To further analyze the Golgi association and subcellular distribution of pUL138 in infected cells, crude membrane fractions from FIXwt-infected MRC5 fibroblasts were prepared over a time course following infection and analyzed by immunoblotting for the presence of pUL138 using the rabbit anti-UL138 antibody and major histocompatibility complex class I (MHC-I) as a control (anti-MHC-I antibody specific to the HLA-B/C haplotype). Golgi membranes are represented primarily in the microsomal (25K) fraction whereas the 3K and 12K pellets represent heavier membrane fractions containing cytoplasmic and nuclear membranes. Consistent with our localization studies (Fig. 8), pUL138 was detected primarily in the microsomal membrane fraction (25K pellet) by 12 hpi (Fig. 9A). Very low levels of pUL138 were present in heavier membrane fractions (3K and 12K pellets) or in the soluble fraction (25K supernatant). In contrast, MHC-I was present in all membrane fractions and the soluble fraction, indicative of its widespread distribution from the endoplasmic reticulum to the plasma membrane. The level of pUL138 in the microsomal membranes was stable over the time course. These data indicate that pUL138 is localized predominantly in the Golgi/microsomal membrane fractions at steady state. However, these data do not exclude the possibility that pUL138 cycles through the secretory pathway.
FIG. 9.

pUL138 is stably and predominantly localized to Golgi membranes. (A) Crude membrane fractions from FIXwt-infected (MOI = 1) MRC5 cells were prepared over a time course following infection and analyzed by immunoblotting for the presence of pUL138 and MHC-I as a control. 25K pellets (pel) represent microsomal membrane fractions, whereas 3K and 12 K pellets represent heavier membrane fractions. The 25K supernatant represents the soluble fraction. Unfractionated membranes from FIXwt- or mock-infected MRC5 cells (far left two lanes) served as positive and negative controls, respectively. (B) To determine if pUL138 is an integral membrane protein, microsomal membranes were stripped with 100 mM sodium carbonate, pelleted at 100,000 × g, and analyzed by immunoblotting for pUL138 and MHC-I. (C) To determine the orientation of pUL138 in Golgi membranes, microsomal membrane fractions were isolated from ARPE-19 cells 48 h after transfection with pEF1-UL138WT. Microsomes untreated or treated with 0.5 ng/μl proteinase K, in the presence or absence of a protease inhibitor (9 mM phenylmethylsulfonyl fluoride [PMSF]), were analyzed by immunoblotting for pUL138 or MHC-I. In all blots, pUL138 and MHC-I were detected using the rabbit anti-UL138 (5484) and mouse anti-MHC-I antibodies, respectively.
We next sought to determine if UL138 was an integral membrane protein in the Golgi apparatus and, if so, the orientation of the protein in the membranes. The predicted transmembrane domain of pUL138 is 3 amino acids from the N terminus of the protein (Fig. 3A). To determine if pUL138 traversed the membrane, microsomal membrane preparations from infected cells at 24 hpi were washed with salt and immunoblotted for MHC-I and pUL138 (Fig. 9B). The salt wash did not remove MHC-I from the membrane, as expected. Like MHC-I, pUL138 was stably associated with membranes. Further, pUL138 could not be solubilized from membranes with 1% digitonin, although stronger detergents such as 1% Triton X-100 or NP-40 will solubilize pUL138 (data not shown). Taken together, these results indicate that pUL138 is an integral membrane protein.
To determine the orientation of pUL138 in the Golgi membranes, microsomal membranes were isolated from ARPE-19 cells transfected with pEF1-UL138WT. Microsomes were subjected to proteinase K treatment in the presence or absence of a protease inhibitor. Untreated and treated microsomes, in addition to an infected cell lysate, were analyzed by immunoblotting for pUL138 and MHC-I as a control. Proteinase K digestion of the type I membrane protein MHC-I in microsomes results in a lower-molecular-weight cleavage product (Fig. 9C, second lane) due to partial cleavage of the short carboxy-terminal tail on the cytoplasmic face of the membranes. However, the large N-terminal domain of MHC-I is protected in the lumen of the Golgi/microsomes. Microsomal pUL138, however, was sensitive to proteinase K digestion. Given the position of the transmembrane domain (Fig. 3A), our results indicate that pUL138 is a type I membrane protein where the large C-terminal domain of pUL138 is exposed on the cytoplasmic face of Golgi/microsomal membranes.
pUL138 is not incorporated into virus particles.
The localization of pUL138 in Golgi membranes prompted us to ask if virus particles acquire pUL138 during secondary envelopment and egress through the Golgi/endosomal compartments. If pUL138 is incorporated into virions, the large C-terminal cytoplasmic domain of pUL138 should extend into the tegument, leaving only three amino acids on the surface of the virion (Fig. 10A). In contrast, lumenal proteins, such as viral glycoprotein B, are exposed on the surface of virus particles following egress (12). We analyzed virions purified on glycerol-tartrate gradients for the presence of pUL138 and the tegument protein pp71. In contrast to the case for pp71, detection of pUL138 in untreated virion preparations was inconsistent, ranging from low to undetectable levels of protein. Results from two representative experiments are shown in Fig. 10B.
FIG. 10.

pUL138 is a type I membrane protein that is not packaged in the virus particle. (A) Schematic of secondary envelopment and egress of HCMV particles. A hypothetical virion particle illustrating the proposed orientation of pUL138 and MHC-I if these Golgi-localized proteins were incorporated into the virus envelope is shown. The chart illustrates the known sensitivities of MHC-I and pUL138 in microsomal membranes, the known sensitivity of pp71 in virions, and the proposed sensitivities of MHC-I and pUL138 in virions to tryptic digestion. (B) Extracellular virions were purified by centrifugation through a sorbitol cushion, followed by banding on glycerol-tartrate gradients. Virions were treated with trypsin at a 20:1 protease-to-protein ratio in the presence or absence of 1% Triton X-100, and 5 μg of protein per lane was analyzed for the presence of pUL138, pp71, and MHC-I by immunoblotting using rabbit anti-UL138 (5484), mouse anti-pp71, and mouse anti-MHC-I antibodies, respectively. Results from two of four independent experiments are shown. A lysate of FIXwt-infected cells was included as a positive control.
The inconsistent detection of pUL138 relative to the consistent detection of pp71 led us to suspect that the pUL138 present in virion preparations may be a contaminant. Over 70 cellular proteins are reported to copurify with virus particles, and it is difficult to distinguish these proteins as bona fide virion components or copurifying contaminants (63). MHC-I protein was analyzed in virus preparations as an indicator of contamination, since this protein is present in Golgi membranes but not virus particles. We detected pUL138 only in virion preparations that also contained high levels of MHC-I (Fig. 10B, experiment 1).
To further test the possibility that pUL138 is a copurifying contaminant in virion preparations, we analyzed the accessibility of pUL138 in purified virions to trypsin digestion in the presence or absence of detergent to disrupt the viral envelope (Fig. 10B). We further analyzed the viral pp71 tegument protein as a control for intact virions and MHC-I as a control for contaminating membrane proteins. The anticipated sensitivity or protection of these proteins to tryptic digestion when associated with microsomes or virus particles is shown in Fig. 10A. As expected, pp71 was protected from proteolytic digestion and became sensitive to digestion only in the presence of detergent. In contrast, pUL138 was susceptible to trypsin proteolysis in the absence of detergent in virion preparations when it was detected (Fig. 10B, experiment 1), whereas MHC-I was susceptible to degradation only in the presence of detergent. Digestion of MHC-I to the lower-molecular-weight species observed in each experiment is consistent with published results analyzing tryptic digestion of membrane-associated MHC-I (39), and full digestion of MHC-I is not expected with trypsin. These results are consistent with the topology of pUL138 and MHC-I in microsomal membranes but not in virus particles (Fig. 10A). The inconsistent detection and the topology of pUL138 in virus preparations are consistent with the supposition that pUL138 and MHC-I are contaminants of virion preparations, perhaps arising from microsomes or exosomes associated with viral infection (1, 44). Based on these analyses, we conclude that it is unlikely that pUL138 is a constituent of the viral envelope. This finding implies that the function of pUL138 during the latent or productive infection requires de novo synthesis of pUL138, as it is not delivered to the cell by the virion.
DISCUSSION
UL138 is the first viral determinant demonstrated to function in promoting HCMV latency (16). UL138 is a previously uncharacterized 510-nucleotide gene encoded within the ULb′ region of the genome unique to clinical or low-passage stains of HCMV. Our study represents an initial characterization of the UL138 gene products. During a productive infection, UL138 is transcribed on two large transcripts expressed with early-late kinetics (Fig. 1). Expression of UL138 is differentially regulated in infected fibroblasts and CD34+ HPCs (Fig. 2), sites of productive and latent infections, respectively. UL138 gives rise to a 21-kDa (Fig. 4), type 1 membrane protein (Fig. 9) that localizes to the Golgi apparatus during viral infection (Fig. 8B) and in the absence of other viral proteins (Fig. 8A). Importantly, the UL138 protein is required for HCMV to efficiently establish and/or maintain a latent infection in CD34+ cells infected in vitro (Fig. 6). However, our results do not preclude a role for the UL138 transcripts or other putative ORFs encoded on the UL138 transcripts (UL133 to UL138b) or deleted in FIX(ur)sub2 (UL136 to UL142). The function of pUL138 in infection is not realized upon viral entry but requires de novo synthesis of pUL138, since pUL138 is not delivered to infected cells as a virion component (Fig. 10).
While the productive HCMV infection has been well characterized, mechanisms contributing to HCMV persistence and latency remain poorly understood. Latent viral coexistence with the host requires the maintenance of viral genomes, suppression of viral replication, inhibition of antiviral responses (i.e., apoptosis and type I interferon response), and evasion the host immune response. Having established a latent infection, the virus retains the ability to reactivate from the latent state given an appropriate stimulus and, ultimately, to replicate productively. Based on this definition, suppression of lytic viral gene expression and replication seems an obvious mechanism by which UL138 might promote HCMV latency. Consistent with this hypothesis, the latency-associated transcript (LAT) of herpes simplex virus (HSV) likely suppresses productive-cycle gene expression by downregulating ICP0 expression (14, 62). However, pUL138 did not directly downregulate expression from the MIEP or interact with IE proteins to suppress expression from the MIEP when expressed transiently (Fig. 7). Consistent with this finding, UL138-null viruses grow with kinetics similar to those of FIXwt in productively infected fibroblasts (Fig. 3C) (16). Our data do not exclude the possibility that pUL138 negatively regulates the IE promoter or proteins in a context dependent manner. The effect of pUL138 on MIEP activity and IE1/IE2 protein expression and function has yet to be explored in the context of infection in HPCs.
UL138 transcripts and protein were detected in both productively infected fibroblasts and latently infected HPCs (Fig. 2 and 4). While the expression of latency-associated genes during the lytic infection is not uncommon among herpesviruses (3, 50, 57), the role of UL138 during the productive infection, if any, is unknown. UL138 is dispensable for viral replication in fibroblasts (Fig. 3C). Similarly, the LATs of HSV are expressed during both the lytic and latent infections, but a role for LATs in viral replication has not been described. However, LATs are differentially transcribed and spliced depending on the context of infection (10, 18). While pUL138 is synthesized in all infected cell types tested, the outcome of infection likely depends on the context-dependent balance of competing viral and cellular factors promoting virus replication or latency.
In fibroblasts, UL138 expression was dependent on IE gene expression (Fig. 1A). It is of interest, then, to speculate how UL138 is expressed in HPCs when IE genes are minimally transcribed (Fig. 2B) and no proteins are detected (Fig. 4C) even at very early times following infection. Several possible scenarios exist to address the possibility that UL138 is expressed in the absence of IE gene expression: (i) pUL138 may be synthesized from RNAs packaged in virions, or (ii) UL138 may be transcribed independently of IE expression in the context of HPC infection. Virion RNAs are packaged nonspecifically and relative to their abundance in the infected cell (60). Given the high levels of expression of UL138 transcripts in infected cells (Fig. 1A and 2A), it is likely that these transcripts are packaged and can perhaps serve as templates for translation in newly infected cells. We have detected UL138 transcripts among DNase-treated RNAs isolated from purified virions treated with RNase prior to lysis, although it is not known if the UL138 transcripts present in virions are full length. However, we have not detected pUL138 expression in fibroblasts infected with HCMV in the presence of actinomycin D (4 μg/ml) by either immunoblotting or immunofluorescence analyses (S. Whitaker and F. Goodrum, unpublished results). These experiments suggest that UL138 transcripts are likely transcribed de novo following infection.
Depending on the context of infection, UL138 may be expressed independently of IE gene expression. During productive infection, herpesvirus gene expression has historically been characterized as a temporally ordered cascade of IE, early, and late gene expression. This dogma is challenged by recent studies investigating CMV infection outside the context of cultured fibroblasts (11, 15, 17, 54). In these studies, viral gene expression was skewed toward genes that are not known to contribute directly to virus replication, was highly specific to the cell type or tissue, and did not reflect conventional viral kinetics. From these findings, we propose that the profile of viral gene expression is dictated by the unique milieu of the particular cellular environment. Accordingly, Epstein-Barr virus infection of B cells results in one of five possible profiles of viral gene expression depending on the differentiation and activation state of the cell (reviewed in reference 61). Early interactions between the cell, virion components, and products of viral gene expression following viral entry likely dictate the resulting viral program of gene expression and ultimately the outcome of the infection.
The localization of pUL138 to the Golgi apparatus (Fig. 8) suggests that pUL138 contributes to latency through a novel mechanism not previously demonstrated for other herpesviruses. In addition to playing a critical role in intracellular protein trafficking and herpesvirus egress, the Golgi apparatus has been implicated in the regulation of apoptosis (9, 21) and the cellular stress response (21). While each of these processes represents candidate mechanisms by which pUL138 may function in viral latency, some are more plausible than others. Infected cell survival is essential to viral latency and is one mechanism by which HSV LATs contribute to latency (7, 38). The localization of the antiapoptotic m41 protein of murine CMV to the Golgi apparatus suggests a role for the Golgi apparatus in mediating apoptosis during viral infection (9). While pUL138 could contribute to latency by promoting cell survival, we consider this possibility unlikely since neither fibroblasts nor CD34+ HPCs infected with UL138-null viruses exhibit decreased viability compared to cells infected with the wild-type virus (data not shown).
HCMV proteins localized throughout the secretory pathway play important roles in viral egress and in modulating intracellular protein trafficking to suppress cell surface presentation of MHC-I (12, 35, 41). It is possible that in its central location in the secretory pathway, pUL138 contributes to latency by blocking late viral glycoprotein trafficking or viral egress (12). While we have no direct evidence to the contrary, transcripts encoding late structural proteins have not been detected during infection of CD34+ HPCs (11, 15, 17). If productive viral gene expression is suppressed in latently infected cells, then it is unlikely that the virus requires a gene function to block the traffic of late proteins or the maturation of virions. Alternatively, pUL138 might act as a “molecular crossing guard” in the Golgi apparatus, regulating the movement and activity of viral and/or cellular proteins to orchestrate the switch from productive to latent infection. The function of pUL138 in the Golgi apparatus is the focus of ongoing investigation in our laboratory.
This work represents the first characterization of the HCMV latency determinant UL138. While UL138 is important to the latent infection in our in vitro system, other viral and cellular components that also contribute to the latent infection likely exist. Indeed, our data [comparing FIX(ur)sub2 and UL138-null viruses in Fig. 6] suggest a requirement for other viral factors in establishing and maintaining the latent infection. The UL138 transcripts as well as other potential ORFs in the ULb′ region may be required for HCMV latency. Future studies will decipher the mechanism by which UL138 promotes the latent infection and will identify additional viral or cellular determinants that contribute to HCMV latency.
Acknowledgments
We gratefully acknowledge T. Shenk for HCMV antibodies, J. Nelson for pSVH-1, J. Wilson for antibodies to Golgi markers, L. Lybarger for anti-MHC-I, and T. Bowden for pRL-SV40. We further acknowledge M. Nevels, S. Campos, and C. Kulesza for critical reading of the manuscript.
This work was supported by grants from the National Institutes of Health (NCI [CA111343] and NIAID [AI079059]), by the American Cancer Society (institution research grant 7400128), and by funding from the University of Arizona and the BIO5 Institute.
Alex Petrucelli designed and performed research, collected and analyzed data, and wrote the paper. Lora Grainger designed and performed research for Fig. 1B, 2, and 4 and contributed considerably to the development of the work. Michael Rak designed and performed research for Fig. 1C and 10C, constructed recombinant viruses, and contributed considerably to the development of the work. Felicia Goodrum designed the research; analyzed the data; performed research for Fig. 1A, 6, and 3C; and wrote the paper.
Footnotes
Published ahead of print on 18 March 2009.
REFERENCES
- 1.Ahmed, K. A., M. A. Munegowda, Y. F. Xie, and J. Xiang. 2008. Intercellular trogocytosis plays an important role in modulation of immune responses. Cell Mol. Immunol. 5261-269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alconada, A., U. Bauer, B. Sodeik, and B. Hoflack. 1999. Intracellular traffic of herpes simplex virus glycoprotein gE: characterization of the sorting signals required for its trans-Golgi network localization. J. Virol. 73377-387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Allen, R. D., III, S. Dickerson, and S. H. Speck. 2006. Identification of spliced gammaherpesvirus 68 LANA and v-cyclin transcripts and analysis of their expression in vivo during latent infection. J. Virol. 802055-2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ausubel, F. M., R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman, J. A. Smith, and K. Struhl (ed.). 1998. Current protocols in molecular biology, vol. I. John Wiley and Sons, Inc., New York, NY.
- 5.Barton, E. S., D. W. White, J. S. Cathelyn, K. A. Brett-McClellan, M. Engle, M. S. Diamond, V. L. Miller, and H. W. Virgin IV. 2007. Herpesvirus latency confers symbiotic protection from bacterial infection. Nature 447326-329. [DOI] [PubMed] [Google Scholar]
- 6.Bego, M., J. Maciejewski, S. Khaiboullina, G. Pari, and S. St. Jeor. 2005. Characterization of an antisense transcript spanning the UL81-82 locus of human cytomegalovirus. J. Virol. 7911022-11034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bloom, D. C. 2004. HSV LAT and neuronal survival. Int. Rev. Immunol. 23187-198. [DOI] [PubMed] [Google Scholar]
- 8.Boeckh, M., W. G. Nichols, G. Papanicolaou, R. Rubin, J. R. Wingard, and J. Zaia. 2003. Cytomegalovirus in hematopoietic stem cell transplant recipients: current status, known challenges, and future strategies. Biol. Blood Marrow Transplant. 9543-558. [DOI] [PubMed] [Google Scholar]
- 9.Brune, W., M. Nevels, and T. Shenk. 2003. Murine cytomegalovirus m41 open reading frame encodes a Golgi-localized antiapoptotic protein. J. Virol. 7711633-11643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen, X., M. C. Schmidt, W. F. Goins, and J. C. Glorioso. 1995. Two herpes simplex virus type 1 latency-active promoters differ in their contributions to latency-associated transcript expression during lytic and latent infections. J. Virol. 697899-7908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheung, A. K., A. Abendroth, A. L. Cunningham, and B. Slobedman. 2006. Viral gene expression during the establishment of human cytomegalovirus latent infection in myeloid progenitor cells. Blood 1083691-3699. [DOI] [PubMed] [Google Scholar]
- 12.Eickmann, M., D. Gicklhorn, and K. Radsak. 2006. Glycoprotein trafficking in virion morphogenesis, p. 245-264. In M. J. Reddehase (ed.), Cytomegaloviruses: molecular biology and immunology. Caister Academic Press, Norfolk, United Kingdom.
- 13.Einsele, H., and H. Hebart. 1999. Cytomegalovirus infection following stem cell transplantation. Haematologica 84(Suppl. EHA-4)46-49. [PubMed] [Google Scholar]
- 14.Garber, D. A., P. A. Schaffer, and D. M. Knipe. 1997. A LAT-associated function reduces productive-cycle gene expression during acute infection of murine sensory neurons with herpes simplex virus type 1. J. Virol. 715885-5893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goodrum, F., C. T. Jordan, S. S. Terhune, K. P. High, and T. Shenk. 2004. Differential outcomes of human cytomegalovirus infection in primitive hematopoietic subpopulations. Blood 104687-695. [DOI] [PubMed] [Google Scholar]
- 16.Goodrum, F., M. Reeves, J. Sinclair, K. High, and T. Shenk. 2007. Human cytomegalovirus sequences expressed in latently infected individuals promote a latent infection in vitro. Blood 110937-945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goodrum, F. D., C. T. Jordan, K. High, and T. Shenk. 2002. Human cytomegalovirus gene expression during infection of primary hematopoietic progenitor cells: a model for latency. Proc. Natl. Acad. Sci. USA 9916255-16260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gussow, A. M., N. V. Giordani, R. K. Tran, Y. Imai, D. L. Kwiatkowski, G. F. Rall, T. P. Margolis, and D. C. Bloom. 2006. Tissue-specific splicing of the herpes simplex virus type 1 latency-associated transcript (LAT) intron in LAT transgenic mice. J. Virol. 809414-9423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hahn, G., R. Jores, and E. S. Mocarski. 1998. Cytomegalovirus remains latent in a common precursor of dendritic and myeloid cells. Proc. Natl. Acad. Sci. USA 953937-3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heineman, T. C., P. Connolly, S. L. Hall, and D. Assefa. 2004. Conserved cytoplasmic domain sequences mediate the ER export of VZV, HSV-1, and HCMV gB. Virology 328131-141. [DOI] [PubMed] [Google Scholar]
- 21.Hicks, S. W., and C. E. Machamer. 2005. Golgi structure in stress sensing and apoptosis. Biochim. Biophys. Acta 1744406-414. [DOI] [PubMed] [Google Scholar]
- 22.Horvath, R., J. Cerny, J. Benedik, Jr., J. Hokl, I. Jelinkova, and J. Benedik. 2000. The possible role of human cytomegalovirus (HCMV) in the origin of atherosclerosis. J. Clin. Virol. 1617-24. [DOI] [PubMed] [Google Scholar]
- 23.Ioudinkova, E., M. C. Arcangeletti, A. Rynditch, F. De Conto, F. Motta, S. Covan, F. Pinardi, S. V. Razin, and C. Chezzi. 2006. Control of human cytomegalovirus gene expression by differential histone modifications during lytic and latent infection of a monocytic cell line. Gene 384120-128. [DOI] [PubMed] [Google Scholar]
- 24.Jenkins, C., A. Abendroth, and B. Slobedman. 2004. A novel viral transcript with homology to human interleukin-10 is expressed during latent human cytomegalovirus infection. J. Virol. 781440-1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kondo, K., H. Kaneshima, and E. S. Mocarski. 1994. Human cytomegalovirus latent infection of granulocyte-macrophage progenitors. Proc. Natl. Acad. Sci. USA 9111879-11883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kondo, K., J. Xu, and E. S. Mocarski. 1996. Human cytomegalovirus latent gene expression in granulocyte-macrophage progenitors in culture and in seropositive individuals. Proc. Natl. Acad. Sci. USA 9311137-11142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Landini, M. P., T. Lazzarotto, J. Xu, A. P. Geballe, and E. S. Mocarski. 2000. Humoral immune response to proteins of human cytomegalovirus latency-associated transcripts. Biol. Blood Marrow Transplant. 6100-108. [DOI] [PubMed] [Google Scholar]
- 28.Loomis, J. S., R. J. Courtney, and J. W. Wills. 2006. Packaging determinants in the UL11 tegument protein of herpes simplex virus type 1. J. Virol. 8010534-10541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maciejewski, J. P., E. E. Bruening, R. E. Donahue, E. S. Mocarski, N. S. Young, and S. C. St. Jeor. 1992. Infection of hematopoietic progenitor cells by human cytomegalovirus. Blood 80170-178. [PubMed] [Google Scholar]
- 30.Maffei, M., F. Ghiotto, M. Occhino, M. Bono, A. De Santanna, L. Battini, G. L. Gusella, F. Fais, S. Bruno, and E. Ciccone. 2008. Human cytomegalovirus regulates surface expression of the viral protein UL18 by means of two motifs present in the cytoplasmic tail. J. Immunol. 180969-979. [DOI] [PubMed] [Google Scholar]
- 31.Meier, J. L., and M. F. Stinski. 2006. Major immediate early enhancer and its gene products, p.151-161. Caister Academic Press, Norfolk, United Kingdom.
- 32.Mendelson, M., S. Monard, P. Sissons, and J. Sinclair. 1996. Detection of endogenous human cytomegalovirus in CD34+ bone marrow progenitors. J. Gen. Virol. 773099-3102. [DOI] [PubMed] [Google Scholar]
- 33.Miller, C. L., and C. J. Eaves. 2002. Long-term culture-initiating cell assays for human and murine cells, p. 123-141. In C. A. Klug and C. T. Jordan (ed.), Hematopoietic stem cell protocols. Humana Press, Totowa, NJ. [DOI] [PubMed]
- 34.Minton, E. J., C. Tysoe, J. H. Sinclair, and J. G. Sissons. 1994. Human cytomegalovirus infection of the monocyte/macrophage lineage in bone marrow. J. Virol. 684017-4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mocarski, E. S., T. Shenk, and R. F. Pass. 2007. Cytomegaloviruses, p. 2701-2773. In D. M. Knipe and P. M. Howley (ed.), Fields virology, 5th ed. Lippincott, Williams & Wilkins, Philadelphia, PA.
- 36.Murphy, E., D. Yu, J. Grimwood, J. Schmutz, M. Dickson, M. A. Jarvis, G. Hahn, J. A. Nelson, R. M. Myers, and T. E. Shenk. 2003. Coding potential of laboratory and clinical strains of human cytomegalovirus. Proc. Natl. Acad. Sci. USA 10014976-14981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Murphy, J. C., W. Fischle, E. Verdin, and J. H. Sinclair. 2002. Control of cytomegalovirus lytic gene expression by histone acetylation. EMBO J. 211112-1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Perng, G. C., C. Jones, J. Ciacci-Zanella, M. Stone, G. Henderson, A. Yukht, S. M. Slanina, F. M. Hofman, H. Ghiasi, A. B. Nesburn, and S. L. Wechsler. 2000. Virus-induced neuronal apoptosis blocked by the herpes simplex virus latency-associated transcript. Science 2871500-1503. [DOI] [PubMed] [Google Scholar]
- 39.Pischel, K. D., and J. R. Little. 1979. Limited trypsin cleavage distinguishes MHC and thymus-leukemia antigens. J. Immunol. 123551-556. [PubMed] [Google Scholar]
- 40.Pourgheysari, B., N. Khan, D. Best, R. Bruton, L. Nayak, and P. A. Moss. 2007. The cytomegalovirus-specific CD4+ T-cell response expands with age and markedly alters the CD4+ T-cell repertoire. J. Virol. 817759-7765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Powers, C., V. DeFilippis, D. Malouli, and K. Fruh. 2008. Cytomegalovirus immune evasion. Curr. Top. Microbiol. Immunol. 325333-359. [DOI] [PubMed] [Google Scholar]
- 42.Reeves, M. B., P. J. Lehner, J. G. Sissons, and J. H. Sinclair. 2005. An in vitro model for the regulation of human cytomegalovirus latency and reactivation in dendritic cells by chromatin remodelling. J. Gen. Virol. 862949-2954. [DOI] [PubMed] [Google Scholar]
- 43.Reeves, M. B., P. A. MacAry, P. J. Lehner, J. G. Sissons, and J. H. Sinclair. 2005. Latency, chromatin remodeling, and reactivation of human cytomegalovirus in the dendritic cells of healthy carriers. Proc. Natl. Acad. Sci. USA 1024140-4145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Simpson, R. J., S. S. Jensen, and J. W. Lim. 2008. Proteomic profiling of exosomes: current perspectives. Proteomics 84083-4099. [DOI] [PubMed] [Google Scholar]
- 45.Sindre, H., G. E. Tjoonnfjord, H. Rollag, T. Ranneberg-Nilsen, O. P. Veiby, S. Beck, M. Degre, and K. Hestdal. 1996. Human cytomegalovirus suppression of and latency in early hematopoietic progenitor cells. Blood 884526-4533. [PubMed] [Google Scholar]
- 46.Slobedman, B., and E. S. Mocarski. 1999. Quantitative analysis of latent human cytomegalovirus. J. Virol. 734806-4812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Soderberg-Naucler, C., K. N. Fish, and J. A. Nelson. 1997. Interferon-gamma and tumor necrosis factor-alpha specifically induce formation of cytomegalovirus-permissive monocyte-derived macrophages that are refractory to the antiviral activity of these cytokines. J. Clin. Investig. 1003154-3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Soderberg-Naucler, C., K. N. Fish, and J. A. Nelson. 1997. Reactivation of latent human cytomegalovirus by allogeneic stimulation of blood cells from healthy donors. Cell 91119-126. [DOI] [PubMed] [Google Scholar]
- 49.Soderberg-Naucler, C., D. N. Streblow, K. N. Fish, J. Allan-Yorke, P. P. Smith, and J. A. Nelson. 2001. Reactivation of latent human cytomegalovirus in CD14+ monocytes is differentiation dependent. J. Virol. 757543-7554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Spivack, J. G., and N. W. Fraser. 1988. Expression of herpes simplex virus type 1 latency-associated transcripts in the trigeminal ganglia of mice during acute infection and reactivation of latent infection. J. Virol. 621479-1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stam, N. J., H. Spits, and H. L. Ploegh. 1986. Monoclonal antibodies raised against denatured HLA-B locus heavy chains permit biochemical characterization of certain HLA-C locus products. J. Immunol. 1372299-2306. [PubMed] [Google Scholar]
- 52.Stenberg, R. M., J. Fortney, S. W. Barlow, B. P. Magrane, J. A. Nelson, and P. Ghazal. 1990. Promoter-specific trans activation and repression by human cytomegalovirus immediate-early proteins involves common and unique protein domains. J. Virol. 641556-1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Streblow, D. N., S. L. Orloff, and J. A. Nelson. 2001. Do pathogens accelerate atherosclerosis? J. Nutr. 1312798S-2804S. [DOI] [PubMed] [Google Scholar]
- 54.Streblow, D. N., K. W. van Cleef, C. N. Kreklywich, C. Meyer, P. Smith, V. Defilippis, F. Grey, K. Fruh, R. Searles, C. Bruggeman, C. Vink, J. A. Nelson, and S. L. Orloff. 2007. Rat cytomegalovirus gene expression in cardiac allograft recipients is tissue specific and does not parallel the profiles detected in vitro. J. Virol. 813816-3826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sylwester, A. W., B. L. Mitchell, J. B. Edgar, C. Taormina, C. Pelte, F. Ruchti, P. R. Sleath, K. H. Grabstein, N. A. Hosken, F. Kern, J. A. Nelson, and L. J. Picker. 2005. Broadly targeted human cytomegalovirus-specific CD4+ and CD8+ T cells dominate the memory compartments of exposed subjects. J. Exp. Med. 202673-685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Talbot, P., and J. D. Almeida. 1977. Human cytomegalovirus: purification of enveloped virions and dense bodies. J. Gen. Virol. 36345-349. [DOI] [PubMed] [Google Scholar]
- 57.Talbot, S. J., R. A. Weiss, P. Kellam, and C. Boshoff. 1999. Transcriptional analysis of human herpesvirus-8 open reading frames 71, 72, 73, K14, and 74 in a primary effusion lymphoma cell line. Virology 25784-94. [DOI] [PubMed] [Google Scholar]
- 58.Taylor-Wiedeman, J., J. G. Sissons, L. K. Borysiewicz, and J. H. Sinclair. 1991. Monocytes are a major site of persistence of human cytomegalovirus in peripheral blood mononuclear cells. J. Gen. Virol. 722059-2064. [DOI] [PubMed] [Google Scholar]
- 59.Taylor-Wiedeman, J., P. Sissons, and J. Sinclair. 1994. Induction of endogenous human cytomegalovirus gene expression after differentiation of monocytes from healthy carriers. J. Virol. 681597-1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Terhune, S. S., J. Schroer, and T. Shenk. 2004. RNAs are packaged into human cytomegalovirus virions in proportion to their intracellular concentration. J. Virol. 7810390-10398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Thorley-Lawson, D. A., and A. Gross. 2004. Persistence of the Epstein-Barr virus and the origins of associated lymphomas. N. Engl. J. Med. 3501328-1337. [DOI] [PubMed] [Google Scholar]
- 62.Umbach, J. L., M. F. Kramer, I. Jurak, H. W. Karnowski, D. M. Coen, and B. R. Cullen. 2008. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature 454780-783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Varnum, S. M., D. N. Streblow, M. E. Monroe, P. Smith, K. J. Auberry, L. Pasa-Tolic, D. Wang, D. G. Camp II, K. Rodland, S. Wiley, W. Britt, T. Shenk, R. D. Smith, and J. A. Nelson. 2004. Identification of proteins in human cytomegalovirus (HCMV) particles: the HCMV proteome. J. Virol. 7810960-10966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vasto, S., G. Colonna-Romano, A. Larbi, A. Wikby, C. Caruso, and G. Pawelec. 2007. Role of persistent CMV infection in configuring T cell immunity in the elderly. Immun. Ageing 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vestergaard, B. F., A. Hornsleth, and S. N. Pedersen. 1972. Occurrence of herpes- and adenovirus antibodies in patients with carcinoma of the cervix uteri. Measurement of antibodies to herpesvirus hominis (types 1 and 2), cytomegalovirus, EB-virus, and adenovirus. Cancer 3068-74. [DOI] [PubMed] [Google Scholar]
- 66.von Laer, D., U. Meyer-Koenig, A. Serr, J. Finke, L. Kanz, A. A. Fauser, D. Neumann-Haefelin, W. Brugger, and F. T. Hufert. 1995. Detection of cytomegalovirus DNA in CD34+ cells from blood and bone marrow. Blood 864086-4090. [PubMed] [Google Scholar]
- 67.Warming, S., N. Costantino, D. L. Court, N. A. Jenkins, and N. G. Copeland. 2005. Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res. 33e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.White, K. L., B. Slobedman, and E. S. Mocarski. 2000. Human cytomegalovirus latency-associated protein pORF94 is dispensable for productive and latent infection. J. Virol. 749333-9337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yu, D., H. M. Ellis, E. C. Lee, N. A. Jenkins, N. G. Copeland, and D. L. Court. 2000. An efficient recombination system for chromosome engineering in Escherichia coli. Proc. Natl. Acad. Sci. USA 975978-5983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhuravskaya, T., J. P. Maciejewski, D. M. Netski, E. Bruening, F. R. Mackintosh, and S. St. Jeor. 1997. Spread of human cytomegalovirus (HCMV) after infection of human hematopoietic progenitor cells: model of HCMV latency. Blood 902482-2491. [PubMed] [Google Scholar]